

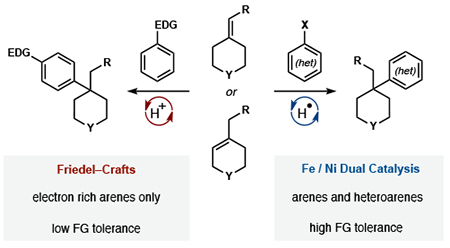
Abstract
Alkene hydroarylation forms carbon–carbon bonds between two foundational building blocks of organic chemistry: olefins and aromatic rings. In the absence of electronic bias or directing groups, only the Friedel–Crafts reaction can engage alkenes with Markovnikov selectivity to generate quaternary carbons. However, the intermediacy of carbocations precludes the use of electron-deficient arenes, including Lewis basic heterocycles. Here we report a highly Markovnikov-selective, dual-catalytic olefin hydroarylation that tolerates arenes and heteroarenes of any electronic character. Hydrogen atom transfer controls the formation of branched products and arene halogenation specifies attachment points on the aromatic ring. Mono-, di-, tri- and tetra-substituted alkenes yield Markovnikov products including quaternary carbons within non-strained rings.
Graphical Abstract
INTRODUCTION
Accelerating interest in high-fraction sp3 (Fsp3) scaffolds has highlighted two major gaps in chemical methodology.1 First, few metal-catalyzed cross-coupling reactions are competent to generate quaternary ring carbons at a non-bridgehead position.2 The products are three-dimensional equivalents of biaryl motifs and represent chemical space that is sparsely populated by synthetic libraries and typically occupied by natural products.3 Second, although nickel complexes have recently reigned supreme as alkyl cross-coupling catalysts, reaction partners can suffer from a triple Achilles heel of low stability, low synthetic efficiency, and low atom economy (high formula weight loss).4 An ideal disconnection would obviate the need for pre-installation of a functional group and open access to a wide range of scaffolds or products inaccessible with other cross-coupling methods. Here we report a cross-coupling reaction with broad scope that forms quaternary carbons from a simple, abundant feedstock: the olefin.
Pioneering nickel-catalyzed arylation methods have used halides,5 organometaloids,6 carboxylic acids,7 and sulfones8 as alkyl radical precursors9 (Figure 1). Olefins offer orthogonal reactivity and the distinct advantage of relative inertness, allowing them to be carried through multistep syntheses. Furthermore, they are among the most prevalent functional groups in natural products and commercially available building blocks. Where Nature is not forthcoming, olefin synthesis or metathesis can merge two building blocks, and the nascent olefin itself can serve as the coupling point (Figure 2). Prior methods for arylative quaternary centers synthesis first form a functionalized tetrasubstituted carbon2a—c, in itself a challenging task; yet the functional group is next ablated during the cross-coupling. A unique benefit of olefins is the ability to form a quaternary center without starting from a preformed tetrasubstituted carbon, circumventing the prefunctionalization step and improving the atom economy of the transformation.
Figure 1.

Prior methods for the formation of arylated quaternary centers using Ni-catalysis.
Figure 2.

Current work combining HAT and Ni catalysis.
Prior methods10 for the hydroarylation of olefins rely on the innate reactivity of arenes. Friedel–Crafts alkylation, a foundational reaction that adds a proton and arene across unbiased α-olefins with high Markovnikov regioselectivity, is generally limited to acid-stable, electron-rich aromatics, whereas electron-deficient and Lewis-basic heteroarenes are not tolerated.11 Similarly, the Minisci reaction is confined to electron-deficient arenes and dependent on substrate bias for its innate regiocontrol.12 In both methods, positional selectivity is controlled by ring electronics and mixtures often result. In contrast, cross-coupling takes advantage of the programmed reactivity of aryl halides, whereby a transition metal enables absolute regiocontrol and the ability to use the full spectrum of arene electronics.4
Here we demonstrate a Markovnikov-selective Fe/Ni dual-catalytic alkene hydroarylation of mono-, di-, tri-, and tetrasubstituted alkenes utilizing electronically diverse arenes and heteroarenes. Furthermore, this cross-coupling embeds quaternary carbons in oxetanes, pyrans, pyrrolidines, piperidines, and thianes in a single step from commercially available materials. Application of this dual catalytic system allowed for improved access to 3 drug leads and high-value building blocks for biomedical application.
RESULTS AND DISCUSSION
Recently we reported a proof-of-principle that olefins could engage in highly branch-selective hydroarylation by merging cobalt-catalyzed hydrogen atom transfer (HAT) and nickel catalysis.13 This reaction was limited to mono-substituted olefins, since substituted olefins (1a) yielded exclusively an allylic isomer (1a’) (Figure 3). This limitation correlated with the tendency for terminal alkenes to form stable organocobalt complexes,14 implicating a transmetalation pathway. Tert-alkylcobalt species have never been observed15 and therefore their participation in this dual catalysis was deemed unlikely, if not mechanistically forbidden.
Figure 3.

Comparison to our prior work with Co(salen) and our current work with Fe(dpm)3: transmetalation vs free radical mechanism.
Early examples of HAT catalysis observed similar collapse to organometallics or rapidly reversible reactions that resulted in inverse kinetic isotope effects.16a More recently, both Carreira16b and Baran16c observed non-inverse (normal) KIEs in reactions where HAT has been implicated, which indicates an irreversible atom transfer and cage escape. Successful engagement of a tert-alkyl radical with a low valent nickel might, therefore, occur with alternative catalyst systems, but would require a complete reconstruction of the dual catalytic cycles.
