

Abstract
Conventional human blood metabolomics employs serum or plasma and provides a wealth of metabolic information therein. However, this approach lacks the ability to measure and evaluate important metabolites such as coenzymes and antioxidants that are present at high concentrations in red blood cells. As an important alternative to serum/plasma metabolomics, we show here that a simple 1H NMR experiment can simultaneously measure coenzymes and antioxidants in extracts of whole human blood, in addition to the nearly 70 metabolites that were shown to be quantitated in serum/plasma recently [Anal. Chem. 2015, 87, 706−715]. Coenzymes of redox reactions: oxidized/reduced nicotinamide adenine dinucleotide (NAD+ and NADH) and nicotinamide adenine dinucleotide phosphate (NADP+ and NADPH); coenzymes of energy including adenosine triphosphate (ATP), adenosine diphosphate (ADP), and adenosine monophosphate (AMP); and antioxidants, the sum of oxidized and reduced glutathione (GSSG and GSH) can be measured with essentially no additional effort. A new method was developed for detecting many of these unstable species without affecting other blood/blood plasma metabolites. The identities of coenzymes and antioxidants in blood NMR spectra were established combining 1D/2D NMR techniques, chemical shift databases, pH measurements and, finally, spiking with authentic compounds. This is the first study to report identification of major coenzymes and antioxidants and quantify them, simultaneously, with the large pool of other metabolites in human blood using NMR spectroscopy. Considering that the levels of coenzymes and antioxidants represent a sensitive measure of cellular functions in health and numerous diseases, the NMR method presented here potentially opens a new chapter in the metabolomics of blood.
Graphical Abstract
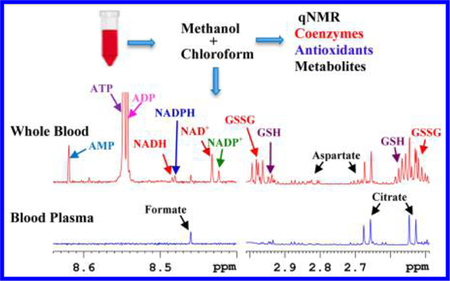
Human blood continues to be the most widely studied biospecimen in the metabolomics field. The association of blood with virtually every living cell in the human body, combined with the ease of its access for routine investigations, imparts clinical relevance for detecting and managing virtually all human diseases. Conventionally, owing to the simplicity of sample handling, blood metabolomics is typically restricted to the analysis of plasma or serum metabolites, while important components of the blood including red blood cells (RBCs), white blood cells, and platelets are discarded during sample processing. In humans, RBCs alone constitute more than 99% of the blood cells and represent nearly 50% of total blood volume. RBCs have high concentrations of many major coenzymes (often referred to as cofactors) of redox reactions and energy, and antioxidants in addition to a large pool of other metabolites that are also common to the widely used serum/plasma metabolome. Redox reactions and energy coenzymes, as well as antioxidants, mediate biochemical reactions fundamental to the functioning of all living cells and hence their levels represent important biomarkers of human health and risk for numerous diseases.1–4 These important species include redox coenzymes: nicotinamide adenine dinucleotide, oxidized (NAD+), nicotinamide adenine dinucleotide, reduced (NADH), nicotinamide adenine dinucleotide phosphate, oxidized (NADP+) and nicotinamide adenine dinucleotide phosphate, reduced (NADPH); energy coenzymes, adenosine triphosphate (ATP) along with its precursors/products adenosine diphosphate (ADP) and adenosine monophosphate (AMP); and antioxidants, glutathione, oxidized (GSSG) and glutathione, reduced (GSH).
