

Abstract
Autophagy is a ubiquitous cellular catabolic process responsive to energy stress status. Research over the last decade has revealed that cardiomyocyte autophagy is a prominent homeostatic pathway, important in adaptation to altered myocardial metabolic demand. The cellular machinery of autophagy involves targeted direction of macromolecules and organelles for lysosomal degradation. Autophagy activation has been identified as cardio-protective in some settings (i.e. ischemia and ischemic preconditioning). In other situations chronically elevated levels of autophagy have been linked with cardiopathology (i.e. sustained pressure overload and failure). Autophagy perturbation in diabetic cardiomyopathy is also observed – and has been associated with both adaptive and maladaptive stress responses. Recent findings indicate that various forms of selective autophagy operate in parallel to manage different catabolic ‘cargo’ types including mitochondria, large proteins, glycogen and lipid stores. In this Review, specific circumstances of autophagy induction associated with cardiac benefit or detriment are considered. The various static and dynamic approaches used for measurement of autophagy are critiqued and current inconsistencies in the understanding of autophagy regulation in the heart are highlighted. The future prospects for pharmacological intervention to achieve therapeutic manipulation of autophagic processes are canvassed.
Introduction
Autophagy, or “self-eating” (a term of Greek derivation: ‘auto’ + ‘phagy’) refers to mechanisms of lysosome-dependent degradation of intracellular components which are recycled (for energy supply or for other purposes). In most contexts, the usage of the term ‘autophagy’ denotes macro-autophagy, a process involving formation of a double membrane phagosomal structure for engulfment of materials prior to degradation effected through lysosome fusion. An understanding of the role of autophagy in the heart is emerging, and autophagy involvement is evident in a number of cardiac disease processes. The rate of cardiomyocyte proliferation in the adult heart is extremely low, resulting in a limited capacity for replacement of myocytes.1 Thus intracellular ‘renewal’ (i.e. recycling) processes play an important role in determining cardiomyocyte functionality and longevity. An appreciation of the reliance of the myocardium on autophagy to prevent accumulation of macromolecular aggregates and dysfunctional organelles in preserving cellular homeostasis, under both baseline conditions and times of stress, is well established.2 Cardiomyocyte autophagy is required for cardiomyocyte survival during the post-natal period of nutrient deprivation,3 and in the adult heart, acute energy stress settings provide a potent stimulus for autophagy activation. Autophagy disturbance has been identified in many cardiac disease states, including hypertrophy, pressure-overload heart failure, ischemic heart disease, diabetes and age-related cardiomyopathy. An understanding of circumstances where autophagy represents a beneficial adaptive response to disease triggers or a maladaptive response involving cell death induction is not yet resolved. New tools for experimental manipulation of autophagy in cardiac disease states are under development, which can potentially allow tissue selective autophagy interventions targeted specifically to macro-autophagy processes. This is important in order to distinguish macro-autophagic processes from other related autophagic pathways also operational in most cell types, namely micro-autophagy (direct recruitment of cytosolic material to the lysosome4), chaperone-mediated autophagy/endosomal micro-autophagy (single protein-recruitment to endosomes/lysosomes5, 6) and mitochondria-derived vesicular autophagy (fusion of mitochondrial outer membranes with endosomes prior to lysosome degradation7). This review focus is on macro-autophagy pathways and processes – and in particular highlights recent advances in defining the role of cargo-selective macro-autophagy subtypes in myocardial stress settings.
The 2016 Nobel Prize for Medicine or Physiology was awarded to Yoshinori Ohsumi for discoveries in autophagy – pioneering work begun with non-mammalian cell investigations in the 1990’s. Given the recognition accorded to this fundamental investigative work of Ohsumi and colleagues, it is timely to evaluate the current state of understanding in relation to cardiac autophagy and the potential opportunities emerging for therapeutic intervention in contexts of deranged myocardial autophagy homeostasis.
Autophagy machinery
Autophagy in the heart, as in other tissues, utilizes a number of protein complexes to carry out selection and degradation of cellular components under tight regulation involving multiple signaling nodes. Two parallel processes are involved in the first stages of autophagy: i) formation of a double-membrane vesicle called an ‘autophagosome’, and ii) recruitment of ‘cargo’ into the autophagosome to be degraded (Figure 1). These steps are then followed by fusion of the autophagosome with a lysosome where acid-activated enzymes breakdown the contents to their constituent molecular building blocks.8 Autophagosome membrane structures are thought to nucleate from endoplasmic reticulum, golgi or mitochondrial membranes, and require a collection of proteins to assemble at the pre-autophagosome structure.9 These proteins include the ULK complex (ULK1/2, Atg13, FIP200, Atg101) and the PI3K complex (PI3K(Class-III), Beclin1, Atg14).8 The dissociation of Beclin1 from Bcl2 is essential for Beclin1 movement into the PI3K complex and is therefore an important regulator of autophagy activity.10 The reciprocal role of Bcl2 in apoptosis suppression and autophagy initiation can mediate reciprocal regulation of these two major cell viability determining processes. Autophagy proceeds through the action of the PI3K complex which produces PI(3)P lipid attracting WIPI, Atg9 and Atg2 to the pre-autophagosome to position these key proteins ready to expand the membrane into an autophagosome.8 Next, the Atg16L complex (Atg16L, Atg12, Atg5) is recruited to the forming autophagosome membrane by Rab33. Incorporation of an Atg8 scaffold protein into the autophagosome membrane requires Atg7-and Atg3-mediated lipidation, anchoring Atg8 proteins into the double-membrane.8 Atg8’s appear to have a dual role in the autophagosome - they are essential for expanding the autophagosome membrane, and also for recruitment of the cargo to be degraded. The lipidation of Atg8’s has been a useful tool for monitoring autophagy activity as the lipid-attachment shifts the protein position by ~2kDa in an SDS-PAGE gel and can therefore be detected by immunoblot to quantify the ‘active’ (lipidated) and the ‘inactive’ (non-lipidated) forms.11 Whilst this autophagosome construction process is generally accepted as canonical, other macro-autophagy like pathways may be operational. An ‘alternative’ form of autophagy has been described11, 12 which is Atg5 and Atg7 independent. This may be a type of chaperone/endosomal mediated autophagy or could rely on non-canonical machinery processes yet to be fully defined.
Figure 1. Autophagy machinery.

The degradation of cellular components by autophagy is coordinated by a number of protein complexes and vesicle fusion events. The pre-autophagosome membrane can originate from the endoplasmic reticulum (ER) and requires the ULK complex (containing ULK1/2, Atg13, FIP200, Atg101) and movement of Beclin1 away from Bcl2 and into the PI3K complex (containing PI3K(III), Beclin1, Atg14). The PI3K complex produces PI(3)P lipid which attracts WIPI, Atg9 and Atg2 to the pre-autophagosome to position these key proteins ready to expand the membrane into an autophagosome. The Atg16L1 complex (containing Atg16L1, Atg12, Atg5) is recruited to the forming autophagosome membrane by Rab33. Atg8 family proteins are incorporated into the autophagosome membrane via Atg7-and Atg3-mediated lipidation (PE), anchoring them into the autophagosome double-membrane. Cellular cargo to be degraded are recruited into the autophagosome by adaptor proteins and bind to an Atg8 family member. The mechanism of autophagosome closure around the cargo is not well established, but may involve Atg2, Atg8 and Atg9. Autophagosomes can traffick along the cytoskeleton and various proteins from the Rab (Rab5, Rab7), UVRAG and SNARE (Stx7, Stx8, Stx17, VTI1b, and Vamp7) families are implicated in lysosome fusion. LAMP1 and LAMP2 have been implicated in various lysosomal functions including maintaining lysosomal membrane integrity, lysosome-autophagosome fusion, and lysosome motility. The vacuolar-type H+ ATPase (V-ATPase) pumps protons across the lysosomal membrane to acidify the lumen. Hydrolases in the lysosome (blue circles) degrade the autophagosome contents. Chloroquine (CQ) and bafilomycin are pharmacological agents used experimentally to block autophagosome/lysosome fusion (by neutralizing or preventing acidification) to assess autophagosome clearance.
Cellular components destined for autophagic degradation can be recruited to the autophagosome in different ways. Ubiquitination of protein aggregates can make them a target for specific adaptor molecules. The adaptor molecules position the ubiquitin-tagged proteins into the autophagosome via binding to a lipidated-Atg8 protein.8 Multiple Atg8 mammalian orthologues have been identified, along with a cohort of interacting (including adaptor) proteins.13 Recent work has demonstrated cargo-specificity of adaptor–Atg8 partners and this is elaborated below. The mechanism of autophagosome closure around the cargo is not well established, but may involve the SNARE complex, Atg2, Atg8 and Atg9. After closure, the next step in the autophagy machinery is the fusion of the autophagosome with a lysosome. Autophagosomes can traffick along the cytoskeleton and various proteins from the Rab (Rab5, Rab7), UVRAG and SNARE (Stx7, Stx8, Stx17, VTI1b, and Vamp7) families are implicated in trafficking and lysosome fusion (Figure 1).14, 15 Autophagolysosome contents are then degraded by lysosomal hydrolases (e.g. esterases, proteases, glycosidases, nucleosidases, and lipases depending on cargo content) and the breakdown products are exported from the autophagolysosome via routes not well characterized but likely involving permease mediated passive transport.
Evaluating a change in autophagy can be tackled in different ways. A snapshot (i.e. ‘static’) quantification of autophagy-related proteins can provide information about comparative levels of participating proteins, which may be useful when evaluating whether there is an increased/decreased autophagic drive in chronic disease settings.11 However, in acute settings of autophagy induction (and in some chronic settings also), autophagy-related proteins may accumulate due to an impairment in lysosome function, rather than an increase in autophagic drive.16 The operation of autophagy machinery requires coordinated regulation to avoid disturbance of cargo clearance, leading to an accumulation of autophagosomes in the cytosol (indicative of impaired processing rather than elevated activity). To evaluate the extent of autophagy trafficking through the system (i.e. a ‘dynamic’ measure), pharmacological tools (e.g. chloroquine, bafilomycin) can be used to induce an acute block of autophagosome-lysosome fusion, and with this maneuver the accumulation of autophagy-related proteins provides a measure of underlying autophagosome through-put (i.e. flux).11, 17 These approaches have provided important insights into the physiological and pathophysiological role of autophagy in the heart, discussed in more detail below.
Cargo-selective autophagy: macro-autophagy subtypes
As autophagy research has evolved, macro-autophagy terminology usage (vs. micro-autophagy) has been primarily relevant to processes involving macro-proteins, large protein aggregates and organelles. With the more recent characterization of cargo-selective autophagy routes, this descriptor may now be understood to represent a collection of degradation processes, each somewhat specialized in relation to the cargo processed. Given this broader understanding, adopting the term ‘macrophagy’ to apply specifically to autophagic processing of protein macromolecules and aggregates seems appropriate, and will be used in this review. Autophagic degradation processes have been described in various tissues for a range of cellular components including macro-proteins/protein aggregates, endoplasmic reticulum, ribosomes, lipid droplets, peroxisomes, glycogen and mitochondria. Cargo-selective machinery (specific adaptors and Atg8 partners) have been identified for some of these processes. In the myocardium, macrophagy processing has been investigated extensively. Understanding of the selective mechanisms involving mitochondrial and glycogen autophagy in the heart has recently advanced significantly, and together with macrophagy, these are dealt with in more detail below (Figure 2).
Figure 2. Cargo-selective autophagy.

A new understanding of cargo-selective autophagy pathways is emerging. Macrophagy involves the breakdown of protein macromolecules with the p62 adaptor protein recruiting the protein aggregates into the autophagosome to bind to LC3B. Negative feedback regulation of macrophagy by proteolysis of the cargo, yielding amino acids - especially branched chain amino acids, is mediated through re-activation of mTOR. Mitophagy involves the recruitment of mitochondria to the autophagosome by a number of different adaptor proteins, including Bnip3, Nix and p62, which bind to LC3B in the autophagosome membrane. Mitophagy is stimulated by reactive oxygen species (ROS), collapse of the mitochondrial membrane potential (mito Ψ) and low ATP levels. Glycophagy involves the tagging of glycogen with the adaptor protein, starch-binding protein domain 1 (STBD1) which recruits glycogen into the autophagosome by binding to GABARAPL1. Glycophagy may be regulated by its breakdown product, glucose.
Macrophagy
Recruitment of cardiac protein aggregates into autophagosomes utilizes ubiquitin-tagging of target proteins, recognized by adaptor molecules which possess both ubiquitin-binding domains and an Atg8-interacting motif, to connect with the forming autophagosome.18 The protein p62 (also known as SQSTM1) is the key macrophagy adaptor and complexes with the Atg8 homologue LC3, which is activated by lipidation and mediates protein cargo incorporation into the forming phagosome. Detection of both p62 and lipidated LC3 (i.e. LC3-I conversion to LC3-II) are relied on experimentally for characterization and localization of macrophagy occurrence in cardiac cell types and tissues.
