

Abstract
Several sulfur-containing platensimycin (PTM) and platencin (PTN) analogues, with activities comparable to the parent natural products, have recently been discovered from microorganisms, implying a biomimetic route to diversify the PTM and PTN scaffolds for structure−activity relationship study. We present here a substrate-directed and scaleable semisynthetic strategy to make PTM and PTN sulfur analogues with excellent diasteroselectivity, without using any chiral catalysts. Most of the sulfur analogues showed strong activities against clinical Staphylococcus aureus isolates, with minimum inhibitory concentrations of 0.5−2 μg mL−1. Density functional theory calculations were in agreement with the observed selectivity for these analogues and suggest that the conformation restraints of the terpene cages of PTM and PTN on the transition states determine the si-face attack selectivity.
Graphical Abstract
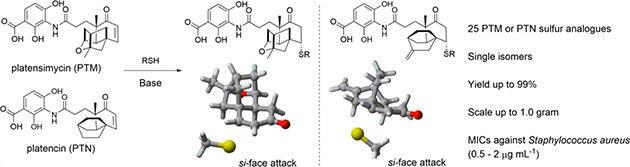
Many natural products or drugs are organosulfur compounds, such as the antibiotics penicillin and cephalosporin, the anticancer agents bleomycin and calicheamicin, and top selling drugs, including rosuvastatin for dyslipidemia and fluticasone for asthma.1,2 Platensimycin (PTM) and platencin (PTN), promising antibiotics or antidiabetic agents, were first isolated from the soil-dwelling bacteria Streptomyces platensis.3–5 Because of their interesting biological activities and unprecedented structures, extensive syntheses of PTM and PTN analogues in the past decade have resulted in a few analogues with comparable or enhanced antibacterial activity, such as adamantaplatensimycin, carboplatensimycin, and 7-phenyl- and 11-methyl 7-phenylplatensimycin.6–10 We have been interested in the biosynthesis of PTM and PTN and recently discovered several sulfur-containing analogues of PTM and PTN from fermentation, such as PTM S1, PTN S1, PTM D1, and PTM ML14, in which PTM D1 showed improved antibacterial activity against Staphylococcus aureus compared to PTM (Figure 1).11,12 However, the poor systemic pharmacokinetic properties of PTM and PTN and the lack of very active PTM or PTN analogues still significantly limit the further clinical development of this fascinating type of molecule.13–15
Figure 1.

Structures of PTM and PTN sulfur analogues.
Carbon−sulfur bond formation plays a prominent role in the generation of many valuable pharmaceuticals. The asymmetric sulfa-Michael addition (SMA) to substituted enones would generate chiral centers, which requires the presence of a chiral auxiliary in the sulfur donor or Michael acceptor or the development of very efficient chiral catalysts.16–22 The natural PTM congener PTM D1 was likely formed by the stereo-specific attack of PTM S1 to the enone of PTM and directed by the rigid terpene cage of the reaction substrate PTM.23 PTM ML14 could be produced in a similar manner. By mimicking the formation of these natural products, we therefore envisioned a versatile approach for the stereoselective semi-synthesis of PTM sulfur analogues. The PTN sulfur analogues may also be synthesized due to the structure similarity between PTM and PTN, although its natural congeners have not been discovered. The availability of bulk amounts of PTM and PTN from fermentation, the many thioacids or thoils available commercially, and the facile conversion of carboxylic acids to thioacids would enable us to rapidly synthesize a series of sulfur-containing PTM and PTN analogues.24–30
RESULTS AND DISCUSSION
We started our study with the reaction of PTM and benzothioic acid 1a or 4-chlorobenzothioic acid 1b, catalyzed by different bases in various solvents (Scheme 1 and Tables S2 and S3). The expected SMA product 2a was conveniently obtained as a single isomer with quantitative yields (Scheme 1, entries 2/3 and 8/9) catalyzed by either organic or inorganic bases. The HMBC correlation between H-7 and C-18 of the purified 2a, as well as the loss of enone, suggested the attachment of sulfur (Table S3 and Figures S1−S4). The stereochemistry at C-7 was confirmed by ROESY NMR analysis, with a correlation between H-7 (δH 4.09) and H-13 (δH 1.83) and a correlation between H-13 (δH 2.24) and CH3-17 (δH 1.32), similar to the stereochemistry found in PTM ML14 and PTM D1, as well as the synthesis of other terpene cage modified PTM analogues (Figures S5 and S6).4,10 In contrast, 2b was produced in yields of 90−98% only when catalyzed by inorganic bases, including Na2CO3, K2CO3, Cs2CO3, and NaOH (Scheme 1, entries 7−10). Among the solvents tested, dichloromethane (DCM) was the best for the synthesis of 2b, while the high-yield synthesis of 2a could be achieved in most solvents, even including water with a yield of 81% (Table S2).
