

Abstract
Two new phenolic glycosides, 1-O-benzyl-[5-O-benzoyl-β-d-apiofuranosyl (1→2)]-β-d-glucopyranoside (1), and 4′-hydroxy-7,3′-dimethoxyflavan-5-O-β-d-gluco-pyranoside (2), together with nine known flavanones 3 −11, have been isolated from the dried whole plants of Viscum articulatum. Their structures were identified by extensive spectral analysis, especially 2D NMR techniques. Compound 9 showed weak anti-HIV-1 activity.
Keywords: Viscum articulatum, Phenolicglycoside, Flavanone
Introduction
The genus Viscum, belonging to the Loranthaceae family, is a group of semi-parasitic shrubby type plants. Some species of this genus have shown to possess medicinal functions. The most well-known is mistletoe (V. album L.), which was frequently used as an alternative cancer treatment in Europe [1]. The plant of V. articulatum Burm. f., which has long been used as a folk herb, is distributed widely in the South and Southwest of China [2]. Previous investigations on V. articulatum showed that flavonoids, triterpenoids and organic acids were the major components of this plant [3,4,5,6,7], and some of them showed the inhibition effect on superoxide anion generation by human neutrophils in response to formyl-L-methionyl-L-leucyl-L-phenylalanin (fMLP)[6].
Figure 1.
The structures of the isolated compounds 1 - 11 from V. articulatum.

3 R1 = -β-D-glucopyranosyl, R2=R3=H
4 R1 = -β-D-glucopyranosyl, R2=H, R3=OH
5R1 = -β-D-glucopyranosyl, R2=R3=OH
6R1 = -β-D-glucopyranosyl, R2=OMe, R3=OH
7 R1 = -[β-D-apiofuranosyl (1→2)]-β-D-glucopyranosyl, R2=R3=H,
8 R1 = -[cinnamoyl (1→5)-β-D-apiofuranosyl (1→2)]-β-D-glucopyranosyl, R2=R3 = H
9 R1 = -β-D-glucopyranosyl, R2=OMe, R3= -O-β-D-apiofuranosyl
10 R1 = -β-D-glucopyranosyl, R2=OMe, R3= -O-cinnamoyl (1→5)-β-D-apiofuranosyl
11 R1 = -[β-D-apiofuranosyl (1→5)-β-D-apiofuranosyl (1→2)]-β-D-glucopyranosyl, R2=R3=H
Aiming at finding bioactive secondary metabolites, we chemically studied the aerial part of V. articulatum, which led to the isolation of two new glycosides: 1-O-benzyl-[5-O-benzoyl-β-d-apiofuranosyl(1→2)]-β-d-glucopyranoside (1) and 4′-hydroxy-7,3′-dimethoxyflavan-5-O-β-d-gluco-pyranoside (2), together with nine known flavonones: pinocembrin 7-O-β-d-glucopyranoside (3) [6], 5,4′-dihydroxyflavanone-7-O-β-d-lucopyranoside (4) [6], 5,3′,4′-trihydroxyflavanone-7-O-β-d-gluco-pyranoside (5) [6], homoeriodictyol-7-O-β-d-glucopyranoside (6) [3], pinocembrin-7-O-β-d-apio-furanosyl(1→2)-β-d-glucopyranoside (7) [4], pinocembrin-7-O-[cinnamoyl (1→5)-β-d-apiofuranosyl (1→2)]-β-d-glucopyranoside (8) [6], homoeriodictyol-7-O-β-d-glucopyranoside-4′-O-β-d-apio-furanoside (9) [3], homoeriodictyol-7-O-β-d-glucopyranoside-4′-O-β-d-(5′′′-cinnamoyl)apiofuranoside (10) [4], and pinocembrin-7-O-β-d-apiofuranosyl-(1→5)-β-D-apiofuranosyl-(1→2)-β-d-glucopyranoside (11) [4]. The structures of the new compounds were determined by means of spectral analysis and the known ones were identified by comparison of their NMR data with those reported in the literature. In addition, compounds 1, 3, 6 – 8 and 10 – 11 were tested for cytotoxicity against the MDA-MB-435 and Hela cell lines and compounds 1 – 4, 6 – 11 were tested for anti-HIV activity.