In non-cardiac cells, emerging evidence suggests that ubiquitin-independent pathways for cargo recruitment may also be important in autophagic clearance of protein aggregates,19 but whether a similar process plays a role in the heart is yet to be established. In an elegant feedback mechanism, amino acid availability is a potent regulator of macrophagy, likely mediated via mTOR signaling.20 Although yet to be confirmed in cardiomyocytes, in non-cardiac cell types, elevated cytosolic amino acid levels activate mTOR complex 1 (mTORC1) by recruitment to the lysosome via Rag family proteins.20 Activated mTOR inhibits the early steps of autophagosome formation (via ULK1/2)21 and lysosomal biogenesis (via TFEB)22 in the heart. In settings of amino acid depletion, mTORC1 dissociates from the lysosome (a process regulated by the translocation of the TSC complex to the lysosome) thus relieving its inhibition of autophagy and autophagy-mediated ‘re-supply’ of amino acids is promoted.20 Macrophagy mediated protein breakdown in the heart may also be important for the regression of cardiac hypertrophy observed following left ventricular assist device implantation, aortic valve replacement or pregnancy. Conversely, augmented autophagy (measured by LC3) is a necessary component of the cardiomyocyte hypertrophic response under stress conditions.23, 24 Mice with cardiomyocyte-specific autophagy deficiency (Atg5 deletion) exhibit impaired regression of cardiac hypertrophy following withdrawal of pressure-overload induced by angiotensin II infusion (withdrawal via osmotic minipump removal).25 Thus protein turnover and synthesis are closely regulated by macrophagy, which is important in myocardial tissue plasticity – although in some contexts macrophagy may be permissive of growth and in others limiting. A more detailed understanding of macrophagy regulation in these contrasting conditions is required in order to consider nuanced interventions in settings of growth perturbation and/or proteotoxicity.
Mitophagy
Perturbations of mitochondrial function in both acute and chronic settings drive adverse remodeling in many cardiac pathologies. Selective degradation of damaged or senescent mitochondria by mitochondrial autophagy (termed ‘mitophagy’) plays an important role in maintaining energy homeostasis and reducing ROS produced by removal of dysfunctional mitochondria. An additional essential role is in removal of mitochondrial DNA, which, through its similarity to bacterial DNA, can trigger immune responses via activation of TLR-9 mediated inflammation. This is exemplified by the cardiomyocyte-specific deletion of lysosomal DNase which results in severe myocarditis and dilated cardiomyopathy in mice when subjected to pressure overload.26 Oxidized mitochondrial DNA can also activate the inflammasome when released into the cytosol, promoting further inflammation.27
For mitophagy to proceed, mitochondria must first be segregated from the network prior to degradation, and it is apparent that proteins involved in mitochondrial fission play an essential role. In mice lacking fission-mediating Drp1, there is an accumulation of elongated dysfunctional mitochondria coincident with impaired mitophagy and left ventricular dysfunction.28 The mitochondrial permeability transition pore (mPTP) may also be important for mitochondrial quality control in addition to its established role in inducing cell death. Low conductance activity or ‘flickering’ gives rise to transient mitochondrial depolarization, which may be important for selection of mitochondria for mitophagy. Indeed, when mPTP is inhibited by cyclophilinD knockout or cyclosporineA treatment there is an accumulation in cardiomyocyte mitochondria coincident with a reduction in autophagosome number.29
The canonical mitophagy pathway involves the proteins PINK1 and Parkin, which trigger the removal of mitochondria in response to a fall in mitochondrial membrane potential. Healthy mitochondria import and degrade PINK1. Collapse of the mitochondrial membrane potential precludes PINK1 from entering the mitochondria, leading to its localization on the outer mitochondrial membrane.30 Subsequent recruitment of the E3-ubiquitin ligase Parkin permits ubiquitination of multiple outer mitochondrial membrane proteins and recruitment of cargo adaptors, e.g. p62, which orchestrate organellar engulfment by the autophagosome.31 The pro-apoptotic BH3-only proteins Bnip3 and Nix have recently been demonstrated to also function as adaptors for recruiting mitochondria to the autophagosome, by interacting with LC3.32 In non-cardiac cell types, the adaptor proteins NDP52 and Optineurin have been found to participate in mitophagy,33 undergoing recruitment to mitochondria in a Pink1-dependent but Parkin-independent manner.34 A role for these specific adaptors in cardiomyocyte mitophagy has not yet been demonstrated. In parallel with mitophagy, mitochondrial integrity may also be preserved by packaging selected damaged components into small vesicles which bud off mitochondria for trafficking directly to the lysosome (as noted earlier).35, 36
The activation of mitophagy may be a significant contributor to the protection mediated by ischemic preconditioning,37 however there are few therapeutic agents which specifically activate mitophagy. The chemical uncouplers FCCP and dinitrophenol have been employed experimentally to demonstrate the dependence of mitophagy activation upon membrane potential; both agents are cardioprotective at mild uncoupling concentrations.38, 39 Activation of mitophagy is also necessary for the cardioprotection mediated by simvastatin.40
Recently developed molecular tools have enabled the visualization of mitochondrial turnover and mitophagy in vivo with the generation of the transgenic MitoTimer, Mito-QC and Mt-Keima transgenic mice.41–43 These tools have facilitated significant advances in the understanding of mitochondrial turnover and of the role of mitophagy in cardiac pathology, particularly in the context of ischemia-reperfusion injury, discussed in more detail below.
Glycophagy
Although observations of glycogen localized to vacuoles and lysosomal structures have been sporadically reported, it is only recently that characterization of a glycogen-specific autophagy pathway and an understanding of the role of myocardial glycogen autophagy has emerged. In 1932, Pompe et al provided the first report of cardiac pathology and premature death linked with apparent glycogen ‘storage’ overload in the heart,44 a condition now known as Pompe’s disease. It was later discovered that cardiac glycogen accumulation in these patients was caused by a deficiency in the lysosomal enzyme for glycogen breakdown, acid α-glucosidase,45 thus highlighting the importance of autophagic-lysosomal processing of glycogen in the heart. Recently, specific autophagy adaptor-Atg8 homologue proteins involved in recruiting glycogen to the autophagosome have been identified. Starch-binding protein domain 1 (STBD1) contains a carbohydrate-binding domain for glycogen, and an Atg8-interacting-motif for binding to the ‘autophagosome-forming’ Atg8 family proteins.46 Proteomics analysis of the human Atg8 subnetwork has identified that STBD1 interacts with Atg8 proteins from the GABARAP subfamily, and not the LC3 subfamily.13 In vitro co-localization studies in non-cardiac cells have confirmed the interaction between STBD1 and GABARAPL1 and identified STBD1 to be a novel cargo binding protein for glycogen recruitment into autophagosomes.47 Collectively these findings demonstrate that glycogen autophagy utilizes specific adaptor-Atg8 scaffold proteins partners, indicative of a selective ‘glycophagy’ process.
Very recent findings demonstrate that in the mammalian myocardium, the glycophagy adaptor-Atg8 partner proteins (STBD1 & GABARAPL1) are present, that glycophagy is operational and is responsive to altered pathophysiologic states. In vivo rodent studies show that cardiac glycophagy is activated in response to fasting, over a 24–48 hour time period.48 Cardiomyocyte exposure to insulin in vitro increases glycogen content and STBD1 protein expression, with no change in GABARAPL1.49 This observation suggests that while insulin-stimulated cardiac glucose access may influence the early stages of glycogen-recruitment to autophagosomes, glucose-induced inhibition of lysosomal acid α-glucosidase activity may occur. In the neonatal heart, elevated acid α-glucosidase activity and visualization of glycogen in autophagic vacuoles by electron microscopy is observed during the postnatal period of nutrient deprivation.50, 51 More detailed dissection of the cardiomyocyte regulatory pathways of insulin signaling, glucose response and glycophagy induction is required.
In addition to STBD1, glycogen synthase (the rate-limiting enzyme of glycogen synthesis) may serve as an alternative/additional adaptor and means for glycogen recruitment to autophagosomes. Glycogen synthase contains an Atg8-interacting motif, and in non-mammalian species molecular interaction has been confirmed. In non-cardiac tissues, Atg8-glycogen synthase co-localization has been reported in response to fasting, indicative of the potential for a nutrient sensing mechanism for glycophagy activation.52 Nutrient regulation of glycophagy is also apparent at the level of the lysosome, evidenced by hyperglycemia-induced inhibition of acid α-glucosidase activity in neonatal rat hepatocytes.53 The existence of a similar nutrient sensing glycophagy trafficking pathway involving glycogen synthase remains to be established in the heart – this route of glycogen shunting may be an important component of energy homeostasis not previously appreciated.
Cross-talk between glycogen and autophagy regulatory signaling pathways may play an important role in synchronizing synthesis and breakdown of glycogen. Although limited literature is available relating to cardiac findings, some insights have been gained from studies in non-cardiac tissues. In neurons, knockdown of glycogen branching enzyme inhibits autophagy, and conversely, activation of autophagy by rapamycin treatment, inhibits glycogen synthase activity.54 In a mouse model of Lafora disease (a genetic neurodegenerative condition characterized by abnormally branched and insoluble intracellular glycogen deposits), the glycogen overload and associated autophagy impairment is mitigated when glycogen synthase deficiency is induced.55 These observations have prompted speculation that glycogen accumulation may be a cause, not a consequence, of autophagy impairment, at least in this genetic context of glycogen storage abnormality.52
Taken together, the new findings which demonstrate functional glycophagy processing in the heart, and the long established recognition of the cardiac pathologies associated with so-called ‘glycogen storage diseases’,56 the importance of understanding the interplay between the mechanisms controlling glycogen cytoplasmic enzymatic degradation and autophagosome-sequestered bulk breakdown becomes apparent. A role for glycophagy in acute and chronic cardiac disease is yet to be elucidated. Given the evidence of potential regulation of glycophagy by nutrient status, further work directed to understanding the glycophagy response in metabolic stress settings of cardiac pathology is an important priority.
Autophagy regulatory signals
Autophagy (in its various forms) is an important cellular process employed in adaptation to altered myocardial metabolic demand to liberate cellular components for energy supply. In vivo fasting and calorie restriction, and in vitro nutrient deprivation, are potent activators of autophagy in cardiomyocytes. Autophagy is essential for survival during the period of nutrient deprivation immediately post-birth,3 and interestingly, timely withdrawal of this initial autophagy burst is also necessary for postnatal cardiac function. Systemic insulin release on re-feeding triggers activation of the insulin signaling pathway to suppress cardiomyocyte autophagy. In mice with impaired cardiac insulin signaling (cardiomyocyte-specific insulin receptor substrate (IRS) deletion), the postnatal autophagic surge is not attenuated by re-feeding and results in cell loss, heart failure and premature death.57 In the adult heart, it is apparent that a similar restrained autophagy response to energy stress is required. While autophagy induction is required for maintenance of heart function with fasting,28 over-activation of autophagy in fasted IGF1-deficient mice was associated with cardiac atrophy.58 The insulin signaling pathway involves activation of PI3K, Akt, Rheb and mTOR, resulting in inhibition of autophagy activity (Figure 3). In vitro, activation of autophagy via downregulation of the Rheb-mTOR signaling pathway is essential for cardiomyocyte survival, as evidenced by accelerated cell death with 18 hours of glucose deprivation in Rheb overexpressing cardiomyocytes.59 Similarly, in response to cardiac ischemia in vivo, Rheb overexpression prevents ischemia-induced autophagy induction and increased infarct size.59 These findings suggest that autophagy activation elicits a pro-survival response in cardiomyocytes, yet in other contexts cardiomyocyte autophagy over-activation has been linked to cell death. A better understanding of the beneficial actions vs. cell death-generating actions of cardiomyocyte autophagy is required to determine what constitutes a cardioprotective intervention in different contexts (see below).
Figure 3. Physiological regulation of autophagy in the heart.