Compelling evidence suggests significant value for metabolite profiling of whole blood in investigations of human health and diseases including effects of age and environmental stress.5–8 The whole blood metabolome provides an added opportunity to discern many aspects of cellular function through access to a wider and complementary pool of metabolites that includes coenzymes and cellular antioxidants with the same effort as used in serum/plasma metabolomics. A challenge to date, however, has been that most of the coenzymes are unstable due to their labile nature9–11 and hence, it had been technically difficult to analyze them straightforwardly. For metabolomics, an additional challenge arises in the analysis of several coenzymes using mass spectrometry (MS) due to their unit mass differences and similar retention times in chromatography. The oxidized forms of the redox coenzymes NAD+ and NADP+ interfere with their reduced forms, NADH and NADPH, respectively, and vice versa. Further, ATP and ADP undergo in-source fragmentation to ADP and/or AMP, which affects the accuracy of their analysis.12
Nuclear magnetic resonance (NMR) spectroscopy is one of the major techniques used in serum/plasma metabolomics. It offers numerous benefits including the ability to reliably identify and simultaneously quantify many compounds in complex biological mixtures with high reproducibility and accuracy.13–17 To date, a limited number of NMR-based studies have focused on the metabolomics of whole blood6,18 and RBCs.8,19–21 However, there have been no studies to show the ability to detect and quantify the redox and energy coenzymes, simultaneously, as a part of blood metabolomics. NMR’s ability to analyze labile metabolites such as the coenzymes is a key characteristic. Recently, using tissue samples from heart, liver, kidney, brain, and skeletal muscle from mouse models, we demonstrated the quantitative analysis of many coenzymes ex vivo, simultaneously, using NMR.10
In the current study, we show that the quantitative serum/plasma-based approach can be extended to whole blood metabolomics to expand the metabolite pool and encompass physiologically sensitive coenzymes of redox reactions and energy, as well as antioxidants, in addition to the nearly 70 blood metabolites that we quantified in serum/plasma.22 An extraction method was first developed for the detection of the unstable redox and energy coenzymes. In the blood NMR spectra, the identities of the redox coenzymes NAD+, NADH, NADP+, and NADPH, the energy coenzymes, ATP along with ADP and AMP, and the antioxidants, GSH and GSSG, were established by combining 1D/2D NMR techniques, chemical shift databases, pH measurements, and spiking with authentic compounds. The measurement of important cellular coenzymes and antioxidants along with many other blood metabolites using the NMR method presented here potentially represents an important alternative to serum/plasma metabolomics. This is the first study to report a simple 1H NMR method to reliably detect, identify, and quantify major coenzymes, in addition to the other metabolites using human whole blood.
MATERIALS AND METHODS
Chemicals and Solvents.
Methanol, chloroform, trichloroacetic acid, hydrochloric acid, sodium hydroxide, monosodium phosphate (NaH2PO4), disodium phosphate (Na2HPO4), sodium salt of 3-(trimethylsilyl)propionic acid-2,2,3,3-d4 (TSP), GSH, GSSG, ATP, ADP, and AMP were obtained from Sigma-Aldrich (St. Louis, MO). NAD+ (β-nicotinamide adenine dinucleotide hydrate), NADH (β-nicotinamide adenine dinucleotide (reduced) disodium salt hydrate), NADP+ (β-nicotinamide adenine dinucleotide phosphate hydrate), and NADPH (β-nicotinamide dinucleotide, 2′-phosphate (reduced) tetra sodium salt hydrate) were generously provided by Dr. Jianhai Du, University of Washington. Deuterium oxide (D2O) was obtained from Cambridge Isotope Laboratories, Inc. (Andover, MA). Deionized (DI) water was purified using an in-house Synergy Ultrapure Water System from Millipore (Billerica, MA). All chemicals were used without further purification.
Phosphate Buffer.
Buffer solution (100 mM) was prepared by dissolving 1124.0 mg of anhydrous Na2HPO4 and 249.9 mg of anhydrous NaH2PO4 in 100 g D2O. A solution of TSP was added to achieve a final concentration of 25 μM. The calculated pH of the buffer solution was 7.4 and measured pH was 7.33. This buffer was used without further pH correction.
Stock Solutions.
One-mL stock solutions (1 mM) of the coenzymes/antioxidants (NAD+, NADH, NADP+, NADPH, ATP, ADP, AMP, GSH, and GSSG) were prepared in D2O by diluting their 50 mM solutions, which were first prepared by weighing each compound and dissolving it in D2O. A 60 μL solution of each compound was mixed with 540 μL of phosphate buffer to obtain solutions of 100 μM concentration, which were transferred to 5 mm NMR tubes. Separately, using the 1 mM solutions, 10 identical mixtures of NAD+, NADH, NADP+, NADPH, ATP, ADP, and AMP were prepared with concentrations approximately matched to their physiological levels;10 the pH for each solution was then adjusted between 2 and 11.25 using hydrochloric acid or sodium hydroxide.
Extraction of Metabolites from Human Whole Blood, Blood Plasma, and Red Blood Cells.
