

Abstract
A safe and simple method for methyl S-arylmercapturate synthesis is described. Thirteen such compounds, to be used afterwards in metabolism studies, have been obtained with yields ranging from 71 to 99.6%. These compounds were obtained using a sulfa-Michael addition and synthesized by adding the corresponding thiophenols to a mixture composed of methyl 2-acetamidoacrylate (MAA), potassium carbonate and a phase transfer catalyst, Aliquat 336. MAA, the initial synthon, was itself isolated in quasi quantitative yield following a fully described synthesis.
Keywords: Mercapturic acids, Sulfa-Michael addition, Phase Transfer Catalysis, Biomarker
Introduction
Aromatic hydrocarbons belong to a broad family of chemical compounds which are widely used in many industrial applications, alone or in mixtures, and as solvents and starting products for chemical synthesis [1]. Along with the ever increasing awareness and knowledge about the toxicity of aromatic hydrocarbons [2,3,4], there is a growing need for assessing reliable and specific biomarkers and for the development of sensitive analytical methods dedicated to occupational exposure biomonitoring [5,6,7,8,9,10].
Mercapturic acids (MA), which are N-acetyl-l-cysteine-S-conjugates (or 2-acetamido-3-sulfanyl-propionic acids according to IUPAC Nomenclature) are end products of the glutathione detoxification pathway (GSH). Since Van Doorn’s preliminary studies [11], increasing attention has been paid to these minor metabolites. Over the last decades, several MA derived from aromatic hydrocarbons have been identified and their corresponding metabolic pathways have been explained [9,10,11,12,13]. In this respect, Angerer et al. [13,14] demonstrated that toluene and xylenes are metabolised into MA using two different oxidation pathways (Scheme 1). As an example, toluene has been shown to interact with GSH by conjugation with reactive electrophilic intermediates. The nature of these intermediates depends on the first oxidation step catalysed by cytochrome P-450 enzymes and which may occur either at the side chain or at the aromatic ring. Side chain oxidation produces benzylic alcohol, which is enzymatically transformed into a sulphate ester [11], then to a S-benzylmercapturic acid (A) once conjugated with GSH. On the other hand, aromatic ring oxidation forms an arene oxide which leads to S-p-tolylmercapturic acid (B) after conjugation with GSH [13].
Scheme 1.

Several research teams have concluded that MAs are more selective and sensitive than traditional biomarkers, which are generally the major metabolites [11,15,16]. During the same period, the American Conference of Govermental Industrial Hygienists (ACGIH) and the Deutsche Forschungs Gemeinschaft (DFG) decided to publish a biological exposure index for S-phenylmercapturic acid as a biomarker of exposure to benzene [17,18].
For the aforementioned reasons, a growing interest in the glutathione pathway and MA synthesis has emerged. While MAs coming from side chain oxidation are well described in the literature [19,20,21], MAs resulting from ring oxidation are still being investigated in order to improve their synthesis. Scheme 2 reports the five main procedures for S-arylmercapturic acids (AMA) (or ester derivative) synthesis: the reaction of aryl diazonium salts with cysteine cuprous mercaptides or with N-acetyl-l-cysteine [13,22,23], the copper-assisted or palladium-assisted nucleophilic substitution of aryliodides [24,25], the nucleophilic substitution of aryl nitrate or nitroarylhalide [26,27,28], a Mitsunobu-type coupling reaction [29], and finally the process of Behringer et al. [30,31]
Scheme 2.

Most of the processes already described gave low yields of complicated mixtures [13,22,23], or needed highly activated aromatics [28,29,31]. Moreover, they generally suffer from different drawbacks (longer reaction time, high temperature reaction, high starting material purity, rigorous dioxygen exclusion, tedious work-up) [30,31].
The aim of the present study was to provide gas chromatographic standards having a methyl S-aryl-mercapturate structure (AME) in order to use them in biological monitoring and metabolism studies. Thirteen compounds have thus been obtained in high yields from the procedure described in Scheme 3. This process is a two-step reaction starting from 2-acetamidoacrylic acid (product 1) and involving a smooth esterification of 1 followed by a sulfa-Michael addition [32] of the adequate thiophenol on the produced acrylate (product 2, MAA) in the presence of a phase-transfer catalyst [33].
Scheme 3.

Results and Discussion
Methyl 2-acetamidoacrylate synthesis (MAA; 2)
Few procedures have been described in the past for MAA synthesis, and conventional acidic esterification attempts to obtain this ester often failed [34,35]. This was probably due to the poor stability of the unsaturated structure. Rothstein et al. [34] first obtained MAA using the nucleophilic action of lead or sodium salts of compound 1 on iodomethane or dimethylsulphate, but yields never exceeded 52%. Later on, Bueno et al. [36], using the potassium salt of 1, produced MAA in yields of up to 80%, but in crude form. Another conventional reagent, diazomethane, is known to be inadequate for unsaturated carboxylic acid esterification, yielding quantitatively pyrazolines structures [37]. Other procedures reported in the literature involve methyl alaninate, cysteinate or serinate in more complicated retrosynthetic mechanisms [38,39,40].
As far as we are concerned, numerous methylation and esterification methods were investigated in the laboratory [41,42], but only one reagent, cesium carbonate, was applied with success. Using experimental conditions described in the literature [43], the yield was gradually improved by increasing the ratio of iodomethane to compound 1 (Table 1). Finally, MAA was obtained quantitatively by carefully controlling the nucleophilic substitution and the time reaction.
Table 1.
MAA (2) synthesis.
| Entry | Cs2CO3 (equiv) | CH3I (equiv) | Time (h) | Yield of 2a (%) |
|---|---|---|---|---|
| 1 | 0.5 | 1.2 | 15 | 50 |
| 2 | 0.5 | 2.0 | 15 | 65 |
| 3 | 0.5 | 2.5 | 15 | 76 |
| 4 | 0.5 | 2.5 | 3 | 99.7 |
a isolated yield
Methyl S-p-toluylmercapturate (3a) synthesis: optimisation
Toluene was the first aromatic hydrocarbon to be examined when our biomonitoring studies began. Optimisation (Scheme 2, step 2) was therefore applied to obtain 3a, a mercapturate stemming from toluene ring oxidation (Scheme 1) [13]. The results concerning this particular optimisation are summarized in Table 2. Tetrabutylammonium hydrogensulfate (TBAHS), which generally performs successfully in all kinds of phase transfer catalysis (PTC) reactions [44,45], was initially chosen as a catalyst. Using liquid-liquid conditions (entry 1), a complete deesterification of 2 unfortunately resulted. This negative result prompted us on the one hand to suppress water and to use a solid-liquid process [46,47,48] and on another hand to replace NaOH by KOH. Three reaction-condition parameters, i.e. the organic phase, the basic anion and the catalyst used, were then successively modified to improve the yields obtained.
Table 2.