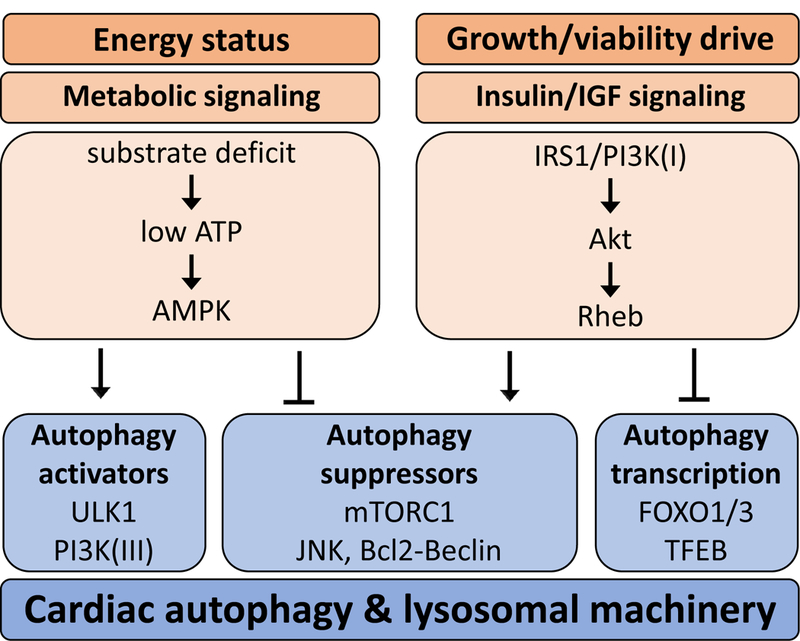
Cardiomyocyte energy status regulates autophagy via metabolic signaling. A substrate deficit (e.g. during nutrient deprivation or hypoxia) leads to low ATP levels which stimulate AMPK activity. AMPK activates autophagy via stimulating ULK1 and PI3K(III) and inhibiting autophagy suppressors such as mTORC1 and JNK which promotes the interaction of Bcl2 and Beclin1, thus preventing Beclin1 from moving to the PI3K(III) complex for autophagy. A growth or cell survival stimulus activates insulin/insulin-like growth factor (IGF) signaling in cardiomyocytes leading to activation of the IRS1/PI3K(I)/Akt pathway. Rheb activation results in inhibition of autophagy via activation of autophagy suppressors (primarily mTORC1) and inhibition of transcription factors FOXO1/3 and TFEB. AMPK: AMP-activated kinase; Atg: autophagy; ULK1: unc-51 like autophagy activating kinase 1; PI3K(III): phosphoinositide-3-kinase class 3, mTORC1: mammalian target of rapamycin complex 1; JNK: c-Jun N-terminal kinase; Bcl2: B-cell lymphoma 2; IGF: insulin growth factor; IRS1: insulin receptor substrate 1; PI3K(I): phosphoinositide-3-kinase class 1; Rheb: Ras homolog enriched in brain; FOXO: Forkhead box; TFEB: transcription factor EB.
Autophagy modulation in response to cardiac energy stress is regulated by a signaling network involving AMPK and insulin signaling pathways (Figure 3). Signaling cross-talk and feedback mechanisms are apparent but the complexities are not well understood. The nutrient-sensing mTOR complex 1 (mTORC1) may play an important role in integrating metabolic signals to trigger autophagy induction, and receives signals from both AMPK and insulin.20 When activated, mTORC1 inhibits both ULK1, a key instigator of autophagosome formation,21 and TFEB, a transcription factor for lysosomal autophagy genes.22 mTORC1 appears particularly sensitive to changes in amino acid availability to regulate macrophagy protein breakdown. Feedback regulation of autophagy via lysosomal breakdown products (amino acids, glucose, lipids, DNA etc.) may provide important regulatory input to upstream autophagic signaling pathways and further understanding of these mechanisms and their role in cardiac metabolic stress settings is required.
Recent evidence suggests that Beclin1 is an important mediator of autophagy regulation in the heart.10 Bcl2 or Bcl-XL proteins bind to Beclin1 to negatively regulate autophagy and increasing evidence suggests that this interaction is regulated by phosphorylation. Phosphorylation of Beclin1 at Thr108 promotes Bcl2-Beclin1 interaction and inhibits cardiomyocyte autophagy.60 Conversely, phosphorylation of Beclin1 at Thr119 promotes Bcl2-Beclin1 dissociation and subsequent activation of autophagy61, 62 - although direct evidence for phosphorylation of Beclin1 in cardiomyocytes is not yet available. In response to fasting, Bcl2 is phosphorylated at Thr69, Ser70, and Ser87 which inhibits the Bcl2-Beclin interaction and promotes autophagy.63 Further investigation of the role of Beclin1 and Bcl2 phosphorylation in cardiac disease states will be informative.
miR signaling involvement
MicroRNAs (miRNAs) are short non-coding RNAs that negatively regulate gene transcription and emerging evidence suggests that miRNAs play an important role in regulating autophagy in the heart. To date, approximately 20 miRNAs have been identified to target autophagy-related genes in the heart. These include miR-30a, which targets Beclin1,64 miR-30e, which targets Atg5,65 and miR212/132, which target FoxO366 a transcription factor targeting autophagy-related genes (Figure 3), all of which act to decrease autophagy. Conversely, miR-99a67 and miR-15368 increase autophagy by targeting mTOR and Mcl-1 (a ‘stress sensor’ which activates autophagy), respectively. The cardioprotective effects of miR-145, which targets FGFR2, have been shown to be mediated by autophagy upregulation in a rodent model of myocardial infarction.69 In non-cardiac tissues, evidence that miRNAs may regulate cargo-selective autophagy pathways has been reported. For example, miR-181a targets Parkin to reduce mitochondria-selective autophagy,70 and miR-195 targets GABARAPL171 which has been implicated in glycogen-selective autophagy.47 The panoply of different miRNA autophagy targets suggests a nuanced approach, whether it is tissue-specific, cargo-selective, restricted to specific signaling pathways, or related to different stages of autophagy processing.
miRNAs are an important component of exosomes, vesicles which are secreted by cells and may be particularly important in stem cell cardiac therapies where miR-146a-3p has been shown to play a role.72, 73 In the heart, cardioprotection elicited by remote preconditioning is reported to be mediated through exosomes,74 although the miRNAs responsible have not yet been identified. It can be confidently asserted that additional miRNAs will be identified as playing a role in the regulation of autophagy in the heart. miRNAs may provide an effective therapeutic approach for modulating autophagy in cardiac disease states. The recent surge in interest in identifying miRNA autophagy targets in the heart provides promise for a clearer understanding of autophagy regulation in the near future.
Cardio-protection mechanisms
Whether upregulated autophagy is cardio-protective or damaging is the subject of considerable debate. As more evidence emerges, it is becoming apparent that both too little, and too much, autophagy can be detrimental, and identifying therapeutic opportunities for autophagy modulation in disease relies on an understanding of ‘how much is too much?’. In many instances, extrapolation of findings from non-cardiac studies is necessary as large knowledge gaps exist in the cardiac autophagy field. Autosis is a type of programmed cell death linked with autophagy. In non-cardiac cells, cell death induced by autophagy activation (via TAT-Beclin1 peptide) is not abrogated by inhibition of apoptosis or necrosis, identifying that an independent cell death pathway exists. In neural cell types, autotic death is evident in response to nutrient-deprivation in vitro and in response to hypoxia-ischemia in vivo.75 Autosis is characterized by fragmented endoplasmic reticulum, focal plasma membrane rupture, nuclear membrane convolution, numerous autophagolysosomes, and swollen mitochondria. Cell death by autosis can be inhibited by autophagy inhibitors but not apoptosis or necrosis inhibitors. Interestingly, autosis has a unique dependence on the Na+K+ATPase evidenced by complete abrogation of TAT-Beclin-induced cell death with cardiac glycosides.75 In cardiomyocytes, direct evidence for autophagy over-activation associated with autotic cell death is not yet available. But evidence that ‘too little’ autophagy is associated with cell death has been reported. Cardiomyocyte death induced by β-adrenergic autoantibodies is linked with decreased autophagy and is rescued by rapamycin-induced autophagy activation.76 Recently, the attachment of small ubiquitin-like modifier proteins (SUMOylation) onto autophagy proteins has been demonstrated, coincident with upregulation of autophagy, improved protein quality control and decreased morbidity in cardiac proteotoxic disease.77
In acute settings of energy stress, autophagy induction is undoubtedly beneficial. Numerous studies have demonstrated that autophagy inhibition during acute fasting, nutrient withdrawal, and cardiac ischemia leads to deleterious outcomes.59, 62 Furthermore, activation of autophagy during ischemia-reperfusion elicits acute cardioprotection.28, 40 Whether sustained autophagy activation in chronic cardiac pathological settings (e.g. cardiomyopathy, heart failure, diabetes) is adaptive or maladaptive has not yet been made clear. The commentary below details the current evidence which supports and refutes a beneficial role for autophagy in different settings of cardiac pathology and highlights opportunities for therapeutic intervention.
Cardiopathology involvement
There is growing evidence that disrupted autophagy is involved in a range of cardiopathologies of diverse nature. Cardiomyocyte autophagy disturbance has been reported in chronic conditions of heart failure,78 hypertrophic and dilated cardiomyopathy (genetic and non-genetic forms),79, 80 cardiac aging,81, 82 and diabetes;83, 84 and in acute conditions of sepsis85, 86 and ischemia-reperfusion injury.87, 88 Cardiomyopathy associated with chemotherapeutic agents such as doxorubicin also involves autophagy disruption with impaired cardiomyocyte autophagosome clearance.89 Cardiac oxidative stress is closely linked with autophagy disturbance in many pathological settings and it has been postulated that cardiomyocyte autophagy induction in sepsis limits ROS production and thus may be an effective therapeutic target.86 Below, the discussion focuses on advances in understanding autophagy involvement in several of these chronic disease states in more detail - highlighting the highly prevalent cardiopathologies of heart failure and hypertrophic remodeling, ischemia-reperfusion injury and diabetic cardiomyopathy.
Heart failure/hypertrophic remodeling
Extensive autophagic vacuoles are evident in human and rodent failing hearts,90, 91 but whether autophagy is cardioprotective, or contributes to pathology in this setting is not established. Cardiac hypertrophy as prelude to failure is a hallmark of remodeling in most settings. AngiotensinII is an important remodeling mediator associated with the hypertrophic response to pressure load and to acute injury, and is involved in cardiomyocyte autophagy regulation via reciprocal actions of the AT1 and AT2 receptor subtypes.92, 93 In rodent models of cardiac hypertrophy (transverse aortic constriction surgery (TAC), isoproterenol), an initial decrease in autophagy markers is evident, followed by autophagy upregulation with longer-term hypertrophic stimulus.94, 95 When a TAC insult is imposed experimentally using a genetic model where autophagosome formation is suppressed, cardiac hypertrophy and systolic dysfunction are prevented.23 These findings are corroborated by a more targeted intervention approach where mice with cardiomyocyte-specific autophagy inhibition (via αMHC-Beclin1 knockdown) are found to exhibit less severe systolic dysfunction following thoracic aortic banding, without affecting cardiac dimensions.96 Conversely, cardiomyocyte-specific autophagy activation (via αMHC-Beclin1 overexpression) exacerbates hypertrophic remodeling and systolic dysfunction.96 These findings suggest that low baseline autophagy sets up the heart for withstanding a hypertrophic insult, and high baseline autophagy makes the heart more susceptible to cardiac remodeling and dysfunction in pressure-overload induced heart failure. But not all studies support this contention. When interventions are used to inhibit cardiomyocyte autophagy just 2 weeks prior to a hypertrophic insult (via cardiomyocyte-specific tamoxifen-induced Atg5 deletion), cell death and systolic dysfunction are accelerated, with no change in hypertrophic remodeling.2 Similarly, cardiomyocyte autophagy inhibition (via αMHC-Beclin1 knockdown) promotes heart failure progression in a mouse model of proteotoxicity (desmin-related cardiomyopathy, CryABR120G mice).91 Conversely, in vivo activation of autophagy (by rapamycin treatment) immediately after aortic banding surgery97 or thyroid hormone treatment,98 prevents hypertrophic remodeling (functional outcomes not evaluated). In patients with dilated cardiomyopathy, the presence of autophagic vacuoles in left ventricular biopsies predict better outcomes over a 12 year follow-up period,79 suggesting that an autophagy response is beneficial for long-term myocyte integrity in pathological settings.
Age-related cardiac remodeling and dysfunction have been linked to autophagy disturbances.81, 99 A role for autophagy in the aging heart is supported by evidence that autophagy activation (via caloric restriction100) slows the progression of age-related cardiac pathology, and autophagy inhibition (via cardiomyocyte-specific Atg5 deletion,101 UVRAG deletion102 or Akt overexpression103) accelerates cardiac dysfunction, hypertrophy and induces premature mortality in aged mice. Loss of ischemic cardioprotection by isoproterenol-preconditioning with age can be restored by antioxidant-induced autophagy upregulation.104 But inconsistent findings showing increased,100, 105 decreased82, 101 or unchanged106 expression of cardiomyocyte autophagy markers with age have been reported. The use of lysosomal inhibitors to measure the extent of autophagy throughput in young vs. aged mice suggests that impaired autophagosome clearance may play a role in age-related cardiac pathology.81, 104 In the aged human heart, although basal cardiac autophagic activity may be lower, the autophagic response to ischemia appears intact,81 thus supporting the viability of autophagy interventions in older patients. Hypermethylation of autophagy genes evident in white blood cells of aged mice107 may be indicative of generalized age-related autophagy impairment - whether a similar epigenetic mechanism plays a role in cardiomyocyte autophagy is yet to be investigated.
