

Abstract
Three new compounds: 2R,3R-pterosin L 3-O-β-D-glucopyranoside (1), β-D-xylopyranosyl(1→2)-7-O-benzoyl-β-D-glucopyranoside (2) and 4-O-benzoyl-β-D-xylo-pyranosyl(1→2)-7-O-benzoyl-β-D-glucopyranoside (3), together with nine known compounds, were isolated from the ethyl acetate extract of Pteris ensiformis. 5-[2-Hydroxyethylidene]-2(5H)-furanone (4), which had been synthesized, was isolated from natural sources for the first time. The structures of all isolated compounds were determined on the basis of mass and spectroscopic evidence. Compound 1 and pterosin B (5) show cytotoxicity against HL 60 cells (human leukemia) with the IC50 values of 3.7 and 8.7 μg/mL, respectively.
Keywords: Pteris ensiformis, pterosin B, human leukemia
Introduction
For hundreds of years Pteris ensiformis Burm. (Pteridaceae) has been one of the most popular constituents of herbal beverages in Taiwan [1]. Some medium polarity extracts of different Pteris species show effective antitumor and antibacterial activity [2]. Chemical investigation of more than 30 species of the Pteridaceae has been reported [3]. Phytochemical investigations on the Pteris genus led to isolation of various phenolic compounds [4], flavonol glycosides [5,6], kauranes [7], and pterosin-sesquiterpenes [8,9]. In bioactivity reports, for example, pterosin B, one of the main pterosins found in the genus Pteris, exhibits potent cytotoxic activity against HL 60 (human leukemia) cells [10,11].
Previously, we identified several antioxidant compounds from the aqueous extract of P. ensiformis and found that caffeic acid derivates and other phenolics attribute to their antioxidant activity [12]. In a large scale screening of crude extracts of Formosan plants, we also found that the whole plant ethyl acetate extract of P. ensiformis showed effective inhibition against HL 60 cells with IC50 = 41.6 μg/mL. Further chemical investigation using bioactivity-guided fractionation led to the isolation of three new compounds: 2R,3R-pterosin L 3-O-β-D-glucopyranoside (1), β-D-xylopyranosyl(1→2)-7-O-benzoyl-β-D-glucopyranoside (2), 4-O-benzoyl-β-D-xylopyranosyl(1→2)-7-O-benzoyl-β-D-gluco-pyranoside (3), together with nine known compounds: 5-[2-hydroxyethylidene]-2(5H)-furanone (4), pterosin B (5), β-D-glucopyranosyl benzoic acid ester (6), benzoic acid (7), 5-O-coumaroylquinic acid (8), coumaric acid (9), cyclolaudenol (10), β-sitosterol-3-O-β-D-glucoside (11), and β-D-sitosterol (12), of which compounds 4, 6, 8–10 are identified for the first time from the genus Pteris (Figure 1). The structural elucidation of 1-4 and the cytotoxicity inhibition properties of the isolates are described herein.
Figure 1.

Structures of compounds 1-10.
Results and Discussion
Structure Determinations of Isolated Compounds
The HR ESI-MS of the new compound 1 showed a sodium adduct molecular ion at m/z 449.1787 [M+Na]+, corresponding to the molecular formula C21H30O9Na (calcd. 449.1782). The IR spectrum indicated the presence of a carbonyl group (1689 cm-1). Characteristic 1H-NMR signals, including one methyl group at δ 1.21 (3H, s, C-10), two aromatic methyl groups attaching to C-5 and C-7 at δ 2.48 (3H, s) and 2.66 (3H, s), two coupled methylenes of a hydroxyethyl group at δ 3.01 (2H, t, J = 8.4 Hz) and 3.61 (2H, t, J = 8.4 Hz ), one hydroxymethyl group at δ 3.37 (2H, AB q, J = 12.1 Hz), one allylic oxygenated methylene at δ 4.97 (1H, s), and one aromatic proton at δ 7.53 (1H, s), indicated the presence of a pterosin-sesquiterpene skeleton [13]. Furthermore, the configuration of the anomeric position was judged to be β from a large 3JH1′,H2′ coupling constant (J = 7.6 Hz) [14].
The assignments of 13C-NMR were confirmed by 2D NMR techniques. The 1H-detected heteronuclear multiple bond connectivity (HMBC) spectrum showed the correlations of H-12/C-6, H-12/C-4, H-15/C-6, H-15/C-8, H-14/C-13, H-13/C-6, H-4/C-9, H-3/C-9, H-3/C-2, H-10/C-1, H-10/C-2, and H-10/C-11, which confirmed 1 as a pterosin-type sesquiterpene. In addition, the HMBC correlation of H-3/C-1′ corresponds to the linkage between the pterosin moiety and the β-glucopyranose (d, J = 7.6 Hz) (Figure 2). The acid hydrolysis of 1 gave the aglycone and D-glucopyranose, which was confirmed by comparison of the 13C-NMR spectra. The absolute configuration at C-2 and C-3 was deduced from the similarity of the CD spectrum {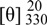 +19800 (MeOH)} of 2R,3R-pterosin L [13]. Additionally, this configuration (2R,3R) was confirmed by NOESY correlations of H-10/H-3 and H-11/H-1′ (Figure 2). Thus, the structure of compound 1, 2R,3R-pterosin L 3-O-β-D-glucopyranoside, was determined.