Scheme 1. Optimization for the Sulfa-Michael Reaction Conditions of PTM and 1a or 1ba.

aOn a 0.1 mmol scale in dichloromethane, PTM:1a or 1b = 1.0:3.0. bDBU: 1,8-diazabicyclo[5.4.0]undec-7-ene; TBA-OH: tetrabutylammonium hydroxide. cYields of 2a or 2b determined by HPLC, and single isomers were obtained.
To examine the generality of this diastereoselective reaction under the optimized reaction conditions, using DCM as the solvent and 50 mol % of Cs2CO3 as the catalyst, sulfur-containing PTM analogues 2 were synthesized from a variety of thioacid or thiol compounds 1 (Scheme 2). Most SMA reactions proceeded smoothly to afford 2 in good to quantitative yields with excellent diastereoselectivities (single isomers were obtained for all cases), for either electron-donating groups (2c, 2d) or electron-withdrawing groups (2e− 2j and 2l−2n) on the benzothioic acids. The SMA adduct 2k was obtained with a 20% yield, which was possibly due to an intramolecular hydrogen bond between the thiolate and the fluoride of the 2-CF3 substituent. Similarly, when sterically demanding naphthalene-1-carbothioic acid or naphthalene-2-carbothioic acid was individually employed as the substrate, only the latter reacted with PTM to generate the naphthalene derivative 2p with a yield of 33%. In addition, the SMA products 2o and 2q−2t were obtained in excellent yields and as single isomers, which demonstrated that this reaction may be extended to heterocyclic and aliphatic thioacids, as well as aromatic and aliphatic thiols, greatly expanding the structure diversity beyond those natural sulfur-containing congeners.
Scheme 2. Scope of the Sulfa-Michael Addition between PTM and Thioacid or Thiol Compounds 1a.

aOn a 0.1 mmol scale, PTM:1:Cs2CO3 = 1.0:3.0:0.5; all products 2 were obtained as single isomers only and shown in isolated yields.
bTen equivalents of EtSH 1t was used.
Interestingly, PTM would react with Cs2CO3 to generate PTM cesium salt in the reaction with ethanethiol (EtSH) 1t in CD3OD, probably due to the stronger acidity of the 2,4-dihydroxy 3-aminobenzoic acid (ADHBA) moiety on PTM than 1t (Figure S7). Therefore, the forming PTM salt might have an unusual dual role in this reaction, to be the substrate and the working catalyst for the synthesis of 2t at the same time (Table S5 and Figures S8−13). In contrast, the formation of PTM salt was prohibitive if the thioacid 1q with stronger acidity than PTM was used (Figure S7).
To further explore the synthetic utility of this SMA reaction, it was conducted on a gram scale by treatment of 2.5 mmol of PTM and 7.5 mmol of commercially available thiobenzoic acid 1a, catalyzed by either DBU or Cs2CO3. The resulting product 2a (1.24 or 1.17 g, 86% or 81% yield, respectively) was obtained as a single isomer, and the remaining small amount of PTM was recovered.
Next we evaluated if PTN and a semisynthesized platensic acid ethylester 4 could be used as alternative substrates under the same reaction conditions (Scheme 3).27 Satisfactorily, four PTN sulfur analogues were obtained as single isomers and in good yields. The attachment of the sulfur and the stereo-chemistry at C-7 were verified by HMBC and ROESY analysis for 3b (Table S6 and Figures S14−19). Similar to the synthesis of 2 catalyzed by Cs2CO3, the sulfur-containing adduct 5 was conveniently obtained as a single isomer by the reaction of 4 and thiobenzoic acid 1a with a yield of 80%, and its stereochemistry was confirmed (Table S7 and Figures S20− S25).
