

Abstract
Traditional approaches to specifying a molecular mechanics force field encode all the information needed to assign force field parameters to a given molecule into a discrete set of atom types. This is equivalent to a representation consisting of a molecular graph comprising a set of vertices, which represent atoms labeled by atom type, and unlabeled edges, which represent chemical bonds. Bond stretch, angle bend, and dihedral parameters are then assigned by looking up bonded pairs, triplets, and quartets of atom types in parameter tables to assign valence terms, and using the atom types themselves to assign nonbonded parameters. This approach, which we call indirect chemical perception because it operates on the intermediate graph of atom-typed nodes, creates a number of technical problems. For example, atom types must be sufficiently complex to encode all necessary information about the molecular environment, making it difficult to extend force fields encoded this way. Atom typing also results in a proliferation of redundant parameters applied to chemically equivalent classes of valence terms, needlessly increasing force field complexity. Here, we describe a new approach to assigning force field parameters terms direct chemical perception that avoids these problems, called the SMIRKS Native Open Force Field (SMIRNOFF) format. Rather than working through the intermediary of the atomtyped graph, direct chemical perception operates directly on the unmodified chemical graph of the molecule to assign parameters. In particular, parameters are assigned to each type of force field term—e.g., bond stretch, angle bend, torsion, and Lennard-Jones—based on standard chemical substructure queries implemented via the industry-standard SMARTS chemical perception language, using SMIRKS extensions that permit labeling of specific atoms within a chemical pattern. We demonstrate the power and generality of this approach using examples of specific molecules that pose problems for indirect chemical perception, and construct and validate a minimalist yet very general force field, SMIRNOFF99Frosst. We find that a parameter definition file only ~300 lines long provides coverage of all but <0.02% of a five million molecule drug-like test set. Despite its simplicity, the accuracy of SMIRNOFF99Frosst for small molecule hydration free energies and selected properties of pure organic liquids, is similar to that of the General Amber Force Field (GAFF), whose specification requires thousands of parameters. This force field provides a starting point for further optimization and refitting work to follow.
Graphical Abstract
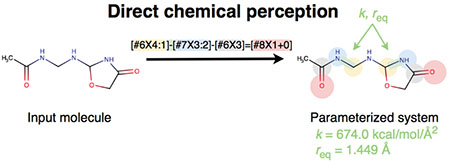
1. Introduction
Classical, all-atom molecular mechanics force fields form the basis of molecular simulations applied in diverse areas of chemistry, biochemistry, biology, drug discovery, and materials science [13, 14, 25, 49, 62, 65, 74, 76, 79]. Often, these are two-body, additive, fixed-charge force fields with the relatively simple Lennard-Jones functional form for dispersion and exclusion interactions [65, 66]. Despite this simplified representation of the physics of intermolecular interactions, such force fields have achieved remarkable successes in the calculation of molecular and material properties far beyond the simple gas phase [66], condensed phase [40, 65], and biomolecular [25, 29, 51, 63] properties used to parameterize them. Examples include blinded predictions of host-guest [58, 59, 92] and protein-ligand [2, 9, 16, 17, 46, 50, 55, 67, 68, 78] binding affinities, small molecule hydration free energies [24, 57], partition and distribution coefficients [4], and ligand binding modes [19]. Extensive retrospective tests for other properties such as dielectric constants [6, 23, 30, 61] and perturbations in protein stability [73] are also worth noting. Despite this, current force fields do not provide consistently high accuracy [11, 28, 41, 67]), including in key applications such as computer-aided drug design [68], resulting in strong interest across many research groups in developing force fields that can deliver more reliable results with comparable computational expense.
The initial development of a new general force field, i.e., one that covers a large segment of common organic chemistry and biomolecules, typically takes years of effort; as a result, subsequent releases tend to be restricted to limited adjustments, rather than full reparameterizations [25, 29, 51, 63, 75]. For example, some systematic errors can be traced to problems with parameters for particular functional groups [43, 53, 54], allowing for targeted improvements via updates to few parameters. For example, the General Amber Force Field (GAFF) parameters yielded systematic errors for alkenes which could be fixed by a minor adjustment to a subset of Lennard-Jones parameters [54]; larger errors for alcohols stemming from issues with underpolarization of the hydroxyl group were corrected via focused parameter modifications [23]. However, such adjustments represent little more than band-aids, rather than the comprehensive refitting that would be needed to fully and self-consistently update a force field based on additional experimental or quantum chemical data, or to extend it to new regions of chemical space. Indeed, core components of many general force fields can be traced to decisions made in the 1980s and early 1990s, which were often based more on chemical intuition than a systematic approach. Furthermore, general force fields have often been developed as relatively simple extensions of previously developed biomolecular force fields [80, 82, 83], instead of being developed de novo from a comprehensive dataset. (One noteworthy example to the contrary is the recent work on hierarchical atom-type definitions [38] and force fields using these definitions.) Finally, only a fraction of the suitable experimental datasets available today have been utilized to produce biomolecular force fields, so valuable opportunities to improve accuracy have gone exploited. As a result, it is far from clear that current general biomolecular force fields are fully and consistently optimized for the range of chemistries for which they provide parameters, leaving significant room to develop improved force fields through comprehensive rebuilding and reoptimization, even within the constraints imposed by current functional forms widely supported by molecular simulation packages.
While our long-term goal is to systematize and automate the force field development process so that human expertise is used to select only the functional form and input data for parameterization, as a first step, we aim to eliminate major obstacles to force field optimization that derive from the common approach of using atom types to assign force field parameters. One problem with this approach is that atom type definitions have, to date, been crafted based on some combination of chemical intuition and analysis, without any rigorous basis for determining how many atom types are necessary and sufficient. Thus, although substantial effort has been invested in developing efficient force field parameter optimization approaches, the science of atom typing is significantly less developed, and does not offer confidence that current atom-types are near optimal or avoid over- or under-fitting in any statistically robust manner. The replacement of human-annotated featurizations with fully automated ones has recently led to advances in both chemistry (e.g., DeepChem [91 ]) and image recognition (e.g., ImageNet [45]), and we see great potential in the use of such methods for force field definition and development.
Another set of problems has to do with the way atom types are used to assign parameters to molecules. In particular, most current approaches use a method we call indirect chemical perception (Figure 1) to assign the parameters to most or all of their energy terms. Indirect chemical perception involves the assignment of a single identifier—an atom type—to each atom in a molecule, based on its local chemical environment, and uses bonded pairs, tuples, or quartets of these identifiers to look up most or all of the required force field parameters (e.g., Lennard-Jones, bond-stretch, angle-bend, and torsions) in parameter tables. Thus, in indirect chemical perception, the set of atom types contains all the information required to parameterize a system.
Figure 1. Direct versus indirect chemical perception.
Indirect chemical perception (top, red arrows) processes a valence representation of the molecule to assign atom types (left red arrow) and typically retains only atom type and connectivity information for final assignment of parameters to the molecule (right red arrow), meaning that atom types must encode all of the requisite information about the chemical environment of each atom. In direct chemical perception (bottom, blue arrow) parameter assignment machinery has access to a valence representation, bond orders, and full information about the chemical environment of each atom, so all of this information can be used in assigning parameters.
This atom typing approach leads to a number of difficulties. First, the creation of a new atom type can lead to undesired proliferation of other force field terms. For example, if new hydration free energy data leads one to add a new atom type to better model Lennard-Jones interactions, this addition immediately requires the creation of parameters for all bond, angle, and torsion types involving this new atom type. Often this leads to guessing or copying from existing “parent” parameters, without any solid basis for these choices (though hierarchical atom typing approaches like those used in the “hierarchical atom type definition” (HAD) scheme can help avoid this issue [38]). Surprisingly, this also creates the potential for human error to inadvertently omit some of these parameters, introducing errors where general valence types are incorrectly used to model interactions that should have been used instead. Furthermore, this addition of many more duplicate parameters dramatically exacerbates the curse of dimensionality should one attempt to further optimize parameters to fit experimental or quantum chemical datasets following this duplication. In principle, this problem might be addressed by constraining some parent and child parameters to be equal, as in one recent study [38], but this is not standard practice, and, since the need for such constraints is not encoded in typical force field files (e.g. such as AMBER-format files), this pedigree is easily lost over time.
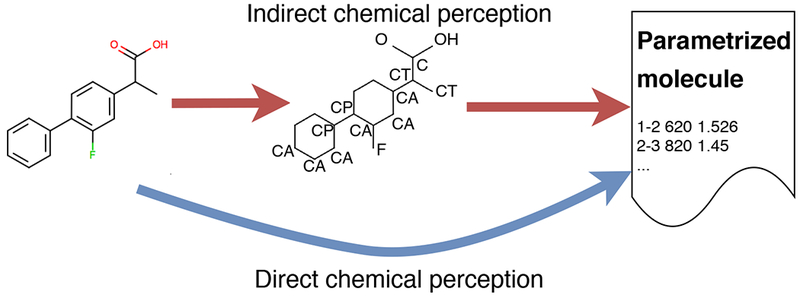
Furthermore, when indirect chemical perception is used, the need to differentiate bond-stretch or bond-torsion parameters can force the creation of new, otherwise needless, atom types, which must either be assigned Lennard-Jones parameters based on parent atom types or left as free parameters to be optimized. For example, in 3-methylenepenta-1,4-diene (Figure 2), the carbon atoms are all assigned the same Lennard-Jones parameters in GAFF, consistent with the fact that they are in similar chemical environments, with one double bond and two single bonds to another carbon or hydrogen atom. However, new atom types (see Figure 2a) had to be introduced merely to encode the fact that some bonds have single-bond character and others double-bond character, since the bond-stretch parameters are inferred from the atom types. Figure 2b,c, and d [81, 83] shows additional cases in which chemically similar atoms with identical Lennard-Jones parameters had to be assigned different atom types to allow assignment of different bond-stretch parameters to single and double bonds. Similarly, in biphenyls and related molecules, atom typing makes it difficult to recognize which bonds should be rotatable without introducing new atom types. These additional atom types, in turn, result in a further proliferation of additional bond, angle and torsion types. To apply automated parameterization machinery when many similar parameters exist, such as the 16 sets of Lennard-Jones parameters for carbon in GAFF/GAFF2 which only have three distinct values [80]), a human expert would have to designate which parameters should be constrained to be identical versus which should be allowed to vary independently.
Figure 2. Simple molecules that present challenging cases for indirect chemical perception via atom typing.
GAFF/GAFF2 atom types are indicated. (a): All carbons are sp2, but they are connected by alternating single and double bonds, forcing introduction of the ce atom type for the inner sp2 carbons to allow single and double carbon-carbon bonds to be distinguished. (b): The bridgehead aromatic carbons in biphenyl must have a single bond joining them, forcing introduction of the cp atom type which is identical to the normal aromatic carbon (ca) except that cp–cp bonds are single and thus rotatable. (c): Introduction of an additional phenyl ring, to make 1,2-diphenylbenzene, forces addition of the cq atom type, purely to prevent creation of a cp–cp (single) bond within the lower aromatic ring; thus, cp–cp and cq–cq bonds are single, but cq–cp bonds (along with bonds involving ca of any sort) are aromatic. This scheme can in principle handle even larger molecules like 1,2,3,4-tetraphenylbenzene (d) though, as discussed in the text, the massive proliferation of torsional parameters resulting from the numerous atom types employed leads to considerable potential for human error. (e): Here, GAFF/GAFF2 assigns the bridgehead bonds between five membered rings as cd–cd or cc–cc, the same as bonds within aromatic rings, and thus incorrectly makes these single bonds non-rotatable (Section 3.2.3). This problem could presumably be fixed by creating still more atom types, as done for the cases in (b) and (c). (f): A molecule that cannot be handled by indirect chemical perception based on atom types [83]; incorrect atom types are shown in boldface and will lead to misassignment of parameters.
In addition to forcing the proliferation of parameters, indirect chemical perception can drive the introduction of errors. For example, while the biphenyl cases just discussed have received careful attention to avoid incorrectly treating the bond between bridgehead carbons, this approach required a human expert to identify and solve problem cases and can fail for other systems which have not received such careful attention. For example, a similar scenario occurs for bonds between the GAFF/GAFF2 types cc–cc, cc–cd, and cd–cd, which have identical Lennard-Jones parameters and are used for carbons in non-pure aromatic systems (Figure 2e). Here, GAFF/GAFF2 give the torsions involving these single bonds a barrier height of 16.00 kcal/mol, which is identical to the aromatic bonds within the five membered rings and even higher than the 14.5 kcal/mol barrier height used for aromatic bonds in biphenyl. Thus, unexpected bridgehead atoms result in a rotatable single bond being treated as aromatic. Finally, for some molecules, it appears that consistent typing is impossible to achieve with indirect chemical perception [83]; while introducing new atom types (cp and cq) can resolve most problems in the biphenyl series of Figure 2b-d, with sufficiently complex molecules, it becomes impossible to avoid assigning a single bond as aromatic (Figure 2f) without introducing a new atom type for essentially every atom of every molecule, which quickly defeats the purpose of producing a generalizable force field.