+19800 (MeOH)} of 2R,3R-pterosin L [13]. Additionally, this configuration (2R,3R) was confirmed by NOESY correlations of H-10/H-3 and H-11/H-1′ (Figure 2). Thus, the structure of compound 1, 2R,3R-pterosin L 3-O-β-D-glucopyranoside, was determined.
Figure 2.

NOESY and HMBC correlations of compound 1.
The molecular formula of 2, C18H24O11, were established by HR ESI-MS ([M+Na]+ m/z 439.1214, calcd. 439,1216). The 1H- and 13C-NMR spectra of 2 and 3 (Table 1) revealed the presence of glucopyranosyl and xylopyranosyl moieties [15]. The β-anomeric configuration for both glucopyranose and xlyopyranose were determined from a large 3JH1′H2′ coupling constant (7.6 Hz). Besides the signals of sugars, the 1H-NMR spectrum of 2 showed a set of proton signals due to a monosubstituted aromatic ring at δH 8.09 (2H, dd, 8.3, 1.4 Hz), 7.50 (2H, td, 8.3, 1.4 Hz) and 7.63 (1H, tt, 8.3, 1.4 Hz), in combination with the signal for a carbonyl carbon at δC 166.3 in the 13C-NMR spectrum, suggesting one benzoyl moiety in 2. The connection of the structural units was determined by HMBC data (Table 1). The β-D-glucopyranose was found to be connected to C-7 of the benzoyl moiety by observation of the HMBC correlation between δH 5.84 (Glc-H-1) and δC 166.3 (benzoyl-C-7). The structure of the sugar chain was assigned by a combination of COSY, HMQC, and HMBC experiments. Starting from the anomeric protons of each sugar unit, all of the proton signals could be identified using a COSY experiment. On the basis of assignments of all proton resonances, the linked carbon atoms were confirmed by HMQC experiment. In 1H-NMR, the anomeric proton of xylose appears at ca. δ 5.0. The high-field anomeric proton signal of the xylose unit at δ 4.50 indicated a sugar-sugar linkage. The down-field shift of C-2′ (δ 83.7) compared to the C-2′ signal of 6 (δ 74.0) indicated that the xylosyl residue was connected to C-2′ of the glucose ring. This linkage was confirmed by the correlation at δ 4.50/83.7 between H-1″ and C-2′ in the HMBC spectrum. Thus, the structure of 2 was determined to be β-D-xylopyranosyl(1→2)-7-O-benzoyl-β-D-glucopyranoside.
Table 1.
1H-NMR (400 MHz) and 13C-NMR (100 MHz) spectral data a of compounds 2 and 3 in CD3OD (δ in ppm, J in Hz).
| Position | 1H-NMR | 13C-NMR | ||
|---|---|---|---|---|
| 2 | 3 | 2 | 3 | |
| 1 | 131.0 | 131.0 | ||
| 2 | 8.09 (1H, dd, 8.3, 1.4) | 8.11 (1H, dd, 8.3, 1.4) | 131.0 | 131.0 |
| 3 | 7.50 (1H, td, 8.3, 1.4) | 7.50 (1H, td, 8.3, 1.4) | 129.6 | 129.5 |
| 4 | 7.63 (1H, tt, 8.3, 1.4) | 7.62 (1H, tt, 8.3, 1.4) | 134.6 | 134.4 |
| 5 | 7.50 (1H, td, 8.3, 1.4) | 7.50 (1H, td, 8.3, 1.4) | 129.6 | 129.5 |
| 6 | 8.09 (1H, dd, 8.3, 1.4) | 8.11 (1H, dd, 8.3, 1.4) | 131.0 | 131.0 |
| 7 | 166.3 | 166.4 | ||
| 7-O-β-glucoside | ||||
| 1′ | 5.84 (1H, d, 8.0) | 5.86 (1H, d, 8.0) | 94.8 | 94.7 |
| 2′ | 3.69 (1H, m) | 3.73 (1H, m) | 83.7 | 83.4 |
| 3′ | 3.68 (1H, m) | 3.72 (1H, m) | 77.5 | 77.6 |
| 4′ | 3.46 (1H, m) | 3.49 (1H, m) | 70.6 | 70.7 |
| 5′ | 3.45 (1H, m) | 3.48 (1H, m) | 78.8 | 78.9 |
| 6′a | 3.86 (1H, dd, 12.3, 2.1) | 3.87 (1H, dd, 12.3, 2.1) | 62.2 | 62.2 |
| 6′b | 3.71 (1H, dd, 12.3, 8.4) | 3.73 (1H, dd, 12.3, 8.4) | ||
| 2′-O-β-xylosyl | ||||
| 1″ | 4.47 (1H, d, 7.6) | 4.63 (1H, d, 7.6) | 106.8 | 106.5 |
| 2″ | 3.15 (1H, dd, 8.8, 7.6) | 3.31 (1H, m) | 75.7 | 75.8 |
| 3″ | 3.43 (1H, t, 8.8) | 3.70 (1H, m) | 77.5 | 74.8 |
| 4″ | 3.30 (1H, m) | 4.77 (1H, ddd,11.2, 9.6, 5.2) | 70.9 | 73.4 |
| 5″ax. | 2.29 (1H, dd, 11.2, 9.6) | 3.16 (1H, dd, 11.2, 9.6) | 67.1 | 63.7 |
| 5″equ. | 3.39 (1H, dd, 11.2, 4.8) | 3.68 (1H, dd, 11.2, 4.8) | ||
| 5″-O-β-benzoyl | ||||
| 1‴ | 130.9 | |||
| 2‴ | 8.00 (1H, dd, 8.3, 1.4) | 130.6 | ||
| 3‴ | 7.46 (1H, td, 8.3, 1.4) | 129.6 | ||
| 4‴ | 7.60 (1H, tt, 8.3, 1.4) | 134.7 | ||
| 5‴ | 7.46 (1H, td, 8.3, 1.4) | 129.6 | ||
| 6‴ | 8.00 (1H, dd, 8.3,1.4) | 130.6 | ||
| 7‴ | 167.3 | |||
| Key HMBC | H-1′/C-7, H-1″/C-2′ | H-1′/C-7, H-1″/C-2′, H-4″/C-7‴ | ||
a Assignments were confirmed by coupling constants,1H-1H COSY, NOESY, HMQC and HMBC analysis.