For the cycles to merge by unstabilized C-centered radical interception, the independent rates of each cycle must be similar. Whereas salen ligands cause rapid alkene consumption,14 alternative ligands decelerate the catalytic cycle.17 Mukaiyama’s pioneering work in alkene radical hydrofunctionaliza tion17,18 indicated that β-diketonate ligands significantly changed reaction rates, but subsequent interception of nickel catalytic cycles lacked precedent. Co(acac)2 consumed the olefin within an hour through hydrogenation and hydration, but yielded none of our desired product (Table 1, entry 1). Mn(dpm)3 similarly yielded only traces of the desired product, however there was little consumption of either starting material (entry 2) and a white heterogeneous slurry resulted. Iron, however, showed no promiscuity with Ni and delivered the first evidence of successful arylation, consistent with the initial cage escape hypothesis.16c,18c,19
Table 1.
Sec-alkyl Optimization
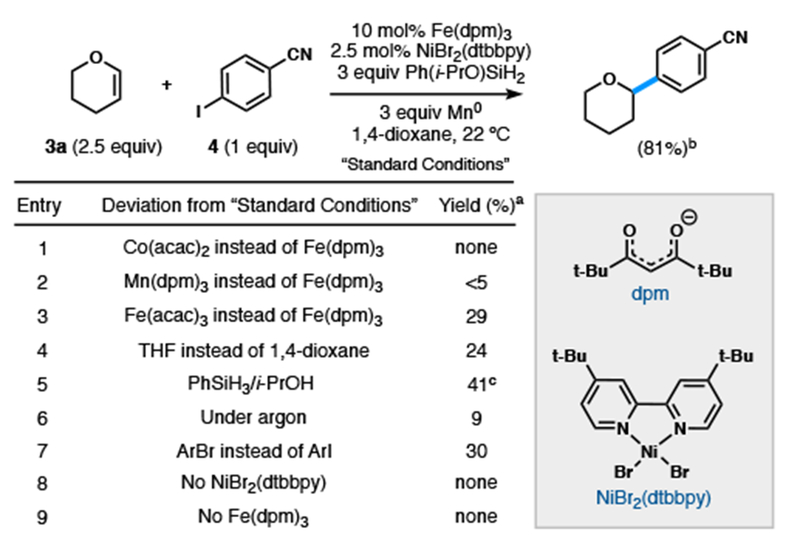 |
0.1 mmol scale, yield determined by FID using dodecane as an internal standard.
0.3 mmol scale, isolated yield.
3 equiv PhSiH3, 6 equiv i-PrOH (anhydrous) instead of Ph(i-PrO)SiH2.
Investigation of olefin 3a and aryl iodide 4 using Fe(acac)316c,18c,19a,b (entry 3) yielded product 3b with low catalyst turnover in the presence of metallic reductants. Whereas Mukaiyama observed no difference between sterically-differentiated complexes of Fe,18c Baran and co-workers correlated increased bulk with reaction efficiency, which they proposed resulted from decreased catalyst decomposition.16c,19b Accordingly, bench stable catalyst, Fe(dpm)3, proved ideal. A brief survey of nickel ligands identified 4,4-di-tert-butylbipyridine (dtbbpy) as optimal, requiring only 2.5 mol% of Ni. The reaction was highly solvent dependent: 1,4-dioxane led to high yields and outperformed similar ethereal solvents (entry 4). Due to the limited solubility of Ni(II) precatalysts in 1,4-dioxane, we observed that precomplexed NiBr2(dtbbpy) allowed for more reproducible yields. Use of Ph(i-PrO)SiH217,20 proved superior to a PhSiH3/i-PrOH21 combination (entry 5) or PhSiH3 alone. Finally, the presence of air was found to be critical for reactivity; no catalytic turnover occurred in its absence (entry 6).16c
Optimal hydroarylation conditions transformed a variety of feedstock olefins—di-, tri- and tetra-substituted (5a-19a)—to branched aromatics (5b-19b) with exceptionally high regioselectivity.22 All olefins were coupled in >20:1 regioselectivity, except where noted. The reaction proved tolerant to many functional groups such as carbamates (7b), strained cyclobutanes (8b), redox-active esters (RAEs) (16b), amides (17b), vinyl thioethers (18b), vinyl boronates (19b), and nitriles (5b-19b). Ring size did not affect reactivity and the arylated products (9b-12b) were obtained in high yield. Although this olefin cross-coupling can be viewed as complementary to decarboxylative or dehalogenative cross-electrophile coupling in some cases, in others there are no options: olefins 8a and 13a are unavailable as the corresponding carboxylate and unprecedented as the halide. Interestingly, [2.2.0]-bicyclohexene 8a, an intermediate in Burns’ scalable ladderane synthesis23 could be hydroarylated in 70% yield where the strained cyclobutyl arene predominated (3:1) over the ring-opened product. Commercially available perfluorinated olefin 13a and other fluorine-containing substrates (14a, 15a) were successfully arylated, offering unique access to these perfluoroalkane and - arene derivatives in moderate to high yields. The propensity of RAEs to cross-couple with iodoarenes or reduce in the presence of silanes is established,24 yet RAEs (16b) emerged unscathed from our reaction conditions: a 95:5 selectivity for cross-coupling via the olefin vs. the ester demonstrated the orthogonality of this coupling to prior work.