Optimisation of the reaction conditions for the addition of p-thiocresol to compound 2.
| Entry | p-Thiocresol (mmol) | Base (mmol) | PTC (mmol) | Solvent | Time (h) | %Yield for 3a |
|---|---|---|---|---|---|---|
| 1 | 1 | NaOH 1M/H2O | TBAHS (0.2) | CH2Cl2/H2O | 48 | 100% of 2 |
| 2 | 1 | NaOH (1.4) | TBAHS (0.2) | THF | 48 | 1 |
| 3 | 1 | KOH (1.2) | TBAHS (0.2) | THF | 18 | 22 |
| 4 | 1 | KOH (1.4) | TBAHS (0.2) | Acetonitrile | 18 | 22 |
| 5 | 1 | KOH (1.2) | TBAHS (0.2) | Toluene | 18 | 33 |
| 6 | 1 | KOH (1.2) | CTAB (0.2) | Toluene | 48 | 31 |
| 7 | 1 | KOH (1.2) | CTAB (0.2) | THF | 18 | 49 |
| 8 | 1 | KOH (1.2) | Aliquat 336 (0.08) | THF | 18 | 6 |
| 9 | 1 | K2CO3 (0.3) | Aliquat 336 (0.08) | THF | 18 | 63 |
| 10 | 1 | K2CO3 (0.3) | Aliquat 336 (0.08) | Toluene | 18 | 61 |
| 11 | 1.4 | K2CO3 (0.3) | Aliquat 336 (0.08) | Toluene | 5 | 99.6 |
Amount of 2 used: 1 mmol
Three solvents were thus investigated and put through the procedure. From the results of runs 3, 4 and 5, toluene may be considered as the reference solvent, other solvents appearing less suitable. This result is not in accordance with Herriott et al. who suggested that the reaction rate in PTC should be directly related to solvent polarity [49]. When the results of runs 9 and 10 were analysed, the relationship between solvent and yield did not appear so simple. The results observed with THF (tetrahydrofuran), which is a polar solvent, were similar to those obtained with toluene.
The investigations then moved on to screening two different bases [50]. The results obtained in runs 2 and 3, and comparatively in runs 8 and 9, clearly demonstrate that K2CO3 is superior to KOH in terms of compound 3a formation. Furthermore, the reactions with this latter base were by far the cleanest, producing only a very minor amounts of by-products.
Two other PTC catalysts, cetyltrimethyl ammonium bromide (CTAB) and methyltrioctyl ammonium chloride (Aliquat 336), were then successively tested to conduct the sulfa-Michael addition (entries 3, 7, 8 and 9). Aliquat 336 emerged as the most active catalyst for our process (lowest quantities, highest rate).
Finally, the reaction was performed with a excess of reagent (entry 11). In fact, it is well known that arylmercaptans are particularly sensitive to oxidation. With an excess of p-cresol a quasi complete conversion was observed. To confirm the catalytic effect, the reaction was then carried out using the conditions described in entry 11 and avoiding the use of the selected PTC catalyst. In contrast with the preceding results, no reaction at all was observed at room temperature over a 24-hour stirring period.
In summary, appropriate solid-liquid phase-transfer catalysis conditions have been determined to carry out the addition of p-thiophenol to MAA, THF and toluene emerging as the solvents of choice. The addition takes place rapidly by successively adding a catalytic quantity of the basic anion (K2CO3) and then the catalyst (Aliquat 336) to the reaction mixture, which yields 3a, the desired product, quasi quantitatively.
Application to the synthesis of various AMEs (3b-m)
Using the optimised conditions established for 3a, the Michael addition was applied to numerous thiophenols (Table 3). As expected from previous results, thiophenols bearing para but also ortho electron donating groups reacted quickly, giving the corresponding AMEs 3b-d in remarkable yields in less than 5 hours. Similarly, thiophenols bearing two electron donating groups afforded AMEs 3e-h in satisfactory yields, but needed generally slightly longer reaction times.
Table 3.
Methyl S-arylmercapturates (AME) synthesis.
| Entry | AMEa | Solvent | Time (h) | RFc | Yieldd (%) | |
|---|---|---|---|---|---|---|
| 1 | 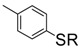 |
3a | Tol | 5 | 0.28 | 99.6 |
| 2 | 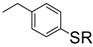 |
3b | Tol | 5 | 0.31 | 99 |
| 3 | 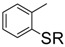 |
3c | Tol | 5 | 0.29 | 94 |
| 4 | 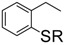 |
3d | Tol | 5 | 0.32 | 96 |
| 5 | 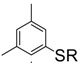 |
3e | Tol | 5 | 0.32 | 95 |
| 6 | 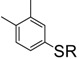 |
3f | Tol | 13 | 0.32 | 90 |
| 7 | 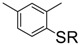 |
3g | Tol | 18 | 0.26 | 82 |
| 8 | 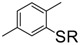 |
3h | Tol | 18 | 0.31 | 98 |
| 9 | 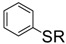 |
3i | Tol | 5 | 0.26 | 96 |
| 10 | 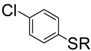 |
3j | Tol/THFb | 18 | 0.27 | 76 |
| 11 | 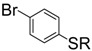 |
3k | Tol | 5 | 0.25 | 88 |
| 12 | 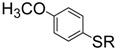 |
3l | Tol | 18 | 0.23 | 76 |
| 13 | 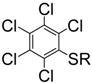 |
3m | Tol/THFb | 18 | 0.26 | 71 |
a R = -CH2-CH(NHCOCH3)(CO2CH3)
b Tol/THF: 1/1; v/v
c The reaction was monitored by thin layer chromatography using diethyl ether as eluent.
d Isolated yield
In order to complete the knowledge of this procedure, several other thiophenols were then investigated. It was successively demonstrated that a donating group was not required (3i), and, that despite the electron withdrawing group presence, 3j-l can be obtained in relatively satisfactory yields with reaction times ranging from 5 to 18 hours. In addition, and unexpectedly, a thiophenol bearing five electron withdrawing groups (3m) gave similar results.
Finally, our procedure is safe, simple, convenient, and compatible with numerous substrates and, by and large, produced AMEs in higher yields compared to other protocols [22,23,24,25,26,27,28,29,30,31]. Furthermore, availability and low cost of Aliquat 336 are an additional favourable factor to consider.
Conclusions
Methyl S-arylmercapturates have been synthesized according to a two-step reaction starting from compound 1 (Scheme 2) and involving a sulfa-Michael addition whose reactivity was enhanced by adding a phase transfer catalyst. A simple and efficient synthesis of MAA (2), the intermediary synthon, is described. The complete method proceeds smoothly with typical yields ranging from 71 to 99.6%, and may be applied to various thiophenols bearing electron donating groups or electron withdrawing groups. These compounds were then used as standards in gas chromatographic analysis and in metabolism studies.