The molecular formula of 3, C25H28O12, was deduced from its HRESIMS (m/z 543.1475 [M+Na]+, calcd. 543.1473). The 1H- and 13C-NMR data of 3 (Table 1) were comparable to those of 2. Extra 13C- NMR signals at 129.6 (2C), 130.6 (2C), 130.9, 134.7, 167.3 suggested an additional benzoyl group. The connection to C-4″ of the β-D-xylopyranosyl moiety was deduced from the HMBC correlation between δH 4.29 (xyl-H-4″) and δC 167.8 (benzoyl-C-7‴). Based on the above results, the structure of 3 was established as 4-O-benzoyl-β-D-xylopyranosyl(1→2)-7-O-benzoyl-β-D-glucopyranoside.
Glycosides with β-D-xylopyranosyl(1→2)-O-β-D-glucopyranoside moieties were isolated from this family plant, Pteridium esculentum [16]. This represents the second time these special disaccharide analogues are found in this family.
In this study 5-[2-hydroxyethylidene]-2(5H)-furanone (4) was isolated from a natural source for the first time, although it had been already synthesized by Siegel and Bruckner [17]. The structure of 4 was readily identified by comparison of its spectroscopic data (1H-NMR, IR, and mass spectrometry data) [15]. We further confirmed its 13C-NMR assignments by 1H-1H COSY, HMQC, and HMBC experiments (Figure 3).
Figure 3.

COSY and HMBC correlations of compound 4.
The structures of the known isolates pterosin B (5) {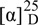 : -16.0o (c 0.20, MeOH)} [18], β-D-glucopyranosyl benzoic acid ester (6) [19], benzoic acid (7) [19], 5-O-coumaroylquinic acid (8) {: -30.5o (c 0.22, MeOH)} [20], coumaric acid (9) [21], cyclolaudenol (10) {: +23.5o (c 0.25, MeOH)} [22], β-sitosterol-3-O-β-D-glucoside (11) {: -50.1° (c 0.10, MeOH)} [23], and β-D-sitosterol (12) {: -20.0o (c 0.18, MeOH)} [23], were determined by comparison with their spectroscopic data as reported in the corresponding literature.
: -16.0o (c 0.20, MeOH)} [18], β-D-glucopyranosyl benzoic acid ester (6) [19], benzoic acid (7) [19], 5-O-coumaroylquinic acid (8) {: -30.5o (c 0.22, MeOH)} [20], coumaric acid (9) [21], cyclolaudenol (10) {: +23.5o (c 0.25, MeOH)} [22], β-sitosterol-3-O-β-D-glucoside (11) {: -50.1° (c 0.10, MeOH)} [23], and β-D-sitosterol (12) {: -20.0o (c 0.18, MeOH)} [23], were determined by comparison with their spectroscopic data as reported in the corresponding literature.
Cytotoxic activity of Isolated Compounds
In previous studies on plants of this genus, illudin-series compounds were reported as precursors of pterosins. In a recent report [24], a cytokinetic experiment with HL 60 cells indicated that illudin S exerts a primary effect on DNA synthesis. Illudin S could cause a complete block at the G1-S phase interface of the cell cycle. However, a series of illudin compounds were regarded as anti-cancer and/or carcinogenic substances [11], and the conflict in these biological results suggested that the safety issue needs to be concerned when using extracts from ferns from the family Pteridaceae. The pterosin metabolites were studied for their bioactivity, e.g. antimicrobial and antitumor activity, and displayed less toxicity than illudins [11,25]. So far, we haven’t yet found any illudin-series compounds in this plant by using a cytotoxicity-guided fractionation method. This might explain why Taiwanese people use this plant extract as one ingredient of combination herbal beverages without serious toxic effects. However, it is an important issue to be clarified. Some pterosin glycosides have been identified [13,25], but 3-O-β-D-glucose substituted pterosins are rare and their activity has not yet been reported. All purified compounds were evaluated for the cytotoxicity toward Hep G2 (human liver cancer), A549 (human lung carcinoma), MDA-MB-231 (breast carcinoma), MCF-7 (breast carcinoma), Ca9-22 (human oral squamous carcinoma), and HL 60 (human leukemia) cell lines. Among them, compound 1 and pterosin B (5) showed selective activity against HL 60 human leukemia cancer cells with the IC50 values of 3.7 and 8.7 μg/mL, respectively. Pterosins are inactive to antioxidant assays, but play an important role to action of cytotoxicity to cancer cell lines.