Human whole blood from four healthy individuals was obtained from Innovative Research Inc. (Novi, MI). Blood samples were kept under 4 °C temperature, shipped, and used for analysis within 24 h. In addition, blood from eight individuals was obtained at the University of Washington Medical School. Blood samples were collected in heparinized BD Vacutainer tubes (BioVision, CA). A part of the whole blood sample was used directly for analysis after extraction. From the remaining part, blood plasma and red blood cells (RBCs) were separated by centrifuging the whole blood at 4 °C for 10 min at 1500 rcf; RBCs were collected from the bottom layer after discarding the upper buffy coat layer containing white blood cells and platelets. Different extraction methods were evaluated, separately, to determine the optimal method for whole blood metabolite extraction. Various methods evaluated included the use of methanol, a mixture of methanol and water, a mixture of methanol and chloroform, and tricholoroacetic acid. All experiments were performed in accordance with human subjects study guidelines from the University of Washington.
Each protein precipitation/metabolite extraction method used 200 to 400 μL of whole blood, plasma, or RBCs. The biospecimens were mixed with cold methanol in a 1:2 sample/methanol (v/v) ratio, 55% methanol in a 1:9 ratio, methanol/chloroform in a 1:2:2 or 1:3.3:6.6 ratio or 4% trichloroacetic acid in a 1:5 ratio (Table S1). All sample solutions were then vortexed for 30 s, sonicated for 2 min at 4 °C, and incubated at −20 °C for 20 min. The mixtures were centrifuged at 13 400 rcf for 30 min to pellet proteins and cell debris. Clear aqueous solutions were transferred to fresh vials and dried using an Eppendorf Vacufuge-Plus vacuum concentrator for 5 h. Dried samples were mixed with 600 μL phosphate buffer containing 25 μM TSP, spun to sediment any residue, and the supernatants were transferred to 5 mm NMR tubes for analysis.
NMR Spectroscopy.
All NMR experiments were performed at 298 K on a Bruker Avance III 800 MHz spectrometer equipped with a cryogenically cooled probe and Z-gradients suitable for inverse detection. The CPMG (Carr–Purcell–Meiboom–Gill) pulse sequence, one pulse sequence, and NOESY pulse sequence, all with water suppression using presaturation, were used for 1H 1D NMR experiments. Unless otherwise noted in the figure captions, each NMR spectrum was obtained with a total data acquisition time of 12 min using the CPMG pulse sequence with presaturation of residual water signal and 128 transients. All 1D spectra were obtained using 32768 time domain data points, 9615 Hz spectral width and 5 s recycle delay, and the data were Fourier transformed by zero-filling the time domain points by a factor of 2. To confirm the identity of peaks for coenzymes and antioxidants in the whole blood NMR spectra, spectra for authentic compounds dissolved in D2O buffer were obtained. Spectra for mixtures of the coenzymes near their physiological concentrations were obtained at different pH values to evaluate the robustness of the coenzyme peak assignments and the sensitivity of chemical shift values to pH changes for the closely spaced peaks. To aid in metabolite identification, homonuclear two-dimensional (2D) experiments, such as 1H−1H double quantum filtered correlation spectroscopy (DQF-COSY) and 1H−1H total correlation spectroscopy (TOCSY) experiments, were performed on whole blood extracts using methanol/chloroform in a (1 (sample):2:2 v/v/v) ratio for extraction. The 2D experiments were performed with suppression of the residual water signal by presaturation during the relaxation delay. For DQF-COSY and TOCSY experiments, sweep widths of 9615 Hz were used in both dimensions; 512 or 400 FIDs were obtained with t1 increments for DQF-COSY or TOCSY, respectively, each with 2048 complex data points. The number of transients and the recycle delay were 16 and 2 s for DQF-COSY and 40 and 1 s for TOCSY, respectively. The resulting 2D data were zero-filled to 1024 points in the t1 dimension. A 90°-shifted squared sine-bell window function was applied to both dimensions before Fourier transformation. Chemical shifts were referenced to the internal TSP signal for 1H 1D or 2D spectra. Bruker Topspin versions 3.1 and 3.2 software packages were used for NMR data acquisition/processing.
Peak Assignment, Unknown Identification, and Metabolite Quantitation.