Here, we show that the problems which result from indirect chemical perception are avoided by what we term direct chemical perception. Rather than assigning valence and nonbonded parameters based on a chemical graph comprising types atoms and undifferentiated edges, direct chemical perception processes the molecular graph, with its varied bond types, to independently assign Lennard-Jones, bond, angle, and torsion parameters directly based on the local chemical environments of the corresponding atoms, bonds, angles, and torsions in the molecule. This paradigm easily accommodates the addition of a new atom with distinct Lennard-Jones parameters without driving the creation of additional valence parameters or types. We furthermore demonstrate the implementation of direct chemical perception using the SMIRKS extension of the widely used SMILES chemical perception language (http://opensmiles.org/opensmiles.html, Figure 3 and Table 1), incorporate this method into a new force field specification format called the SMIRKS Native Open Force Field (SMIRNOFF) format, and show that this format allows the terse yet complete specification of a complete general force field, called SMIRNOFF99Frosst, derived by converting and adapting AMBER parm99 and Merck’s parm@Frosst [5]. We show that SMIRNOFF99Frosst covers comparable chemical space to GAFF/GAFF2, with similar accuracy, in initial tests. Implications for the automated construction of novel force fields are considered in the Discussion.
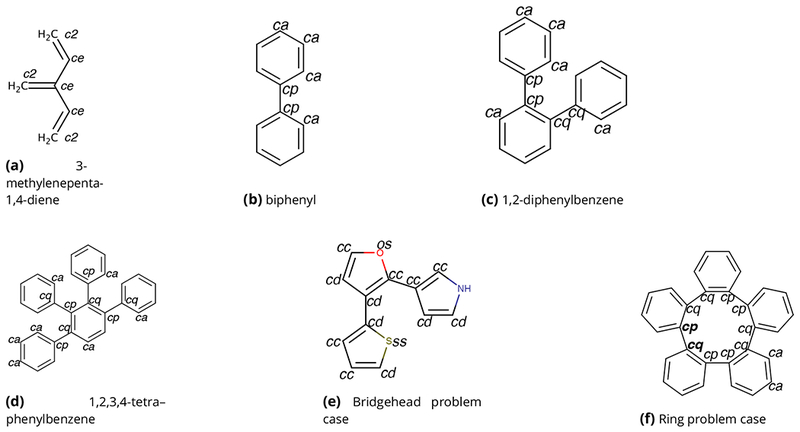
Figure 3. The SMIRKS chemical query language enables direct chemical perception by substructure matching.
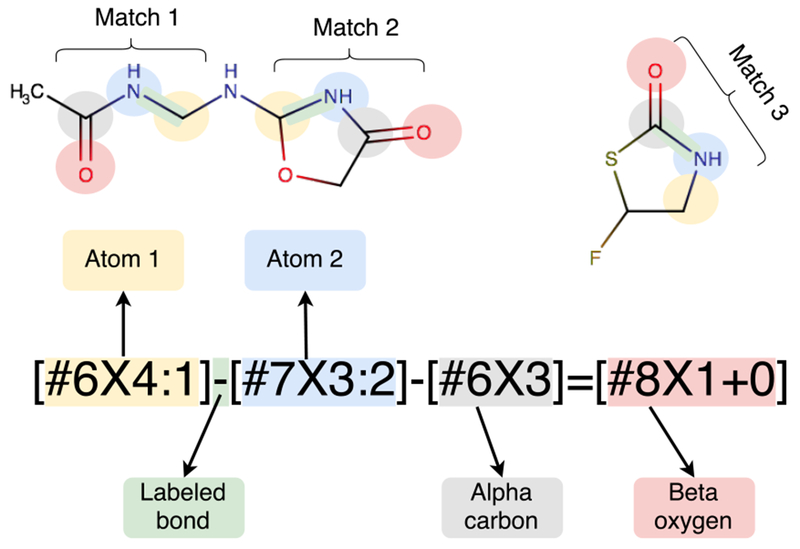
Here, a SMIRKS pattern recognizes the carbon-nitrogen bond in an amide group, so that a bond-stretch parameter can be assigned to it. In the examples, the pattern finds three matches (Matches 1, 2 and 3) in in two different molecules, as indicated by color coding of the atoms and SMIRKS patterns. The relevant pattern is a carbon with four connections ([#6X4:1] (yellow) single bonded (−) to a trivalent nitrogen ([#7X3:2], blue) which is single-bonded to a trivalent carbon ([#6X3], gray) which itself is double bonded (=) to a neutral oxygen with a single connected atom (#8X1+0], red). The first carbon and nitrogen in the pattern are singled out for special treatment by having numerical atom labels (:1 and :2) assigned to them, because the SMIRKS pattern in this case is used to assign a bond parameter to the bond connecting the two labeled atoms.
Table 1. Constructing SMIRKS patterns to match chemical substructures.
Sample SMIRKS patterns suitable for use in SMIRNOFF force fields (top), selected basic building blocks of SMIRKS patterns (Basic Ingredients), and selected decorators used in describing atoms and bonds. SMIRKS patterns are based on SMARTS patterns, which in turn are a modification of SMILES for substructure queries, such that every SMILES is a valid SMARTS pattern (but not every SMARTS pattern is a valid SMILES). For our purposes, SMIRKS patterns are essentially SMARTS patterns supplemented by numerical atom labels that allow later reference to an atom by number, such as in [#8X2H0+0:1], where the atom is labeled 1 with :1. (The ‘:’ character is also used to denote an aromatic bond, but gives a numeric label when used on an atom). SMARTS/SMIRKS patterns can refer to atoms via both atomic numbers and element symbols (e.g., #6 vs. C), though it is important to note that, since it is a subset of SMILES, both c and C refer to carbon atoms, but the former to aromatic and the latter to aliphatic carbon, and similarly for certain other elements. Here, we most frequently use atomic numbers. The OpenSmiles specification (http://opensmiles.org/opensmiles.html) provides a more complete description of SMILES patterns and the operators they can use, beyond those listed here.
| Example SMIRKS patterns | ||
|---|---|---|
| [#1:1] | hydrogen | |
| [#8X2H0+0:1] | neutral divalent oxygen with no connected hydrogens | |
| [#1:1]–[#6X4] ~[*+1,*+2] | hydrogen attached to tetravalent carbon with a +1 or +2 attached atom | |
| [#6:1]#[#7:2] | carbon triple bonded to a nitrogen | |
| Basic ingredients | ||
| [*] | An atom - here, any atom | |
| [Cl] | A chlorine atom | |
| [Cl]–[#6] | Chlorine singly bonded to any carbon | |
| [#6:1]–[#7]([#1])–[#6] | A labeled carbon atom (labeled 1) attached to a nitrogen which is attached to carbon and hydrogen | |
| Decorators for atoms and bonds | ||
| Symbol | Definition | |
| Atoms | #n | Atomic Number |
| * | any atom | |
| A | Aliphatic | |
| a | Aromatic | |
| Hn | Hydrogen count | |
| Xn | Connectivity | |
| ±n | charge | |
| rn | in ring of(smallest size) n | |
| $(*~[a]) | the preceding atom is attached to an aromatic | |
| Bonds | ~ | any bond |
| @ | ring bond | |
| – | single bond | |
| = | double bond | |
| # | triple bond | |
| : | aromatic bond | |
| Booleans for both | , | or |
| & | high priority and | |
| ; | low priority and | |
| ! | Not | |
2. Methods
2.1. The SMIRKS Native Open Force Field (SMIRNOFF) format
Assigning force field parameters via direct chemical perception requires the ability to specify the substructures of a molecule associated with given atom, bond, angle, or torsion parameters. This can be done with SMARTS patterns [32] (http://www.daylight.com/dayhtml/doc/theory/theory.smarts.html), which provide a highly flexible yet compact language for defining chemical substructures and are built on the widely used Simplified Molecular Input Line Entry Specification (SMILES) format (Figure 3 and Table 1). When a substructure is found that will be used to assign parameters for a specific bond stretching term in the force field, for example, it is furthermore necessary to identify the two atoms within the substructure that form the bond to which the parameters must be assigned. To accomplish this, we adopted we adopt the closely related SMIRKS language [33] (http://www.daylight.com/dayhtml/doc/theory/theory.smirks.html), which augments SMARTS patterns with the ability to numerically tag various atoms in the substructure match for use in assigning valence parameters (Figure 3). Although SMIRKS was developed to define chemical reactions, here it is used to match the specific atoms involved in valence terms to assign force field parameters (Figure 4). This SMIRKS variant of SMARTS matching is widely supported as an industry standard, and is available in many cheminformatics toolkits such as the OpenEye toolkit, the RDKit, OpenBabel, CDK, and others.
Figure 4. Application of a SMIRNOFF format force field to methanol.
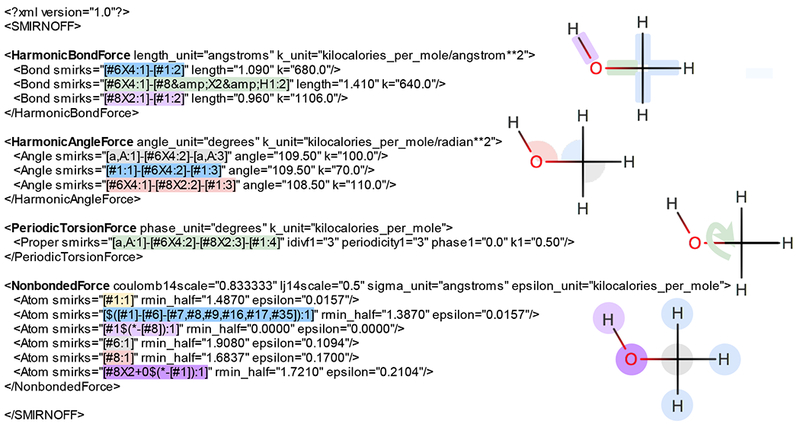
Left: Excerpt of an XML representation of a SMIRNOFF force field designed to cover just the AlkEthOH test set (see main text), showing only lines pertaining to methanol. Right: representation of methanol illustrating how SMIRNOFF provides the necessary force field parameters. For each force type or section in the XML (boldface headers), SMIRNOFF loops over the needed force terms for the molecule, and finds the last (most specialized) SMIRKS match in the XML, applying the parameters indicated there to the relevant force term. Here, the SMIRKS patterns are color coded to the corresponding molecular components, where the color codes correspond to the element(s) involved: by the primary or central atom in the case of NonbondedForce and HarmonicAngleForce parameters (except when there is redundancy, in which case the second occurrence gets a color associated with non-central atoms), and by the central two atoms in the case of HarmonicBondForce and PeriodicTorsionForce parameters. Gray is used for carbon, red for oxygen, light green for oxygen-carbon, yellow for hydrogen, pink for hydrogen-oxygen, and light blue for hydrogen-carbon. Parameterization of some symmetry-equivalent angles is omitted in this diagram for simplicity. The hierarchical nature of parameterization is also illustrated; for example, the NonbondedForce section contains a generic hydrogen SMIRKS pattern ([#1:1], yellow), which is overridden in the case of the hydroxyl hydrogen by a more specialized pattern (pink). Likewise, the generic oxygen ([#8:1], red) is overridden by the more specialized neutral hydroxyl oxygen SMIRKS (magenta). Not shown here are sections for bond charge corrections (BCCs) and constraints, though current specifications for these are provided at https://github.com/open-forcefield-group/openforcefield/blob/master/The-SMIRNOFF-force-field-format.md. It is also worth noting that the XML format is unit-bearing, allowing handling of units utilized by various different force fields.
Below, we describe the SMIRKS Native Open Force Field (SMIRNOFF) format v0.1, which utilizes direct chemical perception principles to type each forcefield energy term independently. While we describe theXML representation of this format here, other representations (such as JSON) could also be created. The format consists of a separate Section describing how parameters are to be applied to each class of interaction terms found in standard widely-supported molecular mechanics force fields, as well as additional sections describing other aspects of the force field that are required to unambiguously define how the potential energy and forces are to be computed from a particular atomic configuration. The specification for each section is described in Tables 2 and 3.
Table 2. SMIRNOFF v0.1 format sections for nonbonded interactions.
