

Abstract
Diseases of marine animals caused by bacteria of the genus Vibrio are on the rise worldwide. Understanding the eco-evolutionary dynamics of these infectious agents is important for predicting and managing these diseases. Yet, compared to Vibrio infecting humans, knowledge of their role as animal pathogens is scarce. Here we ask how widespread is virulence among ecologically differentiated Vibrio populations, and what is the nature and frequency of virulence genes within these populations? We use a combination of population genomics and molecular genetics to assay hundreds of Vibrio strains for their virulence in the oyster Crassostrea gigas, a unique animal model that allows high-throughput infection assays. We show that within the diverse Splendidus clade, virulence represents an ancestral trait but has been lost from several populations. Two loci are necessary for virulence, the first being widely distributed across the Splendidus clade and consisting of an exported conserved protein (R5.7). The second is a MARTX toxin cluster, which only occurs within V. splendidus and is for the first time associated with virulence in marine invertebrates. Varying frequencies of both loci among populations indicate different selective pressures and alternative ecological roles, based on which we suggest strategies for epidemiological surveys.
Introduction
Global change due to rising temperature and ocean acidification but also more directly due to anthropogenic influences, such as widespread use of aquaculture, is predicted to lead to a worldwide increase in Vibrio-associated illness of marine organisms [1–3]. This development is illustrated by recent outbreaks of disease in Crassostrea gigas (oyster) farms in France [4, 5], in which strains related to the Splendidus clade have been implicated [6–10]. The Splendidus clade is a large group of closely related species (e.g., V. splendidus, V. crassostreae, V. tasmaniensis, and V. cyclitrophicus) containing several facultatively pathogenic members that affect diverse marine organisms [11, 12]. Elevated host density due to farming is a likely factor involved in Vibrio-associated disease. However, it is less well understood whether the bacteria are drawn from environmental reservoirs of potential pathogens or have arisen by recent acquisition of virulence traits in response to increased host availability due to aquaculture. The latter appears to be the case in V. crassostreae, which we recently demonstrated to have emerged as an oyster pathogen at least partially as a result of the population-specific spread of a virulence plasmid [6]. However, collections of environmental pathogens are typically biased toward isolates from diseased animals [13, 14]. It is thus difficult to assess how broadly distributed virulence potential is among Vibrio spp., i.e., whether virulence is limited to few strains within populations or is a trait of the entire species. We therefore reasoned that assessment of virulence properties within populations of Vibrio spp. in natural environments where oysters are not farmed, could be used to determine what level of taxonomic resolution has the greatest predictive power for disease risk assessment in oyster-farming areas.
Here we determine the virulence potential of a large collection of Vibrio strains using specific pathogen-free (SPF) oysters as a model. The bacterial strain collection derives from a single location without intense oyster farming, on the Atlantic coast of the United States (Massachusetts, Plum Island). Extensive genetic and microhabitat characterization of these isolates has been previously reported and identified genetically defined groups that correspond to ecologically distinct habitats. This is indicative of different lifestyles (free-living or attached to different types of particulate organic matter and zoo- or phytoplankton) between these groups [15–18]. Subsequent work has found multiple lines of evidence these Vibrio groups represent biologically distinct populations with respect to gene flow [19, 20] and interaction with other organisms (social/behavioral networks) [21–23]. As such, these populations fit species concepts as typically used in plant and animal ecology; however, we note that they are at a finer evolutionary divergence than those traditionally used to separate species in bacterial taxonomy. To assess the virulence potential of these populations, we injected a large number of strains into juvenile SPF oysters and monitored their mortality. These animals are descendants of a pool of genitors that are produced in hatchery under highly controlled conditions to minimize the influence of genetic and environmental parameters that could affect the host sensitivity to the disease [10, 24, 25]. Moreover, these SPF oysters can be used for high-throughput experimental infections with hundreds of individual bacterial isolates [9, 10, 26]. Assaying the virulence of strains by injection has drawbacks, as it fails to capture the importance of initial infection events such as mobility and chemotaxis. However, Vibrio spp. related to Splendidus (both virulent and nonvirulent) naturally colonize the oyster hemolymph [6] and injection in the adductor muscle allows the direct transfer of bacteria in the hemolymph. Here screening Vibrio strains by injection allows the identification of traits that cause death in the animals. It is thus possible to ask whether the functional units of pathogenesis for oysters are either clones that have emerged after recent acquisition of virulence factors by horizontal gene transfer (HGT) or populations of diverse genotypes with virulence as a core function [27].