Peak assignments relied on established literature, specifically, the human metabolome database (HMDB),23 the biological magnetic resonance data bank (BMRB),24 and our own recent investigations of the human serum metabolome.22 Identification of coenzymes and antioxidants and their confirmation used a combination of (1) comprehensive 2D DQF-COSY and TOCSY spectral analyses, (2) matching with chemical shifts, peak multiplicity, and J couplings from the spectra of authentic compounds, and (3) spiking with authentic compounds. The Bruker AMIX software package was used to quantitate metabolites. Integrals of characteristic, isolated peaks for metabolites (including the newly established coenzymes and antioxidants) in whole blood were obtained from the AMIX software relative to the internal reference signal from TSP to enable the determination of absolute concentrations.
RESULTS AND DISCUSSION
Qualitatively, 1H NMR spectra of human whole blood extracts were very similar to spectra of the corresponding blood plasma with the exception that the whole blood spectra exhibited a number of additional signals (Figure 1). In addition, many peaks that were common to both whole blood and plasma differed in their intensities, significantly (Figure 2). Individual peaks in the spectra of whole blood extracts were identified following the protocol established recently in our laboratory for serum,22 except for the new metabolites that were detected only in whole blood. Investigations of the additional peaks in the whole blood spectra established the identities of the coenzymes of redox reactions, NAD+, NADH, NADP+, and NADPH, coenzymes of energy, ATP along with ADP and AMP, and antioxidants, GSH and GSSG. Identification of these metabolites was achieved by developing a new extraction method and comprehensive NMR analyses as described in Materials and Methods. Spectral regions containing the characteristic peaks for the coenzymes of redox reactions, coenzymes of energy, and antioxidants thus identified in human whole blood are shown in Figures 2 and 3. None of these compounds were detected in plasma. We quantified 56 metabolites in plasma and 64 in whole blood, including coenzymes and antioxidants. Metabolites such as acetone, ethanol, methanol, 2-propanol, dimethyl amine, and urea, however, were not detected here due to sample drying or dissolution in deuterated solvent, as described in our previous study.22 An evaluation of the levels of metabolites quantified in both whole blood and plasma, obtained under identical conditions, indicated a vast difference in concentration for many metabolites; many were higher in whole blood and varied by up to a factor of nearly 34 (Table S2). Figure 4 compares absolute concentrations for metabolites in the whole blood and plasma for a typical healthy individual.
Figure 1.

Typical 800 MHz 1H NMR spectra of human blood plasma, whole blood, and red blood cells (RBCs) from the same individual obtained after extraction using methanol and chloroform in a 1:2:2 ratio (v/v/v). Dashed boxes enclose characteristic regions for the identified coenzymes and antioxidants. Specifically, the region (8.4−8.65 ppm) consisting of peaks for redox (NAD+, NADP+, NADH, and NADPH) and energy (ATP, ADP, and AMP) coenzymes is devoid of any interfering peaks (see also Figure 3).
Figure 2.

Portions of 800 MHz 1H NMR spectra of human whole blood and blood plasma from the same individual obtained after extraction using methanol and chloroform in a 1:2:2 ratio (v/v/v) highlighting detection of reduced glutathione (GSH) and oxidized glutathione (GSSG), and significantly high concentrations for certain metabolites such as aspartic acid, creatine, choline, acetylcarnite, and betaine in whole blood compared to blood plasma.
Figure 3.

Portions of typical 800 MHz 1H NMR spectra of human whole blood and blood plasma from the same individual obtained after extraction using methanol and chloroform in a 1:2:2 ratio (v/v/v). The region for whole blood shows characteristic unique peaks for NAD+, NADP+, NADH, NADPH, ATP, ADP, and AMP. Inset: all coenzymes peaks seen in the whole blood spectrum arise from the lone hydrogen atom on the five membered ring of the adenine moiety as indicated by an arrow. Note, none of the coenzymes was detected in plasma.
Figure 4.

Comparison of absolute concentrations of metabolites detected in whole blood with those detected in blood plasma from one healthy individual. Note: GSH was not detected in this sample due to oxidation to GSSG.
Although the coenzymes of redox reactions and energy have high concentrations in human whole blood, the detection of these species was challenged by their unstable nature, which either evaded detection or altered their levels by the use of an extraction method optimized for serum/plasma.22 Hence, in this study, we first explored the development of an alternative method for the detection of the labile compounds. We recently circumvented a similar challenge for the analysis of these coenzymes in animal tissue samples, which led to the development of a simple NMR method for their analysis in one step.10 In the current study, by employing a similar strategy, we evaluated several solvent combinations including those used in a number of previous investigations (Table S1).5,10,22 The results show that a mixture of blood/methanol/chloroform in a ratio of 1:2:2 enabled the detection of all the coenzymes, while the other combinations failed to detect unstable coenzymes, NADH and NADPH in particular (Figure S1). Subsequent analyses of whole blood, plasma as well as RBC metabolites were therefore carried out using the same 1:2:2 mixture ratio.