Scheme 3. Scope of the Sulfa-Michael Addition between PTN (A) or Platensic Acid Ethylester 4 (B) and Thioacid or Thiol Compounds 1a.

aOn a 0.1 mmol scale, PTN or 4:1:Cs2CO3 = 1.0:3.0:0.5; products 3 and 5 were obtained as single isomers only.
Quantum mechanical calculations have been applied successfully to predict the mechanism and sterochemical outcome of asymmetric SMA reactions, catalyzed by a variety of chiral organocatalysts or enzymes, such as cinchona alkaloid-derived urea or thiourea, or lanthionine synthetases.31–35 In the latter cases, the substrates, instead of the enzymes, are in fact the determinants for the re or si face selectivity for the formation of the specific lantipeptides. To gain insight into the stereochemical control of the SMA reactions toward PTM or PTN, directed by themselves and without any chiral catalysts, as well as the biosynthesis of the natural products PTM D1 and PTM ML14, we located the transition states (TSs) for the C−S bond-forming steps using M06–2X/6–31+G(d) with the CPCM continuum solvent model. The single-point energies were further calculated using M06–2X/6–311+G(d,p) with the same solvent model and were used to correct the gas phase energies obtained from the M06–2X/6–31G(d) optimizations, which have been shown to give the closest agreement with the experiments for many SMA reactions.36–38
To simplify our calculation, the previous key synthetic intermediate 6 or 7 in the total synthesis of PTM or PTN and the MeS− anion were used in the model SMA reactions (Figure 2A).13 Our density functional theory (DFT) calculations suggested that there is a large difference in the activation barriers (ΔΔG⧧) calculated for the additions to the re or si faces of PTM mimic 6 (Figure 2B) or PTN mimic 7 (Figure 2C). In both cases, it favors the diastereoselective formation of 7S products by 3.1 kcal mol−1 (>99.5:0.5 dr at 25 °C from TS-1A) or 3.8 kcal mol−1 (>99.8:0.2 dr at 25 °C from TS-2A), in agreement with our experimental observations (Schemes 1−3). The strong influence of the steric hindrance from the terpene cages was also reflected in the large Me−S···C=C dihedral angle in the TSs (−51° for TS-1B and −56° for TS-2B) for the unfavorable re-face attack, in contrast to smaller dihedral angles (14−28°) found in thiol addition to simple substituted enones, since the latter conformation would further allow some favorable electrostatic interaction between the reactants.36
Figure 2.

C−S bond-forming TSs in the asymmetric SMA reactions of MeS− to PTM and PTN mimics. M06–2X/6–31+G(d,p)/CPCM (dichloromethane) was used. All energies are in kcal mol−1 and bond distances in Å.
The thermodynamics parameters were also calculated for the Michael additions of MeSH to enones 6 and 7, and the theoretical values of ΔH and ΔG for the addition of MeSH to both enones were favorable (Table S1). Considering that most Michael donors used in our SMA reactions are significantly larger than methanethiol, this preference for the si-face approach is rationalized due to the bulky cage fragment that is sterically biased to proceed exclusively from the bottom face. The strict stereoselectivity was consistent with the previous semisynthesis of several C-7-substituted PTM adducts using Grignard reagents or the Corey−Chaykovsky cyclopropanation.10
All the sulfur-containing PTM and PTN analogues, except compound 5, which did not show any antibacterial activities due to the lack the essential ADHBA moiety, were tested against S. aureus ATCC 29213, four methicillin-sensitive S. aureus (MSSA) strains, and seven methicillin-resistant S. aureus (MRSA) strains isolated from local hospitals, using the standard agar dilution method with PTM, PTN, and linezolid as positive controls (Table 1).8,27,39 The PTM and PTN sulfur analogues displayed excellent antibacterial activities with minimum inhibitory concentrations (MICs) ranging from 0.5−2 μg mL−1 toward most tested strains, comparable to PTM and PTN. Interestingly, several compounds, such as 2o, 2p, 2s, 2r, 3c, and 3d, showed much higher activities against MSSA strains than MRSA strains, in which the PTN thiophenol adduct 3d had an MIC of 0.5−1 μg mL−1 toward MSSA, even better than its parent compound. Therefore, these new analogues would provide exciting opportunities to address the unfavorable pharmacokinetics of PTM and PTN, while retaining the potent antibacterial activities.13–15,28 For example, 10 of our analogues (2e−2g, 2i−2k, 2m, 2n, 2r, and 3c) contain fluorine or −CF3 substituents, which are known to be highly advantageous to improve the bioavailability of many lead compounds.40
Table 1.