Ischemia-reperfusion
Autophagy plays an important role during energy stress associated with cardiac ischemia and subsequent reperfusion – a common consequence of coronary heart disease, and an increasingly recognized consequence of coronary microvascular dysfunction. Ischemic activation of autophagy is considered to be beneficial because it promotes cell survival and improves cardiac function.88 But whether autophagy is also beneficial during the reperfusion period is yet to be fully established. A complex sequence of shifts in energy intermediates, and autophagy signals are enacted during ischemia-reperfusion (Figure 4). During cardiac ischemia, the rapid depletion of ATP leads to AMPK activation and stimulation of autophagy.87 With reperfusion, ATP levels may be at least partially restored, but reoxygenation of the tissue is associated with a surge in production of reactive oxygen species and AMPK activity can remain elevated.108 Ischemia also triggers inactivation of GTP-binding protein Rheb, which in turn leads to mTORC1 inhibition thus providing a parallel autophagy stimulus. Cardiomyocyte-specific Rheb overexpression promotes mTORC1 inhibition of autophagy during ischemia and increased ischemic injury in mice.59 But whether the Rheb/mTOR signaling changes are sustained/reversed during reperfusion is yet to be established. Interventions which further activate AMPK and inhibit mTOR activity to promote autophagy during ischemia may confer additional protection.109 Indeed, autophagic activation contributes to cardioprotection in established interventions including ischemic preconditioning,110 postconditioning,111 in addition to various pharmacological agents.37, 40, 112, 113 Another important node apparent in the ischemia-reperfusion autophagy response is GSK-3β, which was originally identified as a regulator of glycogen synthesis. More recently GSK-3β has emerged as a critical signaling protein involved in the response to ischemia and hypertrophic remodeling.114 Ischemic activation of GSK-3β (via Ser9 dephosphorylation) promotes autophagy which is subsequently reversed upon reperfusion (via Ser9 phosphorylation). Phasic GSK-3β regulation during IR is assumed to be adaptive since interventions which inhibit the kinase during ischemia or activate during reperfusion are both detrimental to the heart.115
Figure 4. Autophagy and ischemia-reperfusion.

Cartoon depicting the transverse view of the heart showing ischemic ‘at risk’ zone (grey shaded) and post-reperfusion necrotic area (black). During ischemia (blue zone), activation of autophagy is linked with ATP depletion, activation of AMPK and GSK3β, and inhibition of mTORC1. During reperfusion (pink zone), a surge in the production of reactive oxygen species (ROS) is associated with inhibited GSK3β activity and ATP levels are at least partially restored. Question marks highlight areas of uncertainty requiring further investigation. Some indication that sustained mTORC1 inhibition and AMPK activation is linked with elevated autophagy during reperfusion is evident. AMPK: AMP-activated kinase; mTORC1: mammalian target of rapamycin complex 1; GSK3β: glycogen synthase kinase 3β.
The role of autophagy during reperfusion is less clear, and findings of both increased and decreased autophagosome clearance have been reported.116–118 When cardiomyocyte autophagy is inhibited just prior to in vivo myocardial infarction (using cardiomyocyte-specific tamoxifen-induced Atg7 knockdown mice), cardiac remodeling and dysfunction is accentuated post-infarct.119 Cardiac expression of Foxo1, the transcription factor which targets multiple autophagy-related genes (Figure 3), is suppressed following myocardial infarction, only partially recovering after 7 days. Forced expression of the active form of FoxO1 (constitutively deacetylated), attenuates adverse remodeling associated with myocardial infarction, likely associated with upregulated autophagy (although not measured in this study).120 A decrease in lysosome biogenesis may lead to impaired autophagy and accumulation of autophagosomes during reperfusion, evidenced by decreased autophagosome clearance (extent of LC3 increase with chloroquine treatment) and a reduction in the lysosomal membrane protein LAMP2 (a protein implicated in maintaining lysosomal membrane integrity, lysosome-autophagosome fusion, and lysosome motility).118 During reperfusion, increased cardiac expression of the key autophagy protein, Beclin1, is evident, an effect which has been demonstrated to be mediated by reactive oxygen species (ROS).121 Paradoxically, increased Beclin1 during reperfusion has been implicated in downregulation of lysosomal gene transcription (via inhibiting TFEB) - thus supra-basal Beclin1 levels may act to suppress autophagosome clearance. Suppression of Beclin1 by shRNA rescues ROS-induced cell death in cultured neonatal cardiomyocytes via TFEB, and upregulation of Beclin1 (via Adenoviral-Beclin1) suppresses lysosome biogenesis.118, 121 Thus the complexities of feedback loops involved in the regulation of autophagosome/lysosome biogenesis are only just beginning to be understood. It is likely that other autophagy-related proteins may also have dual roles in activating and suppressing autophagy activity depending on expression thresholds.
Diabetic cardiomyopathy
The earliest reports identifying cardiomyocyte autophagy disturbance in experimental diabetic settings proffered discordant findings. Firstly, autophagy upregulation in a type 2 diabetic rodents (fructose diet model) was described122 and almost simultaneously autophagy downregulation was reported in a type 1 diabetic mouse model of pancreatic β-cell ablation (by streptozotocin treatment).123 Despite a rapidly growing experimental literature, which also includes some clinical sample evaluation, a consensus in the field regarding a protective or detrimental role of autophagy in the diabetic heart has not yet been achieved. From detailed examination of all available studies, it is not possible to draw a conclusion on the direction of cardiomyocyte autophagy modulation in response to diabetes, and differences in reported findings cannot be easily attributed to species, diabetes type/model, or disease duration discrepancies (Table 1).
Table 1 |.
Autophagy and the diabetic heart.
| Diabetes type | Model | Cardiac autophagy response | |||
|---|---|---|---|---|---|
| ↑ | ↓ | ↔ | ↑↓? | ||
| T2D | Human | 124 | 131 | ||
| db/db mouse | 124 | 134, 160 | 135 | ||
| GK rat | 136 | ||||
| Diet-induced diabetic rat |
161, 162 | 160 | |||
| Diet-induced diabetic mouse |
122, 163, 164 |
59, 126, 128, 130, 165–168 |
169, 170 | ||
| T1D | STZ rat | 49, 171 | |||
| STZ mouse | 125, 135, 137, 172 |
123, 127, 132, 133, 173–175 |
|||
| OVE26 mouse | 123 | ||||
| Pancreatectomy | 176 | ||||
Row entries relate to reference numbers in bibliography. T1D: type 1 diabetes; T2D: type 2 diabetes; GK: Goto-Kakizaki; STZ: streptozotocin.
When comparing findings from ‘snapshot’ measurements of autophagy markers under basal conditions, approximately equal numbers of studies report increase and decrease in autophagy markers, while fewer investigations report no change (or an ambiguous finding). A modest number of publications report measurements of autophagosome clearance in experimental models of diabetes, determined by the extent of autophagy marker accumulation observed with an acute lysosomal block (via bafilomycin or chloroquine). These studies show an increase,124 or decrease125–130 in autophagosome clearance rate in diabetic hearts relative to control. In human type 2 diabetic patients with ischemic heart disease, right atrial appendage tissue expression of LC3 (lipidated form) and Beclin1 protein is increased, p62 expression decreased, and immuno-visualization of cardiomyocyte LC3 and Beclin1 augmented with accumulation of autophagosomes.124 In contrast, a similar type 2 diabetic patient cohort exhibits decreased cardiac Atg5 protein expression, and no change in LC3.131 The autophagic response to fasting-and ischemia-induced metabolic stress is blunted in obese insulin resistant mice,59, 128 which may underlie exacerbated ischemic injury evident in diabetes. Glycophagy may play an important role in diabetic cardiomyocytes, coincident with disturbances in glycogen metabolism.49 Understanding the progression and nuances of autophagy in the diabetic heart is an important priority, and investigating cargo-selective autophagy pathways may provide crucial revelations in this field. A systematic understanding of differential findings which could be ascribed to type 1 vs type 2 signaling etiology is required.83
Interventions to manipulate cardiomyocyte autophagy in the context of diabetes have been informative, but again, inconsistent findings in the literature have thwarted any solid conclusions regarding whether autophagy is adaptive or maladaptive in this setting. In support of the contention that autophagy plays a beneficial role in the diabetic heart, studies have shown that activation of autophagy via fenofibrate,132 metformin,123, 133 ghrelin,134 or resveratrol,135 rescues diabetes-induced cardiac dysfunction; and inhibition of autophagy via chloroquine135 exacerbates diabetes-induced diastolic dysfunction. In contrast, other studies have shown that autophagy may play a detrimental role in the diabetic heart: inhibition of autophagy via Epigallocatechin-3-gallate,136 chloroquine,137 global Beclin1 haploinsufficiency127 or global Atg16L deficiency127 rescues diabetes-induced cardiac pathology. Given that these studies employ either systemic delivery of autophagy-related pharmacological agents (intraperitoneal injection, oral gavage) or whole-body genetic manipulation, it is likely that systemic and/or non-cardiac tissue changes may influence cardiac outcomes. Only one report to date has directly investigated cardiomyocyte-specific autophagy interventions in the diabetic heart. In streptozotocin-induced type 1 diabetic mice, cardiomyocyte autophagy activation (via αMHC-Beclin1 overexpression) exacerbated diabetes-induced cardiac pathology.127 STZ mouse hearts with Beclin1 overexpression appeared to exhibit enhanced autophagosome clearance (as determined by bafilomycin-induced LC3-II accumulation), thus the previously reported lysosomal biogenesis inhibition actions of Beclin1 were unlikely to be playing a role. These findings suggest that autophagy plays a detrimental role in the diabetic heart, and direct activation of cardiomyocyte autophagy may not a viable therapeutic option in this setting.
Interventional potential
Clinical evaluation of autophagy intervention has to date primarily focused on cancer applications, employing rapamycin (or ‘rapalog’ agents) or involving hydroxy/chlorquine as adjuvant therapy.138 In relation to cardiopathology, autophagy represents a potentially attractive therapeutic target, particularly in the contexts of ischemic preconditioning139 and proteostasis.140 In the experimental setting, it has been possible to identify numerous agents that promote cardiomyocyte autophagy including rapamycin,141–143 sirtuin activators such as NAD,144 hydrogen sulfide,145 various cardioprotective agents (e.g. UTP, diazoxide, and ranolazine),139 and common clinical therapies such as statins40 and metformin.123 A cell-permeable Beclin1 peptide (TAT-Beclin1) has also been shown to promote cardiomyocyte autophagy in vivo.78 HDAC (histone deacetylase) inhibitors have been demonstrated to reduce infarct size in association with a stimulation of autophagic flux,146 as well as exerting anti-hypertrophic effects in experimental models of hypertrophy;24 both effects likely being mediated in part through mTOR inhibition.147 Inhibition of autophagy may be beneficial in a more limited number of settings including diabetic cardiomyopathy127, 148 and possibly some hypertrophic states,149 although more work is required to elucidate appropriate targets, timing, dose and cardiac delivery methods. Autophagy inhibitors are relatively limited, including 3-methyladenine and wortmannin which target PI3K, and chloroquine and bafilomycin which target autophagosome-lysosome fusion. However off-target effects may dominate the outcome, as PI3K signals to various pathways in addition to autophagy regulation, and anti-inflammatory actions of chloroquine have been demonstrated.150 Inhibition of AMP-activated protein kinase (AMPK) with compound C is an indirect means to inhibit autophagy; but its universality remains to be demonstrated.145 As autophagy is essential for cardiac homeostasis, long-term inhibition is likely to be deleterious.101 Indeed suppressed cardiomyocyte autophagy results in an increased susceptibility to cardiac damage induced by acute energy stress settings such as fasting or ischemia.151
Positive and negative regulation of Beclin1, via phosphorylation induced by Mst1 and ROCK1 respectively, may provide a novel tool for autophagy manipulation,60, 62 and a better understanding of these kinases in the heart will be informative. Recently, interest has grown in the potential use of miRNAs as cardiac therapeutic targets. Through an miR library screen, miR-22 is identified as a candidate that is upregulated in the aging heart and which is associated with diminished autophagy.152 Inhibition of miR-22 is associated with upregulated autophagy, mitigation of post-infarction remodeling, and improved cardiac function.152 Of note, this appears to be an age-restricted effect, as inhibition of miR-22 in younger animals has less effect.