Each section corresponds to one nonbonded force type and lists the associated XML nodes and attributes, which are unit-bearing and modify the behavior of the whole section (e.g. the coulombl4scale attribute in the NonbondedForce section controls scaling of 1–4 Coulomb interactions wherever they occur), as well as individual entries which provide details of interactions or other details of the force held (e.g. Atom entries within NonbondedForce give details of specific nonbonded parameters assigned to atoms recognized by specific SMIRKS patterns). Of the sections listed here, only NonbondedForce entries are required, alll entries list as “required” must be present for any section that is included. Full details of the format detailed above are available online at https://github.com/openforcefield/openforcefield/blob/0.1/The-SMIRNOFF-force-field-format.md (visit the main openforcefield repo for links to the current format if it has changed since this writing);Figure 4 shows an example of how it is used.
| NonbondedForce | ||
|---|---|---|
| Section attributes | ||
| attribute | description | required? |
| coulomb14scale | Coulomb scale factor for 1-4 interactions | yes |
| lj14scale | LJ scale factor for 1-4 interactions | yes |
| sigma_unit | Units for sigma or rmin_half | yes |
| epsilon_unit | Units for epsilon or rmin_half | yes |
| Section nodes are Atom entries | ||
| attribute | description | required? |
| SMIRKS pattern with one indexed atom | yes | |
| rmin_half | LJ distance parameter as rmin/2 | yes, or σ |
| sigma | LJ distance parameter as σ | yes, or rmin/2 |
| epsilon | LJ interaction parameter as ϵ | yes |
| GBSAForce | ||
| Section attributes | ||
| attribute | description | required? |
| gb_model | Selected GB model | |
| solvent_dielectric | dielectric constant for solvent | yes |
| solute_dielectric | internal dielectric for solutes | yes |
| radius_units | Units for radii | yes |
| sa_model | surface area model | yes |
| surface_area_penalty | unit-bearing value for surface area penalty | yes |
| solvent_radius | solvent radius with units | yes |
| Section nodes are Atom entries | ||
| attribute | description | required? |
| smirks | SMIRKS pattern with one indexed atom | yes |
| radius | GB radius | yes |
| scale | GB scale parameter | yes |
| BondChargeIncrement | ||
| Section attributes | ||
| attribute | description | required? |
| method | Base charging method, e.g. AM1 | yes |
| increment_unit | units used for bond charge corrections | yes |
| Section nodes are BondChargeIncrement entries | ||
| attribute | description | required? |
| smirks | SMIRKS pattern with two indexed atoms | yes |
| increment | increment to move from first indexed atom to second | yes |
| Constraints | ||
| Section attributes | ||
| None | ||
| Section nodes are Constraint entries | ||
| attribute | description | required? |
| smirks | SMIRKS pattern with two indexed atoms to constrain | yes |
Table 3. SMIRNOFF v0.1 format sections for bonded interactions.
Each section corresponds to one bonded force type and provides the corresponding XML nodes and attributes for bonded forces in the SMIRNOFF format. Each section has overall attributes, which are unit-bearing and control the entire section (e.g. the k_unit attribute in the HarmonicBondForce section controls units for spring constants for bonds), as well as individual entries which provide details of interactions or other details of the force field (e.g. Bond entries within HarmonicBondForce give details of specific bond parameters assigned to bonds recognized by specific SMIRKS patterns). All sections are required, and each must provide all attributes listed as “required”; the others are optional. Full details of the format detailed above are available online at https://github.com/openforcefield/openforcefield/blob/0.1/The-SMIRNOFF-force-field-format.md (visit the main openforcefield repo for links to the current format if it has changed since this writing), and Figure 4 shows an example of how it is used. Note that, although AMBER considers the spring constant k as, effectively, an energy constant, so that , we follow the convention of treating k as a force constant, so that F(r) = k(r−r0) and . SMIRNOFF also differs from AMBER in its treatment of impropertorsions: AMBER picks one particular path through the torsion (which is dependent on atom ordering) and applies a single impropertorsion;we take all paths through the trefoil of the improper and apply all three impropers having the same handedness, after dividing the barrier height by three. For impropers, the second labeled atom is treated as the central atom.
| HarmonicBondForce | ||
|---|---|---|
| Section attributes | ||
| attribute | description | required? |
| length_unit | Unitfor bond lengths | yes |
| k_unit | Units for force constant | yes |
| Section nodes are Bond entries | ||
| attribute | description | required? |
| smirks | SMIRKS pattern with two indexed atoms | yes |
| length | equilibrium bond length | yes |
| k | force constant | yes |
| HarmonicAngleForce | ||
| Section attributes | ||
| attribute | description | required? |
| angle_unit | Unit for angles | yes |
| k_unit | Units for force constant | yes |
| Section nodesare Angle entries | ||
| attribute | description | required? |
| smirks | SMIRKS pattern with three indexed atoms | yes |
| angle | equilibrium angle | yes |
| k | force constant | yes |
| PeriodicTorsionForce | ||
| Section attributes | ||
| attribute | description | required? |
| phase_unit | Unitfor phase angles | yes |
| k_unit | Units for force constants | yes |
| Section nodes are Proper or Improper entries | ||
| attribute | description | required? |
| smirks | SMIRKS pattern with four indexed atoms | yes |
| k1 | force constant for firstterm | yes |
| phase1 | phase angle for first term | yes |
| periodicity1 | periodicity for first term | yes |
| Additionally, for propers only | ||
| idivf1 | AMBER-style idivf factor by which all barriers are divided | no |
| kN | force constant for Nth term | no |
| phaseN | phase angle for Nth term | no |
| periodicityN | periodicity for Nth term | no |
| idivfN | AMBER-style idivf factor by which all barriers are divided | no |
Each section contains a number of entries, each containing a SMIRKS string that matches chemical substructures within the system. These entries are arranged in a hierarchical format, with the last entry to match a substructure taking precendence over earlier ones. Each entry can match many occurrences of the same chemical substructure in the system, and will cause the parameters specified in the entry to applied to all such occurrences (illustrated in Figure 4). The parameters associated with each section are described in Tables 2 and 3, and are generally numerical (unit-bearing) parameters.
This structure allows facile coverage of large swaths of chemical space by first listing general chemical substructure matches and associated parameters, overriding these for chemical substructures where more sophistication is needed. To ensure reproducibility across molecular simulation packages, the SMIRNOFF format parameter set also specifies general parameters associated with the force field necessary for unambiguously determining how the potential energy and forces are to be computed form a given molecular configuration—such as the Coulombic 1–4 scaling terms associated with the forcefield and designated method for assigning partial charges—as well as the aromaticity model used for chemical substructure perception during parameter assignment; here, we primarily use the OpenEye version of the MDL aromaticity model, as discussed in the Supporting Information (Tables 2–3). The optional Constraints section can be used to constrain bonds specified by specific SMIRKS patterns, such as bonds involving a hydrogen atom.
Our hierarchical approach has a good deal of overlap with Jin etal.’s recent work on hierarchical atom-type definitions (HAD) [38]. In addition to both approaches being hierarchical, both allow type definitions to include a tree of connected atoms of arbitrary complexity, allowing definitions to extend out to as many neighbors as necessary. Additionally, both allow different typing to be used for different force terms so that, for example, complex typing could be used for different nonbonded terms, but more simplistic typing could be used for center bonds in torsions. One key difference is that the HAD approach uses sophisticated indirect chemical perception, whereas SMIRNOFF employs direct chemical perception. Another key difference is the use of the widely used SMIRKS chemical perception language in our approach, rather than definition of a new approach to chemical perception in the HAD work.
To illustrate how the SMIRNOFF format can specify a simple small molecule force field, the XML representation of an abbreviated SMIRNOFF force field containing information enabling the assignment of parameters to methanol is shown in Figure 4, for the case where partial charges are provided by the user. The Figure uses color highlighting to demonstrate how each highlighted SMIRKS pattern matches corresponding chemical substructures in methanol.
With SMIRNOFF, charge assignment is handled in one of two ways, with an optional modifier for either. First, input molecules can be provided with charges already assigned and thus user-supplied charges can be employed. Second, charges can be computed via a specified charging engine among those provided by the OpenEye toolkits, such as AM1-BCC [36, 37]. In both cases, bond charge corrections (BCCs, as employed in AM1-BCC) can also be included in the SMIRNOFF format in order to adjust provided or assigned charges. This potentially opens the door for alternate charging schemes and BCCs.
SMARTS patterns have been used previously in assigning force field parameters. Richard Dixon’s OEAntechamber effort provided a proof-of-principle showing that GAFF atom typing could be encoded with SMARTS patterns [20]. More recently, the Foyer effort [1, 7, 31] provides a language to encode existing force fields using SMARTS, and OpenBabel [60] uses SMARTS to encode UFF and GAFF force fields. One key distinction here, however, is our focus on using direct chemical perception: rather than codifying the assignment of atom types with SMARTS/SMIRKS, we assign all parameter classes (e.g., bonds, angles, and torsions) directly based on processing the molecular graph using SMARTS/SMIRKS-based substructure queries.
2.2. Reference implementation of the SMIRNOFF force field parameter assignment engine
We have created a reference implementation of a SMIRNOFF force field parameter assignment engine in Python. This engine parses an XML representation of the SMIRNOFF force field and applies parameters to a molecular topology that specifies that chemical components of the system to generate an OpenMM System object describing the parameterized molecular mechanics force field. These parameterized systems can be exported for use in other popular biomolecular simulation packages (such as AMBER [13], CHARMM [10], GROMACS [3], and NAMD [62]) as well, via ParmEd (http://parmed.github.io/ParmEd). Our reference implementation of a SMIRNOFF force field parameter assignment engine is implemented in the form of a ForceField factory class, which is an extension/replacement of the normal OpenMM ForceField factory. This offers rather similar functionality, though utilizes a SMIRNOFF XML representation (denoted with an .offxml extension) rather than the OpenMM ForceField XML representation utilized by OpenMM. Our reference implementation currently relies on the OpenEye toolkits for handling chemical perception [26, 27, 72], which are free for non-commercial academic use, to process SMIRKS patterns and to execute substructure searches and other chemical tasks. The SMIRNOFF ForceField class and related infrastructure, including examples, are available free and open source on GitHub at http://github.com/open-forcefield-group/openforcefield, and a snapshot of the current release is available in the supporting information (SI).
Assignment of SMIRNOFF parameters to a molecular system is straightforward. First, one loads one or more SMIRNOFF XML files via the ForceField class. Then, one applies the createSystem function, providing it with an OpenMM Topology of the system and OpenEye OEMol objects corresponding to the molecules comprising the system. Various optional arguments allow the user to indicate whether to use provided partial charges or compute new partial charges, and to select options such as cutoffs, periodic boundary conditions, etc. The output is a fully parameterized OpenMM System, which can be converted to other formats via ParmEd. An version of the implementation that uses the fully open source RDKit is under development.
2.3. Validation of the SMIRNOFF format
To verify that a general small molecule force field can be correctly encoded in the SMIRNOFF format, we manually ported a subset of a literature force field into this format. Specifically, we used the SMIRNOFF format to represent the subsets of the AMBER parm99 force field and of Merck’s parm@Frosst [5] force field required to assign parameters to a simple low-complexity compound set. Here, we used the AlkEthOH compound set, a set of 1500 small molecules containing alkane, ether, and hydroxyl functionalities in various combinations (full set available in the SI, selected molecules in Figure 7, Frosst_AlkEthOH_parmAtFrosst.offxml, available on GitHub and in the SI). We then tested whether energies and forces computed with the SMIRNOFF representation matched those based on the original AMBER parameter files. To do this, we first generated reference energies by loading coordinate files for each molecule, along with an an AMBER parameter file with parm@Frosst parameters, into OpenMM, and computing a single-point potential energy. We then assigned assigned parameters from the SMIRNOFF format file and re-evaluated the energy for the same conformer in OpenMM. To further check the format, we also checked every force term applied to each atom in each molecule and ensured that they exactly matched. Code for this is available in the Supporting Information and on GitHub at https://github.com/openforcefield/openforcefield/blob/0.1/examples/SMIRNOFF_comparison/compare_set_energies.py.
Figure 7. Example molecules from the AlkEthOH (alkanes, ethers, and alcohols) 1500 molecule set.
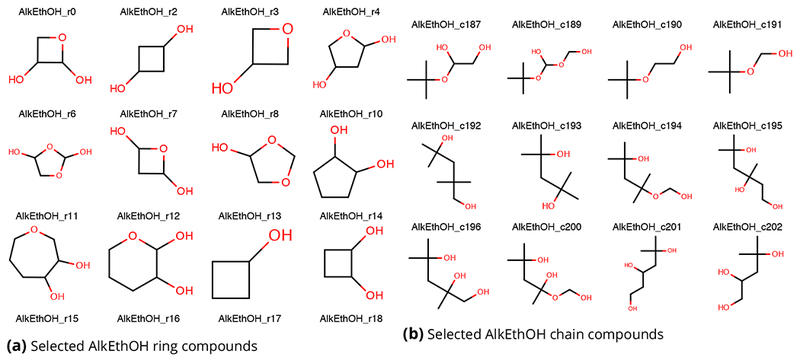
Selected cyclic compounds are shown at left, and selected chain compounds are shown at right. While the set consists only of alkanes, ethers, and hydroxyls, these occur in considerable diversity. The full set of AlkEthOH compounds is provided in the Supporting Information.