In a recent investigation of human serum, we previously showed that using methanol in a 1:2 ratio enabled optimal metabolite extraction, which led to the identification and quantification of nearly 70 metabolites in serum.22 Our efforts to extend this approach for whole blood metabolites proved unsuccessful for the detection of the unstable coenzymes. Recognizing the fact that chloroform plays a critical role in stabilizing these coenzymes,10 one of the methods we evaluated included the introduction of chloroform into the extraction solution without altering the proportion of methanol optimized for serum/plasma metabolites.22 The results show that chloroform is very useful to help detect all the coenzymes in whole blood without affecting the recovery of other blood metabolites. This is substantiated by comparison of metabolite levels for the same blood sample obtained after extraction using methanol and chloroform (1:2:2) or only methanol (1:2) (Figure S2). Clearly, these results indicate that addition of chloroform, while facilitating the detection of the unstable coenzymes, does not affect the recovery of other metabolites deleteriously.
A major challenge for the assignment of the redox and energy coenzymes arose from their similarity in structure, which resulted in similar spectra with either completely overlapped or very closely spaced peaks (Figure S3). The relatively weak signals for some coenzymes such as NADH and NADPH, due to their low concentrations of a few μM (Figures 3 and 4), added to the challenge. The chemical shift region between 8.4 and 8.65 ppm showed characteristic isolated peaks for all the coenzymes, although, peaks for some of the coenzymes were closely spaced. Peaks in this region arise from the lone hydrogen atom of the adenine moiety from the coenzymes. These peaks are all singlets and hence offer an added advantage for reliable identification and analysis. It is also noteworthy that no interfering peaks from blood appear in this region (Figure 3). One concern, however, was the potential pH sensitivity of the chemical shifts, which might cause the closely spaced peaks to interchange positions. For example, the NADH and NADPH peaks are separated by <0.004 ppm (3 Hz). To address this issue, the pH sensitivity of the peaks was assessed using mixtures of authentic compounds with their concentrations matched approximately to their physiological levels. The results indicate that although the peak characteristics, including chemical shifts and line widths, were sensitive at pH < 6.5, physiological pH (pH = ~7.4) provides a highly reproducible fingerprint for all the coenzymes with no interchange of any peak positions (Figure S4).
With regard to molecular stability, it was shown recently in the analysis of coenzymes in animal tissue that the redox coenzymes are unstable in solution, especially, at room temperature.10 Similar investigations here on whole blood metabolites showed that the reduced forms NADH and NADPH become oxidized to NAD+ and NADP+, respectively, relatively quickly. After 24 h at room temperature, a significant reduction in the levels of NADH and NADPH was detected with a proportional increase of corresponding oxidized coenzymes. However, flushing the NMR solution and tubes with an inert gas such as helium reduced the oxidation, significantly (Figure S5).
The ratios of coenzymes provide a measure of cellular redox status and their imbalance can be related to numerous diseases.25 Metabolically, the NADH/NAD+ ratio and pool size are mainly associated with glycolysis and the TCA cycle, while those for NADPH/NADP+ are associated with the pentose phosphate pathway. Because of their high oxygen content, RBCs have a high demand for NADPH to regenerate GSH from GSSG, which is necessary to protect RBCs from oxidative damage.26,27 Further, RBCs lack mitochondria and hence TCA cycle metabolism is absent in these cells. Because of these reasons, the NADPH/NADP+ pool size is anticipated to be higher in blood than in cells that contain mitochondria. Our results do indeed show that the NADPH/NADP+ pool is higher by more than an order of magnitude compared to that found in heart tissue, for example. The ratio between the NADPH/NADP+ pool and NADH/NAD+ pool was 0.58 ± 0.02 in blood vs 0.05 ± 0.005 in heart tissue.10 In addition, the NADP+/NAD+ ratio obtained in our study is similar to that reported for human blood RBCs previously.28
A major advantage of whole blood analysis is that it provides access to both the plasma/serum and RBC metabolome in one step and with little additional effort, as compared to the traditionally used serum or plasma analysis. Thus, whole blood metabolomics offers an added opportunity to gain insights into additional metabolites and metabolic pathways. Of course, RBC metabolomics can be performed separately as well.8,20,28 In addition, whole blood metabolomics enables routine access to important coenzymes and antioxidants, which represent a sensitive measure of cellular function in health and disease. A recent NMR study compared serum and whole blood metabolite profiles and highlighted the benefits of whole blood metabolite analysis.6 Specifically, this study focused on investigations of sepsis highlights the contribution of the RBC metabolome in distinguishing diseases. However, there had been no study so far, either by NMR or MS, which demonstrated the ability to analyze major coenzymes and antioxidants in addition to many of other blood metabolites, simultaneously, in whole blood. The ability to reliably measure these important species by NMR spectroscopy represents an enabling step in blood-based metabolomics.
