

Abstract
Summary
In this report, we introduce an R package KMgene for performing gene-based association tests for familial, multivariate or longitudinal traits using kernel machine (KM) regression under a generalized linear mixed model framework. Extensive simulations were performed to evaluate the validity of the approaches implemented in KMgene.
Availability and implementation
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
The gene-based tests are becoming an attractive complement to the single variant tests used in GWAS (Liu et al., 2010). Compared to single variant tests, gene-based tests are able to identify weak individual signals by combining the effects of variants in the same gene, and greatly reduce the number of multiple testing. One widely used gene-based test is the sequence kernel association test (SKAT) (Wu et al., 2011) based on a Kernel Machine (KM) regression framework. Although SKAT was first developed for testing rare variants, it could be easily applied to common variants. After SKAT was introduced for testing independent samples with continuous and binary traits, a number of methods and corresponding tools have been developed to extend the approach to complex traits (Chen et al., 2013, 2014; Wu et al., 2011; Yan et al., 2015a,b,c), such as familial, multivariate and longitudinal traits. These methods are based on a generalized linear mixed model (GLMM) framework. Since SKAT has imbalanced power performance when the single variant effects are in the same direction (i.e. all rare alleles are risk or protective) or in different directions, the optimal version, SKAT-O (Lee et al., 2012), was developed to balance these two scenarios. However, most of the extended SKAT methods do not consider the optimal tests balancing genetic effects. Several other R packages to support gene-based tests for familial data are available, such as RVFam (https://cran.r-project.org/web/packages/RVFam). They adapt linear mixed model (LMM) to their methods accounting for pedigree. However, none of them can analyze multivariate traits or longitudinal traits. In addition, those programs have different input and output formats, making it difficult to use in practice for bioinformaticians with limited genetics knowledge. Therefore, in this report, we introduce KMgene, a one-stop solution that combines SKAT-type methods for complex traits and extends them to include their corresponding optimal tests. KMgene can perform association tests between a set of genetic variants and familial, multivariate, longitudinal or survival traits (Table 1).
Table 1.
A summary of functions in KMgene package
| Regular (KM) | Optimal (KM-O) | Interaction (KM-Int) | |
|---|---|---|---|
| Continuous family (F-KM) | Chen et al. (2013) | Extended | NA |
| Binary family (Fb-KM) | Yan et al. (2015a) | Extended | NA |
| Continuous multivariate (M-KM) | Maity et al. (2012) | Extended | NA |
| Continuous multivariate family (MF-KM) | Yan et al. (2015b) | Extended | NA |
| Continuous longitudinal (L-KM) | Yan et al. (2015c) | Yan et al. (2015c) | Extended |
| Survival (CoxKM)a | Chen et al. (2014) | NA | NA |
Incorporated from R seqMeta package.
2 Materials and methods
In this study, we describe KMgene methods that use KM regression under a GLMM framework, which can be employed to analyze a large range of traits. In addition, KMgene incorporates survival SKAT functions from R seqMeta package. Specifically, KMgene works in two steps (refer to supplementary material for detailed derivations). The first step with function names, prefix_Null_Model (Supplementary Table S1), fits the model under the null hypothesis (i.e. the genetic effects are zero). The estimates of covariate parameters and covariance matrix are obtained at this step. The covariance matrix can account for relatedness in families, correlation between multivariate traits or between times for longitudinal data. The second step with function names, prefix (Supplementary Table S1), constructs the test statistic and calculates the P-value. We use the parameter estimates from step one to construct the test statistic. Since the parameters are estimated under the null hypothesis and used for all genes, they only need to be calculated once for the whole genome-wide analysis, which greatly reduces the computation time. According to our derivation, the test statistic follows a mixture of distributions and thus we can compute the P-values analytically, also leading to improvement in computation. The KM statistics can be extended to the optimal test by combining with burden statistics (refer to supplementary material for details). Analogously, our optimal tests consist of two steps for fitting null models (prefixO_Null_Model in Supplementary Table S1) and calculating P-values (prefixO in Supplementary Table S1).
Input to the KMgene package are traits, covariates and genotypes pre-grouped in genes and coded as 0, 1, 2 for the number of copies of minor allele (i.e. additive genetic model). The additive genetic model coding can be easily converted from plink (Purcell et al., 2007) format by using option –recodeA. The genotypes should have no missing values. A conservative approach for handling missing genotypes is to assign them to the homozygous reference genotype (i.e. 0), or one can conduct a thorough genotype imputation. It also requires family pedigree when analyzing familial data. The output is gene-level P-values.
3 Performance
3.1 Simulations
In the simulation studies, we evaluate the methods’ validity by checking their type I error rates (refer to supplementary material for each simulation scenario details). The QQ plots indicate that all of the methods in KMgene package retain the correct type I error rates (Supplementary Fig. S1).