This comparison was done for our AlkEthOH parm@Frosst port only, not for SMIRNOFF99Frosst small molecule force field introduced below, as the latter is a distinct force field not intended to exactly reproduce any previous force field.
2.4. SMIRNOFF99Frosst: a general force field expressed in SMIRNOFF format
To further validate and characterize the SMIRNOFF format, and to establish a foundation for future force field optimization, we used SMIRNOFF to define a general force field by porting and adapting parameters drawn from AMBER parm99 and parm@Frosst (with a few subsequent additions as discussed below). The resulting force field, which we call SMIRNOFF99Frosst, is not intended to exactly reproduce prior force fields nor to serve as a final product; rather, it is an adaptation and compression (taking advantage of the simplicity afforded by SMIRKS and direct chemical perception) and extension intended to provide a starting point for future refitting work. As has been noted previously, taking a hierarchical approach to force field fitting and extension can make the refitting task considerably more manageable [38].
Here, we describe how we constructed SMIRNOFF99Frosst. This is intended to be a one-time conversion process into this new format while compressing and adapting the force field. This conversion involved a great deal of human labor and does not represent the procedure we plan to employ in the future for force field development. Instead, the main goals of this construction process were to demonstrate that our new format can capture the needed information, validate the format, and provide a starting point for further optimization. Separate work will address how to automatically extend and refit the force field.
To construct SMIRNOFF99Frosst, we began with an AMBER-format parameter file containing parm99 and parm@Frosst parameters. The parm@Frosst force field is an extension of parm99 which is added via an AMBER-format frcmod file, so our starting point included both the parm99 and parm@Frosst parameters in a single AMBER-format parameter file. From this, an initial mapping was defined from every AMBER atom type to a preliminary SMIRKS string capturing only the element and hybridization; e.g., we mapped atom type CT to [#6X4]; and the set of atom types { C, CA, CM, C*} to [#6X3]. These preliminary SMIRKS strings were substituted for their corresponding AMBER atom names within each force field section of the AMBER parameter file. Thus, for example, the AMBER valence angle CT–OH–HO was converted to the SMIRKS string [#6X4:1]–[#8X2:2]–[#1:3]. Each force field section was then lexically sorted, thus grouping parameter specifications with identical SMIRKS representations. Because of the oversimplified initial SMIRKS representation of the AMBER atom types, the same SMIRKS representation is, at this stage, applied to different parameters of the same type. This degeneracy was repaired manually as follows:
Insertion of correct bond-orders: For example, the carbonyl-containing angle CT–C–O, which had been replaced by [#6X4:1]–[#6X3:2]–[#8X1], was changed to [#6X4:1]–[#6X3:2] = [#8X1]. The SMARTS/SMIRKS bond orders in Table 1 were used, including the wild-card bond order where appropriate (especially for generic parameters, below).
Increasingly detailed SMIRKS strings: Where needed to distinguish between parameters, additional chemical complexity was introduced into the SMIRKS patterns to reflect the chemical complexity in the AMBER atom types that had been oversimplified in the initial SMIRKS assignments.
Merging of degenerate parameters: Many parameter assignments with identical SMIRKS representations were found to correspond to numerically identical, or very similar, parameters. These were collapsed to a single occurrence representing a more generic parameter, leading to a large reduction in the number of parameter assignments.
Each section of parameters (bonds, angles, torsions, etc.) then was manually reordered to provide the correct hierarchical assignment (“last one wins”) behavior, wherein subsequent more specific parameters can overwrite earlier more general parameters, and the resulting file was converted to SMIRNOFF format in XML representation via a custom Python script, convert_frcmod.py available in the SI and on GitHub at https://github.com/openforcefield/openforcefield/tree/0.1/utilities/convert_frosst. The resulting force field uses the OpenEye MDL aromaticity model, as described in the SI.
Finally, the resulting force field, with ~300 lines of parameter specifications (see Section 3.2.1), was slightly modified, as follows. First, to improve its coverage of chemical space, 24 additional entries were adapted from GAFF2. As detailed in the SI, these largely addressed parameters for sulfur, phosphorus and halogens. Second, our previous work (DM, CIB; see SI) had revealed simulation instabilities caused by hydroxyl hydrogens in AMBER-family force fields, which possess partial charges that are not protected from energetic singularities by repulsitve Lennard-Jones sites. In some situations, these allow hydroxyl hydrogens to closely approach polar atoms, such as oxygen atoms in adjacent molecules or residues, resulting in simulation instabilities. To remedy this defect in AMBER force fields, we added a Lennard-Jones site with small epsilon parameter to hydroxyl hydrogens in the SMIRNOFF99Frosst parameter set (see SI)1. The resulting force field specification, termed SMIRNOFF99Frosst v1.0.7, is available in a versioned manner on GitHub at https://github.com/openforcefield/smirnoff99Frosst.
2.5. Evaluation of SMIRNOFF99Frosst v1.0.7
2.5.1. Coverage of chemical space
We examined the ability of SMIRNOFF99Frosst v1.0.7 to assign parameters to four sets of compounds: the FreeSolv hydration free energy database [21, 56]; a subset of the Zinc database [34, 35] that was originally curated to test the parm@Frosst force field [5]; DrugBank [44, 48, 89, 90]; and eMolecules (https://www.emolecules.com). For DrugBank and eMolecules, the compound sets considered excluded those with metals, metalloids, boron, or noble gases (present in DrugBank); those with over 200 heavy atoms or inappropriate valency; entries with more than one compound (present in DrugBank); and any compound for which 3D conformation generation and assignment of AM1-BCC charges [36, 37] with the OpenEye toolkits failed. The resulting compound sets – from FreeSolv, DrugBank, the Zinc subset, and eMolecules – comprise 642, 7,505, 5497, and 5,689,262 molecules, respectively. For eMolecules it was not possible to do parameter assignment with parm@Frosst because no parm@Frosst typer is generally available, whereas for the other two sets, we already had parm@Frosst typed molecules available from the prior work of CIB.
2.5.2. Comparison with experimental physical properties: densities, dielectric constants, and hydration free energies
We evaluated the ability of SMIRNOFF99Frosst v1.0.7 to estimate accurate physical observables by using it to compute the densities and static dielectric constants for neat small molecular liquids previously considered by Beauchamp etal. [6], and the hydration free energies in the FreeSolv database. SMIRNOFF99Frosst results are compared to those from the GAFF force field, which is essentially a sibling force field in the AMBER family and has been widely used in the literature. Thus computed values are expected to differ between the two force fields, as not only is the force field format different but also many of the parameters are different and SMIRNOFF99Frosst is considerably more terse (as noted in Section 2.4). The goal of this validation is to ensure that SMIRNOFF99Frosst indeed provides a reasonable starting point for further force field development work.
Densities and static dielectric constants were computed using a pipeline developed previously for benchmarking physical properties extracted from the NIST TRCThermoML Archive [6] modified to employ SMIRNOFF format force fields. This modified pipeline was used to repeat the full benchmark with both SMIRNOFF99Frosst v1.0.7 and GAFF 1.8 [82, 83] (applied using antechamber from AmberTools (Version 16 patch 16). All calculations and analysis were otherwise as previously described [6]. AM1-BCC charges were employed. [36, 37] All scripts and libraries necessary to reproduce these calculations has been deposited in SI and the GitHub repository (https://github.com/mobleylab/SMIRNOFF_paper_code).
Hydration free energies were computed using the YANK software package [15, 84] v0.16.0, based on the OpenMM GPU-accelerated molecular simulation library [22]. The TIP3P [39] explicit solvent model was used, and simulations were conducted at a temperature of 298.15 K and pressure of 1 atm. Calculations employed Hamiltonian replica exchange over 5000 iterations, with each iteration consisting of 500 steps of Langevin dynamics with 2 femtosecond timestep, using a collision rate of 5/picosecond; pressure was regulated with a Monte Carlo barostat with default settings. An anisotropic dispersion correction was included out to 16 Å [71]. Complete details of the alchemical protocol are given in the SI and on GitHub (https://github.com/MobleyLab/SMIRNOFF_paper_code/blob/master/FreeSolv/scripts/yank_template.yaml). Briefly, the alchemical protocol utilized 20 alchemical states in the solution phase and 5 states in gas phase to annihilate electrostatics and decouple sterics interactions using soft-core interactions, following earlier protocols [56]. Hydration free energies and corresponding statistical uncertainties were estimated with the multistate Bennett acceptance ratio (MBAR) [69] using the standard YANK analysis framework. AM1-BCC [36, 37] charges were employed. Full scripts for conducting and analyzing the calculations are also available on GitHub (https://github.com/MobleyLab/SMIRNOFF_paper_code).
3. Results
Here, we report the results of validation tests of the SMIRNOFF format, as well as an accuracy benchmark of the prototype SMIRNOFF99Frosst v1.0.7 small molecule force field against experimental physical property data. When validating the format, we aim to exactly reproduce energies and forces of an equivalent AMBER-family force field when represented in the new SMIRNOFF format. When validating the SMIRNOFF99Frosst force field, we assess its coverage of chemical space and its accuracy, relative to the related (but not identical) GAFF, for calculations of densities and dielectric constants of neat liquids and hydration free energies of a diverse set of small molecules. We then show how the present approach avoids the proliferation of needless atom types for the conjugated and polyphenyl compounds considered in the Introduction, and explain how SMIRNOFF can be used with simulation codes beyond OpenMM.
3.1. Validation of the SMIRNOFF format for the focused AlkEthOH compound set
We sought to confirm that the SMIRNOFF format could be used to exactly reproduce traditional atom type-based force field parameter assignments—in this case, parm@Frosst [5]. To do this, we constructed a minimal SMIRNOFF force field in which we aimed to replicate traditional AMBER parameter assignments of parm@Frosst [5] for the AlkEthOH set of 1500 alkanes, ethers, and hydroxyl-containing small molecules (see Methods). We then computed the potential energy and forces for a single conformation of each compound with OpenMM, using both the original AMBER-format parameterprmtop file and the new SMIRNOFF-format XML parameter file to assign parameters, and checked to see that all energies and applied forces exactly matched. We found exact agreement for most compounds, but the energies disagreed for molecules involving electron withdrawing groups attached to carbon atoms, and these cases were examined more closely.
We found that all energy/force discrepancies arose from apparent human error in the parm99 (and earlier AMBER versions) parameter files for torsional terms involving atom types H1, H2, and H3, which represent hydrogen atoms connected to carbon atoms bonded in turn to one, two or three, electron-withdrawing groups, respectively. When these atom types were first introduced [18, 77], the new corresponding torsion terms H1–CT–CT–CT, H2–CT–CT–CT and H3–CT–CT–CT introduced were neither intentionally parameterized nor assigned parameters associated with the most similar parent atom type, HC, which would have led to torsion the use of parameters from HC–CT–CT–CT). Instead, they were left to inherit the parameters of the generic X–CT–CT–X torsion. In contrast, the SMIRNOFF rules led to assignment of torsions involving the H1, H2 and H3 the same parameters as those involving HC, leading to assignment of more specific and hence appropriate torsional parameters. The analogous issue arose, also, for other torsions involving H1, H2, H3, such as H1–CT–CT–H1, H1–CT–CT–H2, H1–CT–CT–H3, H1–CT–CT–OH, H2–CT–CT–OH, and H3–CT–CT–OH. Again, parm@Frosst assigns these entirely generic parameters, X–CT–CT–X, rather than those for the closest parent atom type HC, (e.g., HC–CT–CT–HC, HC–CT–CT–OH); Figure 8 provides examples of molecules with these problems. Further investigation revealed that the same issues are also present in GAFF and GAFF2 (Figure 8). While GAFF and GAFF2 are not derived from parm@Frosst, GAFF and parm@Frosst share parm99 as a common ancestor, and considerable similarities—including these apparent bugs—persist to this day.
Figure 8. Example molecules with torsional errors in AMBER force fields uncovered in AlkEthOH.