Antibacterial Activities of PTM and PTN Sulfur Analogues against S. aureus ATCC 29213 and Clinical MSSA and MRSA Strains
| Compounds | ATCC 29213 |
MSSA | MRSA | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | ||
| PTM | 1 | 0.5 | 0.5 | 0.5 | 0.5 | 1 | 1 | 1 | 1 | 1 | 0.5 | 1 |
| PTN | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Linezolid | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| 2a | 1 | 1 | 1 | 1 | 1 | 2 | 1 | 2 | 1 | 1 | 1 | 2 |
| 2b | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 2 |
| 2c | 1 | 1 | 1 | 1 | 1 | 2 | 1 | 2 | 1 | 1 | 1 | 2 |
| 2d | 1 | 1 | 1 | 1 | 1 | 2 | 1 | 2 | 1 | 1 | 1 | 2 |
| 2e | 1 | 1 | 1 | 1 | 1 | 2 | 1 | 1 | 2 | 1 | 1 | 2 |
| 2f | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 2 | 1 | 1 | 1 | 2 |
| 2g | 1 | 1 | 1 | 1 | 1 | 2 | 1 | 2 | 1 | 1 | 1 | 2 |
| 2h | 1 | 1 | 1 | 1 | 1 | 2 | 1 | 1 | 1 | 1 | 1 | 2 |
| 2i | 1 | 0.5 | 1 | 1 | 1 | 2 | 1 | 2 | 1 | 1 | 1 | 2 |
| 2j | 2 | 1 | 1 | 1 | 0.5 | 1 | 1 | 2 | 1 | 1 | 1 | 2 |
| 2k | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 2 | 1 | 1 | 1 | 2 |
| 2l | 1 | 1 | 1 | 1 | 1 | 2 | 1 | 2 | 1 | 1 | 1 | 2 |
| 2m | 2 | 1 | 1 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 |
| 2n | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 2 | 1 | 1 | 1 | 2 |
| 2o | 2 | 1 | 1 | 1 | 1 | 2 | 2 | 8 | 2 | 1 | 1 | 8 |
| 2p | 2 | 1 | 0.5 | 0.5 | 0.5 | 2 | 1 | 4 | 1 | 1 | 1 | 4 |
| 2q | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 4 | 2 | 2 | 2 | 8 |
| 2r | 2 | 2 | 1 | 2 | 1 | 2 | 2 | 4 | 2 | 2 | 2 | 4 |
| 2s | 4 | 2 | 2 | 2 | 2 | 4 | 2 | 4 | 2 | 2 | 2 | 4 |
| 2t | 0.5 | 0.5 | 0.5 | 1 | 1 | 1 | 2 | 1 | 1 | 1 | 1 | 1 |
| 3a | 4 | 1 | 1 | 1 | 1 | 2 | 2 | 4 | 2 | 2 | 1 | 4 |
| 3b | 4 | 1 | 1 | 1 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 4 |
| 3c | 4 | 1 | 1 | 1 | 0.5 | 2 | 2 | 4 | 1 | 1 | 1 | 4 |
| 3d | 2 | 1 | 0.5 | 0.5 | 0.5 | 2 | 1 | 2 | 1 | 1 | 1 | 2 |
| MIC Colour Scale (μg mL−1) | ≤0.5 | 1 | 2 | 4 | 8 | |||||||
Several PTM analogues, 2a, 2c, and 2t, and PTN analogue 3b were further tested for their stability in the phosphate-buffered saline (PBS) buffer or the agar plate assay conditions.39 All the tested compounds were more stable in acidic and neutral PBS, and over 80% of compounds 2a, 2c, and 3b reverted back to the retro-Michael products PTM and PTN under the basic conditions (Figure S72). In the agar plate assay conditions after overnight incubation at 37 °C, about 40−70% of the tested compounds reverted back to PTM or PTN (Figures S73). Under all the conditions tested, compound 2t is the most stable SMA product. Therefore, the instability of these SMA products under our assay conditions suggested that their observed antibacterial activities might be partly due to their parent compound PTM or PTN, probably resulting from the β-elimination of the SMA products. This was consistent with the in vivo stability of PTM D1, since only a small amount of PTM D1 was recovered from the pharmacokinetic study.28 Taken together, the above data might suggest an interesting prodrug strategy to deliver PTM or PTN in vivo, thus improving their poor bioavailability.