Results and Discussion
Compound 1 was obtained as a pale yellow solid. Its molecular formula was determined as C25H29O11 on the basis of HR-ESI-MS data ([M − H]−, found 505.1711, calcd. 505.1709). However, initial observation of the 13C-NMR spectrum of 1 (Table 1) only showed 21 signals, assigned as one carbonyl carbon, four oxymethylene, seven oxymethines, six methines, one monooxygenated quaternary carbon, two quaternary carbons. The 1H-NMR spectrum of 1 (Table 1) displayed two sets of aromatic proton signals at δH 7.27 (d, J = 7.4 Hz, 2H), 7.20 (m, 2H) and 7.15 (m), as well as 7.94 (d, J = 7.6 Hz, 2H), 7.51 (m, 2H) and 7.65 (m). Their analysis in the HMQC spectrum revealed correlations of carbon signals at δC 127.7 (C-2 and C-6) with proton signals at δH 7.27, δC 128.1 (C-3 and C-5) with δH 7.20, δC 129.4 (C-2′′ and C-6′′) with δH 7.94, and δC 128.8 (C-3′′ and C-5′′) with δH 7.51. The chemical shifts and coupling constants of these signals, in combination with the observed 1H−1H COSY and HMBC correlations, suggested the presence of two monosubstituted aromatic rings (Figure 2). Therefore, 25 carbon signals were present in 1, which supported the molecular formula obtained from HR-ESI-MS. Furthermore, the HMBC correlations from H-2 and H-6 to methylene [δC 69.7, C(7)] as well as from H-2′′′ and H-6′′′ to carbonyl carbon [δC 165.7, C(7′′′)] indicated the presence of a benzyl and a benzoyl moiety. Besides above signals, HMQC, 1H−1H COSY and HMBC correlations led us to assign signals at δH 3.10−5.34 and δC 61.1−108.5 as a glucose and an apiose. The HMBC correlations from H−C(1′′) (δH 5.34, brs) to C(2′) (δC 76.6) identified a apiofuranosyl (1→2) glucopyranosyl linkage. The benzyl and the benzoyl moiety were located at the C(1′) of the glucose and the C(5′′) of the apiose, respectively, which was confirmed by the HMBC correlations from H−C(1′) (δ(H) 4.31, d,J = 6.9) to methylene of the benzyl and from H−C(5′′) (H−C(5′′) (δ(H) 4.20 (d, J = 11.2 Hz), 4.29 (d, J = 11.2 Hz)) to C(7′′′) of the benzoyl.
Table 1.
1H- and 13C-NMR Data of 1 (DMSO-d6).
| Position | δ(C) | δ(H) | Position | δ(C) | δ(H) |
|---|---|---|---|---|---|
| C(1) | 137.7 (s) | − | Api: | ||
| H−C(2, 6) | 127.7 (d) | 7.27 (2H, d, J = 7.4) | H−C(1′′) | 108.5 (d) | 5.34 (brs) |
| H−C(3, 5) | 128.1 (d) | 7.20 (2H, m) | H−C(2′′) | 76.3 (d) | 3.82 (d, J = 5.7) |
| H−C(4) | 127.4 (d) | 7.15 (m) | C(3′′) | 77.4 (s) | − |
| CH2(7) | 69.7 (t) | 4.47 (d,J = 11.7) | CH2(4′′) | 73.9 (t) | 3.63 (d,J = 9.4) |
| 4.77 (d,J = 11.7) | 3.91 (d,J = 9.4) | ||||
| Glc: | CH2(5′′) | 67.8 (t) | 4.20 (d, J = 11.2) | ||
| H−C(1′) | 100.3 (d) | 4.31 (d, J = 6.9) | 4.29 (d, J = 11.2) | ||
| H−C(2′) | 76.6 (d) | 3.29−3.30 | C(1′′′) | 129.7 (s) | − |
| H−C(3′) | 76.9 (d) | 3.29−3.30 | H−C(2′′′, 6′′′) | 129.4 (d) | 7.94 (2H, d, J = 7.6) |
| H−C(4′) | 70.4 (d) | 3.10−3.11 | H−C(3′′′, 5′′′) | 128.8 (d) | 7.51 (2H, m) |
| H−C(5′) | 77.2 (d) | 3.10−3.11 | H−C(4′′′) | 133.4 (d) | 7.65 (m) |
| CH2(6′) | 61.1 (t) | 3.45 (dd, 10.1, 5.1) | C(7′′′) | 165.7 (s) | − |
| 3.69 (dd, 10.1, 5.1) |
1H- and 13C-NMR data were obtained at 500 and 100 MHz, respectively.