Experimental
General
Optical rotations were measured on a JASCO DIP-370 digital polarimeter. UV spectra were obtained on a Hitachi 220-20 spectrophotometer. IR spectra were measured on a Hitachi 260-30 spectrophotometer. 1H-NMR and 13C-NMR spectra were recorded on a Varian Inova 500, Varian Unity Plus 400, or Varian Gemini 200 spectrometers using TMS as internal standard. Chemical shifts were reported in parts per million (δ), and coupling constants (J) were expressed in Hertz. LR EI MS were collected on a Bruker APEX II mass or a Quattro GC-MS spectrometer having a direct inlet system. LR ESI-MS and HR ESI-MS were measured on a Bruker APEX II mass spectrometer. Circular dichroism was measured on Jasco J-610. Silica gel 60 (230-400 mesh, Merck) was used for column chromatography. Shimadzu LC-10AT pumps, a SPD-10A UV-vis detector, Hypersil ODS 5 μm (250 x 4.6 mm i.d.), and preparative ODS 5 μm (250 x 21.2 mm i.d.) columns were employed for the HPLC analysis or separation. Pre-coated silica gel plates (Merck, Kieselgel 60 F254, 0.25 mm) were used for preparative thin layer chromatography. TLC was performed on silica gel plates (Merck), spots were detected under UV (254 nm, 366 nm) and by spraying with 50% aqu. H2SO4 followed by heating on a hot plate.
Plant Material
Plant material of Pteris ensiformis Burm. was collected from Taitung District Agricultural Improvement Station in Taitung, Taiwan, in December, 2006. The material was identified by Dr. Ming-Hong Yen (Associate Professor of the Graduate Institute of Natural Products, Kaohsiung Medical University). A voucher specimen (PE001) was deposited in the Graduate Institute of Natural Products, Kaohsiung Medical University, Kaohsiung, Taiwan.
Extraction and Isolation
Dried whole plants of P. ensiformis (2.1 kg) were cut into small pieces, extracted with EtOAc (20 L×3) and concentrated to a volume (ca. 2 L) and partitioned with n-hexane (2L×3). The EtOAc layer was chromatographed on silica gel (2600 g) with different eluents (each 1.5 L, n-hexane-EtOAc, 1:0, 1:1, 9:1 and CHCl3-MeOH, 20:1, 10:1, 8:1, 6:1, 4:1) to get 8 frs (P1-8). Crystalline fr. P4 (2.9 g) was filtered and washed with MeOH to give 12 (90 mg, TLC: n-hexane-EtOAc 1:1, Rf = 0.4). Fr. P5 (2.2 g) was chromatographed over silica gel (20 g) using mixtures of n-hexane-EtOAc (each 0.5 L, n-hexane-EtOAc, 3:2, 1:1, 1:4) to afford 10 (80 mg, TLC: CHCl3 100%, Rf = 0.5) and 9 (13 mg, TLC: CHCl3-MeOH 100:1, Rf = 0.3). Fr. P7 (9.2 g) was rechromatographed over silica gel (1400 g) using mixtures of CHCl3-MeOH (each 1.0 L, CHCl3-MeOH, 30:1, 20:1, 10:1) to obtain subfrs (P7.1-10). Subfr. P7-3 (102 mg) was further purified by repeated silica gel chromatography (CHCl3-MeOH 20:1) to yield 1 (15 mg, TLC: CHCl3-MeOH 20:1, Rf = 0.4), 5 (20 mg, TLC: CHCl3-MeOH 20:1, Rf = 0.5), and 6 (10 mg, TLC, CHCl3-MeOH 10:1, Rf = 0.6). Subfr. P7-5 (2.0 g) using (each 0.5 L, CHCl3-MeOH, 20:1, 10:1) mixtures for elution to afford 11 (120 mg, TLC: CHCl3-MeOH 20:1, Rf = 0.4), 7 (20 mg, TLC: CHCl3-MeOH 20:1, Rf = 0.5), and 8 (9 mg, TLC: CHCl3-MeOH 10:2, Rf = 0.6). Crystalline subfr. P7-7 (2.5 g) was filtered and washed with MeOH-Water (1:1) to give 3 (1.2 g, TLC: CHCl3-MeOH 8:1, Rf = 0.6). Subfr. P7-8 (100 mg) was further separated by preparative reverse-phase HPLC (MeOH-H2O, 50:50, flow rate = 3.5 mL/min) to give 4 (5 mg, tR = 23.0 min), 2 (12 mg, tR = 41.0 min) and 3 (60 mg, tR = 52.0 min).