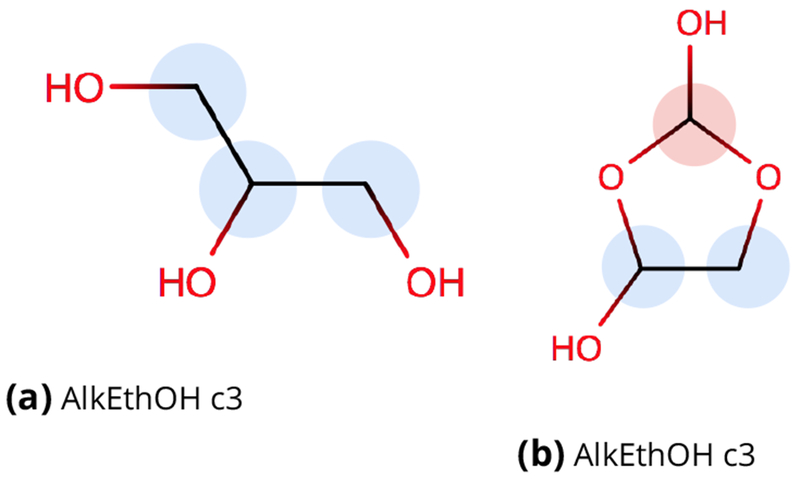
Shown are examples of molecules with torsions which were erroneously assigned generic torsional values in AMBER parm99, parm@Frosst, and GAFF/GAFF2 force fields, as uncovered by our testing of SMIRNOFF for AlkEthOH to ensure it could reproduce parm@Frosst energies. The errors found all involve torsions containing the H1, H2, or H3 parm@Frosst atom types (GAFF/GAFF2 types are equivalent but lowercase), used for hydrogens connected to carbons which are themselves connected to one, two, or three electron withdrawing groups (respectively). Carbons with attached hydrogens having the H1 atom type are highlighted in blue and carbons with hydrogens having the H2 atom type are highlighted in red; carbons here use the CT atom type and oxygens use OH if in hydroxyls and OS otherwise (GAFF/GAFF2 types c3, oh, and os respectively). Specifics of exactly how these errors originated and how they were addressed in our testing are explained in Section 2 of the Supporting Information.
In order to complete the present validation, we brought the SMIRNOFF implementation into agreement with parm@Frosst for these cases by deliberately applying generic torsional parameters to torsions involving atoms of type H1, H2, H3, essentially reintroducing the human errors made in parm99. This required introducing several additional parameters and SMIRKS patterns (Figure 8); thus, the specification became more complex in order to recapitulate the issues in the original force field. Once this was done, all energies and every individual force term applied in every molecule were identical. These results confirm that the SMIRNOFF format can accurately capture—and even help troubleshoot—the information in standard atom type based force fields (Section 3.1). This minimal force field, which we call “Frosst_AlkEthOH_parmAtFrosst” and “Frosst_AlkEthOH” depending on whether it does or does not reproduce parm@Frosst exactly (respectively), was only used for the purposes of validating the format and is not the subject of any subsequent work in this study.
3.2. The SMIRNOFF99Frosst Force Field
To further validate our format, assess the potential of direct chemical perception, and provide a starting point for future force field development work, we tested our new SMIRNOFF99Frosst small molecule force field in several ways. SMIRNOFF99Frosst is an adaptation of the AMBER parm99 and parm@Frosst force fields into SMIRNOFF format, and is a terse new general small molecule force field intended to provide a starting point for subsequent refitting work.
3.2.1. Terse coverage of a large chemical space
The SMIRNOFF99Frosst parameter specification is terse: The SMIRNOFF format XML representation specifying SMIRNOFF99Frosst v1.0.7 has only 335 parameter lines, where each line specifies a single force field term (e.g., one class of bond-stretch or torsion parameters); whereas the files specifying parm@Frosst, GAFF (1.5), and GAFF2 (2.1), have 3613, 6385, and 6794 analogous parameter lines, respectively. There are two reasons for the brevity of the SMIRNOFF99Frosst specification. First, as discussed in the Introduction, by not using atom types to specify valence terms, SMIRNOFF avoids the proliferation of unnecessary redundant parameters that arise in atom type-based specifications. Second, in order to establish a simple starting point for future force field development, we deliberately sought to minimize the complexity of SMIRNOFF99Frosst. For example, there is only a single parameter line for all carbon-carbon single bonds involving two sp2 carbons, and another for all double bonds between sp2 carbons. Angle parameters, too, are highly condensed, in many cases depending only on the SMIRKS specification of the central atom. Additionally, for angles, relatively little attention has been paid to angle parameters within rings, based on a concept that ring geometry may be determined chiefly by on topological constraints rather than the specifics of angle parameters. Torsions require additional complexity, but we reduced their number by grouping torsional assignments with similar parameters in the original force field. More generally, we used relatively generic SMIRKS patterns in order to keep the resulting force field as minimal as possible; our preference was for a simple general force field which might provide a good starting point for further refinement.
The resulting SMIRNOFF99Frosst force field covers a region of chemical space that is slightly larger than that covered by GAFF, and substantially larger than that covered by parm@Frosst, based on comparisons for four compound sets of increasing size and diversity, as follows.
FreeSolv Database All three force fields fully cover the FreeSolv database [21, 56] of 642 small molecules and their hydration free energies (https://github.com/mobleylab/FreeSolv, v0.51).
ZINC subset This set of 7505 compounds drawn from the the ZINC database [34, 35] and made available in the parm@Frosst distribution (http://www.ccl.net/cca/data/parm_at_Frosst/) was originally curated to test the parm@Frosst force field [5]. Five of the compounds proved to have inappropriate valency, such as a carbon or a nitrogen with more than four net bonds, and so were omitted. Of the remaining 7500, SMIRNOFF99Frosstv1.0.7 covers all but 8, while GAFF misses 69 and parm@Frosst misses 3569.
DrugBank Application of the filters described in Section 2.5.1 to DrugBank v5.0.1 (e.g., removal of compounds with metals) left 5497 compounds. Of these, 15 were not covered by SMIRNOFF99Frosstv1.0.7, compared to 32 with GAFF and 2183 for parm@Frosst. Many of the cases not covered by SMIRNOFF99Frosst involve chemistry that is very unusual or perhaps incorrect, such as a di-protonated carboxylic acid, and a protonated nitro group.
eMolecules Here, 5,689,262 molecules remained after filtering eMolecules 2016-09-01. Of these 1,036 molecules were not covered by SMIRNOFF99Frosst v1.0.7, compared to 357,589 for GAFF. Many of the cases not covered by SMIRNOFF99Frosst involve missing torsions parameters around fairly unusual combinations of functional groups, such as sulfur or phosphorous bonds to other non-carbon atoms.
3.2.2. Evaluation of SMIRNOFF99Frosst for physical properties of organic liquids
We carried out an initial evaluation of the accuracy of the SMIRNOFF99Frosst v1.0.7 prototype force field by using it to compute the densities and dielectric constants of 45 pure organic liquids under various experimental conditions, a total of 246 observables [6], along with the 642 small molecule hydration free energies in the FreeSolv database [21] (version 0.51 was used), and comparing the results with matched calculations using GAFF as deposited in the FreeSolv repo [21 ]. SMIRNOFF99Frosst shares common ancestry with the GAFF force field, but is distinct (and distinct from its predecessors parm99/parm@Frosst), so SMIRNOFF99Frosst and GAFF results are expected to differ but have some similarities.
As shown in Figure 5, SMIRNOFF99Frosstv1.0.7 yields densities and dielectric constants similar in accuracy to those afforded by GAFF 1.8, as expected for what are essentially sibling force fields from the same force field family. Thus, densities computed with SMIRNOFF99Frosst have a marginally larger systematic error than those from GAFF, in the direction of being too high relative to experiment, while the dielectric constants computed with SMIRNOFF99Frosst are slightly more accurate than those from GAFF, in terms of average error. The only particularly substantial discrepancy is for flexible force field water, which is not typically used as a water model; in Figure 5(b) this provides the most extreme set of outliers in SMIRNOFF99Frosst, around a predicted density of 1.2 g/mL and an actual density of 1.0 g/ml.
Figure 5. Densities and static dielectric constants of pure solvents, computed with GAFF and SMIRNOFF99Frosst.
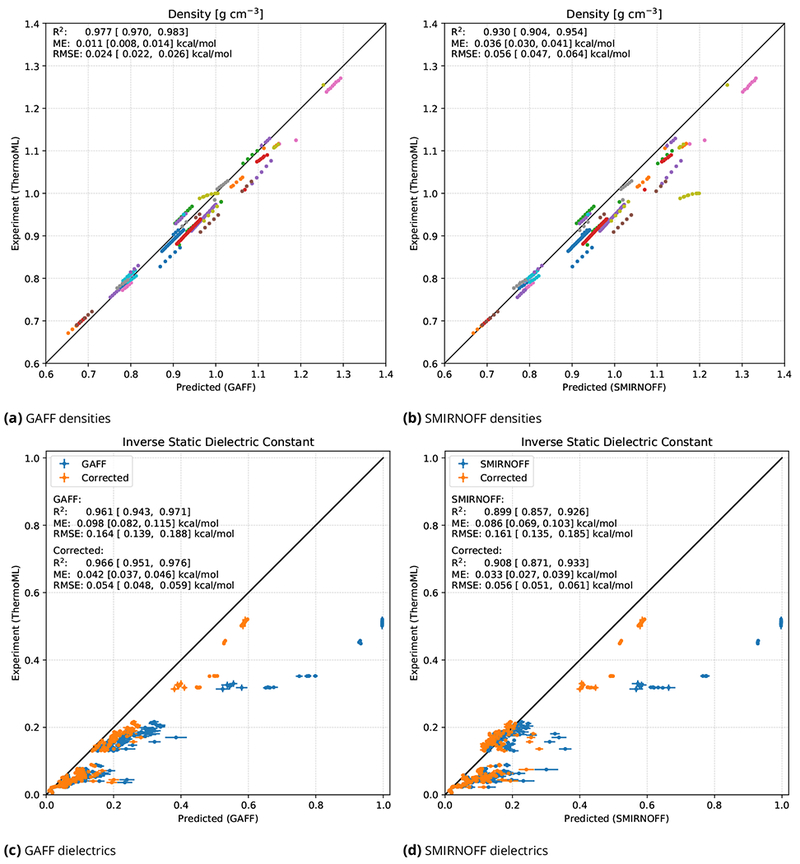
Densities (top) and dielectric constants (bottom) were computed with GAFF (left) and the new SMIRNOFF99Frosst v1.0.7 force field (right) (Section 2.4), for 45 liquids under varied experimental conditions (near 1 atm, various temperatures), leading to 246 data points. All panels include error bars, which are typically smaller than the size of data point markers for densities. Statistics are shown in the inset on each panel, with brackets denoting 95% confidence intervals. In (a) and (b), each compound is shown in a different color, and there are multiple data points for each compound due to variations in experimental conditions (so groups of points of the same color correspond to different experimental conditions). GAFF and SMIRNOFF99Frosst results are expected to differ as they are essentially sibling force fields from the AMBER family, with SMIRNOFF99Frosst being considerably more terse and employing direct chemical perception. A full list of compounds is available in the SI and at https://github.com/MobleyLab/SMIRNOFF_paper_code/blob/master/ThermoML_benchmark/results_GAFF/tables/data_with_metadata.csv
The 642 FreeSolv hydration free energies provide a more extensive benchmark to test the new force field. Figure 6 provides comparisons of SMIRNOFF99Frosst v1.0.7 with previously published GAFF results (panel a), as well as comparisons of both force fields with experiment (panels b, c). We find that SMIRNOFF99Frosst and GAFF agree well, with a Pearson R2 of 0.989 (95% confidence interval (CI) [0.986, 0.992]), mean difference of 0.009 [−0.025,0.044] kcal/mol, and RMS difference of 0.448 [0.378,0.526] kcal/mol; the mean difference is statistically indistinguishable from zero. The small differences in performance relative to experiment are within confidence intervals and depend on the metric examined; e.g., the mean error is smaller with SMIRNOFF99Frosst but the RMS error is smaller with GAFF.
Figure 6. Hydration free energies for FreeSolv from GAFF and SMIRNOFF99Frosst.
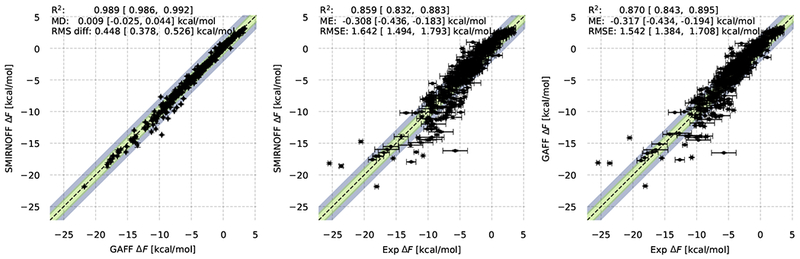
Left: scatter plot of SMIRNOFF99Frosst v1.0.7 versus GAFF results. Middle: Results from the new SMIRNOFF99Frosst v1.0.7 force field versus experiment. Right shows GAFF versus experiment (from prior work [21]). Statistics, with bootstrapped uncertainties representing 95% confidence intervals, are shown at the top of each panel. The x = y line is shown along the diagonal as a guide, and ±1 and ±2 kcal/mol regions are shown in green and blue-gray, respectively. GAFF and SMIRNOFF99Frosst results are expected to differ as they are essentially sibling force fields from the AMBER family. Only some 14 compounds have values differing by more than 2 kcal/mol, as discussed in the text.