More challenging 1,1-disubstituted and trisubstituted olefins probed the capacity of this method to generate quaternary carbons and hindered attached ring motifs. Their reactivity required interception of nickel with a tertiary carbon radical, consistently a more difficult cross-coupling than sec-alkyl counterparts.2,4 Consequently, the conditions described above to couple secondary radicals for olefin 1a and aryl iodide 2 yielded only arylation of the ethereal solvent (Table 3, entry 1).2c An exhaustive ligand screen resulted in, at best, a 1:1 mixture of regioisomers. Instead, the “ligandless” nickel system developed by Fu provided superior results to traditional conditions.25 NiBr2(diglyme) in a mixture of THF/DMA produced the desired product 1b with high regioselectivity (>20:1), although NMP (entry 2) eventually proved superior and 1,2-DCE increased Fe(dpm)3 solubility. Even though air was still critical for reactivity, the addition of MnO2 led to higher catalyst turnover (entry 3).
Table 3.
Formation of Quaternary Center Optimization
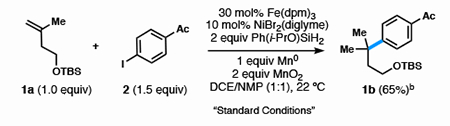
| Entry | Deviation from “Standard Conditions” | Yield (%)a | |
|---|---|---|---|
| 1 | 10 mol% NiBr2(dtbbpy) | none |  |
| 2 | No NMP | none | |
| 3 | No MnO2 | 20 | |
| 4 | Under O2 instead of air | tracec | |
| 5 | 1 equiv H2O | none | |
| 6 | 5 mol% NiBr2(diglyme) | 54 | |
| 7 | 2 equiv Mn | 43 | |
| 8 | 1 equiv Ph(i-PrO)SiH2 | 28 | |
| 9 | No NiBr2(diglyme) | none | |
| 10 | No Fe(dpm)3 | none |
0.1 mmol scale, yield determined by FID using dodecane as an internal standard.
0.3 mmol scale, isolated yield.
Under an O2 balloon, the olefin was predominantly consumed to hydration via Drago—Mukaiyama Hydration.
The significance of this reaction was illustrated by the ease with which quaternary carbons could be generated from simple alkenes, many of which (20a–22a, 24a–30a, 32a–34a) were in turn accessed via one-step olefination reactions from widely available ketones. We focused on cyclic coupling partners, which led to high Fsp3 scaffolds that extend along multiple vectors, yet are uncommon motifs in cross-coupling reactions.3 Embedded in these unsaturated substrates are Lewis basic functionality (20a–29a), carbamates (26a–29a), esters (21a, 30a), silyl ethers (1a, 22a, 32a), phthalimides (38a), carbocycles (30a–34a) and acid sensitive ring motifs (22a, 26a–29a, 32a), all of which were tolerated and left the high branch selectivity largely unaffected (>20:1 regioselectivity except where noted).
Unsaturated heterocycles—oxetanes (20a–22a), tetrahydrofurans (23a), pyrans (24a), thianes (25a), pyrrolidines (26a–28a), and piperidines (29a) —all productively coupled. Of particular note, the compatibility of thianes with this method resulted in the first reported synthesis for this type of attached ring motif. Previously, placement of a quaternary center at C-3 of a pyrrolidine has required the intermediacy of either 2-pyrrolidinone or succinimide, resulting in a 3–4 step sequence that includes strong hydride reductants.26 Fe/ Ni dual-catalysis streamlined access via 3-pyrrolidinone olefination (to 26a–28a) and high-yielding arylation (26b–28b). Olefination via ylides can generate a variety of pyrrolidines for which the corresponding tertiary bromides or boronic acids are unknown, making this method the only viable cross-coupling of these scaffolds.
Acylic olefins have a greater potential to engage in background reductive Heck (anti-Markovnikov) reactivity than their more-hindered cyclic counterparts.2d We nevertheless observed complete regioselectivity; no isomers were detected (1b, 35b–37b, 39b). Allylic substitution was well tolerated yielding branched diols (37b) and phthalimides (38b) in moderate to high yield. The increased steric bulk on tetrasubstituted olefins impedes accessibility by a metal catalyst, making them less reactive than disubstituted olefins.27 Despite this, tetramethylethylene (39b) still served as a compatible olefin, albeit in modest yield. Notably, arylation of 1a could be performed on gram scale without a significant deterioration in yield (52%).
Natural products and their derivatives (40a-46a) could also serve as successful coupling partners (Figure 2). Allyl glycine (40a) was arylated in high yield, opening the door to a range of unnatural amino acids. The terpenes caryophyllene oxide (42a) and β-pinene (43a) were arylated as their ring-opened products; however no evidence of epoxide opening was observed for 42b, a demonstration of the mild, non-acidic conditions.
Although this method was compatible with a broad array of olefins, sterically hindered or unstable olefins (46a–48a) were low yielding and returned substantial starting materials.28 Arylation of tropinone derivative 47a offered access to a previously unexplored vector of phenyltropane derivatives, cocaine receptor ligands that have been pursued for the treatment of drug dependency, Parkinson’s disease, and depression.29 1,3-diaxial interactions across the tropane ring significantly challenged the arylation and the product was obtained in low yield. Similarly, the highly congested core of tetrahydrocanabanoid derivative 48a prevented significant engagement by the Fe catalyst and, while the product could be observed in low yield, the reaction yielded mostly unreacted olefin. For an additional list of less reactive substrates, see the SI.
In contrast to traditional Markovnikov-selective arylation reactions (Friedel-Crafts, Minisci), this dual-catalytic method was compatible with a range of arene electronics. Under the sec-alkyl conditions, the highest yields were obtained with electron-deficient arenes (Table 5A, 49, 50, 53–61), however 4-iodoanisole (52) and an aryl boronate ester (51) were also successfully coupled. Heterocycles (57-61) served as effective coupling partners, although pyridines required substitution at C2.