2R,3R-Pterosin L 3-O-β-D-glucopyranoside (1): brown amorphous powder; : -38.1o (c 0.31, MeOH); CD : +19800 (MeOH); UV: λmax (log ε): 260 (3.90), 219 (4.53) nm; IR (Neat): υmax = 3400, 1689 cm-1; 1H-NMR (CD3OD, 400 MHz): ppm δ 7.53 (1H, s, H-4), 4.97 (1H, s, H-3), 3.61 (2H, t, J = 8.4, H-14), 3.37 (2H, AB q, J = 12.1, H-11), 3.01 (2H, t, J = 8.4, H-13), 2.66 (3H, s, H-15), 2.48 (3H, s, H-12), 1.21 (3H, s, H-10); sugar signals: ppm δ 4.66 (1H, d, J = 7.6, H-1′), 3.93 (1H, dd, J = 12.3, 2.1 H-6′a), 3.75 (1H, dd, J = 12.3, 8.1, H-6′b), 3.40 (1H, m, H-3′), 3.38 (1H, m, H-2′), 3.31 (1H, m, H-4′), 3.22 (1H, m, H-5′); 13C-NMR (CD3OD, 100 MHz): ppm δ 208.5 (C-1), 151.3 (C-9), 145.1 (C-4), 137.4 (C-5), 136.7 (C-6), 131.6 (C-7), 125.2 (C-8), 83.8 (C-3), 65.9 (C-11), 60.3 (C-14), 56.1 (C-2), 31.8 (C-13), 20.2 (C-12), 17.5 (C-10), 12.9 (C-15), sugar signals: ppm δ 104.4 (C-1′), 77.0 (C-5′), 76.0 (C-3′), 74.3 (C-2′), 70.4 (C-4′), 61.5 (C-6′); LR ESI-MS m/z 449.0 [M+Na]+; HR ESI-MS m/z 449.1787 (calcd. for C21H30O9Na, 449.1789).
β-D-Xylopyranosyl(1→2)-7-O-benzoyl-β-D-glucopyranoside (2): white amorphous powder; 1H-NMR (400 MHz, CD3OD) and 13C-NMR (CD3OD, 100 MHz) see Table 1; LR ESI-MS m/z: 439 [M+Na]+, HR ESI-MS m/z: 439.1214 (calcd. for C18H24O11Na+, 439.1216).
4-O-Benzoyl-β-D-xylopyranosyl(1→2)-7-O-benzoyl-β-D-glucopyranoside (3): white amorphous powder; 1H-NMR (400 MHz, CD3OD) and 13C-NMR (CD3OD, 100 MHz) see Table 1; LR ESI-MS m/z: 439 [M+Na-benzoyl]+, 543 [M+Na]+; HR ESI-MS m/z: 543.1475 (calcd. for C25H28O12Na+, 543.1473).
5-[2-Hydroxyethylidene]-2(5H)-furanone (4): brown amorphous powder; : -20.7o (c 0.31, MeOH); UV (MeOH): λmax (log ε): 264 (3.83) nm; IR (Neat): υmax = 1745 cm-1; 1H-NMR (CDCl3, 400 MHz): ppm δ 7.39 (1H, d, J = 5.0 Hz, H-3), 6.24 (1H, d, J = 5.0 Hz, H-4), 5.47 (1H, t, J = 5.0 Hz, H-6), 4.53 (2H, d, J = 5.0 Hz, H-7); 13C-NMR (CDCl3, 100 MHz): ppm δ 169.5 (C-2), 120.6 (C-3), 143.6 (C-4),149.5 (C-5), 114.1 (C-6), 57.3 (C-7); LR ESI-MS m/z: 149 [M+Na]+.
Pterosin B (5): white amorphous powder; : -16.0o (c 0.20, MeOH); 1H-NMR (200 MHz, CD3OD): ppm; δ 7.09 (1H, s, H-4), 3.76 (2H, t, J = 7.4 Hz, H-13), 3.02 (2H, t, J = 7.4 Hz, H-12), 2.68 (3H, s, H-14), 2.55-2.65 (2H, m, H-3), 2.43 (3H, s, H-11), 1.27 (3H, d, J = 6.3 Hz, H-10); 13C-NMR (50 MHz, CD3OD): ppm δ 210.3 (C-1), 152.6 (C-9), 144.4 (C-5), 138.0 (C-7), 134.8 (C-6), 132.5 (C-8), 125.8 (C-4), 61.6 (C-12), 42.8 (C-2), 34.7 (C-3), 32.8 (C-13), 21.4 (C-11), 16.8 (C-10), 13.7 (C-14); LR ESI-MS m/z: 241 [M+Na] +.
β-D-Glucopyranosyl benzoic acid ester (6): white amorphous powder; 1H-NMR (200 MHz, CD3OD): ppm δ 8.09 (2H, dd, 8.3, 1.4 Hz, H-2,6), 7.50 (2H, td, 8.3, 1.4 Hz, H-3,4), 7.63 (1H, tt, 8.3, 1.4 Hz, H-1), sugar signals: ppm δ 5.73 (1H, d, J = 7.6, H-1′), 3.86 (1H, dd, J = 12.1, 2.1, H-6′a), 3.71 (1H, dd, J = 12.1, 8.1, H-6′b), 3.52 (1H, m, H-2′), 3.49 (1H, m, H-3′), 3.44 (1H, m, H-4′), 3.43 (1H, m, H-5′); 13C NMR (CD3OD, 100 MHz): ppm δ 131.0 (C-1), 130.7 (C-2), 129.6 (C-3), 134.8 (C-4), 129.6 (C-5), 130.7 (C-6), 166.7 (C-7), sugar signals: ppm δ 96.3 (C-1′), 78.0 (C-3′), 78.9 (C-5′), 74.0 (C-2′), 71.0 (C-4′), 62.3 (C-6′); LR ESI-MS: m/z 307 [M+Na]+.