On the hydration free energy test, only some 14 compounds have values differing (between the sibling force fields GAFF and SMIRNOFF99Frosst) by more than 2 kcal/mol. These include four carboxylic acids (acetic acid, diflunisal, 2-(2,3-dimethylphenyl)aminobenzoic acid and dicamba) where slow sampling of a torsional barrier [42] could cause convergence problems; three unusual phosphorous containing compounds where parameters in both force fields likely need further refinement (ethion, diethyl (2R)-2-dimethoxyphosphinothioylsulfanylbutanedioate, and (1R)-2,2,2-trichloro-1-dimethoxyphosphoryl-ethanol)); three unusual sulfur-containing compounds which have previously been observed to be somewhat problematic with GAFF (sulfolane, methylsulfonylmethane, and endosulfan alpha) [53], two other very flexible and highly polar compounds which could have sampling problems (diethyl butanedioate and (2R,3R,4R,5R)-Hexan-1,2,3,4,5,6-hexol), and two others (1-(2-hydroxyethylamino)-9,10-anthraquinone and naphthalen-1-yl N-methylcarbamate. Since GAFF and SMIRNOFF99Frosst, while related, are distinct force fields some differences are not surprising, though they may warrant further exploration.
In summary, then, SMIRNOFF99Frosstv1.0.7 provides a level of accuracy on these tests that is comparable to that of its sibling GAFF despite its relative simplicity.
3.2.3. Parameter assignment for conjugated and polyphenyl compounds
The SMIRNOFF format easily solves the problems encountered when bond parameters are assigned based on atom types for molecules with alternating single and double or aromatic bonds; see Introduction and Figure 2. This is because direct chemical perception can assign parameters based simply on whether a bond is single, aromatic, double or triple, without the need to introduce complex atom types. This simplicity not only leads to a more terse force field specification (see above) but also reduces the risk of human error.
To illustrate this, we assigned parameters to 1,2,3,4-tetraphenylbenzene (Figure 2e) with GAFF, GAFF2, and SMIRNOFF99Frosst v1.0.7, and ran brief, gas-phase molecular dynamics simulations. For GAFF and GAFF2, the central ring is found to adopt improbable distorted conformations (Figure 9a, b), due to the assignment of single bond character to what are actually aromatic bonds within the central ring. This problem arises because GAFF and GAFF2 lack parameters for a number of the torsions in the complex ring system, notably ca–cp–cq–cq, ca–cp–cq–cp, cp–cp–cq–ca, cq–cp–cq–ca, cp–cp–cq–cq, cp–cp–cq–cp, cq–cp–cq–cq, and cq–cp–cq–cp (Figure 2e). However, the single bond ca–cp–cp–ca is present in the force field and has a small barrier height of 0.795 kcal/mol, and the parmchk2 program, which estimates missing parameters, assigns single-bond parameters to all of the missing torsions, even when they correspond to aromatic bonds within a ring. Thus, aromatic bonds within rings in this system end up with parameters which should only be associated with a rotatable single bond between aromatic rings, which ca–cp–cp–ca is intended for. Torsional distributions (probability distribution functions from molecular dynamics) for this case are shown in the Supporting Information (SI) (SI Figure 1). In contrast, with SMIRNOFF99Frosst, use of the SMIRKS pattern [*:i]~[#6X3:2]:[#6X3:3]~[*:4] to assign parameters for aromatic carbon-carbon bonds results in correct assignment of torsion parameters with a high barrier to rotation for all aromatic torsions in the system, and thus to an appropriately flat ring geometry (Figure 9c).
Figure 9. Representative geometries for 1,2,3,4-tetraphenylbenzene (top row) and the bridgehead problem case of Figure 2e (bottom row).
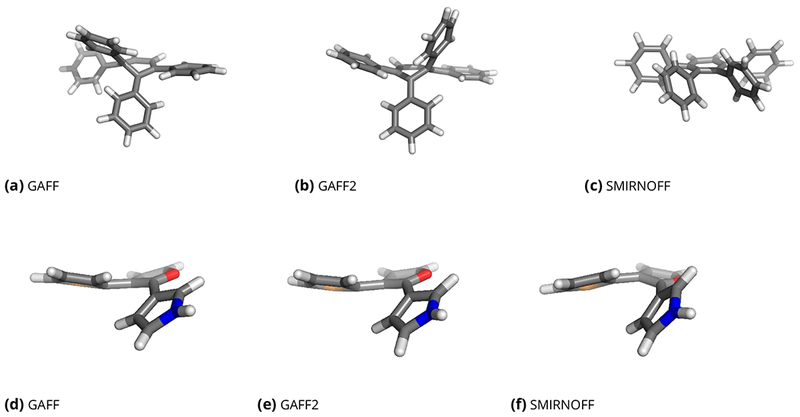
Here, we show representative geometries from short gas-phase simulations with each force field for each molecule. Depending on what force field is applied, the typical geometry varies considerably. In the case of 1,2,3,4-tetraphenylbenzene, GAFF and GAFF2 (a-b) lead to incorrect buckling of the central aromatic ring, whereas the prototype force field introduced here, SMIRNOFF99Frosst, keeps the aromatic ring remains planar, as expected (c). For the molecule in the bottom row, GAFF and GAFF2 (d, e) make the bond connecting the five-membered aromatic rings be non-rotatable, resulting in a slight buckling of the aromatic rings. In SMIRNOFF99Frosst (f), the connecting bonds are rotatable so no buckling occurs.
The GAFF and GAFF2 force fields also lead to incorrect parameter assignments and nonphysical geometries for the molecule of Figure 2e. As noted in the Introduction, GAFF and GAFF2 treat the bridgehead single bond between these rings as aromatic, and hence essentially non-rotatable, so the entire ring system tries to remain planar and is forced by steric strain to buckle (Figure 9d,e top left). In contrast, SMIRNOFF99Frosst recognizes the bridgehead bond as a rotatable single bond, allowing it to rotate and thus avoid nonphysical steric strain and consequent geometric distortion (Figure 9f). The rotatable bonds also allow both rings to flip between rotamers, while GAFF and GAFF2 maintain the geometry shown in Figure 9d and e throughout our simulations due to the high torsional barrier. Torsional probability distributions from MD for this case are shown in SI Figure 2.
3.3. Using SMIRNOFF-parameterized systems in major molecular simulation packages
The ForceField class used to interpret the SMIRNOFF XML files provides what is essentially a drop-in replacement for the OpenMM ForceField class. It loads and parses a SMIRNOFF format force field specification file, and the createSystem function then applies that force field to a specific molecular system. This step requires an OpenMM Topology describing the system to be parameterized, as well as a set of OpenEye OEMol objects for the molecules comprising the system, and various arguments concerning charging method, cutoff, and other choices for the system to be set up. (An RDKit-based implementation, which will make the OpenEye toolkit unnecesssary, is in development.) The final result is an OpenMM System object containing all the information on how to compute energies and forces, which then can be used for molecular simulation or modeling applications. This System object can be readily converted for use in other codes via ParmEd (http://parmed.github.io/ParmEd) or InterMol (http://intermol.readthedocs.io/en/latest/), allowing export to GROMACS [3], AMBER [13], DESMOND [8], CHARMM [10], and LAMMPS [64] formats [70] for use in a wide variety of packages. Note, however, that the existing procedure is not facile for long polymers, like proteins and nucleic acids, and plans to improve handling of such molecules are considered in the Discussion section.
4. Discussion
In this paper, we have introduced the concept of direct chemical perception for the assignment of force field parameters and demonstrated the advantages of this approach over traditional atom typing. We have furthermore shown that direct chemical perception can be implemented with SMIRKS strings, defined a new SMIRKS-based force field format (SMIRNOFF), and used this to create a terse expression of a new, AMBER-based, general force field, SMIRNOFF99Frosst v1.0.7. This novel approach creates a robust infrastructure and starting point for further advances in classical force field development, such as refitting and optimization work. Below, we summarize key conclusions and touch on directions for future work.
As detailed above, the use of atom types to classify force field terms for bonds, angles and torsions, introduces a variety of complications to the force field development process. In particular, by creating complex dependencies among the specifications required for the various force field terms, it expands the number of adjustable parameters and needlessly creates opportunities for human error. In contrast, separate assignment of atom, bond, angle and torsion parameters by direct chemical perception avoids such dependencies and thus does not generate these problems. Indeed, as shown here, shifting to direct chemical perception surfaced old errors and fixed several problems encountered by even modern fixed charge force fields. Importantly, direct chemical perception still has the expressive power of atom type-based methods, if not more, as confirmed by the present validation studies.
We have illustrated and validated the capabilities of direct chemical perception by using it, in the context of the SMIRNOFF specification format defined above, to specify the force field SMIRNOFF99Frosst v1.0.7 for general organic molecules. This is a logical descendant of the AMBER family force field parm@frosst [5] but covers a vast chemical space with fewer than 350 lines of parameters, as opposed to the thousands of lines needed by GAFF and other modern force fields. Although we view SMIRNOFF99Frosst v1.0.7 as a prototype, the tests reported here, based on the densities and dielectric constants of organic liquids and the hydration free energies of small molecules, indicate roughly comparable performance with GAFF, so we believe it represents a strong starting point for future development, especially in view of its relative simplicity.
We plan to develop improved versions of SMIRNOFF99Frosst using experimental data primarily to tune nonbonded interactions, quantum data to tune bond-stretch, bond-angle, and torsional terms, and potentially a combination of both to address electrostatic interactions. We envision two aspects of this process. The first is a straightforward adjustment of parameters associated with the existing set of SMIRKS strings. Note that, because SMIRNOFF99Frosst has far fewer adjustable parameters than, e.g., GAFF and parm99@Frosst, it should be more amenable to automated parameter fitting methods, such as ForceBalance [47, 85–88] or the approach used for fitting in the HAD scheme [38]. The second aspect is to include automated optimization of the SMIRKS classifications of Lennard-Jones, bond-stretch, angle-bend, and torsional terms. Our goal is to escape the traditional reliance on potentially idiosyncratic chemical insight in assigning atom types. This process, which has not hitherto been amenable to automation, will be facilitated by our parallel development of the SMIRKY tool, which automatically carries out stochastic sampling and optimization over the SMIRKS strings used in the SMIRNOFF format [93]. It is this work on automated inference of the relevant chemistry which will allow us to move away from atom or parameter types decided by human experts and chemical intuition, and towards a more rigorous, data-driven approach for determining how many terms are needed in a force field. Until such automated tools are integrated into the force field development process, human expertise and chemical intuition will likely continue to play a major role in force field development.
Direct chemical perception can provide critical advantages when applied to proteins and nucleic acids as well as small molecules. While the SMIRNOFF format already supports application to biopolymers, the real promise of the approach is that it can provide a systematic and consistent way to handle biopolymers which include nonnatural amino acids, posttranslational modifications, and covalent inhibitors, along with ligands, cofactors, and other covalent modifications. The main obstacle to this to change how electrostatic parameters are assigned; currently they are assigned either via charges provided with the input molecule(s), or via a supported charging scheme like AM1-BCC [36, 37]. To consistently handle biopolymers will require the ability to specify a molecular fragmentation scheme, in order to efficiently handle large molecules consisting of repeating units, thereby enabling the full range of applications to modifed biopolymers. However, until this capability is in place, SMIRNOFF force fields may be used in much the same manner as GAFF and GAFF2. Here, one splits the molecular system into components, applies an established force field to any biopolymers present, applies the SMIRNOFF force field to all other components, then merges the components again. This can currently be done with the ParmEd software; see example in the openforcefield GitHub repository at https://github.com/openforcefeld/openforcefeld/tree/master/examples/mixedFF_structure, as well as in the present SI.
Finally, it is worth noting that the SMIRNOFF format is intended to allow the specifcation of force fields with more detailed functional forms. For example, additional SMIRKS strings could be used to assign atom-centered point polarizabilities, or to position off-atom charges or multipoles. Here, too, setting aside the artificial reliance on atom types and instead using direct chemical perception to assign the parameters for each separate force field term will keep the number of adjustable parameters to a minimum, reduce the chances of human error, and maximize flexibility.
Supplementary Material
5. Acknowledgements
DLM and CCB appreciate the financial support from the National Science Foundation (CHE 1352608) and the National Institutes of Health (1R01GM108889-01) and computing support from the UCI GreenPlanet cluster, supported in part by NSF Grant CHE-0840513. JDC appreciates support from the Sloan Kettering Institute, the National Institutes of Health (National Cancer Institute Cancer Center grant P30 758 CA008748), and the National Science Foundation (CHE 1738979). MKG appreciates support from NIGMS grant R01GM061300. MRS appreciates the support of the National Science Foundation (CHE-1738975). MKG acknowledges funding from National Institute of General Medical Sciences (GM061300). The contents of this paper are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. MKG has an equity interest in and is a cofounder and scientific advisor of VeraChem LLC. DLM serves on the scientific advisory board of OpenEye Scientific Software, and JDC serves on the advisory board of Schrödinger.