Table 5.
Aryl Halide Scope
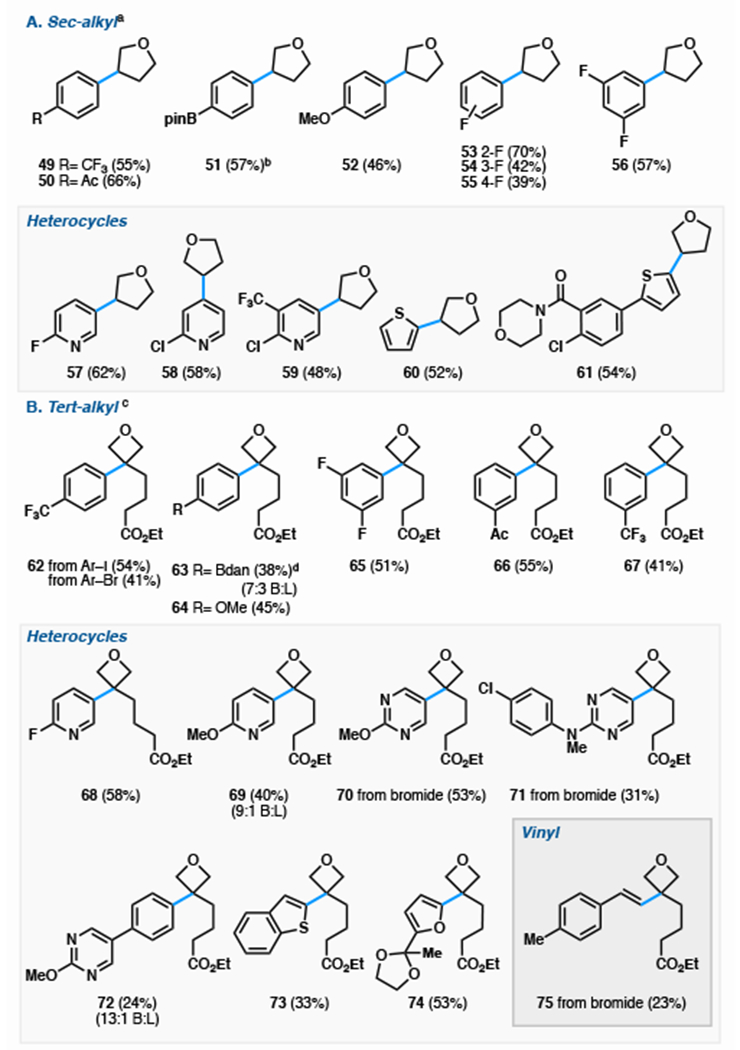 |
A) Aryl halide scope for sec-alkyl conditions starting from olefin 6a.
0.3 mmol scale, isolated yield, reaction time: 12—48 h. 1 mL 1,4-dioxane, N2 atmosphere for 2 h, then open to air. All substrates gave >20:1 branched:linear ratio except where notated
2:1 ratio of arylation at C3 vs C2.
(B) Aryl halide scope for tert-alkyl conditions using olefin 21a.
0.3 mmol scale, isolated yield, reaction time 16 h. 1.5 mL 1,2-DCE and 1.5 mL NMP. N2 atmosphere for 1.5 h then open to air.
Electron-deficient, -neutral, and -rich arenes successfully coupled in moderate to good yield, providing a library of C3-quaternary oxetanes (Table 5B, 62–75). Iodopyridines (68, 69) and bromopyrimidines (70, 71) were successful coupling partners independent of electronic character, as was electron-rich 2-iodo-5-furan-dioxolane, which coupled in high yield with the acid-labile acetal fully intact. Other electron-rich heterocycles, such as benzothiophene proved to be more difficult, yet still were compatible—crucial for structure-activity interrogation campaigns. As proof-of-principle for the broader generality of this reaction engine, we found that a vinyl bromide could replace the haloarene and engage in cross-coupling with no subsequent hydrogenation of the styrene product (75). Interestingly, bromoarenes coupled effectively with tert-alkyl partners, but were significantly less efficient with sec-alkyl reactants (62 vs Table 1, entry 7). The capacity of bromoarenes to cross-couple offers a significant benefit due to their breadth of commercial availability compared to iodoarenes (ca. 78,000 (hetero)aryl bromides versus ca. 17,000 (hetero)aryl iodides).30
During the assessment of the haloarene scope (Table 5), we focused on arenes in which Friedel–Crafts or Minisci reactions would be unsuccessful.11,12 The position of aryl halide substitution perfectly translated to alkylated product, whereas Friedel–Crafts alkylations (e.g. with anisole) generate mixtures of o- and p- regioisomers. Friedel–Crafts reactions are also unfavorable with electron-rich heterocycles such as furan and benzothiophene due to catalyst-induced polymerization or polyalkylation;31 dihydrofuran 6a has never been reported in a Friedel–Crafts, presumably due to decomposition. Minisci-type reaction with 2-chloropyridine would yield a mixture of 58 and its C6 isomer. Furthermore, radical addition into C3 of pyridine and C5 of a pyrimidine is unfavorable, and products 57, 59, and 68–71 are not viable using Minisci chemistry.