Benzoic acid (7): white amorphous powder, 1H-NMR (200 MHz, CD3OD): ppm δ 8.09 (2H, dd, 8.3, 1.4 Hz, H-2,6), 7.50 (2H, td, 8.3, 1.4 Hz, H-3,4), 7.63 (1H, tt, 8.3, 1.4 Hz, H-1); LR EI-MS: m/z 145 [M+Na]+.
5-O-Coumaroylquinic acid (8): yellow amorphous powder; : -30.5o (c 0.22, MeOH): 1H-NMR (400 MHz, CD3OD): ppm δ 7.65 (d, J 16.0 Hz, H-7′), 7.48 (d, J 8.6 Hz, H-2′,6 ′), 6.84 (d, J 8.6 Hz, H-3′,5 ′), 6.34 (d, J = 16.0 Hz, H-8′), 5.33 (m, H-5), 3.90 (d, J = 3.1 Hz, H-3), 3.77 (dd, J = 8.6, 3.0 Hz, H-4), 2.01-2.29 (m, H-2, -6); 13C-NMR (100 MHz, CD3OD): ppm δ 177.64 (C-7), 169.38 (C-9′), 161.48 (C-4′), 147.34 (C-7′), 131.69 (C-2′,6′), 127.70 (C-1′), 117.31 (C-3′,5 ′), 115.68 (C-8′), 76.60 (C-1), 73.82 (C-5), 72.43 (C-3), 72.43 (C-3), 71.66 (C-4), 39.14 (C-6), 38.56 (C-2); LR ESI-MS: m/z 339 [M+H]+.
Coumaric acid (9): yellow amorphous powder; 1H-NMR (200 MHz, CD3OD): ppm δ 7.57 (1H, d, J =16.0 Hz, H-7) , 6.28 (1H, d, J =16.0 Hz, H-8), 7.44 (2H, d, J =8.0 Hz, H-2, -6), 6.80 (2H, d, J = 8.0 Hz, H-3,-5); LR ESI-MS: m/z 165 [M+H]+.
Cyclolaudenol (10): amorphous powder; : +23.5o (c 0.25, MeOH); 1H-NMR (400 MHz, CDCl3): ppm δ 4.67 (2H, m, H-26), 3.28 (1H, dd, J = 11.5, 4.5 Hz, H-3), 2.10 (1H, pseudosextet, J = 7.0 Hz, H-24), 1.96 (1H, m, H-11α), 1.87 (1H, m, H-7β), 1.75 (1H, m, H-2α), 1.64 (3H, brs, H-27), 1.60 (2H, m, H-12), 1.58 (1H, m, H-6α), 1.56 (m, H-1α, H-2β, H-17,), 1.50 (1H, m, H-8), 1.42 (1H, m, H-23a), 1.36 (1H, m, H-20), 1.33 (2H, m, H-16a, H-22a), 1.28 (4H, m, H-5, H-7α, H-15a,b), 1.24 (1H, m, H-1β), 1.10 (H, m, H-11β, H-16b, H-23a), 1.00 (3H, d, J = 7.0 Hz, H-31), 0.97 (3H, s, H-28), 0.96 (3H, s, H-18), 0.92 (H, m, H-22b), 0.89 (3H, s, H-30), 0.86 (3H, d, J = 6.5 Hz, H-21), 0.81 (3H, s, H-29), 0.80 (1H, m, H-6β), 0.55, 0.33 (each 1H, d, J = 4.5 Hz, H-19); 13C-NMR (CDCl3, 100 MHz): δ 150.2 (C-25), 109.4 (C-26), 78.9 (C-3), 52.3 (C-17), 48.8 (C-13), 48.0 (C-8), 47.1 (C-5), 45.3 (C-14), 41.6 (C-24), 40.5 (C-4), 36.0 (C-20), 35.6 (C-15), 33.9 (C-22), 32.9 (C-12), 32.0 (C-1), 31.5 (C-23), 30.4 (C-2), 29.9 (C-19), 28.1 (C-7), 26.5 (C-11), 26.1 (C-10), 26.0 (C-16), 25.4 (C-28), 21.1 (C-6), 20.1 (C-31), 20.0 (C-9), 19.3 (C-30), 18.7 (C-27), 18.3 (C-21), 18.0 (C-18), 14.0 (C-29); LR EI-MS: m/z 440 [M]+.
β-Sitosterol-3-O-β-D-glucoside (11): white amorphous powder; : -20.0o (c 0.18, MeOH); 1H-NMR (200 MHz , CDCl3): δ 5.25 (1H, d, J = 4.5 Hz, H-6), 5.12 (1H, dd, J = 12.3, 8.3 Hz, H-23), 5.02 (1H, dd, J = 12.3, 8.3 Hz, H-22), 4.35 (1H, d, J = 7.6 Hz, aromeric H), 3.45 (1H, m, H-3), 3.10-3.90 (5H, m, sugar moiety H), 1.01 (3H, s, H-19), 0.68 (3H, s, H-18), 0.94 (3H, d, J = 6.5 Hz, H-21), 0.86 (3H,t, J = 7.1 Hz, H-29), 0.84 (3H, s, H-27); LR EI-MS: m/z 576 [M]+.