Although clinical studies designed to assess cardiac endpoints relating to autophagy interventions have yet to be undertaken, various agents already in clinical use have been identified to elicit cardiomyocyte autophagy-effects, including verapamil,153 metformin,123 and rapamycin.143, 154 Nonselective beta-adrenergic receptor agonists isoproterenol and norepinephrine as well as the selective beta2 adrenergic receptor agonist salbutamol were reported to increase autophagic flux which was blocked by propranolol.155 Confusingly, it has also been reported that the beta-blocker propranolol156 stimulates autophagy. Statins stimulate autophagy and mitophagy,40 but it is unknown whether the cardioprotective benefit of statins is dependent upon autophagy. Early results from Statin for Acute Myocardial Infarction Trial (SAMIT) show improved left ventricular ejection fraction 6 months after infarction in the patients who were assigned to receive atorvastatin on admission, followed by percutaneous coronary intervention for the affected vessel, suggesting a cardioprotective effect of statins.157 However, a similar trial in which higher dose atorvastatin (but maintained for a shorter period) failed to show functional benefit by imaging at 3 months.158 The HDAC inhibitor suberoylanilide hydroxamic acid, which is an FDA-approved cancer therapeutic, has been reported to attenuate myocardial reperfusion injury by inducing cardiomyocyte autophagy.146
A major limitation in translation of autophagy interventions to treatment of human cardiac pathologies is the difficulty of measuring autophagy in heart tissue in clinical settings. Noninvasive imaging of autophagy is limited,141 requiring tissue sampling to determine autophagy, and autophagosome clearance measurements in patients may be challenging. Development of a noninvasive imaging modality for autophagy will represent a major advance, allowing assessment of efficacy of therapeutic interventions.
Conclusions
Cardiomyocyte autophagy disturbances have now been identified in a plethora of cardiac pathologic states – of both genetic and non-genetic etiology. Whether autophagy is beneficial or detrimental in each case has been difficult to establish, and appears dependent on context. Indeed a differential role for autophagy in acute vs. chronic disease states is evident. Short-term autophagy activation may confer cardiac benefit, while the sustained effects of elevated autophagy may have detrimental consequences. Much of the variability reported in altered autophagic activity in disease conditions may reflect diversity of disease status including disease type, severity and longevity. Careful longitudinal experimental investigations are required to map altered autophagic activity through the progression of acute and chronic disease states to establish the adaptive or non-adaptive value of autophagic activity. With accumulation of this information, it will be possible to more rationally identify optimal intervention timepoints and to determine whether autophagy suppression or augmentation may be beneficial. Further work is required to discern how interventions may be designed to titrate autophagy activity for optimal cardioprotection – and more extensive usage of assay methods to evaluate autophagy throughput in different circumstances is necessary. Understanding how disrupted autophagy progresses to cardiomyocyte death by autosis is also an important research priority.
With the identification of cargo-selective autophagy subtypes (macrophagy, mitophagy, glycophagy and others), and with expanding knowledge of complex autophagy regulatory and signaling networks, development of targeted intervention tools (including small molecules) may provide useful therapeutic avenues. Given the new discoveries that subpopulations of autophagosomes are relatively (but not exclusively) cargo selective it could be anticipated that subpopulations of lysosomal structures with matched enzyme complement might exist. These latter structures could represent unique therapeutic intervention sites. A largely neglected area of autophagy research, is the investigation of the routes of autophagolysosome efflux of degraded autophagy substrates. This endpoint of the cell recycling machinery could be an important locus of therapeutic intervention. The challenges of intervening in any cellular process of fundamental homeostatic importance, and avoiding damaging off-target effects, are considerable. Harnessing knowledge of autophagy mechanisms for interventional potential is an ambitious undertaking – but if lessons learned from apoptosis-directed cancer therapeutics provides a guide,159 the coming decade will be a time of major translational cardiomyocyte autophagy advance.
Key Points.
Autophagic breakdown of cellular components is tightly regulated by multiple signaling nodes including insulin and AMPK pathways, and is essential for cardiomyocyte survival in response to energy stress.
A new understanding of cargo-selective autophagy in the heart is emerging, relating to specific pathways for degradation of proteins (macrophagy), mitochondria (mitophagy) and glycogen (glycophagy).
Disturbances in cardiomyocyte autophagy are evident in numerous cardiac pathological settings including cardiac hypertrophy, pressure-overload heart failure, ischemic heart disease, diabetes, and age-related cardiomyopathy.
Cardioprotective actions of autophagy in acute energy stress settings are clear, but whether sustained elevation of autophagy in chronic disease states is beneficial or detrimental is not yet resolved.
In cardiopathology, autophagy represents a potentially attractive therapeutic target, and this review identifies new areas for future investigation related to cardiomyocyte-specific, and cargo-specific autophagy interventions.
Author biography
Prof Lea MD Delbridge gained her PhD from the University of Melbourne, Australia in 1993 and had training positions at Loyola University (Chicago, USA) and Dalhousie University (Halifax, Canada) as an American Heart Association fellow. Currently, Dr Delbridge is a Professor in the Department of Physiology, University of Melbourne, Australia where she leads the Cardiac Phenomics Laboratory. She has published extensively and is recognized internationally for leading preclinical research in the field of cardiac disease mechanism. Prof Delbridge described the role of GPCR in the regulation of autophagy, and has made novel discoveries relating to cardiac autophagy in diabetes and in hypertrophy development.
Dr Kimberley M Mellor is a Faculty member in the Department of Physiology at the University of Auckland and leads the Cellular and Molecular Cardiology laboratory. She obtained her PhD from the University of Melbourne, Australia in 2011. Dr Mellor, together with Prof Delbridge, provided the first report that autophagy is modulated in the diabetic heart and recently identified glycophagy as a specific form of pathology in cardiomyocyte energy/metabolic stress.
Dr David J Taylor obtained his PhD in 2009 from the University of Hull, UK where he studied mitochondrial function in uremic cardiomyopathy. Currently Dr Taylor is a postdoctoral fellow at Cedars-Sinai Medical Center investigating therapeutic opportunities related to cargo-selective autophagy pathways in cardiac ischemia-reperfusion injury.
Prof Roberta A Gottlieb gained her MD from The Johns Hopkins University School of Medicine in 1984 and completed her clinical training in Houston, USA. She completed postdoctoral fellowships at the University of California San Diego and The Scripps Research Institute, USA. Currently, Prof Gottlieb holds the Dorothy and E. Phillip Lyon Chair in Molecular Cardiology and is Director of the Molecular Cardiobiology group in the Heart Institute at Cedars-Sinai Medical Center. Prof Gottlieb is an international leader in the field of autophagy and mitochondrial regulation in cardioprotection.
References
- 1.Senyo SE, Lee RT and Kühn B Cardiac regeneration based on mechanisms of cardiomyocyte proliferation and differentiation. Stem Cell Research 13, 532–541 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, Nishida K, Hori M, Mizushima N and Otsu K The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med 13, 619–624 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T and Mizushima N The role of autophagy during the early neonatal starvation period. Nature 432, 1032–1036 (2004). [DOI] [PubMed] [Google Scholar]
- 4.Li WW, Li J and Bao JK Microautophagy: lesser-known self-eating. Cell Mol Life Sci 69, 1125–1136 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jackson, Matthew P. and Hewitt, Eric W. Cellular proteostasis: degradation of misfolded proteins by lysosomes. Essays in Biochemistry 60, 173–180 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sahu R, Kaushik S, Clement CC, Cannizzo ES, Scharf B, Follenzi A, Potolicchio I, Nieves E, Cuervo AM and Santambrogio L Microautophagy of cytosolic proteins by late endosomes. Dev Cell 20, 131–139 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cadete VJ, Deschenes S, Cuillerier A, Brisebois F, Sugiura A, Vincent A, Turnbull D, Picard M, McBride HM and Burelle Y Formation of mitochondrial-derived vesicles is an active and physiologically relevant mitochondrial quality control process in the cardiac system. J Physiol 594, 5343–5362 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tanida I Autophagy basics. Microbiol Immunol 55, 1–11 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Tooze SA and Yoshimori T The origin of the autophagosomal membrane. Nat Cell Biol 12, 831–835 (2010). [DOI] [PubMed] [Google Scholar]
- 10.Maejima Y, Isobe M and Sadoshima J Regulation of autophagy by Beclin 1 in the heart. J Mol Cell Cardiol 95, 19–25 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, Adhihetty PJ, Adler SG, Agam G, Agarwal R, Aghi MK et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12, 1–222 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, Komatsu M, Otsu K, Tsujimoto Y and Shimizu S Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 461, 654–658 (2009). [DOI] [PubMed] [Google Scholar]
- 13.Behrends C, Sowa ME, Gygi SP and Harper JW Network organization of the human autophagy system. Nature 466, 68–76 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huber LA and Teis D Lysosomal signaling in control of degradation pathways. Curr Opin Cell Biol 39, 8–14 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Itakura E, Kishi-Itakura C and Mizushima N The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 151, 1256–1269 (2012). [DOI] [PubMed] [Google Scholar]
- 16.Gottlieb RA, Andres AM, Sin J and Taylor DP Untangling Autophagy Measurements: All Fluxed Up. Circ Res 116, 504–514 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gurney MA, Huang C, Ramil JM, Ravindran N, Andres AM, Sin J, Linton PJ and Gottlieb RA Measuring cardiac autophagic flux in vitro and in vivo. Methods Mol Biol 1219, 187–197 (2015). [DOI] [PubMed] [Google Scholar]
- 18.Martins-Marques T, Ribeiro-Rodrigues T, Pereira P, Codogno P and Girao H Autophagy and ubiquitination in cardiovascular diseases. DNA Cell Biol 34, 243–251 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Korac J, Schaeffer V, Kovacevic I, Clement AM, Jungblut B, Behl C, Terzic J and Dikic I Ubiquitin-independent function of optineurin in autophagic clearance of protein aggregates. J Cell Sci 126, 580–592 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tan VP and Miyamoto S Nutrient-sensing mTORC1: Integration of metabolic and autophagic signals. J Mol Cell Cardiol 95, 31–41 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Egan D, Kim J, Shaw RJ and Guan KL The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy 7, 643–644 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martina JA, Chen Y, Gucek M and Puertollano R MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 8, 903–914 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rifki OF, Bodemann BO, Battiprolu PK, White MA and Hill JA RalGDS-dependent cardiomyocyte autophagy is required for load-induced ventricular hypertrophy. J Mol Cell Cardiol 59, 128–138 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cao DJ, Wang ZV, Battiprolu PK, Jiang N, Morales CR, Kong Y, Rothermel BA, Gillette TG and Hill JA Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc Natl Acad Sci U S A 108, 4123–4128 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oyabu J, Yamaguchi O, Hikoso S, Takeda T, Oka T, Murakawa T, Yasui H, Ueda H, Nakayama H, Taneike M, Omiya S, Shah AM, Nishida K and Otsu K Autophagy-mediated degradation is necessary for regression of cardiac hypertrophy during ventricular unloading. Biochem Biophys Res Commun 441, 787–792 (2013). [DOI] [PubMed] [Google Scholar]