Figure 2.

Key COSY ( ) and HMBC (→) correlations of 1.
) and HMBC (→) correlations of 1.
The 1H- and 13C-NMR data of 1 were similar to those of 2-O-β-d-apiosyl-d-glucose-1β,5′-dibenzoate [8,9]. The difference is that a methylene (δH 4.47 (d, J = 11.7 Hz), 4.77 (d, J = 11.7 Hz), δC 69.7) replaced one of the two carbonyl carbons in 1, which was determined by HMBC correlation between H−C(1′) (δH 4.31 (d, J = 6.9 Hz) and methylene C(7). The β-anomeric configuration for the glucose was determined from a large 3JH1′, H2′ coupling constant value (6.9 Hz). The β-anomeric configuration for the apiose was indicated from the anomeric signals at δC 108.5 and a small 3JH1′′, H2′′ coupling constant [10]. Since only the d-configuration is known to exist in naturally occurring glucose and apiose [11], the sugars in 1 were tentatively assigned the d-configuration. Thus, the structure of 1 was established as 1-O-benzyl [5-O-benzoyl-β-d-apiofuranosyl (1→2)]-β-d-glucopyranoside.
Compound 2 was obtained as pale yellow amorphous powder. Its molecular formula was determined as C23H27O10 on the basis of HR-ESI-MS data ([M − H]−, found 463.1615, calcd. 463.1604). The 13C-NMR spectrum (Table 2) showed the presence of twelve aromatic carbons (δC 93.6 −159.3), an oxymethine (δC 78.4), and two methylenes (δC 29.8, 20.1), indicating a flavane skeleton. In addition, the 13C-NMR spectrum displayed two methoxyl carbon signals at δC 55.8 and 56.3. Besides above data, a glucose moiety was established by the presence of signals at δH 3.41 − 4.90 and δC 71.4 − 102.0, in combination with analysis of HMQC and 1H−1H COSY spectrum. The connectivity of the glucose moiety to the flavane was designated by HMBC correlation from H−C(1′′) (δ(H) 4.90, d, J = 7.9 Hz) to C(5) (δC 158.5), which was further confirmed by NOESY correlation of anomeric proton H−C(1′′) with H−C(6).
Table 2.
1H- and 13C-NMR Data of 2 (acetone-d6).
| Position | δ(C) | δ(H) | Position | δ(C) | δ(H) |
|---|---|---|---|---|---|
| H−C(2) | 78.4 (d) | 4.87 (m) | C(4′) | 147.1 (s) | − |
| CH2(3) | 29.8 (t) | 1.95 (m) | H−C(5′) | 115.5 (d) | 6.81 (d, J = 8.1) |
| 2.13 (m) | H−C(6′) | 119.8 (d) | 6.87 (d, J = 8.1) | ||
| CH2(4) | 20.1 (t) | 2.59 (m) | MeO−(7) | 55.8 (q) | 3.78 (3H, s) |
| 2.65 (m) | MeO−(3′) | 56.3 (q) | 3.84 (3H, s) | ||
| C(5) | 158.5 (s) | − | Glc: | ||
| H−C(6) | 97.9 (d) | 6.18 (s) | H−C(1′′) | 102.0 (d) | 4.90 (d, J = 7.9) |
| C(7) | 159.3 (s) | − | H−C(2′′) | 74.7 (d) | 3.41−3.44 |
| H−C(8) | 93.6 (d) | 6.28 (s) | H−C(3′′) | 77.7 (d) | 3.48−3.51 |
| C(9) | 157.2 (s) | − | H−C(4′′) | 71.4 (d) | 3.41−3.44 |
| C(10) | 105.4 (s) | − | H−C(5′′) | 78.0 (d) | 3.48−3.51 |
| C(1′) | 134.2 (s) | − | CH2(6′′) | 62.7 (t) | 3.67 (dd, J = 10.2, 5.1) |
| H−C(2′) | 110.8 (d) | 7.03 (s) | 3.86 (m) | ||
| C(3′) | 148.2 (s) | − |
1H- and 13C-NMR data were obtained at 500 and 125 MHz, respectively.