β-D-Sitosterol (12): white amorphous powder; : -50.1° (c 0.10, MeOH); 1H-NMR (200 MHz, CDCl3): δ 5.36 (1H, br s, H-6), 5.12 (1H, dd, J = 16.1, 8.3 Hz, H-23), 5.02 (1H, dd, J = 16.1, 8.3 Hz, H-22), 1.01 (3H, s, H-19), 0.92 (3H, d, J = 6.4 Hz, H-21), 0.86 (3H, t, J = 7.0 Hz, H-29), 0.84 (3H, d, J = 6.8 Hz, H-27), 0.81 (3H, d, J = 6.8 Hz, H-26), 0.68 (3H, s, H-18); LR EI-MS: m/z 412 [M]+.
Cytotoxicity Assays
Compounds were assayed for cytotoxicity against Hep G2, A549, MCF-7, MDA-MB-231, Ca9-22 and HL 60 cell lines using the MTT method [26]. Freshly trypsinized cell suspensions were seeded in 96-well microtitre plates at densities of 5,000-10,000 cells per well and tested compounds were added from a DMSO stock solution. After 3 days in culture, attached cells were incubated with MTT (0.5 μg/ml, 1 h) and subsequently dissoluble in DMSO. The absorbance was measured at 550 nm using a microplate reader. The IC50 is the concentration of agent reduced cell growth by 50% under the experimental conditions. Results represent the mean two to three separate experiments, each performed in triplicate.
Acid hydrolysis of compound 1
A solution of compound 1 (5.0 mg) in 6% aqueous HCl (3.5 mL) was refluxed for 2 h. The reaction mixture was diluted with water and then extracted with ethyl acetate. The resulting glucose (in water) and 2R,3R pterosin L (in ethyl acetate) were identified by their corresponding 13C-NMR spectra.
Acknowledgements
We are gratefully acknowledged the financial support for the project from National Science Council and the center for resources, research & development of Kaohsiung Medical University, Taiwan, ROC. We also thank Ms. Chyi-Jia Wang and Mr. Wen-Hsiung Lu for technical assistance in NMR operation. We thank to the National Center for High-performance Computing for data survey.
Footnotes
Sample availability: Contact the authors.
References
- 1.Wu M.J., Wang L., Weng C.Y., Lian T.W. Immunomodulatory mechanism of the aqueous extract of sword brake fern (Pteris ensiformis Burm.) J. Ethnopharmacol. 2005;98:73–81. doi: 10.1016/j.jep.2004.12.031. [DOI] [PubMed] [Google Scholar]
- 2.Gong X.L., Chen Z.H., Liang N.C. Advances in study on chemical constituents and pharmacological activities of plants of genus Pteris. Zhongguo Zhong Yao Za Zhi. 2007;32:1382–1387. [PubMed] [Google Scholar]
- 3.Castillo U.F., Ojika M., Alonso-Amelot M., Sakagamia Y., Ptaquiloside Z. a new toxic unstable sesquiterpene glucoside from the neotropical bracken fern Pteridium Aquilinum Var. Caudatum. Bioorg. Med. Chem. 1998;6:2229–2233. doi: 10.1016/S0968-0896(98)00168-0. [DOI] [PubMed] [Google Scholar]
- 4.Jesudass L.L., Manickam V.S., Gopalakrishnan S. Polyphenolic glycosides of Pteris confusa T.G. walker of kothayar hills of the western ghats of south India. Res. J. Chem. Environ. 2000;4:77–78. [Google Scholar]
- 5.Salatino M.L., Prado F. Flavonoid glycosides of Pteridaceae from Brazil. J. Bio. System. Eco. 1998;26:761–769. doi: 10.1016/S0305-1978(98)00032-5. [DOI] [Google Scholar]
- 6.Tanaka N., Murakami T., Saiki Y., Chen C.M., Gomez L.D. Chemical and chemotaxonomical studies of the Pteris family and related families (Pteridaceae). XXII. Chemical studies of Pteris grandifolia L. Chem. Pharm. Bull. 1978;26:3580–3582. doi: 10.1248/cpb.26.3580. [DOI] [Google Scholar]
- 7.Woerdenbag H.J., Lutke L.R., Bos R., Stevens J.F. Isolation of two cytotoxic diterpenes from the fern Pteris multifida. Zeitschrift Naturforsch C: Biosci. 1996;51:635–638. doi: 10.1515/znc-1996-9-1006. [DOI] [PubMed] [Google Scholar]
- 8.Saito K., Nagao T., Takatsuki S., Koyama K., Natori S. The sesquiterpenoid carcinogen of bracken fern, and some analogs, from the Pteridaceae. Phytochemistry. 1990;29:1475–1479. doi: 10.1016/0031-9422(90)80104-O. [DOI] [Google Scholar]