Experimental
General
2-Acetamidoacrylic acid (1), thiophenol, chlorothiophenol, pentachlorothiophenol, 4-toluenethiol, 2-ethylthiophenol, 2-toluenethiol, cesium carbonate, tetrabutylammonium hydrogensulfate (TBAHS), cetyltrimethyl ammonium bromide (CTAB) and methyltrioctyl ammonium chloride (Aliquat 336) were purchased from Sigma-Aldrich-Fluka. 4-bromothiophenol, 2,4-dimethylthiophenol, 2,5-dimethyl-thiophenol, 3,4-dimethylthiophenol and 3,5-dimethylthiophenol were all available from Acros Organics (Fisher Bioblock, France). Finally, 4-ethylthiophenol was obtained from Avocado (Interchim, France). All the other chemicals were of analytical grade. 1H-NMR and 13C-NMR spectra were obtained on a Bruker AM 400 spectrometer operated at 400 and 100 MHz, respectively. The chemical shifts are reported in parts per million relative to TMS. The melting points (m.p.) were determined with an Electrothermal digital melting point apparatus and were uncorrected. The elemental analyses (EA) were performed by the Central Analysis Department of CNRS (Vernaison, France). The mass spectra (MS) were collected in electronic impact mode (70 eV) on a Varian 1200 MS/MS mass spectrometer coupled to a Varian 3800 gas chromatograph (GC). A Varian DB-5MS column (30 m; 0.25 mm; 0.25 μm) was used for the AME chromatography. The carrier gas was helium (1 mL/min). The injector (split ratio = 30) was held at 250 °C. The GC oven program was: 70 °C initial temperature, isothermal for 1.5 min, then 15 °C/min to 250 °C, and finally isothermal for 7 min. Thin-layer chromatography (TLC) was carried out on Merck 60 F254 precoated silica gel plates (0.25mm) using diethyl ether (100%) as eluent (RF in Table 3). Spots were visualized successively with a UV lamp (λ=254 nm) and iodine vapors.
Methyl 2-acetamidoacrylate synthesis (MAA; 2)
Cs2CO3 (4.2 mmol) was added to 1 (1368 mg, 8.4 mmol) dissolved in MeOH (20 mL), and the mixture was stirred at room temperature for one hour. The methanolic solution was concentrated until dryness. Iodomethane (42 mmol; 2.7 mL), in controlled portions and undergoing constant stirring, was added over a two-hour period to the residue dissolved in DMF (30 mL). The reaction mixture was finally concentrated to dryness, and the crude residue, purified over a silica gel column using diethyl ether (Et2O 100%, RF = 0.55) as eluent, gave 1199 mg of 2 as white needles, i.e. a yield of 99.7 %
Methyl 2-acetamidoacrylate (2). White solid, mp: 52-53°C; 1H-NMR (CDCl3): δ (ppm) 2.11 (s, 3 H, COCH3), 3.82 (s, 3 H, OCH3), 5.85 (s, 1H, CH2), 6.57 (s, 1H, CH2), 7.72 (s, 1H, NH); 13C-NMR (CDCl3): δ (ppm) 24.56 (CH3), 52.88 (CH3), 108.63 (CH2), 130.85 (C=CH2), 164.56 (CO2), 168.74 (CO); IR: ν (cm-1) 3364, 3155, 3057, 2963, 1813, 1708, 1678, 1629, 1512, 1428, 1369, 1327, 1246, 1208, 1172, 1038, 995, 977, 905, 856, 809; MS (m/z, EI): 143 (M+, 66), 111 (44), 101 (100), 71 (27), 43 (88), 42 (63); Anal. Calcd for C6H9NO3: C, 50.35; H, 6.34; N, 9.79; O, 33,53; Found: C, 50.41; H, 6.37; N, 9.39; O, 33,30.
General procedure for methyl S-arylmercapturate (AME) synthesis: compounds 3a-m
Compounds 3b to 3m were synthesized using the parameters defined for 3a in Table 2 entry 11 and in Table 3. Briefly, adequate thiophenol (1.4 mmol), Aliquat 336 (32 mg, 0.08 mmol) and finally powdered potassium carbonate (41 mg, 0.3 mmol) were added respectively to 2 (143 mg, 1 mmol) dissolved in toluene (30 mL, except if otherwise indicated in Table 3). The reaction mixture was stirred at room temperature and regularly monitored by thin layer chromatography using Et2O as eluent (RF, Table 3). The mixture filtered, the crude product, isolated after solvent removal, was subjected to silica gel column chromatography using Et2O as eluent. Yields are given in Table 3.
2-Acetamido-3-p-tolylsulfanylpropionic acid methyl ester (3a). White solid, mp: 84-85°C; 1H-NMR (CD3OD): δ (ppm) 1.88 (s, 3H, COCH3), 2.31 (s, 3H, CH3-Ar), 3.28-3.44 (m, 2H, SCH2), 3.56 (s, 3H, O-CH3), 4.60-4.63 (m, 1H, CH-CH2), 4.81-4.85 (m, 1H), 7.09-7.32 (m, 4H, ArH); 13C-NMR (CD3OD): δ (ppm) 20.1 (CH3), 21.4 (CH3), 36.2 (CH2), 51.8 (CH3), 52.7 (CH), 129.9 (CHAr), 131.4 (CHAr), 131.6 (C), 137.5 (C), 171.4 (CO), 172.2 (CO); IR: ν (cm-1) 3322, 3075, 2984, 2944, 2931, 2843, 1727, 1645, 1543, 1495, 1423, 1344, 1321, 1248, 1179, 1089, 1036, 803; MS (m/z, EI): 267 (M+, 15), 208 (80), 149 (30), 43 (100); Anal. Calcd for C12H17NO3S; C 58.40, H 6.41, N 5.24, O 17.95, S 11.99; Found: C 58.66, H 6.48, N 5.14, O 18.40, S 12.16.
2-Acetamido-3-(4-ethylphenylsulfanyl)-propionic acid methyl ester (3b). White solid, mp: 64-65°C; 1H-NMR (CD3OD): δ (ppm) 1.19 (t, 3J = 7.6 Hz, 3 H, CH3), 1.92 (s, 3 H, COCH3), 2.59 (q, 3J = 7.6 Hz, 2 H, CH2-CH3), 3.18 (dd, 2J = 14 Hz, 3J = 7.8 Hz, 1 H, SCH2), 3.34 (dd, 2J = 14 Hz, 3J = 4.8 Hz, 1 H, SCH2), 3.60 (s, 3 H, OCH3), 4.55 (dd, 3J = 7.8 Hz, 3J = 4.8 Hz, 1 H, CH2-CH), 4.84 (s, 1 H, NH), 7.13-7.16 (m, 2 H, 2-Ar-H), 7.32-7.35 (m,2 H, 2 Ar-H); 13C-NMR (CD3OD): δ (ppm) 16.2 (CH3), 22.4 (CH3), 29.5 (CH2), 37.3 (CH2), 53.0 (CH3), 53.8 (CH), 129.8 (CHAr), 131.0 (CAr), 132.7 (CHAr), 145.0 (CAr), 172.4 (CO), 173.2 (CO); IR: ν (cm-1) 3318, 3076, 2969, 2946, 2934, 1723, 1637, 1546, 1496, 1438, 1422, 1347, 1321, 1248, 1219, 1180, 1091, 1036, 1017, 814, 696; MS (m/z, EI): 281 (M+, 26), 222 (100), 207 (10), 190 (6), 163 (53), 151 (55), 136 (78), 123 (29), 105 (25), 88 (52), 77 (60), 59 (51); Anal. Calcd for C14H19NO3S; C 59.76, H 6.81, N 4.98, O 17.06, S 11.40; Found: C 59.82, H 6.79, N 4.90, O 17.35, S 11.46.