An altogether different advantage of whole blood metabolite analysis is that, unlike with serum or plasma, whole blood is devoid of confounding effects potentially associated with hemolysis. Separating serum/plasma from whole blood involves sample preprocessing, which is a critical step and carries the risk of cellular metabolites seeping into the serum/plasma due to hemolysis. RBCs are rich with metabolites and transport, apart from oxygen, many metabolites including amino acids to various parts of the human body.5,29 As noted above, nearly 50% of whole blood is constituted by RBCs, and contamination of serum/plasma with the RBC metabolome due to hemolysis is common. Hemoglobin in serum/plasma is a marker of hemolysis and its concentration represents the degree to which hemolysis has occurred.30 Previous studies have found varying amounts of hemoglobin even in normal serum and the hemoglobin correlated positively with many metabolites including ATP, which is a product of hemolysis;6,31 these results indicate the deleterious confounding effects of hemolysis on serum/plasma metabolite profiling. Apart from hemolysis, even differing levels of oxygen in RBCs can cause release of RBC metabolites into plasma,32,33 which further contaminates the serum/plasma metabolome. It is often difficult to exclude hemolyzed samples from analysis and hence a suggestion to list metabolites that are unreliable to measure in hemolyzed samples was made recently.34 Our investigations show that virtually all NMR detected metabolites in plasma are common to RBCs; and many are highly enriched in RBCs (see, for example, Figure S6 and Table S2). Hence, excluding such potentially unreliable metabolites in the analysis of serum/plasma would eliminate many important metabolites.
It is important to note that RBCs are metabolically very active cells. Glycolysis is the major metabolic pathway in RBCs to meet the energy demand and to protect cells from reactive oxygen species using GSH as the antioxidant. Under hypoxic conditions RBCs exhibit accelerated glucose consumption leading to accumulation of lactate.35,36 On the basis of nontargeted metabolomics, it was shown that metabolic changes in plasma separated from whole blood and kept on ice for up to 4 h may be minor when compared to freshly prepared plasma.37 In our study, whole blood stored at 4 °C for 24 h, before extraction, exhibited a decrease in the levels of glucose with a concomitant increase in lactate levels (Figure S7). These results are in accordance with the anticipated accelerated glycolysis in RBCs under storage and indicate the deleterious effects of storing whole blood on ice or at 4 °C for longer time periods before extraction or separating plasma.
The antioxidants, GSH and GSSG have been previously identified and characterized by 1H NMR in intact whole RBCs, to assess redox status.21,38,39 However, owing to the relatively poor spectral resolution, this approach has not found wider utility. On the other hand, the challenge for ex vivo analysis of GSH is that it is extremely labile and it readily oxidizes to GSSG. Owing to its unstable nature, inconsistent results for GSH and GSSG have been reported in a large number of published studies.40 To overcome this challenge, it is suggested that the unstable thiol group of GSH be blocked by derivatization using agents such as N-ethylmaleimide before extraction/protein precipitation. In our study, as a result of the lability of GSH, high levels of GSSG were detected in whole blood extracts arising from the conversion of GSH to GSSG (Figures 2 and 4). Thus, in the absence of detectable GSH, the measured GSSG level represents the sum of GSH and GSSG.