3.2 Computation
The optimal KM test takes more computational time than regular KM test due to a more complex model. In KMgene, the model fitting continuous multivariate familial (MF) traits has the most complex form. Thus, we used MF-KM and MFO-KM to estimate the computation. For MF-KM, analysis of a region of 60 variants on 300 trios took 147.61 s (147.33 s for fitting the null model) on a single computing node with a 3 GHz CPU and 4 GB memory, and 183.92 s (182.49 s for fitting the null model) for MFO-KM. Based on this simulation, it could take approximately 1.6 h for MF-KM and 8.0 h for MFO-KM to analyze the whole genome (assuming 20 000 genes with an average of 60 variants each, for 1 200 000 total variants). The GLMM based tests are more reliable with larger sample size, but larger sample size costs dramatic computation increase. The computation time increases faster as the sample size increases than as the gene size increases (Supplementary Fig. S2). Although large genes take much more time to process than small genes, we anticipate that using multiple CPUs, genome-wide data analysis could be completed within hours using all the methods in KMgene.
3.3 Real data example
The functions in KMgene have been applied to several real data studies (Chen et al., 2013; Maity et al., 2012; Yan et al., 2015b,c). Here, as an illustrative example, we apply MFKM_Null_Model() and MFKM() to carry out a gene-based genome wide association test of the correlated lung function phenotypes FEV1 (Forced Expiratory Volume in One Second) and FEV1/FVC (Forced Vital Capacity) ratio (Yan et al., 2015). We identified COL6A6 associated with these two traits (Fig. 1) and COL6A6 is known to be in the chronic obstructive pulmonary disease related regions based on Rat Genome Database (RGD) (Shimoyama et al., 2015).
Fig. 1.
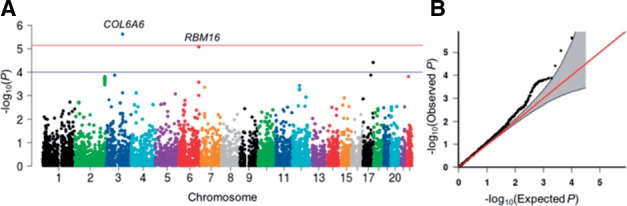
(A) Genome wide gene-based results of MFKM on lung function data. Each dot represents P-value of a gene. (B) QQ plot of P-values from the lung function analysis, with 95% pointwise confidence band (gray area)
4 Conclusion
In conclusion, this R package adapts GLMM to conduct gene-based tests for complex traits and uses Cox model for survival trait. KMgene can handle genome-wide genotypic datasets with reasonable computational time. KMgene currently uses the linear kernel that is the most commonly used kernel in genetic studies. Moreover, to speed up computation for large datasets, we can implement our package in C++ with the help of R libraries, for example, ‘Rcpp’ and ‘RcppParallel’. We will add more kernel options (e.g. quadratic and IBS kernels) in the package and hope to incorporate our ongoing method for analyzing multiple types of omics data in this package in near future.
Supplementary Material
Acknowledgements
We thank Dr Daniel E. Weeks and Dr Nianjun Liu for helpful suggestions on the method development.
Funding
This work was supported by the National Institutes of Health [R01HG007358, R01EY024226].
Conflict of Interest: none declared.
References
- Chen H. et al. (2014) Sequence kernel association test for survival traits. Genet. Epidemiol., 38, 191–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H. et al. (2013) Sequence kernel association test for quantitative traits in family samples. Genet. Epidemiol., 37, 196–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. et al. (2012) Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am. J. Hum. Genet., 91, 224–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.Z. et al. (2010) A versatile gene-based test for genome-wide association studies. Am. J. Hum. Genet., 87, 139–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maity A. et al. (2012) Multivariate phenotype association analysis by marker-set kernel machine regression. Genet. Epidemiol., 36, 686–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S. et al. (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet., 81, 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimoyama M. et al. (2015) The Rat Genome Database 2015: genomic, phenotypic and environmental variations and disease. Nucleic Acids Res., 43, D743–D750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M.C. et al. (2011) Rare-variant association testing for sequencing data with the sequence kernel association test. Am. J. Hum. Genet., 89, 82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Q. et al. (2015a) A sequence kernel association test for dichotomous traits in family samples under a generalized linear mixed model. Hum. Hered., 79, 60–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Q. et al. (2015b) Associating multivariate quantitative phenotypes with genetic variants in family samples with a novel kernel machine regression method. Genetics, 201, 1329–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Q. et al. (2015c) Rare-variant kernel machine test for longitudinal data from population and family samples. Hum. Hered., 80, 126–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.