2-Acetamido-3-o-tolylsulfanylpropionic acid methyl ester (3c). White solid, mp: 66-67°C; 1H-NMR (CD3OD): δ (ppm) 1.95 (s, 3 H, COCH3), 2.38 (s, 3 H, CH3-Ar), 3.21 (dd, 2J = 14.0 Hz, 3J = 8.0 Hz, 1 H, SCH2), 3.39 (dd, 2J = 14.0 Hz, 3J = 4.9 Hz, 1 H, SCH2), 3.64 (s, 3 H, OCH3), 4.58 (dd, 3J = 8.0 Hz, 3J = 4.9 Hz, 1 H, CH-CH2), 7.12 – 7.22 (m, 3 H, Ar-H), 7.41 – 7.43 (m, 1 H, Ar-H); 13C-NMR (CD3OD): δ (ppm) 19.6 (CH3), 21.2 (CH3), 34.8 (CH2), 52.0 (CH3), 52.7 (CH), 126.9 (CHAr), 127.4 (CHAr), 130.5 (CHAr), 130.7 (CHAr), 134.1 (CAr), 139.1 (CAr), 171.3 (CO), 172.2 (CO); IR: ν (cm-1) 3311, 3069, 2952, 2940, 2838, 1736, 1650, 1542, 1432, 1335, 1223, 1168, 1036, 744; MS (m/z, EI): 267 (M+, 13), 208 (100), 149 (56), 137 (44), 134 (61), 91 (18), 43 (26); Anal. Calcd for C12H17NO3S; C 58.40, H 6.41, N 5.24, O 17.95, S 11.99; Found: C 58.28, H 6.45, N 5.08, O 18.19, S 12.19.
2-Acetamido-3-(2-ethylphenylsulfanyl)propionic acid methyl ester (3d). White solid, mp: 55-56°C; 1H-NMR (CD3OD): δ (ppm) 1,18 (t, 3J = 7.5 Hz, 3H, CH3-CH2), 1.94 (s, 3H, COCH3), 2.78 (q, 3J = 7.5 Hz, 2H, CH2-CH3), 3.19 (dd, 2J = 13.9 Hz, 3J = 8.1 Hz, 1 H, SCH2), 3.39 (dd, 2J = 13.9 Hz, 3J = 5,0 Hz, 1 H, SCH2), 3.62 (s, 3H, CH3-Ar), 4.55 (dd, 3J = 8.1 Hz, 3J = 5.0 Hz, 1 H, CH-CH2), 7.15 – 7.21 (m, 3 H, Ar-H), 7.41 – 7.44 (m, 1 H, Ar-H); 13C-NMR (CD3OD): δ (ppm) 15.7 (CH3), 22.4 (CH3), 28.2 (CH2), 36.5 (CH2), 53.0 (CH3), 53.7 (CH), 127.8 (CHAr), 128.4 (CHAr), 130.1 (CHAr), 131.9 (CHAr), 134.6 (CAr), 146.3 (CAr), 172.5 (CO), 173.3 (CO); IR: ν (cm-1) 3280, 3058, 2964, 2872, 1747, 1655, 1541, 1437, 1373, 1214, 1128, 1027, 752; MS (m/z, EI): 281 (M+, 42), 222 (88), 151 (30), 148 (100), 144 (25), 136 (45), 91 (12), 43 (21); Anal. Calcd for C14H19NO3S; C 59.76, H 6.81, N 4.98, O 17.06, S 11.40; Found: C 59.39, H 6.76, N 4.88, O 17.46, S 11.74.
2-Acetamido-3-(3,5-dimethylphenylsulfanyl)propionic acid methyl ester (3e). White solid, mp: 73-74°C; 1H-NMR (CD3OD): δ (ppm) 1.92 (s, 3 H, COCH3), 2.25 (s, 6 H, 2 CH3-Ar), 3.18 (dd, 2J = 14.2 Hz, 3J = 7.6 Hz, 1 H, SCH2), 3.35 (dd, 2J = 14.2 Hz, 3J = 4.8 Hz, 1 H, SCH2), 3.62 (s, 3 H, OCH3), 4.57 (dd, 3J = 7.6 Hz, 3J = 4.8 Hz, 1 H, CH2-CH), 4.84 (s, 1H, NH), 6.85 (m, 1 H, 1 Ar-H), 7.01 (m, 2 H, 2 Ar-H); 13C-NMR (CD3OD): δ (ppm) 21.4 (2 x CH3), 22.4 (CH3), 34.6 (CH2), 53.0 (CH3), 52.4 (CH), 129.3 (CHAr), 131.1 (CHAr), 131.5 (CAr), 134.2 (CAr), 170.1 (CO), 171.1 (CO); IR: ν (cm-1) 3269, 3072, 2953, 2914, 2856, 1757, 1733, 1652, 1549, 1430, 1376,1249, 1216, 1125, 1031, 841; MS (m/z, EI): 281 (M+, 25), 222 (45), 189 (8), 163 (100), 151 (42), 135 (19), 122 (14), 105 (51), 91 (36), 77 (34), 59 (35); Anal. Calcd for C14H19NO3S; C 59.76, H 6.81, N 4.98, O 17.06, S 11.40; Found: C 59.72, H 6.96, N 4.81, O 16.94, S 11.42.
2-Acetamido-3-(3,4-dimethylphenylsulfanyl)propionic acid methyl ester (3f). White solid, mp: 84-85°C; 1H-NMR (CD3OD): δ (ppm) 1.92 (s, 3 H, COCH3), 2.20 (s, 3 H, CH3-Ar(4)), 2.21 (s, 3 H, CH3-Ar (3)), 3.14 (dd, 2J = 14.0 Hz, 3J = 8.0 Hz, 1 H, SCH2), 3.31 (dd, 2J = 14.0 Hz, 3J = 4.8 Hz, 1 H, SCH2), 3.61 (s, 3 H, OCH3), 4.53 (dd, 3J = 8.0 Hz, 3J = 4.8 Hz, 1 H, CH2-CH), 4.84 (s, 1H, NH), 7.05 (m, 1 H, Ar-H), 7.13 (m, 1 H, Ar-H), 7.19 (m, 1 H, Ar-H); 13C-NMR (CD3OD): δ (ppm) 19.5 (CH3), 19.9 (CH3), 22.4 (CH3),37.3 (CH2), 52.9 (CH3), 53.8 (CH3), 130.2 (CHAr), 131.4 (CHAr), 132.6 (CAr), 133.7 (CHAr), 137.2 (CAr), 138.7 (CAr), 172.5 (CO), 173.2 (CO); IR: ν (cm-1) 3284, 3061, 3015, 2951, 2928, 2867, 1747, 1659, 1539, 1488, 1436, 1372, 1213, 1129, 1022, 814; MS (m/z, EI): 281 (M+, 18), 222 (65), 191 (7), 163 (100), 151 (55), 137 (16), 105 (53), 91 (38), 77 (49), 59 (28); Anal. Calcd for C14H19NO3S; C 59.76, H 6.81, N 4.98, O 17.06, S 11.40; Found: C 59.87, H 6.98, N 4.93, O 16.84, S 11.19.