In conclusion, we present a simple NMR method for metabolite profiling of human whole blood. The method is complementary to the widely used plasma and serum based metabolomics and offers access to an expanded pool of metabolites. Importantly, the method enables access to major coenzymes of redox reactions (NADH, NAD+, NADPH, and NADP+), coenzymes of energy (ATP, ADP, and AMP), and antioxidants (the sum of GSH and GSSG). The ability to measure these classes of compounds routinely along with a large pool of other metabolites in blood provides new opportunities for investigations of human diseases. Owing to numerous challenges including the instability of many coenzymes, thus far many of these compounds had not been analyzed simultaneously in human blood by any method. The present study thus represents the first, simple method to detect all these important molecules along with a large pool of other blood metabolites. The redox and energy coenzymes and antioxidants play a fundamental role in virtually all cellular functions and are normally measured using tissue specimens. Here, the ability to measure them in the blood, instead, opens a new avenue for routine use in health and disease association studies. In general, whole blood metabolomics enables access to a wider and complementary pool of metabolites but also avoids the confounding effect of hemolysis in serum/plasma, when present. The NMR method presented here for whole blood metabolite analysis potentially opens a new chapter in the metabolomics of blood.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge financial support from the NIH (National Institute of General Medical Sciences Grant R01GM085291).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.anal-chem.7b00171.
Sample information, ratios between whole blood and plasma metabolite concentrations, whole blood metabolite concentrations with different extractions, plasma and RBC metabolite concentrations, and 1H NMR spectra (PDF)
The authors declare the following competing financial interest(s): Daniel Raftery reports holding equity and an executive position at Matrix Bio, Inc.
REFERENCES
- (1).Ying W Antioxid. Redox Signaling 2008, 10 (2), 179–206. [DOI] [PubMed] [Google Scholar]
- (2).Nakamura M; Bhatnagar A; Sadoshima J Circ. Res 2012, 111 (5), 604–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Oka S; Hsu CP; Sadoshima J Circ. Res 2012, 111 (5), 611–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Mischley LK; Standish LJ; Weiss NS; Padowski JM; Kavanagh TJ; White CC; Rosenfeld ME Oxid. Med. Cell. Longevity 2016, 2016, 9409363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Chaleckis R; Murakami I; Takada J; Kondoh H; Yanagida M Proc. Natl. Acad. Sci. U. S. A 2016, 113 (16), 4252–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Stringer KA; Younger JG; McHugh C; Yeomans L; Finkel MA; Puskarich MA; Jones AE; Trexel J; Karnovsky A Shock 2015, 44 (3), 200–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Chaleckis R; Ebe M; Pluskal T; Murakami I; Kondoh H; Yanagida M Mol. BioSyst 2014, 10 (10), 2538–51. [DOI] [PubMed] [Google Scholar]
- (8).Catalán Ú; Rodríguez MÁ; Ras MR; Maciá A; Mallol R; Vinaixa M; Fernández-Castillejo S; Valls RM; Pedret A; Griffin JL; Salek R; Correig X; Motilva MJ; Sola R Mol. BioSyst 2013, 9 (6), 1411–1422. [DOI] [PubMed] [Google Scholar]
- (9).Gil A; Siegel D; Permentier H; Reijngoud DJ; Dekker F; Bischoff R Electrophoresis 2015, 36, 2156–2169. [DOI] [PubMed] [Google Scholar]
- (10).Nagana Gowda GA; Abell L; Lee CF; Tian R; Raftery D Anal. Chem 2016, 88 (9), 4817–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Liang X; Yang L; Qin AR; Ly J; Liederer BM; Messick K; Ma S; Zak M; Dragovich PS; Dean BJ; Hop CE; Deng Y Bioanalysis 2014, 6 (11), 1445–1457. [DOI] [PubMed] [Google Scholar]