In conclusion we obtained 25 PTM and PTN sulfur analogues by a highly stereoselective and atom-economical synthesis. The SMA reactions could be scaled-up without loss of diastereoselectivity, the stereoselectivity of which could be predicted based on DFT calculations. The conformation of the rigid terpene cages of PTM and PTN has a strong influence on the TS geometry, a remarkable effect different from SMA reactions with less steric constraints. Most of the sulfur analogues displayed strong antibacterial activity against clinical MRSA strains. The study not only established the optimal conditions to rapidly make PTM and PTN sulfur analogues useful antimicrobial agents but showed how the stereo-chemistry of the SMA reactions was elegantly controlled through the rigid terpene cage preinstalled on the substrates in the biosynthesis of PTM D1 and PTM ML14. This chemistry raises the possibility to identify the SMA products of PTN from fermentation, thus shedding new light on its biosynthesis.
EXPERIMENTAL SECTION
General Experimental Procedures.
All commercially available reagents were directly used as received from vendors, unless otherwise stated. 1H and 13C NMR spectra were recorded on a Bruker 400 or 500 MHz spectrometer. Chemical shifts are reported in ppm relative to the internal standard tetramethylsilane (δ = 0 ppm) for 1H NMR and deuteriochloroform (δ = 77.00 ppm) for 13C NMR spectroscopy. The following abbreviations were used to designate chemical shift multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad. HRMS spectra were recorded on a Bruker ULTRAFLEX III TOF/TOF 200 instrument. All compounds examined possessed a purity of at least 95%. Details are available in the Supporting Information.
General Procedures for the Sulfa-Michael Addition Reaction.
To a stirred solution of PTM or PTN (0.10 mmol) in DCM (2.0 mL) were added Cs2CO3 (16.2 mg, 0.05 mmol) and thiol compound 1(0.30 mmol). The mixture was stirred under room temperature for 8h. TLC indicated the reaction was complete. HCl (2 M, 5.0 mL) was added. The resulting mixture was extracted with DCM (3 × 5.0 mL), and the combined organic portions were dried over anhydrous sodium sulfate. Concentration followed by flash column chromatography (eluent: EtOAc/MeOH, 30:1−5:1) afforded analogues 2 or 3.
Procedure for Preparation of 5 from PTMA Ester 4.
To a stirred solution of 4 (29.1 mg, 0.10 mmol) in DCM (2.0 mL) were added Cs2CO3 (16.2 mg, 0.05 mmol) and 4-chlorobenzothioic acid 1b(51.6 mg, 0.30 mmol). The mixture was stirred under room temperature for 8 h. TLC indicated the reaction was complete. HCl (2 M, 5.0 mL) was added. The resulting mixture was extracted with DCM (3 × 5.0 mL), and the combined organic portions were dried over anhydrous sodium sulfate. Concentration followed by flash column chromatography (eluent: light petroleum ether/EtOAc, 30:1− 5:1) afforded 5.
Scale-up Synthesis of 2a.
To a stirred solution of PTM (1.10 g,2.50 mmol) in DCM (50 mL) were added DBU (190 μL, 1.25 mmol) or Cs2CO3 (406 mg, 1.25 mmol) and benzothioic acid 1a (1.04 g, 7.50 mmol). The mixture was stirred under room temperature for 48 h and monitored by TLC. Upon completion of the reaction, 2 M HCl (50 mL) was added. The organic layer was separated, and the aqueous layer was extracted with DCM (3 × 50 mL). The combined organic portions was dried over anhydrous sodium sulfate, concentrated under vacuum, and purified with flash column chromatography (eluent: EtOAc/MeOH, 30:1−5:1) to afford 1.24 g for DBU and 1.17 g for Cs2CO3 of the PTM analogue 2a.