2-Acetamido-3-(2,4-dimethylphenylsulfanyl)propionic acid methyl ester (3g). White solid, mp: 84-85°C; 1H-NMR (CD3OD): δ (ppm) 1.95 (s, 3 H, COCH3), 2.27 (s, 3 H, CH3-Ar (4)), 2.36 (s, 3 H, CH3-Ar (2)), 3.13 (dd, 2J = 14.0 Hz, 3J = 8.0 Hz, 1 H, SCH2), 3.30 (dd, 2J = 14.0 Hz, 3J = 5.0 Hz, 1 H, SCH2), 3.62 (s, 3 H, OCH3), 4.52 (dd, 3J = 8.0 Hz, 3J = 5.0 Hz, 1 H, CH2-CH), 4.86 (s, 1H, NH), 6.98 (m, 1 H, Ar-H), 7.04 (m, 1 H, Ar-H), 7.31 (m, 1 H, Ar-H); 13C-NMR (CD3OD): δ (ppm) 20.8 (CH3), 21.2 (CH3), 22.4 (CH3),36.5 (CH2), 53.0 (CH3), 53.7 (CH), 128.5 (CHAr), 131.3 (CAr), 132.4 (CHAr), 132.9 (CHAr), 138.6 (CAr), 140.7 (CAr), 172.5 (CO), 173.2 (CO); IR: ν (cm-1) 3293, 3063, 2959, 2916, 1749, 1652, 1546, 1435, 1411, 1376, 1292, 1258, 1218, 1170, 1127, 821; MS (m/z, EI): 281 (M+, 48), 222 (91), 207 (11), 190 (10), 163 (70), 151 (63), 138 (29), 122 (18), 105 (100), 91 (52), 77 (74), 59 (54); Anal. Calcd for C14H19NO3S; C 59.76, H 6.81, N 4.98, O 17.06, S 11.40; Found: C 59.76, H 6.86, N 4.82, O 17.40, S 11.38.
2-Acetamido-3-(2,5-dimethylphenylsulfanyl)propionic acid methyl ester (3h). White solid, mp: 84-85°C; 1H-NMR (CD3OD): δ (ppm) 1.95 (s, 3 H, COCH3), 2.29 (s, 3 H, CH3-Ar (5)), 2.33 (s, 3 H, CH3-Ar (2)), 3.18 (dd, 2J = 14.0 Hz, 3J = 8.0 Hz, 1 H, SCH2), 3.35 (dd, 2J = 14,0 Hz, 3J = 4.8 Hz, 1 H, SCH2), 3.63 (s, 3 H, OCH3), 4.57 (dd, 3J = 8.0 Hz, 3J = 4.8 Hz, 1 H, CH2-CH), 4.86 (s, 1H, NH), 6.95 (d, 3J = 7.8 Hz, 1 H, 1 Ar-H), 7.07 (d, 3J = 7.8 Hz, 1 H, 1 Ar-H), 7.24 (s, 1 H, 1 Ar-H); 13C-NMR (CD3OD): δ (ppm) 20.3 (CH3), 21.1 (CH3), 22.4 (CH3), 35.9 (CH2), 53.0 (CH3), 53.7 (CH), 129.0 (CHAr), 131.4 (CHAr), 132.2 (CHAr), 134.7 (CAr), 137.0 (CAr), 137.4 (CAr), 172.4 (CO), 173.3 (CO); IR: ν (cm-1) 3319, 3004, 2952, 2915, 1769, 1651, 1530, 1489, 1432, 1379, 1251, 1216, 1189, 1171, 1029, 976, 800; MS (m/z, EI): 281 (M+, 25), 222 (73), 190 (10), 163 (100), 149 (98), 135 (25), 122 (15), 105 (84), 91 (58), 77 (78), 59 (58); Anal. Calcd for C14H19NO3S; C 59.76, H 6.81, N 4.98, O 17.06, S 11.40; Found: C 59.91, H 6.84, N 4.95, O 17.41, S 11.52.
2-Acetamido-3-phenylsulfanylpropionic acid methyl ester (3i). White solid, mp: 64-65°C; 1H-NMR (CD3OD): δ (ppm) 1.96 (s, 3 H, COCH3), 3.23 (dd, 2J = 14.0 Hz, 3J = 8.0 Hz, 1 H, SCH2), 3.40 (dd, 2J = 14,0 Hz, 3J = 5.2 Hz, 1 H, SCH2), 3.62 (s, 3 H, OCH3), 4.68 (dd, 3J = 8.0 Hz, 3J = 5.2 Hz, 1 H, CH2-CH), 4.85 (s, 1H, NH), 7.20-7.24 (m, 1 H, 1 Ar-H), 7.28-7.32 (m, 2 H, 2 Ar-H), 7.39-7.43 (m, 2 H, 2 Ar-H); 13C-NMR (CD3OD): δ (ppm) 22.4 (CH3), 36.6 (CH2), 53.0 (CH3), 53.8 (CH), 128.1 (CHAr), 130.3 (CHAr), 131.8 (CHAr), 136.3 (CAr), 172.4 (CO), 173.3 (CO); IR: ν (cm-1) 3283, 3058, 2952, 2849, 1747, 1653, 1539, 1482, 1438, 1373, 1306, 1214, 1177, 1128, 1025, 743; MS (m/z, EI): 253 (M+,10), 194 (84), 163 (14), 135 (100), 123 (73), 109 (45), 88 (42), 77 (35), 65 (47); Anal. Calcd for C12H15NO3S; C 56.90, H 5.97, N 5.53, O 18.95, S 12.66; Found: C, 57.35; H, 5.91; N, 5.32; O, 18.75; S, 12.66.