A survey of compounds containing arylated quaternary centers identified the importance of oxetanes, a small ring that alters scaffold lipophilicity and acts as a ketone bioisostere with increased metabolic stability.32 Oxetanes may be prepared via transition metal-catalyzed conjugate addition, requiring extraneous functional groups (cyano, ester, sulfonates) that are often excised in the desired target.32 Alternatively, oxetanes can be prepared through ring closing dehydration that requires 3 or more steps and varies dramatically in yield (19-81%).33 A Vertex patent detailing the use of small molecules for the modulation of ATP-binding cassette transporters investigated oxetane 80 (Figure 4),33c whose aryl bromide precursor (79) has been employed in 9 patents33b—j and one discovery article.33k Oxetane 79 is commercially available at $368/g34 or can be prepared in 4 steps (8% overall yield) through Mitsunobu dehydration of the corresponding diol (77).33c
Figure 4.

Application of Fe/Ni Dual catalysis to various biological intermediates containing oxetanes and piperidines.
Employment of our Fe/Ni dual catalysis allowed for the cross-coupling of olefin 20a with 1,4-dibromobenzene, resulting in a 3 step synthesis of 79 from the inexpensive ketone 20’ in an overall yield of 27%. Furthermore, no dialkylation of the dihaloarene was observed. This method bypasses designer diol intermediates and cyclodehydration, and obviates the need for potentially explosive diazo reagents, providing a route that is more amenable to intermediate diversification.
A 2014 Allergan patent documented a library of tert-alkyl substituted arylthioethers for treatment of sphingosine-1 phosphate-related ocular diseases.35 The patent identifies 82 as an intermediate en route to 84, however there is no reported synthesis in the patent or elsewhere. Access may be attained through esterification of acid 83, which is priced at $3990/100 mg from the sole commercial vendor.36 Alternatively, olefin hydroarylation simplifies access to 82 through oxetane olefination (Figure 4) and cross-coupling with 4-fluoroiodobenzene (51% yield).
JDTic and its analogs (87) are potent kappa-opioid receptor antagonists that contain arylated quaternary carbons embedded in a piperidine.37 Prior access to 86 required a 5-step sequence for the construction of the quaternary center.37 Olefin cross-coupling, however, retrosynthetically signals the Wittig olefination of N-Boc 4-piperidone (85b’) followed by arylation/ deprotection to yield intermediate 86, resulting in an 883% improvement in overall yield (53% versus 6%) and a 40% reduction in step count compared to the prior route. As the olefin coupling partner is prepared by a Wittig olefination of N-Boc 4-piperidone, this sequence offers the ability to expansively derivatize similar heterocyclic ketones—oxetanones, piperidones, pyrrolidinones, pyranones—all of which are inexpensive.
Mechanistic analysis of this hydroarylation is complicated by the number of reaction components, but a thumbnail sketch is suggested in Figure 5A. A metal hydride formed from the iron precatalyst and silane has been proposed to undergo hydrogen atom transfer (HAT) with alkenes to generate carbon-centered radicals, consistent with the ring opening observed in 42b and 43b.16c,17 To further probe the intermediacy of a carbon-centered radical, a competition between our hydroarylation reaction and previously reported Fe-catalyzed Giese reaction (which has been postulated to occur via a radical mechanism)15c was conducted (Figure 5B). In the absence of nickel, only Giese product was observed in a moderate yield. In the presence of nickel, our desired product and Giese product were formed in comparable yields. Additionally, in the presence of TEMPO, the reaction was completely inhibited and the TEMPO-adduct of the olefin starting material was isolated (Figure 5c).
Figure 5.

(A) Potential mechanism for sec- and tert- alkyl arylation. (B) Competition experiment to probe for intermediacy of radical reaction. (C) Radical trapping experiment with TEMPO.
Underscored by similarity in reaction conditions, coupling with sec-alkyl fragments may overlap in mechanism with Weix’s reductive electrophile coupling, i.e. capture of the alkyl radical by an aryl—Ni(II) complex.5a However, a 50% reduction in yield occurs when a bromoarene replaces an iodoarene under sec-alkyl coupling conditions, whereas tert-alkyl coupling conditions can use these haloarenes interchangeably with only slight variation in yield. Such insensitivity by the tert-alkyl conditions may indicate a reordering of steps, where an electron-rich alkyl Ni(I) species engages the haloarene, as suggested by Molander and Kozlowski.38 Even though both tert- and sec-alkyl coupling reactions derive from identical transition metal nuclei, differences in optimal ligand, co-oxidant, solvent and rate indicate significant mechanistic disparities between both dual-catalytic cycles.
CONCLUSION
In conclusion, olefins may now be considered viable and broadly applicable cross-coupling partners for branch-selective hydroarylation, without the regioselectivity and chemoselectivity restrictions of Minisci and Friedel–Crafts reactions. Arylations of polysubstituted alkenes are enabled by iron-catalyzed hydrogen atom transfer and interception of traditional nickel catalytic cycles; two cycles that, to the best of our knowledge, have not been combined previously. These dual catalytic cycles enable cross-coupling of chemical feedstocks in a fundamentally new way, connecting unsaturated building blocks and arenes to generate privileged medicinal chemistry scaffolds and validated drug leads. Hydroarylation of partly saturated rings resembles an sp3 equivalent of biaryl coupling, replacing one aromatic ‘hub’ with a high Fsp3 motif. Such cross-couplings hasten synthetic, ideally combinatorial, approaches to natural product space and answer the call to escape from flatland.1
Supplementary Material
Table 2.