The 1H−NMR spectrum showed that one of the aromatic rings was tetrasubstituted with two meta-coupled protons at δH 6.18 (s) and 6.28 (s) and the other trisubstituted, whose protons coupled in an ABX system at δH 6.81 (d, J=8.1 Hz), 6.87 (d, J=8.1 Hz) and 7.03 (s). The 1H- and 13C-NMR data of 2 were similar to those of 7, 3′, 4′-dimethoxyflavan-5-O-β-D-glucopyranoside [12]. The difference was a hydroxyl replaced the methoxyl at C(4′) in 2. In addition, two MeO-atoms located at C(7) and C(3′) were determined by HMBC correlations (Figure 3)of δH 3.78 (s, 3H) and 3.84 (s, 3H) with C(7) and C(3′) (δC 159.3 and 148.2), which was further confirmed by the NOESY correlations of δH 3.78 (s, 3H) with H−C(6) (6.18, s) and H−C(8) (6.28, s), and 3.84 (s, 3H) with H−C(2′) (7.03, s), respectively.
Figure 3.

Key COSY () and HMBC (→) correlations of 2.
Acid hydrolysis of 2 afforded d-glucose, which was identified by comparison of their Rf with authentic sample. The β-anomeric configuration of the glucose was determined from a large 3JH1′′, H2′′ coupling constant value (7.9 Hz). Thus, the structure of 2 was established as 4′-hydroxy-7,3′-dimethoxyflavan 5-O-β-d-glucopyranoside.
Compounds 1, 3, 6 – 8 and 10 – 11 were tested for their anti-MDA-MB-435 and anti-Hela cell line activity by the MTT method, with cisplatin as positive control, but none of these compounds exhibited activity. Compounds 1 – 4, 6 – 11 were tested for cytotoxicity against C8166 cells (CC50), and anti-HIV-1 activity was evaluated by the inhibition assay for the cytopathic effects of HIV-1 (EC50), using AZT as a positive control. Compound 9 showed weak anti-HIV activity with CC50 > 200 μg/mL, EC50 = 18.09 μg/mL. Compound 9 exerted its weak protection of HIV-1ШB inducted MT-4 host cells lytic effects with a TI > 11.06.
Table 3.
Cytotoxicity and Anti-HIV-1 Activity of Compounds 1 - 4, 6 - 11.
| Compound | Cytotoxicity, CC50 (ĉg/ml) | Anti-HIV-1 activity, EC50 (ĉg/ml) | TI |
|---|---|---|---|
| 1 | >200 | 112.05 | >1.78 |
| 2 | 170.17 | 83.57 | >2.04 |
| 3 | >200 | 78.53 | >2.55 |
| 4 | >200 | 80.54 | >2.48 |
| 6 | >200 | 79.08 | >2.53 |
| 7 | >200 | 98.96 | >2.02 |
| 8 | >200 | 116.31 | >1.72 |
| 9 | >200 | 18.09 | >11.06 |
| 10 | >200 | 100.47 | >1.99 |
| 11 | >200 | 93.09 | >2.15 |
Experimental
General
Column chromatography (CC) was performed on silica gel (100 – 200 mesh; Qingdao Marine Chemical, Inc., P.R. China) and silica gel H (10 – 40 μm, Qingdao). Fractions were monitored by TLC, and spots were visualized by heating plates spraying with 10% H2SO4 in EtOH. UV Spectra: Shimadzu 210A double-beam spectrophotometer; λmax log (ε) in nm. IR Spectra: Bio-Rad FTS-135 spectrophotometer, KBr discs; in νmax cm–1. Optical rotations: Horiba SEPA-300 spectro-polarimeter 1D- and 2D-NMR Spectra: Bruker AM-400 and DRX-500 instruments; chemical shifts δ in ppm rel. to residual solvent signals, J in Hz. ESIMS and HRESIMS: VG AutoSpec-3000 spectrometers.
Plant material
The whole plants of V. articulatum, semiparasites on the tree branches of Lithocarpus variolosus Chun, were collected in Lijiang county of Yunnan province in 2007 and verified by Prof. Yongping Yang and Prof. Zhekun Zhou. A voucher specimen (CJH 20070502-01) was deposited at Kunming Institute of Botany, Chinese Academy of Sciences, P. R. China.