- 9.Murakami T., Satake T., Ninomiya K., Iida H., Yamauchi K., Tanaka N., Saiki Y., Chen C.M. Chemical and chemotaxonomic studies of ferns. Part 25. Pterosin derivatives from the family Pteridaceae. Phytochemistry. 1980;19:1743–1746. doi: 10.1016/S0031-9422(00)83806-6. [DOI] [Google Scholar]
- 10.Qin B., Zhu D.-y. Review on the sesquiterpenoids from the spices of Pteridaceae. (II) - Chemical synthesis, transformation and biological activities of 1H-inden-1-one sesquiterpenoids. Huaxue Yanjiu. 2004;15:66–70. [Google Scholar]
- 11.McMorris T.C., Kelner M.J., Wang W., Estes L.A., Montoya M. A., Taetlel R. Structure-activity relationships of illudins: analogs with improved therapeutic index. J. Org. Chem. 1992;57:6876–6883. doi: 10.1021/jo00051a037. [DOI] [Google Scholar]
- 12.Chen Y.H., Chang F.R., Lin Y.J., Wang L., Chen J.F., Wu Y.C., Wu M.J. Identification of phenolic antioxidants from Sword Brake fern (Pteris ensiformis Burm.) Food Chem. 2007;105:48–56. doi: 10.1016/j.foodchem.2007.03.055. [DOI] [Google Scholar]
- 13.Kuraishi T., Murakami T., Taniguchi T., Kobuki Y., Maehashi H., Tanaka N., Saiki Y., Chen C. M. Chemical and chemotaxonomical studies of ferns. LIV. Pterosin derivatives of the genus Microlepia (Pteridaceae) Chem. Pharm. Bull. 1985;33:2305–2312. doi: 10.1248/cpb.33.2305. [DOI] [Google Scholar]
- 14.Roen A., Padron J.I., Vazquez J.T. Hydroxymethyl rotamer populations in disaccharides. J. Org. Chem. 2003;68:4615–4630. doi: 10.1021/jo026913o. [DOI] [PubMed] [Google Scholar]
- 15.Cabrita L., Andersen Q.M. Anthocyanins in blue berries of vaccinium padifolium. Phytochemistry. 1999;52:1693–1696. doi: 10.1016/S0031-9422(99)00281-2. [DOI] [Google Scholar]
- 16.Tanaka N., Yuhara H., Wada H., Murakami T., Cambie R. C., Braggins J. E. Chemical and chemotaxonomical studies of ferns. Part 82. Phenolic constituents of Pteridium esculentum. Phytochemistry. 1993;32:1037–1039. doi: 10.1016/0031-9422(93)85251-L. [DOI] [Google Scholar]
- 17.Siegel K., Bruckner R. First total synthesis of dihydroxerulin, a potent inhibitor of the biosynthesis of cholesterol. Chem. Eur. J. 1988;11:16–22. [Google Scholar]
- 18.Tanaka N., Satake T., Takahashi A., Mochizuki M., Murakami T., Saiki Y., Yang J.Z., Chen C.M. Chemical and chemotaxonomical studies of Ferns XXXIX. Chemical studies on the constituents of Pteris bella Tagawa and Pteridium aquilinum subsp wightianum (wall) Shich. Chem. Pharm. Bull. 1982;30:3640–3646. doi: 10.1248/cpb.30.3640. [DOI] [Google Scholar]
- 19.Nahrstedt A., Rockenbach J., Wray V. Phenylpropanoid glycosides, a furanone glucoside and geniposidic acid from members of the rubiaceae. Phytochemistry. 1995;39:375–378. doi: 10.1016/0031-9422(94)00906-A. [DOI] [Google Scholar]
- 20.Bergman M., Varshavsky L., Gottlieb H., Grossman S. The antioxidant activity of aqueous spinach extract: chemical identification of active fractions. Phytochemistry. 2001;58:143–52. doi: 10.1016/S0031-9422(01)00137-6. [DOI] [PubMed] [Google Scholar]
- 21.Norbæk R., Nielsen K., Kondo T. Anthocyanins from flowers of Cichorium intybus. Phytochemistry. 2002;60:357–359. doi: 10.1016/S0031-9422(02)00055-9. [DOI] [PubMed] [Google Scholar]
- 22.Cantillo-Ciau Z., Brito-Loeza W., Quijano L. Triterpenoids from Tillandsia fasciculate. J. Nat. Prod. 2001;64:953–955. doi: 10.1021/np0100744. [DOI] [PubMed] [Google Scholar]
- 23.Chang Y.C., Chang F.R., Wu Y.C. The constituents of Lindera glauca. J. Chin. Chem. Soc. 2000;47:913–920. [Google Scholar]
- 24.Kelner M.J., McMorris T.C., Beck W.T., Zamora J.M., Taetle R. Preclinical evaluation of illudins as anticancer agents. Cancer Res. 1987;47:3186–3190. [PubMed] [Google Scholar]
- 25.Kobayashi A., Egawa H., Koshimizu K., Mitsui T. Antimicrobial constituents in Pteris inaequalis. Agric. Biol. Chem. 1975;39:1851–1856. doi: 10.1271/bbb1961.39.1851. [DOI] [Google Scholar]
- 26.Sladowski D., Steer S., Clothier R.H., Balls M., Chihiro I. An improved MTT assay. J. Immunol. Meth. 1993;157:203–207. doi: 10.1016/0022-1759(93)90088-O. [DOI] [PubMed] [Google Scholar]