Sec-alkyl Olefin Scopea
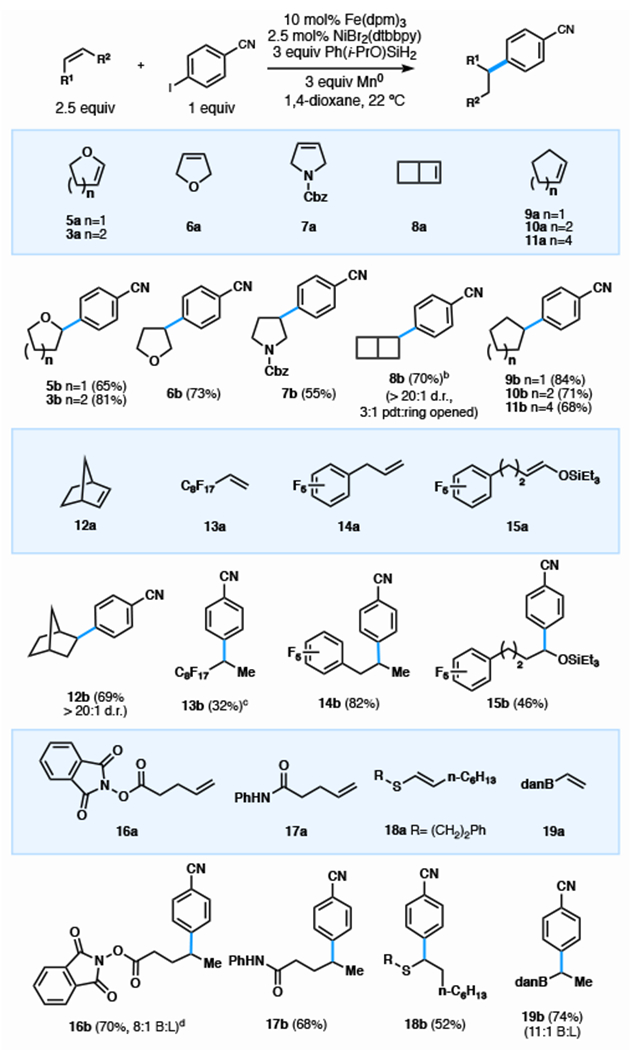 |
0.3 mmol scale, isolated yield, reaction time: 12—48 h. 1 mL 1,4-dioxane, N2 atmosphere for 2 h, then open to air. All substrates gave >20:1 branched:linear ratio except where notated
NMR yield.
5 mol% NiBr2(dtbbpy), 800 μL 1,4-dioxane.
5 mol% NiBr2(dtbbpy).
Table 4.
Formation of Quaternary Centers: Olefin Scopea
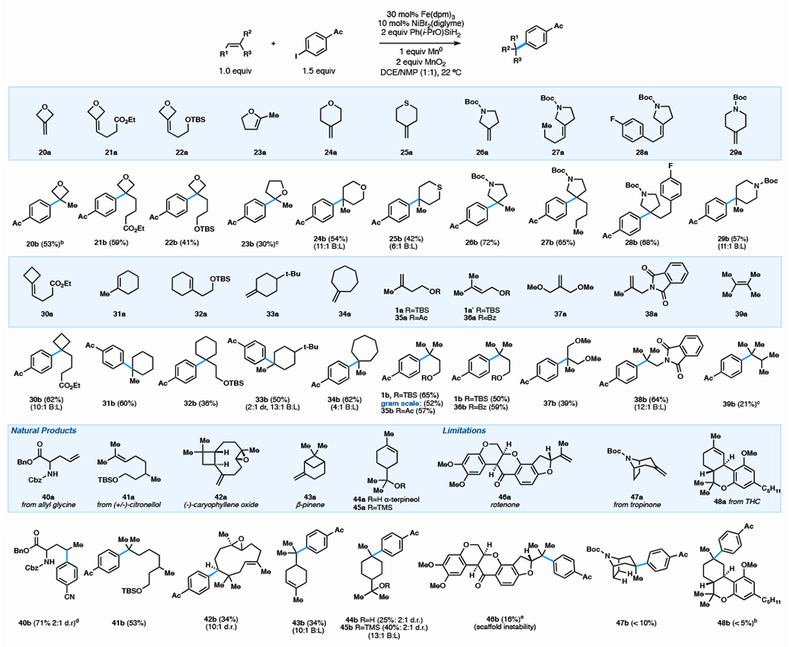 |
0.3 mmol scale, isolated yield, reaction time 16 h. 1.5 mL 1,2-DCE and 1.5 mL NMP. N2 atmosphere for 1.5 h then open to air. All substrates gave >20:1 branched:linear (B:L) ratio except where notated
0.1 mmol scale, isolated yield.
run under an air balloon.
0.3 mmol scale using reaction conditions from Table 2. Isolated yield.
0.1 mmol scale, Fe(dibm)3 (50 mol%), NiBr2(diglyme) (30 mol%), 2.5 h, NMR yield.
ACKNOWLEDGMENT
We would like to thank the following individuals for graciously providing the following starting materials: N. Z. Burns (8a), J. C. Lo (29a), R. M. Martinez (33a), and C. L. Obradors (36a). Generous support was provided by the National Science Foundation (GRFP to S. A. G) and the National Institutes of Health (R35 GM122606).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Detailed experimental procedures, compound characterization, and spectral data (PDF).
The authors declare no competing financial interest.