DFT Calculations on Transition States.
All the DFT calculations were carried out in the Gaussian09 software package (revision D.01).41The geometric structures of intermediates and transition states were fully optimized using the M06–2X functional method combined with the 6–31G(d) basis set. Solvent effects were considered implicitly during optimization and energy evaluation through the CPCM polarizable continuum model as implemented in Gaussian 09.42,43 Single-point energies were also calculated using M06–2X, the same solvation model, and the 6–311++G(d,p) basis set. This DFT function was recently demonstrated to have a superior performance in Michael-type addition reactions when sulfur nucleophiles were used.35–38 The frequency analysis was also performed based on the optimized structures to verify the energy minimum and transition states.
All discussed energy values are the Gibbs free energies calculated at the temperature 298 K and the distances in angstrom. Gibbs free energies (ΔG) were used for the discussion on the relative stabilities of the considered structures. The theoretical ratio of reaction products was obtained through the energy of the different transition states using a Maxwell−Boltzmann distribution at 298 K at which thermal and entropic corrections to energy were calculated.44 Computed structures were illustrated in GaussView or CYLview.45
Antibacterial Activity Assay.
The antibacterial activities of the 24 compounds were tested against S. aureus strains. MIC values of tested compounds were determined using an agar dilution assay. In brief, the S. aureus strains were cultivated on LB broth overnight and diluted. Approximate 104 colony forming units (CFU) of each strain were spotted (2 μL) onto LB agar plates containing different concentrations of the test compounds, ranging from 0.5 to 8 μg mL−1 LB agar. Then the plates were incubated in 37 °C overnight and monitored. The LB agar containing different concentrations of the tested compounds were prepared previously.
Stability Test of Selected PTM or PTN Sulfur Analogues.
The compounds 2a, 2c, 2t, and 3b (0.1 mg mL−1) were dissolved in 1 mM PBS (pH = 5.8, 7.1, or 8.0), incubated at 37 °C for 16 h, and then analyzed by HPLC. Alternatively, the compounds 2a, 2c, 2t, and 3b(0.1 mg mL−1) were diluted in 5 mL of LB agar (1.5% v/v) to 0.008 mg mL−1, and the resulting LB agar plates were incubated at 37 °C for 16 h. The LB agar with the tested compounds were then extracted by DCM (3 × 10 mL), dried by anhydrous Na2SO4, concentrated in vacuum, and analyzed by HPLC.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by NSFC grants 81473124 (to Y.H.) and 21502238 (to L.Q.), the Chinese Ministry of Education 111 Project B0803420 (to Y.D.), China Postdoctoral Science Foundation 2015M582353 and the Postdoctoral Science Foundation of Central South University (CSU) 155220 (to L.Q.), and NIH grant GM114353 (to B.S.). We thank the Center for Advanced Research in CSU for the HRMS and NMR experiments and China National Supercomputing Center in Shenzhen for the computation resources.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jnat-prod.7b00745.