2-Acetamido-3-(4-chlorophenylsulfanyl)propionic acid methyl ester (3j). White solid, mp: 106-107°C; 1H-NMR (DMSO-d6): δ (ppm) 1.83 (s, 3 H, COCH3), 3.19 (dd, 2J = 13.8 Hz, 3J = 8.0 Hz, 1 H, SCH2), 3.32 (dd, 2J = 13.8 Hz, 3J = 5.4 Hz, 1 H, SCH2), 3.57 (s, 3 H, OCH3), 4.40 (dd, 3J = 8.0 Hz, 3J = 8.0 Hz, 3J = 5.4 Hz, 1 H, CH-CH2), 7.36 (s, 4 H, Ar-H), 8.50 (d, 3J = 8.0 Hz, 1H, NH); 13C-NMR (DMSO-d6): δ (ppm) 22.4 (CH3), 34.6 (CH2), 51.8 (CH3), 52.4 (CH), 129.3 (CHAr), 131.1 (CHAr), 131.5 (CAr), 134.2 (CAr), 170.1 (CO), 171.1 (CO); IR: ν (cm-1) 3308, 3075, 2951, 2930, 1730, 1643, 1546, 1478, 1425, 1369, 1349, 1247, 1219, 1192, 1177, 1099, 1037, 1011, 808; MS (m/z, EI): 287 (M+,11), 230 (41), 228 (100), 197 (10), 169 (50), 157 (22), 143 (15), 108 (12), 88 (29); Anal. Calcd for C12H14ClNO3S; C 50.09, H 4.90, Cl 12.32, N 4.87, O 16.68, S 11.14; Found: C 50.80, H 5.23, Cl 11.93, N 4.68, O 16.20, S 10.54.
2-Acetamido-3-(4-bromophenylsulfanyl)propionic acid methyl ester (3k). White solid, mp: 121-122°C; 1H-NMR (CDCl3): δ (ppm) 1.80 (s, 3H, COCH3), 3.21 (dd, 2J = 14.2 Hz, 3J = 4.7 Hz, 1 H, SCH2), 3.35 (dd, 2J = 14.2 Hz, 3J = 4.7 Hz, 1 H, SCH2), 3.49 (s, 3H, OCH3), 4.73 (m, 1 H, CH-CH2), 6.16 (s, 1H, NH), 7,14-7,16 (m, 2 H, Ar-H), 7.29 – 7.31 (m, 2 H, Ar-H); 13C-NMR (CDCl3): δ (ppm) 22.9 (CH3), 36.5 (CH2), 52.2 (OCH3), 52.5 (CH), 121.0 (CAr), 132.1 (CHAr), 132.3 (CHAr), 134.0 (CAr), 169.6 (CO), 170.6 (CO); IR: ν (cm-1) 3307, 3047, 2950, 1729, 1641, 1546, 1474, 1425, 1350, 1324, 1245, 1176, 1092, 1039, 1006, 806; MS (m/z, EI): 333 (18), 331 (M+, 17), 274 (99), 272 (100), 215 (10), 213 (20), 203 (13), 201 (13), 189 (11), 187 (11), 144 (18), 134 (53), 122 (67), 108 (50), 88 (74), 75 (25), 60 (60); Anal. Calcd for C12H14BrNO3S: C, 43.38; H, 4.25; Br, 24.05; N, 4.22; O, 14.45; S, 9.65. Found: C,44.56, H, 4.41, Br, 23.59, N, 4.04, O, 14.38, S, 9.29.
2-Acetamido-3-(4-methoxyphenylsulfanyl)propionic acid methyl ester (3l). Colorless oil; 1H-NMR (CDCl3): δ (ppm) 1.89 (s, 3H, COCH3), 3.22 (dd, 2J = 14.2 Hz, 3J = 5.0 Hz, 1 H, SCH2), 3.32 (dd, 2J = 14.2 Hz, 3J = 4.7 Hz, 1 H, SCH2), 3.54 (s, 3H, OCH3), 3.76 (s, 3 H, Ar-OCH3), 4.77 (m, 1 H, CH-CH2), 6.81 – 6.83 (m, 2 H, Ar-H), 7.35 – 7.37 (m, 2 H, Ar-H); 13C-NMR (CDCl3): δ (ppm) 22.9 (CH3), 38.0 (CH2), 52.2 (OCH3), 52.3 (CH), 55.3 (OCH3), 114.7 (CHAr), 124.7 (CAr), 134.2 (CHAr), 159.4 (CAr), 169.7 (CO), 170.8 (CO); IR: ν (cm-1) 3289, 3065, 2953, 2838, 1746, 1656, 1592, 1541, 1494, 1437, 1373, 1286, 1246, 1175, 1029, 827; MS (m/z, EI): 283 (47), 224 (100), 165 (27), 153 (22), 144 (25), 165 (27), 109 (9), 84 (7); Anal. Calcd for C13H17NO4S: C, 55.11; H, 6.05; N, 4.94; O, 22.59; S, 11.32. Found: C, 55.27; H, 6.32; N, 4.87; O, 22.79; S, 11.30.
2-Acetamido-3-pentachlorophenylsulfanylpropionic acid methyl ester (3m). White solid, mp: 145-146°C; 1H-NMR (DMSO-d6): δ (ppm) 11.75 (s, 3 H, COCH3), 3.17 (dd, 2J = 14.0 Hz, 3J = 8.8 Hz, 1 H, SCH2), 3.40 (dd, 2J = 14.0 Hz, 3J = 4.8 Hz, 1 H, SCH2), 3.57 (s, 3 H, OCH3), 4.36 (ddd, 3J = 8.8 Hz, 3J = 8.0 Hz, 3J = 4.8 Hz, 1 H, CH-CH2), 8.36 (d, 3J = 8.0 Hz, 1H, NH); 13C-NMR (DMSO-d6): δ (ppm) 22.4 (CH3), 36.0 (CH2), 52.5 (CH), 52.8 (CH3), 131.4 (CAr), 133.5 (CAr), 134.4 (CAr), 138.2 (CAr), 170.0 (CO), 170.8 (CO); IR: ν (cm-1) 3293, 2956, 1758, 1651, 1532, 1435, 1378, 1334, 1305, 1259, 1215, 1190, 1170, 1130, 688; MS (m/z, EI): 425 (M+, 5), 366 (100), 324 (13), 246 (18), 167 (12), 144 (37), 117 (48), 102 (18), 88 (90), 69 (34); Anal. Calcd for C12H10Cl5NO3S; C 33.87, H 2.37, Cl 41.66, N 3.29, O 11.28, S 7.54; Found: C 34.24, H 2.58, Cl 42.01, N 2.97, O 10.75, S 6.96.
Acknowledgements
The authors gratefully acknowledge Caroline Bastien and Stéphane Grossmann for their technical assistance.
Footnotes
Sample Availability: Samples of the compounds mentioned in this paper are available from the corresponding author (B.C.)