We thank Drs. Christopher Fennell (Oklahoma State), Bryce Manubay (University of Colorado Boulder), Paul Nerenberg (California State University, Los Angeles), and Patrick Grinaway (MSKCC), for helpful discussions. The authors are grateful to members of the computational chemistry community producing high-quality open source software, especially Jason Swails (ParmEd), without which this project would not be possible. We also particularly thank OpenEye Scientific Software for granting us an academic software license, and for helpful discussions and suppory. including CIB’s sabbatical in the Mobley lab at UCI to help initiate this work.
Footnotes
6 Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at (details) and includes a PDF containing a 2D pictures of the full AlkEthOH compound set, probability distributions (measured from simulations) for the problematic torsions highlighted in Figure 2e, a discussion of the MDL and OpenEye MDL aromaticity models, a discussion of parameters imported from GAFF2; and an extensive set of supporting files, including a full archive copy of the openforcefield GitHub repository including our code, an archive copy of the smirnoff99Frosst GitHub repository, an archive copy of our SMIRNOFF_paper_code GitHub repository containing code used specifically for this paper (such as for FreeSolv hydration calculations, and for torsional distribution calculations), and a full set of AlkEthOH molecules and AMBER parameter and coordinate files.
The GitHub repository provides examples applying SMIRNOFF force fields to set up and, in some cases, to conduct, simulations of a variety of different situations. These are found in the examples directory on the repository (a copy of which is archived here in the SI). Specifically, our examples include setup of a simple simulation of a small molecule in the gas phase, setup of a simulation of a mixed force field system where the ligand uses a SMIRNOFF force field and the protein uses an AMBER-family force field, and setup of a simulation of host-guest binding in water. In our examples, simulation preparation is done entirely in Python, except in the case of systems using mixed force fields (such as an AMBER force field for a protein) where use of AmberTools [12] is also necessary (via a Python wrapper).
A more detailed explanation of the development process is available elsewhere [52].
References
- [1].Foyer: A Package for Atom-Typing as Well as Applying and Disseminating Forcefields; 2018. https://github.com/mosdef-hub/foyer.
- [2].Abel R, Wang L, Harder ED, Berne BJ, Friesner RA. Advancing Drug Discovery through Enhanced Free Energy Calculations. Acc Chem Res. 2017. July; 50(7):1625–1632. [DOI] [PubMed] [Google Scholar]
- [3].Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX. 2015. September; 1–2:19–25. [Google Scholar]
- [4].Bannan CC, Burley KH, Chiu M, Shirts MR, Gilson MK, Mobley DL. Blind Prediction of Cyclohexane–Water Distribution Coefficients from the SAMPL5 Challenge. JComput Aided Mol Des. 2016. September; 30(11):1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bayly C, McKay D, Truchon J, An Informal AMBER Small Molecule Force Field: Parm@ Frosst; 2010. http://www.ccl.net/cca/data/parm_at_Frosst/. [Google Scholar]
- [6].Beauchamp KA, Behr JM, Rustenburg AS, Bayly CI, Kroenlein K, Chodera JD. Towards Automated Benchmarking of Atomistic Forcefields: Neat Liquid Densities and Static Dielectric Constants from the ThermoML Data Archive. J Phys Chem B. 2015. September; 119(40):12912–12920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Black JE, Silva GMC, Klein C, Iacovella CR, Morgado P, Martins LFG, Filipe EJM, McCabe C. Perfluoropolyethers: Development of an All-Atom Force Field for Molecular Simulations and Validation with New Experimental Vapor Pressures and Liquid Densities. J Phys Chem B. 2017. July; 121(27):6588–6600. [DOI] [PubMed] [Google Scholar]
- [8].Bowers K, Chow E, Xu H, Dror R, Eastwood M, Gregersen B, Klepeis J, Kolossvary I, Moraes M, Sacerdoti F, Salmon J, Shan Y, Shaw D. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In: Proceedings of the ACM/IEEE Conference on Supercomputing (SC06) IEEE; 2006. p. 43–43. [Google Scholar]
- [9].Boyce SE, Mobley DL, Rocklin GJ, Graves AP, Dill KA, Shoichet BK. Predicting Ligand Binding Affinity with Alchemical Free Energy Methods in a Polar Model Binding Site. J Mol Biol. 2009. December; 394(4):747–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Brooks BR, Brooks CL, Mackerell AD, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, et al. CHARMM: The Biomolecular Simulation Program. Journal of Computational Chemistry. ; 30(10):1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Caleman C, van Maaren PJ, Hong M, Hub JS, Costa LT, van der Spoel D. Force Field Benchmark of Organic Liquids: Density, Enthalpy of Vaporization, Heat Capacities, Surface Tension, Isothermal Compressibility, Volumetric Expansion Coefficient, and Dielectric Constant. J Chem Theory Comput. 2012. January; 8(1):61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Case DA, Cerutti DS, Cheatham TE III, Darden TA, Duke RE, Giese TJ, Gohlke H, Goetz AW, Greene D, Homeyer N, Izadi S, Kovalenko A, Lee TS, LeGrand S, Li P, Lin C, Liu J, Luchko T, Luo R, Mermelstein D, et al. , AmberTools17. San Francisco, CA; 2017. [Google Scholar]
- [13].Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, Onufriev A, Simmerling C, Wang B, Woods RJ. The Amber Biomolecular Simulation Programs. J Comp Chem. 2005. December; 26(16):1668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chipot C, Pohorille A, editors. Free Energy Calculations: Theory and Applications in Chemistry and Biology Springer Series in Chemical Physics, Berlin Heidelberg: Springer-Verlag; 2007. [Google Scholar]
- [15].Chodera JD, Rizzi A, Naden LN, Beauchamp KA, Grinaway P, Rustenburg AS, Albanese SK, Saladi S. Choderalab/Yank: 0.16.0 Full API and Python 3.6. Zenodo; 2017. [Google Scholar]
- [16].Christ CD, Binding Affinity Prediction from Molecular Simulations: A New Standard Method in Structure-Based Drug Design?; 2016. dx.doi.org/10.7490/f1000research.1112651.1. [Google Scholar]
- [17].Christ CD, Fox T. Accuracy Assessment and Automation of Free Energy Calculations for Drug Design. J Chem Inf Model. 2014. January; 54(1):108–120. [DOI] [PubMed] [Google Scholar]
- [18].Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J Am Chem Soc. 1995. May; 117(19):5179–5197. [Google Scholar]
- [19].Deng N, Forli S, He P, Perryman A, Wickstrom L, Vijayan RSK, Tiefenbrunn T, Stout D, Gallicchio E, Olson AJ, Levy RM. Distinguishing Binders from False Positives by Free Energy Calculations: Fragment Screening Against the Flap Site of HIV Protease. J Phys Chem B. 2015. January; 119(3):976–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Dixon R, SimTK: OEAntechamber Assign and Generate AMBER Atom Types and Structural Parameters: Project Home; 2010. https://simtk.org/projects/oeante. [Google Scholar]
- [21].Duarte Ramos Matos G, Kyu DY, Loeffler HH, Chodera JD, Shirts MR, Mobley DL. Approaches for Calculating Solvation Free Energies and Enthalpies Demonstrated with an Update of the FreeSolv Database. J Chem Eng Data. 2017. May; 62(5):1559–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Eastman P, Swails J, Chodera JD, McGibbon RT, Zhao Y, Beauchamp KA, Wang LP, Simmonett AC, Harrigan MP, Stern CD, Wiewiora RP, Brooks BR, Pande VS. OpenMM 7: Rapid Development of High Performance Algorithms for Molecular Dynamics. PLOS Comput Biol. 2017. July; 13(7):e1005659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Fennell CJ, Wymer KL, Mobley DL. A Fixed-Charge Model for Alcohol Polarization in the Condensed Phase, and Its Role in Small Molecule Hydration. J Phys Chem B. 2014. June; 118(24):6438–6446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Geballe MT, Guthrie JP. The SAMPL3 Blind Prediction Challenge: Transfer Energy Overview. J Comput Aided Mol Des. 2012. April; 26(5):489–496. [DOI] [PubMed] [Google Scholar]
- [25].Harder E, Damm W, Maple J, Wu C, Reboul M, Xiang JY, Wang L, Lupyan D, Dahlgren MK, Knight JL, Kaus JW, Cerutti DS, Krilov G, Jorgensen WL, Abel R, Friesner RA. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J Chem Theory Comput. 2016. January; 12(1):281–296. [DOI] [PubMed] [Google Scholar]
- [26].Hawkins PCD, Nicholls A. Conformer Generation with OMEGA: Learning from the Data Set and the Analysis of Failures. - PubMed - NCBI. J Chem Inf Model. 2012. November; 52(11):2919–2936. [DOI] [PubMed] [Google Scholar]
- [27].Hawkins PCD, Skillman AG, Warren GL, Ellingson BA, Stahl MT. Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures from the Protein Databank and Cambridge Structural Database. J Chem Inf Model. 2010. April; 50(4):572–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Henriksen NM, Fenley AT, Gilson MK. Computational Calorimetry: High-Precision Calculation of Host–Guest Binding Thermodynamics. J Chem Theory Comput. 2015. September; 11(9):4377–4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Comparison of Multiple Amber Force Fields and Development of Improved Protein Backbone Parameters. Proteins. 2006. November; 65(3):712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Horta BAC, Merz PT, Fuchs PFJ, Dolenc J, Riniker S, Hünenberger PH. A GROMOS-Compatible Force Field for Small Organic Molecules in the Condensed Phase: The 2016H66 Parameter Set. J Chem Theory Comput. 2016. August; 12(8):3825–3850. [DOI] [PubMed] [Google Scholar]
- [31].Iacovella CR, Sallai J, Klein C, Ma T. Idea Paper: Development of a Software Framework for Formalizing Force1eld Atom-Typing for Molecular Simulation. 4th Workshop on Sustainable Software for Science: Practice and Experiences (WSSSPE4). 2016. June; p. 8. [Google Scholar]
- [32].Inc DCIS, Daylight Theory: SMARTS - A Language for Describing Molecular Patterns; 2018. http://www.daylight.com/dayhtml/doc/theory/theory.smarts.html.
- [33].Inc DCIS, Daylight Theory: SMIRKS - A Reaction Transform Language; 2018. http://www.daylight.com/dayhtml/doc/theory/theory.smirks.html.