REFERENCES
- (1).Lovering F; Bikker J; Humblet C J. Med. Chem. 2009, 52, 6752. [DOI] [PubMed] [Google Scholar]
- (2).(a) Zultanski SL; Fu GC J. Am. Chem. Soc. 2013, 135, 624. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang X; Wang S; Xue W; Gong HJ Am. Chem. Soc. 2015, 137, 11562. [DOI] [PubMed] [Google Scholar]; (c) Primer DN; Molander GA J. Am. Chem. Soc. 2017, 139, 9847. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Mei T-S; Patel HH; Sigman MS Nature 2014, 508, 340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Brown DG; Bostrom JJ Med. Chem. 2016, 59, 4443. [DOI] [PubMed] [Google Scholar]
- (4).Tasker SZ; Standley EA; Jamison TF Nature 2014, 509, 299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).For selected examples see; (a) Weix DJ, Acc. Chem. Res. 2015, 48, 1767. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhou J; Fu GC J. Am. Chem. Soc. 2004, 126, 1340. [DOI] [PubMed] [Google Scholar]; (c) Kadunce NT; Reisman SE J. Am. Chem. Soc. 2015, 137, 10480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).For selected examples see; (a) Tellis JC; Primer DN; Molander GA Science 2014, 345, 433. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jouffroy M; Primer DN; Molander GA J. Am. Chem. Soc. 2016, 138, 475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).For selected examples see; (a) Cornella J; Edwards JT; Qin T; Kawamura S; Wang J; Pan C-M; Gianatassio R; Schmidt M; Eastgate MD; Baran PS J. Am. Chem. Soc. 2016, 138, 2174. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zuo Z; Ahneman DT; Chu L; Terrett JA; Doyle AG; MacMillan DWC Science 2014, 345, 437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).For selected examples see; (a) Denmark SE; Cresswell AJ J. Org. Chem. 2013, 78, 12593. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ariki ZT; Maekawa Y; Nambo M; Crudden CM J. Am. Chem. Soc. 2018, 140, 78. [DOI] [PubMed] [Google Scholar]; (c) Miao W; Zhao Y; Ni C; Gao B; Zhang W; Hu JJ Am. Chem. Soc. 2018, 140, 880. [DOI] [PubMed] [Google Scholar]; (d) Merchant RR; Edwards JT; Qin T; Kruszyk MM; Bi C; Che G; Bao D-H; Qiao W; Sun L; Collins MR; Fadeyi OO; Gallego GM; Mousseau JJ; Nuhant P; Baran PS Science 2018, 360, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Yan M; Lo JC; Edwards JT; Baran PS J. Am. Chem. Soc. 2016, 138, 12692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Nickel is known to participate in the hydroarylation of olefins via reductive heck reactions giving the anti-Markovnikov (or linear) product. For an example of such reactivity see Lu X; Xiao B; Zhang Z; Gong T; Su W; Yi J; Fu Y; Liu L Nature Comm. 2016, 7, article no. 11129. [Google Scholar]
- (11).Naredla RR; Klumpp DA Chem. Rev. 2013, 113, 6905. [DOI] [PubMed] [Google Scholar]
- (12).For selected examples see Ma X; Dang H; Rose JA; Rablen P; Herzon SB J. Am. Chem. Soc. 2017, 139, 5998. [DOI] [PubMed] [Google Scholar]
- (13).Green SA; Matos JLM; Yagi A; Shenvi RA J. Am. Chem. Soc. 2016, 138, 12779. [DOI] [PubMed] [Google Scholar]
- (14).Crossley SWM; Barabe F; Shenvi RA J. Am. Chem. Soc. 2014, 136, 16788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Puxeddu A; Costa G; Marsich NJ Chem. Soc. Dalton Trans. 1980, 9, 1489. [Google Scholar]
- (16).(a) Halpern J Pure & Appl. Chem. 1986, 58, 575. [Google Scholar]; (b) Waser J; Gaspar B; Nambu H; Carreira EM J. Am. Chem. Soc. 2006, 128, 11693. [DOI] [PubMed] [Google Scholar]; (c) Lo JC; Kim D; Pan C-M; Edwards JT; Yabe Y; Gui J; Qin T; Gutierrez S; Giacoboni J; Smith MW; Holland PL; Baran PS J. Am. Chem. Soc. 2017, 139, 2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Crossley SWM; Obradors CL; Martinez RM; Shenvi RA Chem. Rev. 2016, 116, 8912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).(a) Mukaiyama T; Isayama S; Inoki S; Kato K; Yamada T; Takai T Chem. Lett. 1989, 18, 449. [Google Scholar]; (b) Inoki S; Kato K; Isayama S; Mukaiyama T Chem. Lett. 1990, 19, 1869. [Google Scholar]; (c) Kato K; Mukaiyama T Chem. Lett. 1992, 21, 1137. [Google Scholar]
- (19).(a) Lo JC; Yabe Y; Baran PS J. Am. Chem. Soc. 2014, 136, 1304. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lo JC; Gui J; Yabe Y; Pan C-M; Baran PS Nature 2014, 516, 343. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Leggans EK; Barker TJ; Duncan KK; Boger DL Org. Lett. 2012, 14, 1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Obradors CL; Martinez RM; Shenvi RA J. Am. Chem. Soc. 2016, 138, 4962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).PhSiH3/EtOH at 60 °C is commonly employed in Fe-HAT reactions and has been proposed to form Ph(OEt)SiH2 under the reaction conditions (ref 15c). In our reaction, this combination yields the product in 31% yield. Instead, PhSiH3/i-PrOH could be used at room temperature to provide 41% of the desired coupling product (Table 1). A significant amount of protodehalogenation of the aryl iodide was seen with PhSiH3/i-PrOH, which is likely the cause for the deterioration in yield. For a further discussion on silanes, please see the SI.
- (22).Reactions performed with styrene yielded product, albeit in low yield (< 20%).