- 26.Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, Oyabu J, Murakawa T, Nakayama H, Nishida K, Akira S, Yamamoto A, Komuro I and Otsu K Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 485, 251–255 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM, Rentsendorj A, Vargas M, Guerrero C, Wang Y, Fitzgerald KA, Underhill DM, Town T and Arditi M Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome During Apoptosis. Immunity 36, 401–414 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ikeda Y, Shirakabe A, Maejima Y, Zhai P, Sciarretta S, Toli J, Nomura M, Mihara K, Egashira K, Ohishi M, Abdellatif M and Sadoshima J Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res 116, 264–278 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Carreira RS, Lee Y, Ghochani M, Gustafsson AB and Gottlieb RA Cyclophilin D is required for mitochondrial removal by autophagy in cardiac cells. Autophagy 6, 462–472 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eiyama A and Okamoto K PINK1/Parkin-mediated mitophagy in mammalian cells. Curr Opin Cell Biol 33, 95–101 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Gottlieb RA and Bernstein D Mitochondrial remodeling: Rearranging, recycling, and reprogramming. Cell Calcium 60, 88–101 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saito T and Sadoshima J The Molecular Mechanisms of Mitochondrial Autophagy/Mitophagy in the Heart. Circulation research 116, 1477–1490 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heo J-M, Ordureau A, Paulo, Joao A, Rinehart J and Harper JW The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Molecular Cell 60, 7–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI and Youle RJ The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roberts RF, Tang MY, Fon EA and Durcan TM Defending the mitochondria: The pathways of mitophagy and mitochondrial-derived vesicles. Int J Biochem Cell Biol 79, 427–436 (2016). [DOI] [PubMed] [Google Scholar]
- 36.Cadete V, Deschênes S, Cuillerier A, Picard M, McBride H and Burelle Y Mitochondrial-Derived Vesicle Formation in Cardiac Mitochondrial Quality Control. The FASEB Journal 29, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang C, Andres AM, Ratliff EP, Hernandez G, Lee P and Gottlieb RA Preconditioning involves selective mitophagy mediated by Parkin and p62/SQSTM1. PLoS One 6, e20975 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brennan JP, Southworth R, Medina RA, Davidson SM, Duchen MR and Shattock MJ Mitochondrial uncoupling, with low concentration FCCP, induces ROS-dependent cardioprotection independent of KATP channel activation. Cardiovasc Res 72, 313–321 (2006). [DOI] [PubMed] [Google Scholar]
- 39.Minners J, van den Bos EJ, Yellon DM, Schwalb H, Opie LH and Sack MN Dinitrophenol, cyclosporin A, and trimetazidine modulate preconditioning in the isolated rat heart: support for a mitochondrial role in cardioprotection. Cardiovasc Res 47, 68–73 (2000). [DOI] [PubMed] [Google Scholar]
- 40.Andres AM, Hernandez G, Lee P, Huang C, Ratliff EP, Sin J, Thornton CA, Damasco MV and Gottlieb RA Mitophagy is required for acute cardioprotection by simvastatin. Antioxid Redox Signal 21, 1960–1973 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stotland A and Gottlieb RA alpha-MHC MitoTimer mouse: In vivo mitochondrial turnover model reveals remarkable mitochondrial heterogeneity in the heart. J Mol Cell Cardiol 90, 53–58 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McWilliams TG and Prescott AR mito-QC illuminates mitophagy and mitochondrial architecture in vivo 214, 333–345 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sun N, Yun J, Liu J, Malide D, Liu C, Rovira II, Holmstrom KM, Fergusson MM, Yoo YH, Combs CA and Finkel T Measuring In Vivo Mitophagy. Mol Cell 60, 685–696 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pompe JC Over idiopathische hypertrophie van het hart. . Ned. Tijdschr. Geneeskd 76, 304–312 (1932). [Google Scholar]
- 45.Hers HG alpha-Glucosidase deficiency in generalized glycogenstorage disease (Pompe’s disease). Biochem J 86, 11–16 (1963). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jiang S, Heller B, Tagliabracci VS, Zhai L, Irimia JM, DePaoli-Roach AA, Wells CD, Skurat AV and Roach PJ Starch binding domain-containing protein 1/genethonin 1 is a novel participant in glycogen metabolism. J Biol Chem 285, 34960–34971 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jiang S, Wells CD and Roach PJ Starch-binding domain-containing protein 1 (Stbd1) and glycogen metabolism: Identification of the Atg8 family interacting motif (AIM) in Stbd1 required for interaction with GABARAPL1. Biochem Biophys Res Commun 413, 420–425 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reichelt ME, Mellor KM, Curl CL, Stapleton D and Delbridge LM Myocardial glycophagy - A specific glycogen handling response to metabolic stress is accentuated in the female heart. J Mol Cell Cardiol 65, 67–75 (2013). [DOI] [PubMed] [Google Scholar]
- 49.Mellor KM, Varma U, Stapleton D and Delbridge LMD Cardiomyocyte glycophagy is regulated by insulin and exposure to high extracellular glucose. Am J Physiol Heart Circ Physiol 306, H1240–1245 (2014). [DOI] [PubMed] [Google Scholar]
- 50.Kondomerkos DJ, Kalamidas SA and Kotoulas OB An electron microscopic and biochemical study of the effects of glucagon on glycogen autophagy in the liver and heart of newborn rats. Microsc Res Tech 63, 87–93 (2004). [DOI] [PubMed] [Google Scholar]
- 51.Kondomerkos DJ, Kalamidas SA, Kotoulas OB and Hann AC Glycogen autophagy in the liver and heart of newborn rats. The effects of glucagon, adrenalin or rapamycin. Histol Histopathol 20, 689–696 (2005). [DOI] [PubMed] [Google Scholar]
- 52.Singh PK and Singh S Changing shapes of glycogen-autophagy nexus in neurons: perspective from a rare epilepsy. Front Neurol 6, 14 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kalamidas SA and Kondomerkos DJ The administration of nonmetabolizable glucose analogues fails to suppress the development of glycogen autophagy in newborn rat hepatocytes. Microsc Res Tech 73, 1009–1014 (2010). [DOI] [PubMed] [Google Scholar]
- 54.Kakhlon O, Glickstein H, Feinstein N, Liu Y, Baba O, Terashima T, Akman HO, Dimauro S and Lossos A Polyglucosan neurotoxicity caused by glycogen branching enzyme deficiency can be reversed by inhibition of glycogen synthase. J Neurochem 127, 101–113 (2013). [DOI] [PubMed] [Google Scholar]
- 55.Duran J, Gruart A, Garcia-Rocha M, Delgado-Garcia JM and Guinovart JJ Glycogen accumulation underlies neurodegeneration and autophagy impairment in Lafora disease. Hum Mol Genet 23, 3147–3156 (2014). [DOI] [PubMed] [Google Scholar]
- 56.Chandramouli C, Varma U, Stevens EM, Xiao RP, Stapleton DI, Mellor KM and Delbridge LM Myocardial glycogen dynamics - new perspectives on disease mechanisms. Clin Exp Pharmacol Physiol (2015). [DOI] [PubMed]
- 57.Riehle C, Wende AR, Sena S, Pires KM, Pereira RO, Zhu Y, Bugger H, Frank D, Bevins J, Chen D, Perry CN, Dong XC, Valdez S, Rech M, Sheng X, Weimer BC, Gottlieb RA, White MF and Abel ED Insulin receptor substrate signaling suppresses neonatal autophagy in the heart. J Clin Invest 123, 5319–5333 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Troncoso R, Vicencio JM, Parra V, Nemchenko A, Kawashima Y, Del Campo A, Toro B, Battiprolu PK, Aranguiz P, Chiong M, Yakar S, Gillette TG, Hill JA, Abel ED, Leroith D and Lavandero S Energy-preserving effects of IGF-1 antagonize starvation-induced cardiac autophagy. Cardiovasc Res 93, 320–329 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sciarretta S, Zhai P, Shao D, Maejima Y, Robbins J, Volpe M, Condorelli G and Sadoshima J Rheb is a critical regulator of autophagy during myocardial ischemia: pathophysiological implications in obesity and metabolic syndrome. Circulation 125, 1134–1146 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maejima Y, Kyoi S, Zhai P, Liu T, Li H, Ivessa A, Sciarretta S, Del Re DP, Zablocki DK, Hsu CP, Lim DS, Isobe M and Sadoshima J Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat Med 19, 1478–1488 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zalckvar E, Berissi H, Mizrachy L, Idelchuk Y, Koren I, Eisenstein M, Sabanay H, Pinkas-Kramarski R and Kimchi A DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep 10, 285–292 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gurkar AU, Chu K, Raj L, Bouley R, Lee SH, Kim YB, Dunn SE, Mandinova A and Lee SW Identification of ROCK1 kinase as a critical regulator of Beclin1-mediated autophagy during metabolic stress. Nat Commun 4, 2189 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wei Y, Pattingre S, Sinha S, Bassik M and Levine B JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell 30, 678–688 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yin X, Peng C, Ning W, Li C, Ren Z, Zhang J, Gao H and Zhao K miR-30a downregulation aggravates pressure overload-induced cardiomyocyte hypertrophy. Mol Cell Biochem 379, 1–6 (2013). [DOI] [PubMed] [Google Scholar]
- 65.Lai L, Wang N, Zhu G, Duan X and Ling F MiRNA-30e mediated cardioprotection of ACE2 in rats with doxorubicin-induced heart failure through inhibiting cardiomyocytes autophagy. Life Sci (2016). [DOI] [PubMed]
- 66.Ucar A, Gupta SK, Fiedler J, Erikci E, Kardasinski M, Batkai S, Dangwal S, Kumarswamy R, Bang C, Holzmann A, Remke J, Caprio M, Jentzsch C, Engelhardt S, Geisendorf S et al. The miRNA-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nat Commun 3, 1078 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li Q, Xie J, Li R, Shi J, Sun J, Gu R, Ding L, Wang L and Xu B Overexpression of microRNA-99a attenuates heart remodelling and improves cardiac performance after myocardial infarction. J Cell Mol Med 18, 919–928 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zou Y, Liu W, Zhang J and Xiang D miR-153 regulates apoptosis and autophagy of cardiomyocytes by targeting Mcl-1. Mol Med Rep 14, 1033–1039 (2016). [DOI] [PubMed] [Google Scholar]
- 69.Higashi K, Yamada Y, Minatoguchi S, Baba S, Iwasa M, Kanamori H, Kawasaki M, Nishigaki K, Takemura G, Kumazaki M, Akao Y and Minatoguchi S MicroRNA-145 repairs infarcted myocardium by accelerating cardiomyocyte autophagy. Am J Physiol Heart Circ Physiol 309, H1813–1826 (2015). [DOI] [PubMed] [Google Scholar]
- 70.Cheng M, Liu L, Lao Y, Liao W, Liao M, Luo X, Wu J, Xie W, Zhang Y and Xu N MicroRNA-181a suppresses parkin-mediated mitophagy and sensitizes neuroblastoma cells to mitochondrial uncoupler-induced apoptosis. Oncotarget (2016). [DOI] [PMC free article] [PubMed]
- 71.Mo J, Zhang D and Yang R MicroRNA-195 regulates proliferation, migration, angiogenesis and autophagy of endothelial progenitor cells by targeting GABARAPL1. Biosci Rep 36, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Barile L, Moccetti T, Marban E and Vassalli G Roles of exosomes in cardioprotection. Eur Heart J (2016). [DOI] [PubMed]
- 73.Ibrahim AG, Cheng K and Marban E Exosomes as critical agents of cardiac regeneration triggered by cell therapy. Stem Cell Reports 2, 606–619 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Giricz Z, Varga ZV, Baranyai T, Sipos P, Paloczi K, Kittel A, Buzas EI and Ferdinandy P Cardioprotection by remote ischemic preconditioning of the rat heart is mediated by extracellular vesicles. J Mol Cell Cardiol 68, 75–78 (2014). [DOI] [PubMed] [Google Scholar]
- 75.Liu Y, Shoji-Kawata S, Sumpter RM Jr., Wei Y, Ginet V, Zhang L, Posner B, Tran KA, Green DR, Xavier RJ, Shaw SY, Clarke PG, Puyal J and Levine B Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc Natl Acad Sci U S A 110, 20364–20371 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang L, Hao H, Wang J, Wang X, Zhang S, Du Y, Lv T, Zuo L, Li Y and Liu H Decreased autophagy: a major factor for cardiomyocyte death induced by beta1-adrenoceptor autoantibodies. Cell Death Dis 6, e1862 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gupta MK, McLendon PM, Gulick J, James J, Khalili K and Robbins J UBC9-Mediated Sumoylation Favorably Impacts Cardiac Function in Compromised Hearts. Circ Res 118, 1894–1905 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shirakabe A, Zhai P, Ikeda Y, Saito T, Maejima Y, Hsu CP, Nomura M, Egashira K, Levine B and Sadoshima J Drp1-Dependent Mitochondrial Autophagy Plays a Protective Role Against Pressure Overload-Induced Mitochondrial Dysfunction and Heart Failure. Circulation 133, 1249–1263 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Saito T, Asai K, Sato S, Hayashi M, Adachi A, Sasaki Y, Takano H, Mizuno K and Shimizu W Autophagic vacuoles in cardiomyocytes of dilated cardiomyopathy with initially decompensated heart failure predict improved prognosis. Autophagy 12, 579–587 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schlossarek S, Mearini G and Carrier L Cardiac myosin-binding protein C in hypertrophic cardiomyopathy: mechanisms and therapeutic opportunities. J Mol Cell Cardiol 50, 613–620 (2011). [DOI] [PubMed] [Google Scholar]