- (12).Trammell SAJ; Brenner C Comput. Struct. Biotechnol. J 2013, 4, e201301012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Nagana Gowda GA; Raftery DJ Magn. Reson 2015, 260, 144–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Powers RJ Med. Chem 2014, 57 (14), 5860–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Bingol K; Bruschweiler R Anal. Chem 2014, 86 (1), 47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Clendinen CS; Stupp GS; Ajredini R; Lee-McMullen B; Beecher C; Edison AS Front. Plant Sci 2015, 6, 611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Fan TW; Lane AN Prog. Nucl. Magn. Reson. Spectrosc 2016, 92–93, 18–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Serkova NJ; Zhang Y; Coatney JL; Hunter L; Wachs ME; Niemann CU; Mandell MS Transplantation 2007, 83 (4), 517–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Teng R; Junankar PR; Bubb WA; Rae C; Mercier P; Kirk K NMR Biomed. 2009, 22 (3), 292–302. [DOI] [PubMed] [Google Scholar]
- (20).Pertinhez TA; Casali E; Lindner L; Spisni A; Baricchi R; Bernim P Blood Transfus. 2014, 12 (4), 548–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Rabenstein DL J. Biochem. Biophys. Methods 1984, 9 (4), 277–306. [DOI] [PubMed] [Google Scholar]
- (22).Nagana Gowda GA; Gowda YN; Raftery D Anal. Chem 2015, 87 (1), 706–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Wishart DS; Jewison T; Guo AC; Wilson M; Knox C; Liu Y; Djoumbou Y; Mandal R; Aziat F; Dong E; Bouatra S; Sinelnikov I; Arndt D; Xia J; Liu P; Yallou F; Bjorndahl T; Perez-Pineiro R; Eisner R; Allen F; Neveu V; Greiner R; Scalbert A Nucleic Acids Res 2013, 41 (D1), D801–D807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Ulrich EL; Akutsu H; Doreleijers JF; Harano Y; Ioannidis YE; Lin J; Livny M; Mading S; Maziuk D; Miller Z; Nakatani E; Schulte CF; Tolmie DE; Kent Wenger R; Yao H; Markley JL Nucleic Acids Res. 2007, 36 (Database), D402–D408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Bar-Or D; Bar-Or R; Rael LT; Brody EN Redox Biol. 2015, 4, 340–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).D’Alessandro A; Geviz F; Zolla L Mol. BioSyst 2013, 9, 1196–1209. [DOI] [PubMed] [Google Scholar]
- (27).Thompson RH Clin. Chim. Acta 1965, 12 (2), 198–205. [DOI] [PubMed] [Google Scholar]
- (28).Teng R; Lehane AM; Winterberg M; Shafik SH; Summers RL; Martin RE; van Schalkwyk DA; Junankar PR; Kirk K Biosci. Rep 2014, 34 (6), 685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Tunnicliff G Comp. Biochem. Physiol. Comp. Physiol 1994, 108 (4), 471–478. [DOI] [PubMed] [Google Scholar]
- (30).Malinauskas RA Artif. Organs 1997, 21, 1255–1267. [DOI] [PubMed] [Google Scholar]
- (31).Gorman MW; Feigl EO; Buffington CW Clin. Chem 2007, 53 (2), 318–325. [DOI] [PubMed] [Google Scholar]
- (32).Bergfeld GR; Forrester T Cardiovasc. Res 1992, 26 (1), 40–47. [DOI] [PubMed] [Google Scholar]
- (33).Ellsworth ML; Forrester T; Ellis CG; Dietrich HH Am. J. Physiol 1995, 269 (6), H2155–H2161. [DOI] [PubMed] [Google Scholar]
- (34).Denihan NM; Walsh BH; Reinke SN; Sykes BD; Mandal R; Wishart DS; Broadhurst DI; Boylan GB; Murray DM Clin. Biochem 2015, 48, 534–537. [DOI] [PubMed] [Google Scholar]
- (35).Murphy JR J. Lab. Clin. Med 1960, 55, 286–302. [PubMed] [Google Scholar]
- (36).Hamasaki N; Asakura T; Minakami SJ Biochem 1970, 68 (2), 157–161. [DOI] [PubMed] [Google Scholar]
- (37).Yin P; Peter A; Franken H; Zhao X; Neukamm SS; Rosenbaum L; Lucio M; Zell A; Haring HU; Xu G; Lehmann R Clin. Chem 2013, 59 (5), 833–845. [DOI] [PubMed] [Google Scholar]
- (38).Reglinski J; Smith WE; Wilson R; Buchanan LM; McKillop JH; Thomson JA; Brzeski M; Marabani M; Sturrock RD Clin. Chim. Acta 1991, 201, 45–58. [DOI] [PubMed] [Google Scholar]
- (39).Reglinski J; Smith WE; Brzeski M; Marabani M; Sturrock RD J. Med. Chem 1992, 35 (11), 2134–7. [DOI] [PubMed] [Google Scholar]
- (40).Rossi R; Milzani A; Dalle-Donne I; Giustarini D; Lusini L; Colombo R; Di Simplicio P Clin. Chem 2002, 48 (5), 742–753. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.