References and Notes
- 1.Lauwerys R.R., Haufroid V., Hoet P., Lison D. In: Toxicologie industrielle et intoxications professionnelles. Elsevier Masson S.A.S., editor. Issy-les-Moulineaux; (France): 2007. pp. 528–557. [Google Scholar]
- 2.Lataye R., Campo P., Pouyatos B., Cossec B., Blachere V., Morel G. Solvent ototoxicity in the rat and guinea pig. Neurotox. Teratol. 2003;25:39–50. doi: 10.1016/S0892-0362(02)00326-4. [DOI] [PubMed] [Google Scholar]
- 3.Gagnaire F., Marignac B., Langlais C., Bonnet P. Ototoxicity in rats exposed to ortho-, meta- and para-xylene vapours for 13 weeks. Pharmacol. Toxicol. 2001;89:6–14. doi: 10.1034/j.1600-0773.2001.d01-129.x. [DOI] [PubMed] [Google Scholar]
- 4.Korsak Z., Rydzynski K. Neurotoxic effects of acute and subchronic inhalation exposure to trimethylbenzene isomers (pseudocumene, mesitylene, hemimellitene) in rats. Int. J. Occup. Med. Environ. Health. 1996;9:341–349. [PubMed] [Google Scholar]
- 5.Vermeulen N.P.E. Analysis of mercapturic acids as a tool in biotransformation, biomonitoring and toxicological studies. Trends Pharmacol. Sci. 1989;10:177–181. doi: 10.1016/0165-6147(89)90232-0. [DOI] [PubMed] [Google Scholar]
- 6.Nelson E.D. Determination of mercapturic acid and excretions in exposure control to toxicants. Crit. Rev. Toxicol. 1992;22:371–389. doi: 10.3109/10408449209146313. [DOI] [PubMed] [Google Scholar]
- 7.De Rooij B.M., Commandeur J.N.M., Vermeulen N.P.E. Mercapturic acids as biomarkers of exposure to electrophilic chemicals: applications to environmental and industrial chemicals. Biomarkers. 1998;3:239–303. doi: 10.1080/135475098231101. [DOI] [PubMed] [Google Scholar]
- 8.Angerer J., Ewers U., Wilhelm M. Human biomonitoring: State of the art. Int. J. Hyg. Environ. Health. 2007;210:201–228. doi: 10.1016/j.ijheh.2007.01.024. [DOI] [PubMed] [Google Scholar]
- 9.Perbellini L., Veronese N., Princivalle A. Mercapturic acids in the biological monitoring of occupational exposure to chemicals. J. Chrom. B Analyt. Technol. Biomed. Life Sci. 2002;781:269–290. doi: 10.1016/S1570-0232(02)00501-9. [DOI] [PubMed] [Google Scholar]
- 10.Haufroid V., Lison D. Mercapturic acids revisited as biomarkers of exposure to reactive chemicals in occupational toxicology: a minireview. Int. Arch. Occup. Environ. Health. 2005;78:343–354. doi: 10.1007/s00420-005-0620-z. [DOI] [PubMed] [Google Scholar]
- 11.Van Doorn R., Leijdekkers C.M., Bos R.P., Brouns R.M.E., Henderson P.T. Alcohol and sulphate intermediates in the metabolism of toluene and xylenes to mercapturic acids. J. Appl. Toxicol. 1981;1:236–242. doi: 10.1002/jat.2550010411. [DOI] [PubMed] [Google Scholar]
- 12.Zheng J., Hanzlik R.P. Bromo(monohydroxy)phenyl mercapturic acids from bromobenzene-treated rats. Drug Metabol. Disp. 1992;20:688–694. [PubMed] [Google Scholar]
- 13.Angerer J., Schildbach M., Kramer A. S-p-Toluylmercapturic acid in the urine of workers exposed to toluene: a new biomarker for toluene exposure. Arch. Toxicol. 1998;72:119–123. doi: 10.1007/s002040050478. [DOI] [PubMed] [Google Scholar]
- 14.Gonzalez-Reche L.M., Schettgen T., Angerer J. New approaches to the metabolism of xylenes: verification of the formation of phenylmercapturic acid metabolites of xylenes. Arch. Toxicol. 2003;77:80–85. doi: 10.1007/s00204-002-0419-6. [DOI] [PubMed] [Google Scholar]
- 15.Boogaard P.J., Van Sittert N.J. Biological monitoring of exposure to benzene – A comparison between S-phenylmercapturic acid, trans,trans-muconic acid and phenol. Occup. Environ. Med. 1995;52:611–620. doi: 10.1136/oem.52.9.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Inoue O., Kanno E., Kasai K., Ukai H., Okamoto S., Ikeda M. Benzylmercapturic acid is superior to hippuric acid and o-cresol as a urinary marker of occupational exposure to toluene. Toxicol. Lett. 2004;147:177–186. doi: 10.1016/j.toxlet.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 17.American Conference of Governmental Industrial Hygienists. Threshold Limit Values and Biological Exposure Indices. 1998.
- 18.Deutsche Forschungsgemeinschaft . List of MAK and BAT values. Commission for the investigation of health hazards of chemical compounds in the work area, Report N° 32. VCH; New-York: 1998. [Google Scholar]
- 19.Van Bladeren P.J., Buys W., Breimer D.D., Van der Gen A. The Synthesis of Mercapturic Acids and their Esters. Eur. J. Med. Chem. 1980;15:495–497. [Google Scholar]
- 20.Hayden P., Schaeffer V.H., Larsen G., Stevens J.L. Cysteine S-conjugates. Meth. Enzymol. 1987;143:228–234. doi: 10.1016/0076-6879(87)43043-7. [DOI] [PubMed] [Google Scholar]
- 21.Buijs W., Eid M.I.A., Onkenhout W., Vermeulen N.P.E. The use of sulfenyl halides in the synthesis of mercapturic acids and their esters. Recl. Trav. Chim. Pays-Bas. 1986;105:449–455. doi: 10.1002/recl.19861051022. [DOI] [Google Scholar]
- 22.West H.D., Mathura G.R. Synthesis of some aryl-substituted L-cysteines and their fate in the animal body. J. Biol. Chem. 1954;208:315–318. [PubMed] [Google Scholar]
- 23.Parke D.V., Williams R.T. Studies in detoxication 38. The metabolism of benzene: the determination of phenylmercapturic acid in urine; mercapturic acid excretion by rabbits receiving benzene. Biochem. J. 1951;48:624–629. doi: 10.1042/bj0480624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ciattini P.G., Morera E., Ortar G. A new, palladium-catalyzed synthesis of aromatic mercapturic acid derivatives. Tetrahedron Lett. 1995;36:4133–4136. doi: 10.1016/0040-4039(95)00677-5. [DOI] [Google Scholar]
- 25.Hickman R.J.S., Christie B.J., Guy R.W., White T.J. Synthesis of aromatic S-substituted derivatives of N-acetyl-L-cysteine. Aust. J. Chem. 1985;38:899–904. doi: 10.1071/CH9850899. [DOI] [Google Scholar]