- 81.Linton PJ, Gurney M, Sengstock D, Mentzer RM Jr. and Gottlieb RA This old heart: Cardiac aging and autophagy. J Mol Cell Cardiol 83, 44–54 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ceylan-Isik AF, Dong M, Zhang Y, Dong F, Turdi S, Nair S, Yanagisawa M and Ren J Cardiomyocyte-specific deletion of endothelin receptor A rescues aging-associated cardiac hypertrophy and contractile dysfunction: role of autophagy. Basic Res Cardiol 108, 335 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 83.Mellor KM, Reichelt ME and Delbridge LM Autophagy anomalies in the diabetic myocardium. Autophagy 7, 1263–1267 (2011). [DOI] [PubMed] [Google Scholar]
- 84.Zhang Y and Ren J Epigenetics and obesity cardiomyopathy: From pathophysiology to prevention and management. Pharmacol Ther 161, 52–66 (2016). [DOI] [PubMed] [Google Scholar]
- 85.Ceylan-Isik AF, Zhao P, Zhang B, Xiao X, Su G and Ren J Cardiac overexpression of metallothionein rescues cardiac contractile dysfunction and endoplasmic reticulum stress but not autophagy in sepsis. J Mol Cell Cardiol 48, 367–378 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yuan H, Perry CN, Huang C, Iwai-Kanai E, Carreira RS, Glembotski CC and Gottlieb RA LPS-induced autophagy is mediated by oxidative signaling in cardiomyocytes and is associated with cytoprotection. Am J Physiol Heart Circ Physiol 296, H470–479 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gustafsson AB and Gottlieb RA Autophagy in ischemic heart disease. Circ Res 104, 150–158 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B and Sadoshima J Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res 100, 914–922 (2007). [DOI] [PubMed] [Google Scholar]
- 89.Li DL, Wang ZV, Ding G, Tan W, Luo X, Criollo A, Xie M, Jiang N, May H, Kyrychenko V, Schneider JW, Gillette TG and Hill JA Doxorubicin Blocks Cardiomyocyte Autophagic Flux by Inhibiting Lysosome Acidification. Circulation 133, 1668–1687 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kostin S, Pool L, Elsasser A, Hein S, Drexler HC, Arnon E, Hayakawa Y, Zimmermann R, Bauer E, Klovekorn WP and Schaper J Myocytes die by multiple mechanisms in failing human hearts. Circ Res 92, 715–724 (2003). [DOI] [PubMed] [Google Scholar]
- 91.Tannous P, Zhu H, Johnstone JL, Shelton JM, Rajasekaran NS, Benjamin IJ, Nguyen L, Gerard RD, Levine B, Rothermel BA and Hill JA Autophagy is an adaptive response in desmin-related cardiomyopathy. Proc Natl Acad Sci U S A 105, 9745–9750 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Porrello ER, D’Amore A, Curl CL, Allen AM, Harrap SB, Thomas WG and Delbridge LM Angiotensin II type 2 receptor antagonizes angiotensin II type 1 receptor-mediated cardiomyocyte autophagy. Hypertension 53, 1032–1040 (2009). [DOI] [PubMed] [Google Scholar]
- 93.Porrello ER and Delbridge LM Cardiomyocyte autophagy is regulated by angiotensin II type 1 and type 2 receptors. Autophagy 5, 1215–1216 (2009). [DOI] [PubMed] [Google Scholar]
- 94.Li B, Chi RF, Qin FZ and Guo XF Distinct changes of myocyte autophagy during myocardial hypertrophy and heart failure: association with oxidative stress. Exp Physiol 101, 1050–1063 (2016). [DOI] [PubMed] [Google Scholar]
- 95.Pfeifer U, Fohr J, Wilhelm W and Dammrich J Short-term inhibition of cardiac cellular autophagy by isoproterenol. J Mol Cell Cardiol 19, 1179–1184 (1987). [DOI] [PubMed] [Google Scholar]
- 96.Zhu H, Tannous P, Johnstone JL, Kong Y, Shelton JM, Richardson JA, Le V, Levine B, Rothermel BA and Hill JA Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest 117, 1782–1793 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ha T, Li Y, Gao X, McMullen JR, Shioi T, Izumo S, Kelley JL, Zhao A, Haddad GE, Williams DL, Browder IW, Kao RL and Li C Attenuation of cardiac hypertrophy by inhibiting both mTOR and NFkappaB activation in vivo. Free Radic Biol Med 39, 1570–1580 (2005). [DOI] [PubMed] [Google Scholar]
- 98.Kuzman JA, O’Connell TD and Gerdes AM Rapamycin prevents thyroid hormone-induced cardiac hypertrophy. Endocrinology 148, 3477–3484 (2007). [DOI] [PubMed] [Google Scholar]
- 99.Shirakabe A, Ikeda Y, Sciarretta S, Zablocki DK and Sadoshima J Aging and Autophagy in the Heart. Circ Res 118, 1563–1576 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wohlgemuth SE, Julian D, Akin DE, Fried J, Toscano K, Leeuwenburgh C and Dunn WA Jr. Autophagy in the heart and liver during normal aging and calorie restriction. Rejuvenation Res 10, 281–292 (2007). [DOI] [PubMed] [Google Scholar]
- 101.Taneike M, Yamaguchi O, Nakai A, Hikoso S, Takeda T, Mizote I, Oka T, Tamai T, Oyabu J, Murakawa T, Nishida K, Shimizu T, Hori M, Komuro I, Takuji Shirasawa TS, Mizushima N and Otsu K Inhibition of autophagy in the heart induces age-related cardiomyopathy. Autophagy 6, 600–606 (2010). [DOI] [PubMed] [Google Scholar]
- 102.Song Z, An L, Ye Y, Wu J, Zou Y, He L and Zhu H Essential role for UVRAG in autophagy and maintenance of cardiac function. Cardiovasc Res 101, 48–56 (2014). [DOI] [PubMed] [Google Scholar]
- 103.Hua Y, Zhang Y, Ceylan-Isik AF, Wold LE, Nunn JM and Ren J Chronic akt activation accentuates aging-induced cardiac hypertrophy and myocardial contractile dysfunction: role of autophagy. Basic Res Cardiol (2011). [DOI] [PubMed]
- 104.Ma L, Zhu J, Gao Q, Rebecchi MJ, Wang Q and Liu L Restoring Pharmacologic Preconditioning in the Aging Heart: Role of Mitophagy/Autophagy. J Gerontol A Biol Sci Med Sci (2016). [DOI] [PubMed]
- 105.Boyle AJ, Shih H, Hwang J, Ye J, Lee B, Zhang Y, Kwon D, Jun K, Zheng D, Sievers R, Angeli F, Yeghiazarians Y and Lee R Cardiomyopathy of aging in the mammalian heart is characterized by myocardial hypertrophy, fibrosis and a predisposition towards cardiomyocyte apoptosis and autophagy. Exp Gerontol 46, 549–559 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Inuzuka Y, Okuda J, Kawashima T, Kato T, Niizuma S, Tamaki Y, Iwanaga Y, Yoshida Y, Kosugi R, Watanabe-Maeda K, Machida Y, Tsuji S, Aburatani H, Izumi T, Kita T and Shioi T Suppression of phosphoinositide 3-kinase prevents cardiac aging in mice. Circulation 120, 1695–1703 (2009). [DOI] [PubMed] [Google Scholar]
- 107.Khalil H, Tazi M, Caution K, Ahmed A, Kanneganti A, Assani K, Kopp B, Marsh C, Dakhlallah D and Amer AO Aging is associated with hypermethylation of autophagy genes in macrophages. Epigenetics 11, 381–388 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Russell RR 3rd, Li J, Coven DL, Pypaert M, Zechner C, Palmeri M, Giordano FJ, Mu J, Birnbaum MJ and Young LH AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J Clin Invest 114, 495–503 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Huang L, Dai K, Chen M, Zhou W, Wang X, Chen J and Zhou W The AMPK Agonist PT1 and mTOR Inhibitor 3HOI-BA-01 Protect Cardiomyocytes After Ischemia Through Induction of Autophagy. J Cardiovasc Pharmacol Ther 21, 70–81 (2016). [DOI] [PubMed] [Google Scholar]
- 110.Huang C, Yitzhaki S, Perry CN, Liu W, Giricz Z, Mentzer RM Jr. and Gottlieb RA Autophagy induced by ischemic preconditioning is essential for cardioprotection. J Cardiovasc Transl Res 3, 365–373 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wei C, Li H, Han L, Zhang L and Yang X Activation of autophagy in ischemic postconditioning contributes to cardioprotective effects against ischemia/reperfusion injury in rat hearts. J Cardiovasc Pharmacol 61, 416–422 (2013). [DOI] [PubMed] [Google Scholar]
- 112.Yitzhaki S, Huang C, Liu W, Lee Y, Gustafsson ÅB, Mentzer RM and Gottlieb RA Autophagy is required for preconditioning by the adenosine A1 receptor-selective agonist CCPA. Basic research in cardiology 104, 157–167 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yao H, Han X and Han X The cardioprotection of the insulin-mediated PI3K/Akt/mTOR signaling pathway. Am J Cardiovasc Drugs 14, 433–442 (2014). [DOI] [PubMed] [Google Scholar]
- 114.Lal H, Ahmad F, Woodgett J and Force T The GSK-3 family as therapeutic target for myocardial diseases. Circ Res 116, 138–149 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhai P and Sadoshima J Glycogen synthase kinase-3beta controls autophagy during myocardial ischemia and reperfusion. Autophagy 8, 138–139 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hariharan N, Zhai P and Sadoshima J Oxidative stress stimulates autophagic flux during ischemia/reperfusion. Antioxid Redox Signal 14, 2179–2190 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hamacher-Brady A, Brady NR and Gottlieb RA Enhancing Macroautophagy Protects against Ischemia/Reperfusion Injury in Cardiac Myocytes. Journal of Biological Chemistry 281, 29776–29787 (2006). [DOI] [PubMed] [Google Scholar]
- 118.Ma X, Liu H, Foyil SR, Godar RJ, Weinheimer CJ, Hill JA and Diwan A Impaired autophagosome clearance contributes to cardiomyocyte death in ischemia/reperfusion injury. Circulation 125, 3170–3181 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Li S, Liu C, Gu L, Wang L, Shang Y, Liu Q, Wan J, Shi J, Wang F, Xu Z, Ji G and Li W Autophagy protects cardiomyocytes from the myocardial ischaemia-reperfusion injury through the clearance of CLP36. Open Biol 6, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kappel BA, Stöhr R, De Angelis L, Mavilio M, Menghini R and Federici M Posttranslational modulation of FoxO1 contributes to cardiac remodeling in post-ischemic heart failure. Atherosclerosis 249, 148–156 (2016). [DOI] [PubMed] [Google Scholar]
- 121.Ma X, Liu H, Murphy JT, Foyil SR, Godar RJ, Abuirqeba H, Weinheimer CJ, Barger PM and Diwan A Regulation of the transcription factor EB-PGC1alpha axis by beclin-1 controls mitochondrial quality and cardiomyocyte death under stress. Mol Cell Biol 35, 956–976 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mellor KM, Bell JR, Young MJ, Ritchie RH and Delbridge LM Myocardial autophagy activation and suppressed survival signaling is associated with insulin resistance in fructose-fed mice. J Mol Cell Cardiol 50, 1035–1043 (2011). [DOI] [PubMed] [Google Scholar]
- 123.Xie Z, Lau K, Eby B, Lozano P, He C, Pennington B, Li H, Rathi S, Dong Y, Tian R, Kem D and Zou MH Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes 60, 1770–1778 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Munasinghe PE, Riu F, Dixit P, Edamatsu M, Saxena P, Hamer NS, Galvin IF, Bunton RW, Lequeux S, Jones G, Lamberts RR, Emanueli C, Madeddu P and Katare R Type-2 diabetes increases autophagy in the human heart through promotion of Beclin-1 mediated pathway. Int J Cardiol 202, 13–20 (2016). [DOI] [PubMed] [Google Scholar]
- 125.Wang B, Yang Q, Sun YY, Xing YF, Wang YB, Lu XT, Bai WW, Liu XQ and Zhao YX Resveratrol-enhanced autophagic flux ameliorates myocardial oxidative stress injury in diabetic mice. J Cell Mol Med 18, 1599–1611 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Trivedi PC, Bartlett JJ, Perez LJ, Brunt KR, Legare JF, Hassan A, Kienesberger PC and Pulinilkunnil T Glucolipotoxicity diminishes cardiomyocyte TFEB and inhibits lysosomal autophagy during obesity and diabetes. Biochim Biophys Acta 1861, 1893–1910 (2016). [DOI] [PubMed] [Google Scholar]
- 127.Xu X, Kobayashi S, Chen K, Timm D, Volden P, Huang Y, Gulick J, Yue Z, Robbins J, Epstein PN and Liang Q Diminished autophagy limits cardiac injury in mouse models of type 1 diabetes. J Biol Chem 288, 18077–18092 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Andres AM, Kooren JA, Parker SJ, Tucker KC, Ravindran N, Ito BR, Huang C, Venkatraman V, Van Eyk JE, Gottlieb RA and Mentzer RM Jr. Discordant signaling and autophagy response to fasting in hearts of obese mice: Implications for ischemia tolerance. Am J Physiol Heart Circ Physiol 311, H219–228 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Xu X, Hua Y, Nair S, Zhang Y and Ren J Akt2 knockout preserves cardiac function in high-fat diet-induced obesity by rescuing cardiac autophagosome maturation. J Mol Cell Biol 5, 61–63 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Jaishy B, Zhang Q, Chung HS, Riehle C, Soto J, Jenkins S, Abel P, Cowart LA, Van Eyk JE and Abel ED Lipid-induced NOX2 activation inhibits autophagic flux by impairing lysosomal enzyme activity. J Lipid Res 56, 546–561 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Montaigne D, Marechal X, Coisne A, Debry N, Modine T, Fayad G, Potelle C, El Arid JM, Mouton S, Sebti Y, Duez H, Preau S, Remy-Jouet I, Zerimech F, Koussa M et al. Myocardial contractile dysfunction is associated with impaired mitochondrial function and dynamics in type 2 diabetic but not in obese patients. Circulation 130, 554–564 (2014). [DOI] [PubMed] [Google Scholar]