- (23).Mercer JAM; Cohen CM; Shuken SR; Wagner AM; Smith MW; Moss FR; Smith MD; Vahala R; Gonzalez-Martinez A; Boxer SG; Burns NZ J. Am. Chem. Soc. 2016, 138, 15845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).(a) Huihui KMM; Caputo JA; Melchor Z; Olivares AM; Spiewak AM; Johnson KA; DiBenedetto TA; Kim S; Ackerman LKG; Weix DJ J. Am. Chem. Soc. 2016, 138, 5016. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Qin T; Malins LR; Edwards JT; Merchant RR; Novak AJE; Zhong JZ; Mills RB; Yan M; Yuan C; Eastgate MD; Baran PS Angew. Chem. Int. Ed. 2017, 56, 260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Chu CK; Liang Y; Fu GC J. Am. Chem. Soc. 2016, 138, 6404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).(a) Otani G; Yamada S-I Chem. Pharm. Bull. 1973, 21, 2119. [DOI] [PubMed] [Google Scholar]; (b) Oda K; Meyers AI Tetrahedron Lett. 2000, 41, 8193. [Google Scholar]
- (27).Cui X; Burgess K Chem. Rev. 2005, 105, 3272. [DOI] [PubMed] [Google Scholar]
- (28).α-methyl styrene was found to give only traces of product with mostly dimerization of the α-methyl styrene observed.
- (29).Kuhar MJ; Carrol F; Boja JW; Lewin AH; Abraham P US Patent US20080153870A1.2008. [Google Scholar]
- (30).Number of commercially available compounds from Reaxys as of May 31st, 2018.
- (31).Joule JA; Mills K Heterocyclic chemistry fifth edition (Wiley, 2010). [Google Scholar]
- (32).(a) Bull JA; Croft RA; Davis OA; Doran R; Morgan KF Chem. Rev. 2016, 116, 12150. [DOI] [PubMed] [Google Scholar]; (b) Wuitschik G; Carreira EM; Wagner B; Fischer H; Parrilla I; Schuler F; Rogers-Evans M; Muller KJ Meet. Chem. 2010, 53, 3227. [DOI] [PubMed] [Google Scholar]
- (33).(a) Llona-Minguez S; Hoglund A; Ghassemian A; Desroses M; Calderon-Montano JM; Moron EB; Valerie NCK; Wita E; Almlof I; Koolmeister T; Mateus A; Cazares-Korner C; Sanjiv K; Homan E; Loseva O; Baranczewski P; Darabi A; Mehdizadeh M; Fayezi S; Jemth A-S; Berglund UW; Sigmundsson K; Lundback T; Jensen AJ; Artursson P; Scobie M; Helleday T J. Med. Chem. 2017, 60, 4279. [DOI] [PubMed] [Google Scholar]; (b) Brodney MA International patent WO2006106416A1. 2006. [Google Scholar]; (c) Ruah SS, Miller MT; Bear B; McCartney J; Grootenhuis PD J. International patent WO2007087066A2. 2007. [Google Scholar]; (d) Klar U; Schwede W; Moeller C; Rotgeri A; Bone W; International patent WO2011009534A2. 2011. [Google Scholar]; (e) Apgar JM; Srasappan A; Biftu T; Ping C; Danqing F; Guidry E; Hicks J; Kekec A; Wei L; Wilkeniing R; Wu Z International patent WO2014031515A1. 2014. [Google Scholar]; (f) Scott JD; Stamford AW; Gilbert EJ; Cumming JN; Iserloh U; Wang L; Li W International patent WO2011044187A1. 2011 [Google Scholar]; (g) Siegrist R; Heidmann B; Stamm S; Gatfield J; Bezencon O International patent WO2015186056A1. 2015. [Google Scholar]; (h) Becker-Pelster EM; Buchgraber P; Buchmuller A; Engel K; Gnoth MJ; Himmel H; Kast R; Klar J; Knorr A; Lang D; Lindner N; Schmeck C; Schohe-Loop R; Tinel H; Trubel H; Wunder F; Keldenich J International patent WO2015091415A1. 2015. [Google Scholar]; (i) Li B; Breinlinger E; Davis H; Hoemann M; Li B; Somal G; Van ES; Wang L International patent WO2014169473A1. 2014. [Google Scholar]; (j) Bhagirath N; Dominique R; Kennedy-Smith J;Lopez-Tapia FJ; Mertz E; Qiao Q; So S-S. International patent WO2014064131A2. 2014. [Google Scholar]; (k) Bezenjon O; Hedimann B; Siegrist R; Stamm S; Richard S; Pozzi D; Corminboeuf O; Roch C; Kessler M; Ertel EA; Reymond I; Pfeifer T; Kanter R; Toeroek-Schafroth M; Moccia LG; Mawet J; Moon R; Rey M; Capeleto B; Fournier EJ Med. Chem. 2017, 60, 9769. [DOI] [PubMed] [Google Scholar]
- (34).The price of 3-(4-bromophenyl)-3-methyloxetane (79) is $3680/10g from Pharmablock as of May 31st, 2018.
- (35).Nguyen PX; Cappiello JR; Heidelbaugh TM United States Patent US2014171393A1. 2014. [Google Scholar]
- (36).The price of 85 is from Aurora Fine Chemicals ($3990/100 mg) as of May 31st, 2018.
- (37).(a) Carroll FI; Gichinga MG; Kormos CM; Maitra R; Runyon SP; Thomas JB; Mascarella SW; Decker AM; Navarro HA Bioorg. Med. Chem. 2015, 23, 6379. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nagai Y; Hino K; Uno H; Minami S Chem. Pharm. Bull. 1980, 28, 1387. [DOI] [PubMed] [Google Scholar]
- (38).Gutierrez O; Tellis JC; Primer DN; Molander GA; Kozlowski MC J. Am. Chem. Soc. 2015, 137, 4896. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.