- 26.Kondoh A., Yorimitsu H., Oshima K. Nucleophilic aromatic substitution reaction of nitroarenes with alkyl- or arylthio groups in dimethyl sulfoxide by means of cesium carbonate. Tetrahedron. 2006;62:2357–2360. doi: 10.1016/j.tet.2005.11.073. [DOI] [Google Scholar]
- 27.Pombrio J.M., Giangreco A., Li L.Q., Wempe M.F., Anders M.W., Swett D.H., Pritchard J.B., Ballatori N. Mercapturic acids (N-acetylcysteine S-conjugates) as endogenous substrates for the renal organic anion transporter-1. Mol. Pharmacol. 2001;60:1091–1099. doi: 10.1124/mol.60.5.1091. [DOI] [PubMed] [Google Scholar]
- 28.Saxena M., Henderson G.B. MOAT4, a novel multispecific organic-anion transporter for glucuronides and mercapturates in mouse L1210 cells and human erythrocytes. Biochem. J. 1996;320:273–281. doi: 10.1042/bj3200273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakamura S., Goto K., Kondo M., Naito S., Tsuda Y., Shishido K. Enantioselective total synthesis and structure determination of the mercapturic acid sulfoxide conjugate. Bioorg. Med. Chem. Lett. 1997;7:2033–2036. doi: 10.1016/S0960-894X(97)00363-6. [DOI] [Google Scholar]
- 30.Behringer H., Fackler E. Eine einfache synthese der racemischen Mercaptursäuren. Justus Liebigs Ann. Chem. 1949;564:73–78. doi: 10.1002/jlac.19495640111. [DOI] [Google Scholar]
- 31.Hanzlik R.P., Weller P.E., Desai J., Zheng J., Hall L.R., Slaughter D.E. Synthesis of mercapturic acid-derivatives of putative toxic metabolites of bromobenzene. J. Org. Chem. 1990;55:2736–2742. doi: 10.1021/jo00296a034. [DOI] [Google Scholar]
- 32.Enders D., Lüttgen K., Narine A.A. Asymmetric sulfa-Michael additions. Synthesis. 2007;7:959–980. doi: 10.1055/s-2007-965968. [DOI] [Google Scholar]
- 33.Montanari F., Landini D., Rolla F. Phase transfer catalyzed reactions. Top. Curr. Chem. 1982;101:147–200. [Google Scholar]
- 34.Rothstein E. Experiments in the synthesis of derivatives of α,α-aminoacrylic acid from serine and N-substituted serines. J. Chem. Soc. 1949:1968–1971. doi: 10.1039/jr9490001968. [DOI] [Google Scholar]
- 35.Cardillo G., Gentilucci L., Tolomelli A., Tomasini C. Asymmetric 1,4 addition of Grignard reagents to chiral alpha,beta-unsaturated esters in the presence of Lewis acids. Tetrahedron. 1999;55:6231–6242. doi: 10.1016/S0040-4020(99)00267-7. [DOI] [Google Scholar]
- 36.Bueno M.P., Cativiela C., Finol C., Mayoral J.A., Jaime C. Diels-Alder reactions of methyl-N-acyl-alpha,beta-dehydroalaninates with cyclopentadiene. Can. J. Chem. 1987;65:2182–2186. doi: 10.1139/v87-365. [DOI] [Google Scholar]
- 37.Roper R., Ma T.S. Diazomethane as a reagent for microsynthesis. Microchemical J. 1957;1:245–260. doi: 10.1016/0026-265X(57)90070-X. [DOI] [Google Scholar]
- 38.Kolar A.J., Olsen R.K. Convenient, large-scale preparation of 2-acetamidoacrylic acid and its methyl ester. Synthesis. 1977;457:457–459. doi: 10.1055/s-1977-24439. [DOI] [Google Scholar]
- 39.Rich D.H., Tam J.P. Synthesis of didehydropeptides from peptides containing 3-alkylthio-amino acid residues. Tetrahedron Lett. 1975:211–212. doi: 10.1016/S0040-4039(00)71824-7. [DOI] [Google Scholar]
- 40.Srivastava V.P., Roberts M., Holmes T., Stammer C.H. Synthesis of (+/-) 2,3-methanovaline and (+/-)-2,3-methanoleucine. J. Org. Chem. 1989;54:5866–5870. doi: 10.1021/jo00286a015. [DOI] [Google Scholar]
- 41.Placidi-Rampont V. PhD Thesis. Henri Poincaré University Nancy 1; Nancy, France: 1997. Synthèse, séparation et analyse de métabolites soufrés du toluène. [Google Scholar]
- 42.Bastien C. Synthèse et identification de métabolites soufrés du toluène. Henri Poincaré University Nancy 1; Nancy, France: 2000. DEA report. [Google Scholar]
- 43.Wang S.S., Gisin B.F., Winter D.P., Makofske R., Kulesha I.D., Tzougraki C., Meienhofer J. Facile synthesis of amino-acid and peptide esters under mild conditions via cesium salts. J. Org. Chem. 1977;42:1286–1290. doi: 10.1021/jo00428a004. [DOI] [PubMed] [Google Scholar]
- 44.Chidambaran M., Sonavane S.U., de la Zerda J., Sasson Y. Didecyldimethylammonium bromide (DDAB): a universal, robust, and highly potent phase-transfer catalyst for diverse organic transformations. Tetrahedron. 2007;63:7696–7701. doi: 10.1016/j.tet.2007.05.017. [DOI] [Google Scholar]
- 45.Makosza M. Phase-transfer catalysis. A general green methodology in organic synthesis. Pure Appl. Chem. 2000;72:1399–1403. doi: 10.1351/pac200072071399. [DOI] [Google Scholar]
- 46.Sirovski F., Reichardt C., Garokhova M., Ruban S., Stoikova E. Solid liquid phase-transfer catalysis. Some models and solvent influence. Tetrahedron. 1999;55:6363–6374. doi: 10.1016/S0040-4020(99)00212-4. [DOI] [Google Scholar]
- 47.Yang H.M., Lin C.L. Phase-transfer catalyzed benzylation of sodium benzoate using aliquat 336 as catalyst in liquid-liquid system. J. Mol. Catal. A: Chem. 2003;206:67–76. doi: 10.1016/S1381-1169(03)00446-1. [DOI] [Google Scholar]
- 48.Lopez A., Moreno-Manas M., Pleixats R., Roglans A., Ezquerra J., Pedregal C. Ethyl N-(diphenylmethylene)glycinate as anionic glycine equivalent. Monoalkylation, dialkylation and Michael additions under solid-liquid phase-transfer catalysis. Tetrahedron. 1996;52:8385–8386. doi: 10.1016/0040-4020(96)00395-X. [DOI] [Google Scholar]
- 49.Herriott A.W., Picker D. Phase transfer catalysis – evaluation of catalysts. J. Am. Chem. Soc. 1975;97:2345–2349. doi: 10.1021/ja00842a006. [DOI] [Google Scholar]
- 50.Superchi S., Nardiello M., Donnoli M.I., Scafato P., Menicagli R., Rosini C. Enantioselective synthesis of the fragrance trans-magnolione under asymmetric phase transfer catalysis. C.R. Chimie. 2005;8:867–874. doi: 10.1016/j.crci.2005.02.017. [DOI] [Google Scholar]