- 132.Zhang J, Cheng Y, Gu J, Wang S, Zhou S, Wang Y, Tan Y, Feng W, Fu Y, Mellen N, Cheng R, Ma J, Zhang C, Li Z and Cai L Fenofibrate increases cardiac autophagy via FGF21/SIRT1 and prevents fibrosis and inflammation in the hearts of Type 1 diabetic mice. Clin Sci (Lond) 130, 625–641 (2016). [DOI] [PubMed] [Google Scholar]
- 133.He C, Zhu H, Li H, Zou MH and Xie Z Dissociation of Bcl-2-Beclin1 complex by activated AMPK enhances cardiac autophagy and protects against cardiomyocyte apoptosis in diabetes. Diabetes 62, 1270–1281 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Pei XM, Yung BY, Yip SP, Chan LW, Wong CS, Ying M and Siu PM Protective effects of desacyl ghrelin on diabetic cardiomyopathy. Acta Diabetol (2014). [DOI] [PubMed]
- 135.Kanamori H, Takemura G, Goto K, Tsujimoto A, Mikami A, Ogino A, Watanabe T, Morishita K, Okada H, Kawasaki M, Seishima M and Minatoguchi S Autophagic adaptations in diabetic cardiomyopathy differ between type 1 and type 2 diabetes. Autophagy 11, 1146–1160 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Liu J, Tang Y, Feng Z, Liu J, Liu J and Long J (−)-Epigallocatechin-3-gallate attenuated myocardial mitochondrial dysfunction and autophagy in diabetic Goto-Kakizaki rats. Free Radic Res 48, 898–906 (2014). [DOI] [PubMed] [Google Scholar]
- 137.Yuan X, Xiao YC, Zhang GP, Hou N, Wu XQ, Chen WL, Luo JD and Zhang GS Chloroquine improves left ventricle diastolic function in streptozotocin-induced diabetic mice. Drug Des Devel Ther 10, 2729–2737 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Levine B, Packer M and Codogno P Development of autophagy inducers in clinical medicine. J Clin Invest 125, 14–24 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Huang C, Yitzhaki S, Perry CN, Liu W, Giricz Z, Mentzer RM and Gottlieb RA Autophagy induced by ischemic preconditioning is essential for cardioprotection. Journal of Cardiovascular Translational Research 3, 365–373 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Bhuiyan MS, Pattison JS, Osinska H, James J, Gulick J, McLendon PM, Hill JA, Sadoshima J and Robbins J Enhanced autophagy ameliorates cardiac proteinopathy. J Clin Invest 123, 5284–5297 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chen HH, Mekkaoui C, Cho H, Ngoy S, Marinelli B, Waterman P, Nahrendorf M, Liao R, Josephson L and Sosnovik DE Fluorescence tomography of rapamycin-induced autophagy and cardioprotection in vivo. Circ Cardiovasc Imaging 6, 441–447 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Gurusamy N, Lekli I, Mukherjee S, Ray D, Ahsan MK, Gherghiceanu M, Popescu LM and Das DK Cardioprotection by resveratrol: a novel mechanism via autophagy involving the mTORC2 pathway. Cardiovasc Res 86, 103–112 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wang LQ, Cheng XS, Huang CH, Huang B and Liang Q Rapamycin protects cardiomyocytes against anoxia/reoxygenation injury by inducing autophagy through the PI3k/Akt pathway. J Huazhong Univ Sci Technolog Med Sci 35, 10–15 (2015). [DOI] [PubMed] [Google Scholar]
- 144.Yamamoto T, Byun J, Zhai P, Ikeda Y, Oka S and Sadoshima J Nicotinamide mononucleotide, an intermediate of NAD+ synthesis, protects the heart from ischemia and reperfusion. PLoS One 9, e98972 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Xie H, Xu Q, Jia J, Ao G, Sun Y, Hu L, Alkayed NJ, Wang C and Cheng J Hydrogen sulfide protects against myocardial ischemia and reperfusion injury by activating AMP-activated protein kinase to restore autophagic flux. Biochem Biophys Res Commun 458, 632–638 (2015). [DOI] [PubMed] [Google Scholar]
- 146.Xie M, Kong Y, Tan W, May H, Battiprolu PK, Pedrozo Z, Wang ZV, Morales C, Luo X, Cho G, Jiang N, Jessen ME, Warner JJ, Lavandero S, Gillette TG, Turer AT and Hill JA Histone deacetylase inhibition blunts ischemia/reperfusion injury by inducing cardiomyocyte autophagy. Circulation 129, 1139–1151 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Morales CR, Li DL, Pedrozo Z, May HI, Jiang N, Kyrychenko V, Cho GW, Kim SY, Wang ZV, Rotter D, Rothermel BA, Schneider JW, Lavandero S, Gillette TG and Hill JA Inhibition of class I histone deacetylases blunts cardiac hypertrophy through TSC2-dependent mTOR repression. Sci Signal 9, ra34 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Riehle C and Abel ED Insulin regulation of myocardial autophagy. Circ J 78, 2569–2576 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Rothermel BA and Hill JA Autophagy in load-induced heart disease. Circ Res 103, 1363–1369 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Yasuda H, Leelahavanichkul A, Tsunoda S, Dear JW, Takahashi Y, Ito S, Hu X, Zhou H, Doi K, Childs R, Klinman DM, Yuen PS and Star RA Chloroquine and inhibition of Toll-like receptor 9 protect from sepsis-induced acute kidney injury. Am J Physiol Renal Physiol 294, F1050–1058 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ridout RM, Wildenthal K and Decker RS Influence of agents that alter lysosomal function on fetal mouse hearts recovering from anoxia and substrate depletion. J Mol Cell Cardiol 18, 867–876 (1986). [DOI] [PubMed] [Google Scholar]
- 152.Gupta SK, Foinquinos A, Thum S, Remke J, Zimmer K, Bauters C, de Groote P, Boon RA, de Windt LJ, Preissl S, Hein L, Batkai S, Pinet F and Thum T Preclinical Development of a MicroRNA-Based Therapy for Elderly Patients With Myocardial Infarction. Journal of the American College of Cardiology 68, 1557–1571 (2016). [DOI] [PubMed] [Google Scholar]
- 153.Salabei JK, Balakumaran A, Frey JC, Boor PJ, Treinen-Moslen M and Conklin DJ Verapamil stereoisomers induce antiproliferative effects in vascular smooth muscle cells via autophagy. Toxicol Appl Pharmacol 262, 265–272 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Chiao YA, Kolwicz SC, Basisty N, Gagnidze A, Zhang J, Gu H, Djukovic D, Beyer RP, Raftery D, MacCoss M, Tian R and Rabinovitch PS Rapamycin transiently induces mitochondrial remodeling to reprogram energy metabolism in old hearts. Aging (Albany NY) 8, 314–327 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Aranguiz-Urroz P, Canales J, Copaja M, Troncoso R, Vicencio JM, Carrillo C, Lara H, Lavandero S and Diaz-Araya G Beta(2)-adrenergic receptor regulates cardiac fibroblast autophagy and collagen degradation. Biochim Biophys Acta 1812, 23–31 (2011). [DOI] [PubMed] [Google Scholar]
- 156.Bahro M, Dammrich J and Pfeifer U Short-term inhibition of cellular autophagy by isoproterenol in the submandibular gland. Virchows Arch B Cell Pathol Incl Mol Pathol 53, 32–36 (1987). [DOI] [PubMed] [Google Scholar]
- 157.Shimomura M, Oyama J, Takeuchi M, Shibata Y, Yamamoto Y, Kawasaki T, Komoda H, Kodama K, Sakuma M, Toyoda S, Inoue Y, Mine D, Natsuaki M, Komatsu A, Hikichi Y, Yamagishi S, Inoue T and Node K Acute effects of statin on reduction of angiopoietin-like 2 and glyceraldehyde-derived advanced glycation end-products levels in patients with acute myocardial infarction: a message from SAMIT (Statin for Acute Myocardial Infarction Trial). Heart Vessels 31, 1583–1589 (2016). [DOI] [PubMed] [Google Scholar]
- 158.Post S, Post MC, van den Branden BJ, Eefting FD, Goumans MJ, Stella PR, van Es HW, Wildbergh TX, Rensing BJ and Doevendans PA Early statin treatment prior to primary PCI for acute myocardial infarction: REPERATOR, a randomized placebo-controlled pilot trial. Catheter Cardiovasc Interv 80, 756–765 (2012). [DOI] [PubMed] [Google Scholar]
- 159.Delbridge AR, Grabow S, Strasser A and Vaux DL Thirty years of BCL-2: translating cell death discoveries into novel cancer therapies. Nat Rev Cancer 16, 99–109 (2016). [DOI] [PubMed] [Google Scholar]
- 160.Marsh SA, Powell PC, Dell’italia LJ and Chatham JC Cardiac O-GlcNAcylation blunts autophagic signaling in the diabetic heart. Life Sci 92, 648–656 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Gao H, Yang Q, Dong R, Hou F and Wu Y Sequential changes in autophagy in diabetic cardiac fibrosis. Mol Med Rep 13, 327–332 (2016). [DOI] [PubMed] [Google Scholar]
- 162.Lai CH, Tsai CC, Kuo WW, Ho TJ, Day CH, Pai PY, Chung LC, Huang CC, Wang HF, Liao PH and Huang CY Multi-Strain Probiotics Inhibit Cardiac Myopathies and Autophagy to Prevent Heart Injury in High-Fat Diet-Fed Rats. Int J Med Sci 13, 277–285 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Chou IP, Chiu YP, Ding ST, Liu BH, Lin YY and Chen CY Adiponectin receptor 1 overexpression reduces lipid accumulation and hypertrophy in the heart of diet-induced obese mice--possible involvement of oxidative stress and autophagy. Endocr Res 39, 173–179 (2014). [DOI] [PubMed] [Google Scholar]
- 164.Russo SB, Baicu CF, Van Laer A, Geng T, Kasiganesan H, Zile MR and Cowart LA Ceramide synthase 5 mediates lipid-induced autophagy and hypertrophy in cardiomyocytes. J Clin Invest 122, 3919–3930 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Guo R, Zhang Y, Turdi S and Ren J Adiponectin knockout accentuates high fat diet-induced obesity and cardiac dysfunction: role of autophagy. Biochim Biophys Acta 1832, 1136–1148 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Cui M, Yu H, Wang J, Gao J and Li J Chronic caloric restriction and exercise improve metabolic conditions of dietary-induced obese mice in autophagy correlated manner without involving AMPK. J Diabetes Res 2013, 852754 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.He C, Bassik MC, Moresi V, Sun K, Wei Y, Zou Z, An Z, Loh J, Fisher J, Sun Q, Korsmeyer S, Packer M, May HI, Hill JA, Virgin HW et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 481, 511–515 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Xu X and Ren J Macrophage migration inhibitory factor (MIF) knockout preserves cardiac homeostasis through alleviating Akt-mediated myocardial autophagy suppression in high-fat diet-induced obesity. Int J Obes (Lond) 39, 387–396 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Hsu HC, Liu CH, Tsai YC, Li SJ, Chen CY, Chu CH and Chen MF Time-dependent cellular response in the liver and heart in a dietary-induced obese mouse model: the potential role of ER stress and autophagy. Eur J Nutr 55, 2031–2043 (2016). [DOI] [PubMed] [Google Scholar]
- 170.Lancel S, Montaigne D, Marechal X, Marciniak C, Hassoun SM, Decoster B, Ballot C, Blazejewski C, Corseaux D, Lescure B, Motterlini R and Neviere R Carbon monoxide improves cardiac function and mitochondrial population quality in a mouse model of metabolic syndrome. PLoS One 7, e41836 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Lee Y, Hong Y, Lee SR, Chang KT and Hong Y Autophagy contributes to retardation of cardiac growth in diabetic rats. Lab Anim Res 28, 99–107 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Eguchi M, Kim YH, Kang KW, Shim CY, Jang Y, Dorval T, Kim KJ and Sweeney G Ischemia-reperfusion injury leads to distinct temporal cardiac remodeling in normal versus diabetic mice. PLoS One 7, e30450 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Sun S, Zhang M, Lin J, Hu J, Zhang R, Li C, Wei T, Sun D, Wei J and Wang H Lin28a protects against diabetic cardiomyopathy via the PKA/ROCK2 pathway. Biochem Biophys Res Commun 469, 29–36 (2016). [DOI] [PubMed] [Google Scholar]
- 174.Zhao Y, Zhang L, Qiao Y, Zhou X, Wu G, Wang L, Peng Y, Dong X, Huang H, Si L, Zhang X, Zhang L, Li J, Wang W, Zhou L and Gao X Heme oxygenase-1 prevents cardiac dysfunction in streptozotocin-diabetic mice by reducing inflammation, oxidative stress, apoptosis and enhancing autophagy. PLoS One 8, e75927 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Zhang M, Zhang L, Hu J, Lin J, Wang T, Duan Y, Man W, Feng J, Sun L, Jia H, Li C, Zhang R, Wang H and Sun D MST1 coordinately regulates autophagy and apoptosis in diabetic cardiomyopathy in mice. Diabetologia 59, 2435–2447 (2016). [DOI] [PubMed] [Google Scholar]
- 176.Lee JH, Lee JH, Jin M, Han SD, Chon GR, Kim IH, Kim S, Kim SY, Choi SB and Noh YH Diet control to achieve euglycemia induces significant loss of heart and liver weight via increased autophagy compared with ad libitum diet in diabetic rats. Exp Mol Med 46, e111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]