Extraction and Isolation
The air-dried and powdered whole plants (1.5 kg) were extracted with 95% ethanol (3 × 10 L) for 24 h at room temperature and concentrated in vacuo to give a crude extract (85 g), which was suspended in H2O, and extracted successively with AcOEt. The AcOEt solution was evaporated, and the residue was directly subjected to column chromatography over MCI-gel CHP-20P eluting with 95% ethanol. The elute from 95% ethanol (67.5 g) was concentrated in vacuo and subjected to column chromatography over silica gel (200 − 300 mesh) eluting with petroleum ether and acetone step gradients to afford fractions A − E. Fraction C was repeatedly subjected to Sephadex LH − 20 and column chromatography over silica gel. Further purification with RP-18 yielded compound 1 (6 mg), and repeated column chromatography over silica gel eluting with CHCl3−MeOH (9:1) yielded compound 2 (11 mg), 3 (300 mg), 4 (5 mg), 5 (2 mg), 6 (2.1 g), 7 (1.5 g), 8 (30 mg), 9 (14 mg), 10 (30mg), 11 (38mg).
Compound
1: pale yellow solid; 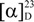 −50.0 (c 1.50, MeOH); UV (MeOH): 201 (0.58), 227(0.29), 274(0.04). IR (KBr): 3396, 2917, 1717, 1453, 1365, 1276, 1026, 825, 762, 718cm−1; 1H- and 13C-NMR: see Table 1. HR-ESI-MS (neg.) m/z 505.1711 (calcd for C25H29O11 [M − H]−, 505.1709).
−50.0 (c 1.50, MeOH); UV (MeOH): 201 (0.58), 227(0.29), 274(0.04). IR (KBr): 3396, 2917, 1717, 1453, 1365, 1276, 1026, 825, 762, 718cm−1; 1H- and 13C-NMR: see Table 1. HR-ESI-MS (neg.) m/z 505.1711 (calcd for C25H29O11 [M − H]−, 505.1709).
Compound
2: pale yellow amorphous powder; −31.4 (c 0.18, MeOH); UV (MeOH): 204(0.64), 280(0.05). IR (KBr): 3424, 2925, 1616, 1597, 1494, 1454, 1109, 821 cm−1; 1H- and 13C-NMR: see Table 2. HR-ESI-MS (neg.) m/z 463.1615 (calcd for C23H27O10 [M − H]−, 463.1604).
Acid Hydrolysis of 2.
A solution of 2 (8 mg) in 2 M HCl (3 mL) was heated in a water bath at 70 °C for 6 h. After cooling, the reaction mixture was neutralized with NaHCO3 and extracted with CHCl3. Through TLC comparison with an authentic sample using CHCl3-MeOH (8:2) as a developing system, D-glucose was detected in the water layer (Rf = 0.16).
Cytotoxicity Assay
Cytotoxic activity was tested by MTT method with cisplatin as positive control [13] [14]. MDA-MB-435 and Hela cells were plated in the 96-well plate at a cell density of 5,000 cells per well and incubate at 37 °C for 24 h before treatment and continuously exposed to various concentrations of compounds. After 72 hours incubation, cell proliferation was analyzed by Cell Proliferation Kit I (MTT) according to the manufacturer’s instructions. The optical density of the wells was measured with a microplate reader at 570 nm. All assays were done in triplicate.
Anti-HIV-1 Assay
Cytotoxicity against C8166 (CC50) was assessed using the MTT method, and anti-HIV-1 activity was evaluated by the inhibition assay for the cytopathic effects of HIV-1 (EC50), with AZT as a positive control [15]. The assays included cytotoxicity in C8166 and MT-4 cells, inhibition of syncytium formation in HIV-1ШB-infected C8166 cells, and effect in protecting HIV-1ШB-infected MT-4 host cells from lytic effects in vitro.
Acknowledgements
This research is supported by Ford Foundation (600704601411) and the West Light Foundation of The Chinese Academy of Sciences (awarded to Xiao-Li Li).
Footnotes
Sample Availability: Samples of the compounds 1−11 are available from the authors.