Experimental details and characterization data (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Chauhan P; Mahajan S; Enders D Chem. Rev 2014, 114, 8807–8864. [DOI] [PubMed] [Google Scholar]
- (2).Jacob C Nat. Prod. Rep 2006, 23, 851–863. [DOI] [PubMed] [Google Scholar]
- (3).Wang J; Soisson SM; Young K; Shoop W; Kodali S; Galgoci A; Painter R; Parthasarathy G; Tang YS; Cummings R; Ha S; Dorso K; Motyl M; Jayasuriya H; Ondeyka J; Herath K; Zhang CW; Hernandez L; Allocco J; Basilio A; Tormo JR; Genilloud O; Vicente F; Pelaez F; Colwell L; Lee SH; Michael B; Felcetto T; Gill C; Silver LL; Hermes JD; Bartizal K; Barrett J; Schmatz D; Becker JW; Cully D; Singh SB Nature 2006, 441, 358–361. [DOI] [PubMed] [Google Scholar]
- (4).Wang J; Kodali S; Lee SH; Galgoci A; Painter R; Dorso K; Racine F; Motyl M; Hernandez L; Tinney E; Colletti SL; Herath K; Cummings R; Salazar O; Gonzalez I; Basilio A; Vicente F; Genilloud O; Pelaez F; Jayasuriya H; Young K; Cully DF; Singh SB Proc. Natl. Acad. Sci. U. S. A 2007, 104, 7612–7616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Singh SB; Kang L; Nawrocki AR; Zhou D; Wu M; Previs S; Miller C; Liu H; Hines CDG; Madeira M; Cao J; Herath K; Spears LD; Semenkovich CF; Wang L; Kelley DE; Li C; Guan HP PLoS One 2016, 11, e0164133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Nicolaou KC; Tang Y; Wang J; Stepan AF; Li A; Montero AJ Am. Chem. Soc 2007, 129, 14850–14851. [DOI] [PubMed] [Google Scholar]
- (7).Nicolaou KC; Lister T; Denton RM; Montero A; Edmonds DJ Angew. Chem., Int. Ed 2007, 46, 4712–4714. [DOI] [PubMed] [Google Scholar]
- (8).Nicolaou KC; Stepan AF; Lister T; Li A; Montero A; Tria GS; Turner CI; Tang Y; Wang J; Denton RM; Edmonds DJ J. Am. Chem. Soc 2008, 130, 13110–13119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Jang KP; Kim CH; Na SW; Kim H; Kang H; Lee E Bioorg. Med. Chem. Lett 2009, 19, 4601–4602. [DOI] [PubMed] [Google Scholar]
- (10).Shen HC; Ding F-X; Singh SB; Parthasarathy G; Soisson SM; Ha SN; Chen X; Kodali S; Wang J; Dorso K; et al. Bioorg. Med. Chem. Lett 2009, 19, 1623–1627. [DOI] [PubMed] [Google Scholar]
- (11).Rudolf JD; Dong L-B; Manoogian K; Shen BJ Am. Chem. Soc 2016, 138, 16711–16721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Dong L-B; Rudolf JD; Shen B Bioorg. Med. Chem 2016, 24, 6348–6353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Nicolaou KC; Chen JS; Edmonds DJ; Estrada AA Angew. Chem., Int. Ed 2009, 48, 660–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Martens E; Demain AL J. Antibiot 2011, 64, 705–710. [DOI] [PubMed] [Google Scholar]
- (15).Rudolf JD; Dong L-B; Shen B Biochem. Pharmacol 2017, 133, 139–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Hiemstra H; Wynberg HJ Am. Chem. Soc 1981, 103, 417–430. [Google Scholar]
- (17).Nishimura K; Ono M; Nagaoka Y; Tomioka KJ Am. Chem. Soc 1997, 119, 12974–12975. [Google Scholar]
- (18).Nishide K; Ozeki M; Kunishige H; Shigeta Y; Patra PK; Hagimoto Y; Node M Angew. Chem., Int. Ed 2003, 42, 4515–4517. [DOI] [PubMed] [Google Scholar]
- (19).Li W; Zu LS; Wang J; Wang W Tetrahedron Lett. 2006, 47, 3145–3148. [Google Scholar]
- (20).Enders D; Luttgen K; Narine AA Synthesis 2007, 2007, 959–980. [Google Scholar]
- (21).Rosenker CJ; Krenske EH; Houk KN; Wipf P Org. Lett 2013, 15, 1076–1079. [DOI] [PubMed] [Google Scholar]
- (22).Farley AJM; Sandford C; Dixon DJ J. Am. Chem. Soc 2015, 137, 15992–15995. [DOI] [PubMed] [Google Scholar]