- [34].Irwin JJ, Shoichet BK. ZINC–a Free Database of Commercially Available Compounds for Virtual Screening. J Chem Inf Model. 2005. Jan-Feb; 45(1):177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Irwin JJ, Sterling T, Mysinger MM, Bolstad ES, Coleman RG. ZINC: A Free Tool to Discover Chemistry for Biology. J Chem Inf Model. 2012. July; 52(7):1757–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Jakalian A, Bush BL, Jack DB, Bayly CI. Fast, Efficient Generation of High-Quality Atomic Charges. AM1-BCC Model: I. Method. J Comput Chem. 2000. January; 21(2):132–146. [DOI] [PubMed] [Google Scholar]
- [37].Jakalian A, Jack DB, Bayly CI. Fast, Efficient Generation of High-Quality Atomic Charges. AM1-BCC Model: II. Parameterization and Validation. J Comput Chem. 2002. December; 23(16):1623–1641. [DOI] [PubMed] [Google Scholar]
- [38].Jin Z, Yang C, Cao F, Li F, Jing Z, Chen L, Shen Z, Xin L, Tong S, Sun H. Hierarchical Atom Type Definitions and Extensible All-Atom Force Fields. J Comput Chem. 2016. March; 37(7):653–664. [DOI] [PubMed] [Google Scholar]
- [39].Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of Simple Potential Functions for Simulating Liquid Water. J Chem Phys. 1983. July; 79(2):926–935. [Google Scholar]
- [40].Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. Journal of the American Chemical Society. 1996. January; 118(45):11225–11236. [Google Scholar]
- [41].Kenney IM, Beckstein O, Iorga BI. Prediction of Cyclohexane-Water Distribution Coefficients for the SAMPL5 Data Set Using Molecular Dynamics Simulations with the OPLS-AA Force Field. J Comput Aided Mol Des. 2016. August; 30(11):1–14. [DOI] [PubMed] [Google Scholar]
- [42].Klimovich PV, Mobley DL. Predicting Hydration Free Energies Using All-Atom Molecular Dynamics Simulations and Multiple Starting Conformations. J Comput Aided Mol Des. 2010. April; 24(4):307–316. [DOI] [PubMed] [Google Scholar]
- [43].Knight JL, Yesselman JD, Brooks CL III. Assessing the Quality of Absolute Hydration Free Energies among CHARMM-Compatible Ligand Parameterization Schemes. J Comput Chem. 2013. January; 34(11):893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Knox C, Law V, Jewison T, Liu P, Ly S, Frolkis A, Pon A, Banco K, Mak C, Neveu V, Djoumbou Y, Eisner R, Guo AC, Wishart DS. DrugBank 3.0: A Comprehensive Resource for ‘omics’ Research on Drugs. Nucleic Acids Res. 2011. January; 39(Database issue):D1035–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Krizhevsky A, Sutskever I, Hinton GE. ImageNet Classification with Deep Convolutional Neural Networks In: Pereira F, Burges CJC, Bottou L, Weinberger KQ, editors. Advances in Neural Information Processing Systems 25 Curran Associates, Inc; 2012. p. 1097–1105. [Google Scholar]
- [46].Kuhn B, Tichý M, Wang L, Robinson S, Martin RE, Kuglstatter A, Benz J, Giroud M, Schirmeister T, Abel R, Diederich F, Hert J. Prospective Evaluation of Free Energy Calculations for the Prioritization of Cathepsin L Inhibitors. J Med Chem. 2017. March; 60(6):2485–2497. [DOI] [PubMed] [Google Scholar]
- [47].Laury ML, Wang LP, Pande VS, Head-Gordon T, Ponder JW. Revised Parameters for the AMOEBA Polarizable Atomic Multipole Water Model. J Phys Chem B. 2015. July; 119(29):9423–9437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Law V, Knox C, Djoumbou Y, Jewison T, Guo AC, Liu Y, Maciejewski A, Arndt D, Wilson M, Neveu V, Tang A, Gabriel G, Ly C, Adamjee S, Dame ZT, Han B, Zhou Y, Wishart DS. DrugBank 4.0: Shedding New Light on Drug Metabolism. Nucleic Acids Res. 2014. January; 42(Database issue):D1091–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Li C, Strachan A. Molecular Scale Simulations on Thermoset Polymers: A Review. J Polym Sci Part B: Polym Phys. 2015. January; 53(2):103–122. [Google Scholar]
- [50].Lovering F, Aevazelis C, Chang J, Dehnhardt C, Fitz L, Han S, Janz K, Lee J, Kaila N, McDonald J, Moore W, Moretto A, Papaioannou N, Richard D, Ryan MS, Wan ZK, Thorarensen A. Imidazotriazines: Spleen Tyrosine Kinase (Syk) Inhibitors Identi1ed by Free-Energy Perturbation (FEP). ChemMedChem. 2016. January; 11(2):217–233. [DOI] [PubMed] [Google Scholar]
- [51].Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J Chem Theory Comput. 2015. August; 11(8):3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Mobley D, Bannan CC, Rizzi A, Bayly CI, Chodera JD, Lim VT, Lim NM, Beauchamp KA, Shirts MR, Gilson MK, Eastman PK. Open Force Field Consortium: Escaping Atom Types Using Direct Chemical Perception with SMIRNOFF v0.1. bioRxiv. 2018. March; p. 286542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Mobley DL, Bayly CI, Cooper MD, Dill KA. Predictions of Hydration Free Energies from All-Atom Molecular Dynamics Simulations. J Phys Chem B. 2009. January; 113:4533–4537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Mobley DL, Bayly CI, Cooper MD, Shirts MR, Dill KA. Small Molecule Hydration Free Energies in Explicit Solvent: An Extensive Test of Fixed-Charge Atomistic Simulations. J Chem Theory Comput. 2009. February; 5(2):350–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Mobley DL, Graves AP, Chodera JD, McReynolds AC, Shoichet BK, Dill KA. Predicting Absolute Ligand Binding Free Energies to a Simple Model Site. J Mol Biol. 2007. August; 371(4):1118–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Mobley DL, Guthrie JP. FreeSolv: A Database of Experimental and Calculated Hydration Free Energies, with Input Files. J Comput Aided Mol Des. 2014. July; 28(7):711–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Mobley DL, Liu S, Cerutti DS, Swope WC, Rice JE. Alchemical Prediction of Hydration Free Energies for SAMPL. J Comput Aided Mol Des. 2012. January; 26(5):551–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Muddana HS, Fenley AT, Mobley DL, Gilson MK. The SAMPL4 Host–Guest Blind Prediction Challenge: An Overview. J Comput Aided Mol Des. 2014. March; 28(4):305–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Muddana HS, Varnado CD, Bielawski CW, Urbach AR, Isaacs L, Geballe MT, Gilson MK. Blind Prediction of Host–Guest Binding Affinities: A New SAMPL3 Challenge. J Comput Aided Mol Des. 2012. February; 26(5):475–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: An Open Chemical Toolbox. J Cheminformatics. 2011. October; 3:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Paranahewage SS, Gierhart CS, Fennell CJ. Predicting Water-to-Cyclohexane Partitioning of the SAMPL5 Molecules Using Dielectric Balancing of Force Fields. J Comput Aided Mol Des. 2016. August; 30(11):1059–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K. Scalable Molecular Dynamics with NAMD. J Comput Chem. 2005. December; 26(16):1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Piana S, Klepeis JL, Shaw DE. Assessing the Accuracy of Physical Models Used in Protein-Folding Simulations: Quantitative Evidence from Long Molecular Dynamics Simulations. Curr Opin Struct Biol. 2014. February; 24:98–105. [DOI] [PubMed] [Google Scholar]
- [64].Plimpton S Fast Parallel Algorithms for Short-Range Molecular Dynamics. Journal of Computational Physics. 1995. March; 117(1):1–19. [Google Scholar]
- [65].Ponder JW, Case DA. Force Fields for Protein Simulations In: Advances in Protein Chemistry, vol. 66 of Protein Simulations Academic Press; 2003. p. 27–85. [DOI] [PubMed] [Google Scholar]
- [66].Riniker S Fixed-Charge Atomistic Force Fields for Molecular Dynamics Simulations in the Condensed Phase: An Overview. J Chem Inf Model. 2018. March;. [DOI] [PubMed] [Google Scholar]
- [67].Rocklin GJ, Boyce SE, Fischer M, Fish I, Mobley DL, Shoichet BK, Dill KA. Blind Prediction of Charged Ligand Binding Affinities in a Model Binding Site. Journal of Molecular Biology. 2013. November; 425(22):4569–4583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Sherborne B, Shanmugasundaram V, Cheng AC, Christ CD, DesJarlais RL, Duca JS, Lewis RA, Loughney DA, Manas ES, McGaughey GB, Peishoff CE, van Vlijmen H. Collaborating to Improve the Use of Free-Energy and Other Quantitative Methods in Drug Discovery. J Comput Aided Mol Des. 2016. December; 30(12):1139–1141. [DOI] [PubMed] [Google Scholar]
- [69].Shirts MR, Chodera JD. Statistically Optimal Analysis of Samples from Multiple Equilibrium States. J Chem Phys. 2008. September; 129(12):124105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Shirts MR, Klein C, Swails JM, Yin J, Gilson MK, Mobley DL, Case DA, Zhong ED. Lessons Learned from Comparing Molecular Dynamics Engines on the SAMPL5 Dataset. J Comput Aided Mol Des. 2017. January; 31(1):147–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Shirts MR, Mobley DL, Chodera JD, Pande VS. Accurate and Efficient Corrections for Missing Dispersion Interactions in Molecular Simulations. J Phys Chem B. 2007. November; 111(45):13052–13063. [DOI] [PubMed] [Google Scholar]
- [72].Software OS, OEChem Toolkit. Santa Fe, NM, USA; 2018. [Google Scholar]
- [73].Steinbrecher T, Zhu C, Wang L, Abel R, Negron C, Pearlman D, Feyfant E, Duan J, Sherman W. Predicting the Effect of Amino Acid Single-Point Mutations on Protein Stability—Large-Scale Validation of MD-Based Relative Free Energy Calculations. J Mol Biol. 2017. April; 429(7):948–963. [DOI] [PubMed] [Google Scholar]
- [74].Sun H, Jin Z, Yang C, Akkermans RLC, Robertson SH, Spenley NA, Miller S, Todd SM. COMPASS II: Extended Coverage for Polymer and Drug-like Molecule Databases. J Mol Model. 2016. February; 22(2):47. [DOI] [PubMed] [Google Scholar]
- [75].Tan D, Piana S, Dirks RM, Shaw DE. RNA Force Field with Accuracy Comparable to State-of-the-Art Protein Force Fields. PNAS. 2018. January; p. 201713027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].van der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJC. GROMACS: Fast, Flexible, and Free. J Comp Chem. 2005; 26(16):1701–1718. [DOI] [PubMed] [Google Scholar]
- [77].Veenstra DL, Ferguson DM, Kollman PA. How Transferable Are Hydrogen Parameters in Molecular Mechanics Calculations? J Comput Chem. 1992. October; 13(8):971–978. [Google Scholar]
- [78].van Vlijmen H, Desjarlais RL, Mirzadegan T. Computational Chemistry at Janssen. J Comput Aided Mol Des. 2017. March; 31(3):267–273. [DOI] [PubMed] [Google Scholar]
- [79].Wang CC, Pilania G, Boggs SA, Kumar S, Breneman C, Ramprasad R. Computational Strategies for Polymer Dielectrics Design. Polymer. 2014. February; 55(4):979–988. [Google Scholar]
- [80].Wang J A Snapshot of GAFF2 Development; 2017. [Google Scholar]
- [81].Wang J, Cieplak P, Kollman PA. How Well Does a Restrained Electrostatic Potential (RESP) Model Perform in Calculating Conformational Energies of Organic and Biological Molecules? J Comput Chem. 2000. September; 21(12):1049–1074. Parm99. [Google Scholar]
- [82].Wang J, Wang W, Kollman PA, Case DA. Automatic Atom Type and Bond Type Perception in Molecular Mechanical Calculations. J Mol Graph Model. 2006. October; 25(2):247–260. [DOI] [PubMed] [Google Scholar]
- [83].Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and Testing of a General AMBER Force Field. J Comp Chem. 2004. July; 25(9):1157–1174. [DOI] [PubMed] [Google Scholar]
- [84].Wang K, Chodera JD, Yang Y, Shirts MR. Identifying Ligand Binding Sites and Poses Using GPU-Accelerated Hamiltonian Replica Exchange Molecular Dynamics. J Comput Aided Mol Des. 2013. December; 27(12):989–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Wang LP, Chen J, Van Voorhis T. Systematic Parametrization of Polarizable Force Fields from Quantum Chemistry Data. J Chem Theory Comput. 2013. January; 9(1):452–460. [DOI] [PubMed] [Google Scholar]
- [86].Wang LP, Head-Gordon T, Ponder JW, Ren P, Chodera JD, Eastman PK, Martinez TJ, Pande VS. Systematic Improvement of a Classical Molecular Model of Water. J Phys Chem B. 2013. August; 117(34):9956–9972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Wang LP, Martinez TJ, Pande VS. Building Force Fields: An Automatic, Systematic, and Reproducible Approach. J Phys Chem Lett. 2014. June; 5(11):1885–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Wang LP, McKiernan KA, Gomes J, Beauchamp KA, Head-Gordon T, Rice JE, Swope WC, Martínez TJ, Pande VS. Building a More Predictive Protein Force Field: A Systematic and Reproducible Route to AMBER-FB15. J Phys Chem B. 2017. April; 121(16):4023–4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Wishart DS, Knox C, Guo AC, Cheng D, Shrivastava S, Tzur D, Gautam B, Hassanali M. DrugBank: A Knowledgebase for Drugs, Drug Actions and Drug Targets. Nucleic Acids Res. 2008. January; 36(Database issue):D901–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Wishart DS, Knox C, Guo AC, Shrivastava S, Hassanali M, Stothard P, Chang Z, Woolsey J. DrugBank: A Comprehensive Resource for in Silico Drug Discovery and Exploration. Nucleic Acids Res. 2006. January; 34(Database issue):D668–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Wu Z, Ramsundar B, N Feinberg E, Gomes J, Geniesse C, S Pappu A, Leswing K, Pande V. MoleculeNet: A Benchmark for Molecular Machine Learning. Chemical Science. 2018; 9(2):513–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Yin J, Henriksen NM, Slochower DR, Shirts MR, Chiu MW, Mobley DL, Gilson MK. Overview of the SAMPL5 Host–Guest Challenge: Are We Doing Better? J Comput Aided Mol Des. 2017; 31(1):1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Zanette C, Bannan CC, Bayly CI, Fass J, Gilson MK, Shirts MR, Chodera J, Mobley DL. Toward Learned Chemical Perception of Force Field Typing Rules. chemRxiv. 2018. May;. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.