References
- 1.Seifert G., Jesse P., Laengler A. Molecular mechanisms of mistletoe plant extract-induced apoptosis in acute lymphoblastic leukemia in vivo and in vitro. Cancer Lett. 2008;264:218–228. doi: 10.1016/j.canlet.2008.01.036. [DOI] [PubMed] [Google Scholar]
- 2.Kiu H. S., Ling Y. R. Flora Reipublicae Popularis Sinicae. Volume 24. Science Press; Beijing, P.R. China: 1988. pp. 147–156. [Google Scholar]
- 3.Wang X. L., Li M. R., Li L. Q. The constituents of Viscum articulatum (I) Huaxi Yaoxue Zazhi. 1990;5:63–68. [Google Scholar]
- 4.Wang X. L., Li L. Q., Li M. R. The constituents of Viscum articulatum (II) Huaxi Yaoxue Zazhi. 1992;7:71–76. [Google Scholar]
- 5.Wang X. L., Li L. Q., Li M. R. The constituents of Viscum articulatum (III) Huaxi Yaoxue Zazhi. 1995;10:1–3. [Google Scholar]
- 6.Leu Y. L., Kuo S. M., Hwang T. L., Chiu S. T. The Inhibition of Superoxide Anion Generation by Neutrophils from Viscum articulactum. Chem. Pharm. Bull. 2004;52:858–860. doi: 10.1248/cpb.52.858. [DOI] [PubMed] [Google Scholar]
- 7.Ray S., Thakur T. N., Ghosh A., Barua A. K. Chemical Investigation of Viscum articulatum. J. Indian Chem. Soc. 1984;61:727–728. [Google Scholar]
- 8.Bowden B. F., Collins D. J. Confirmation of the Structure of an Apiosylglucose Dibenzoate from Daviesia latifolia by Two-Dimensional NMR Techniques. J. Nat. Prod. 1988;51:311–313. doi: 10.1021/np50056a020. [DOI] [Google Scholar]
- 9.Hansson B., Johansson I., Lindberg B. A Disaccharide Dibenzoate from Daviesia latifolia. Acta Chem. Scand. 1966;20:2358–2362. doi: 10.3891/acta.chem.scand.20-2358. [DOI] [Google Scholar]
- 10.Kitagawa I., Hori K., Sakagami M., Hashiuchi F., Yoshikawa M., Ren J. Saponin and Sapogenol. XLIX. On the Constituents of the Roots of Glycyrrhiza inflate Batalin from Xinjiang, China. Characterization of Two Sweet Oleanane-Type Triterpene Oligoglycosides, Apioglycyrrhizin and Araboglycyrrhizin. Chem. Parm. Bull. 1993;41:1350–1357. doi: 10.1248/cpb.41.1350. [DOI] [PubMed] [Google Scholar]
- 11.Hou C. C., Lin S. J., Cheng J. T., Hsu F. L. Bacopaside III, Bacopasaponin G, and Bacopasides A, B, and C from Bacopa monniera. J. Nat. Prod. 2002;65:1759–1763. doi: 10.1021/np020238w. [DOI] [PubMed] [Google Scholar]
- 12.Ghosal S., Jaiswal D. K., Singh S. K., Srivastava R. S. Dichotosin and Dichotosinin, Two Adaptogenic Glucosyloxy Flavans from Hoppea dichotoma. Phytochemistry. 1985;24:831–833. doi: 10.1016/S0031-9422(00)84903-1. [DOI] [Google Scholar]
- 13.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Meth. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 14.Huang Y. J., Wang J. F., Li G. L., Zheng Z. H., Su W. J. Antitumor and antifungal activities in endophytic fungi isolated from pharmaceutical plants Taxus mairei, Cephalataxus fortunei and Torreya grandis. FEMS Immunol. Med. Microbiol. 2001;31:163–167. doi: 10.1111/j.1574-695X.2001.tb00513.x. [DOI] [PubMed] [Google Scholar]
- 15.Wang J. H., Tam S. C., Huang H., Ouyang D. Y., Wang Y. Y., Zheng Y. T. Site-directed PEGylation of trichosanthin retained its anti-HIV activity with reduced potency in vitro. Biochem. Biophys. Res. Commun. 2004;317:965–971. doi: 10.1016/j.bbrc.2004.03.139. [DOI] [PubMed] [Google Scholar]