- (23).Hoveyda AH; Evans DA; Fu GC Chem. Rev 1993, 93, 1307–1370. [Google Scholar]
- (24).Smanski MJ; Peterson RM; Rajski SR; Shen B Antimicrob. Agents Chemother 2009, 53, 1299–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Hindra; Huang T; Yang D; Rudolf JD; Xie P; Xie G; Teng Q; Lohman JR; Zhu X; Huang Y; Zhao L-X; Jiang Y; Duan Y; Shen B J. Nat. Prod 2014, 77, 2296–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Shi J; Pan J; Liu L; Yang D; Lu S; Zhu X; Shen B; Duan Y; Huang YJ Ind. Microbiol. Biotechnol 2016, 43, 1027–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Qiu L; Tian K; Pan J; Jiang L; Yang H; Zhu X; Shen B; Duan Y; Huang Y Tetrahedron 2017, 73, 771–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Dong LB; Rudolf JD; Lin L; Ruiz C; Cameron MD; Shen B Bioorg. Med. Chem 2017, 25, 1990–1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Rao Y; Li X; Nagorny P; Hayashida J; Danishefsky SJ Tetrahedron Lett. 2009, 50, 6684–6686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Kang MT; Meng M; Tan YN; Cheng T; Liu CY Chem. - Eur. J 2016, 22, 3115–3126. [DOI] [PubMed] [Google Scholar]
- (31).Hamza A; Schubert G; Soós T; Pápai IJ Am. Chem. Soc 2006, 128, 13151–13160. [DOI] [PubMed] [Google Scholar]
- (32).Zuend SJ; Jacobsen EN J. Am. Chem. Soc 2007, 129, 15872–15883. [DOI] [PubMed] [Google Scholar]
- (33).Zhu J-L; Zhang Y; Liu C; Zheng A-M; Wang WJ Org. Chem 2012, 77, 9813–9825. [DOI] [PubMed] [Google Scholar]
- (34).Grayson MN; Houk KN J. Am. Chem. Soc 2016, 138, 9041–9044. [DOI] [PubMed] [Google Scholar]
- (35).Tang W; Jiménez-Osés G; Houk KN; van der Donk WA Nat. Chem 2015, 7, 57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Krenske EH; Petter RC; Zhu Z; Houk KN J. Org. Chem 2011, 76, 5074–5081. [DOI] [PubMed] [Google Scholar]
- (37).Krenske EH; Petter RC; Houk KN J. Org. Chem 2016, 81, 11726–11733. [DOI] [PubMed] [Google Scholar]
- (38).Simon L; Goodman JM Org. Biomol. Chem 2011, 9, 689–700. [DOI] [PubMed] [Google Scholar]
- (39).Wiegand I; Hilpert K; Hancock RE W. Nat. Protoc 2008, 3, 163–175. [DOI] [PubMed] [Google Scholar]
- (40).Müller K; Faeh C; Diederich F Science 2007, 317, 1881–1886. [DOI] [PubMed] [Google Scholar]
- (41).Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Mennucci B; Petersson GA; Nakatsuji H; Caricato M; Li X; Hratchian HP; Izmaylov AF; Bloino J; Zheng G; Sonnenberg JL; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Montgomery JA Jr.; Peralta JE; Ogliaro F; Bearpark M; Heyd JJ; Brothers E; Kudin KN; Staroverov VN; Kobayashi R; Normand J; Raghavachari K; Rendell A; Burant JC; Iyengar SS; Tomasi J; Cossi M; Rega N; Millam JM; Klene M; Knox JE; Cross JB; Bakken V; Adamo C; Jaramillo J; Gomperts R; Stratmann RE; Yazyev O; Austin AJ; Cammi R; Pomelli C; Ochterski JW; Martin RL; Morokuma K; Zakrzewski VG; Voth GA; Salvador P; Dannenberg JJ; Dapprich S; Daniels AD; Farkas Ö ; Foresman JB; Ortiz JV; Cioslowski J; Fox DJ Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, 2009. [Google Scholar]
- (42).Barone V; Cossi MJ Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar]
- (43).Cossi M; Rega N; Scalmani G; Barone VJ Comput. Chem 2003, 24, 669–681. [DOI] [PubMed] [Google Scholar]
- (44).Carey FA; Sunderg RJ Advanced Organic Chemistry, Part A: Structure and Mechanisms; Kluwer Academic/Plenum Publisher: New York, 2000. [Google Scholar]
- (45).Legault CY CYLview, 1.0b; Université de Sherbrooke, 2009. (http://www.cylview.org).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.