We show that the ability to trigger oyster mortality is an ancestral trait of populations within the Splendidus clade and depends on at least two loci. While in some populations all members are potentially virulent, the trait occurs intermittently in other populations and some populations have lost it altogether. The ancestrally acquired gene R5.7 is necessary for full virulence and while it has co-diversified with populations, it was lost in the non-virulent populations. Across populations, additional genes may, however, play a role in virulence. This is illustrated by the occurrence of several different types of MARTX gene clusters in V. splendidus for which frequency-dependent selection is suggested. The widespread occurrence of virulence genes across environmental Vibrio populations suggests an important biological role but their different frequency also indicates that the role is population specific.
Materials and methods
Genome sequencing, assembly, and annotation
In the present study, a total of 37 strains (Table S1) were sequenced (Plateforme génomique, Institut Pasteur, Paris) using the Illumina HiSeq2000 technology with ~50-fold coverage as described previously [9]. Contigs were assembled de novo using Velvet [28]. Computational prediction of coding sequences together with functional assignments was performed using the automated annotation pipeline implemented in the MicroScope platform [29]. Some gene annotations were manually curated using InterPro, FigFam, PRIAM, COGs, PsortB, TMHMM, and synteny group computation.
Protein family identification
The proteome of each strain was first compared by performing a Blastp all-vs-all. The SiLiX software [30] was used to reconstruct protein families based on reciprocal alignment length (at least 80% of sequence length) and different thresholds of identity depending on the analysis. A threshold of 60% identity was used when comparing two populations, while a threshold of 80% was used to reconstruct families within a population.
Phylogenetic tree construction
Species trees were reconstructed based on core genes present in a single copy. Protein sequences of each family were first aligned with Muscle, filtered using Gblocks with relaxed parameters [31] and concatenated using a custom script (available at https://github.com/mbruto/OPOPOP). Phylogenetic reconstruction was done using RAxML [32] on this concatemer using a LG model of evolution with a gamma model and default parameters. For single-marker gene trees, we aligned the sequences with Muscle and performed the phylogenetic reconstruction with RAxML using a GTR model of evolution with a gamma model and default parameters.
Phylogenetic logistic regression analysis
The correlation between gene family presence/absence and virulence (percentage of induced mortality) was computed using the phylogenetic logistic regression implemented in the R package “phylolm” [33]. Virulence was first standardized to have a mean equal to 0 and standard deviation equal to 1, as suggested in previous work [34]. The following command was used to infer a correlation coefficient: phyloglm(Fam ~ Mort, phy = Tree, data = Corr_data, method = “logistic_MPLE”). The variable Corr_data is a table containing the presence/absence of a single family in the first column (named Fam), while the second column contains virulence values that were standardized to have a mean of 0 and a standard deviation of 1 (named Mort). The Tree variable is the species tree of the strains considered. We used the logistic_MPLE model as it appears to be the most general one and can be applied on a wider range of data sets [33].
Ancestral character reconstruction
Ancestral character reconstructions were performed with the version 3.31 of Mesquite (http://mesquiteproject.org) using the maximum parsimony method. The presence or absence of the Splendidus specific locus R5.7/8 was coded as 1 or 0 in a character matrix. Together with the species tree, this matrix was used as an input to reconstruct the presence or absence of the cluster on each node using the unordered parsimony model, in which acquisitions and losses have an equal cost.
Molecular microbiology
The Vibrio collection used for the high-throughput infection assays (population F1–18), i.e., Plum Island collection, has been described previously [15]. The Vibrio strains isolated from a French oyster farming area have been described by Bruto et al. [6]. Strains and plasmids used or constructed in the present study are described in Table S2 and S3. Vibrio isolates were grown at 20 °C in Zobell broth or agar (4 g l−1 bactopeptone and 1 g l−1 yeast extract in artificial seawater, pH 7.6), Luria-Bertani (LB), or LB-agar (LBA) + 0.5 M NaCl. Escherichia coli strains were grown at 37 °C in LB or on LBA for conjugation experiments and in ZYP 5052 medium [35] for induction of protein expression in E. coli, strain BL21 (DE3). Chloramphenicol (Cm, 5 or 25 μg ml−1 for Vibrio and E. coli, respectively), carbenicillin (100 μg ml−1), thymidine (0.3 mM), and diaminopimelate (0.3 mM) were added as supplements when necessary. Induction of the Pbad promoter was achieved by the addition of 0.2% l-arabinose to the growth media, and conversely, was repressed by the addition of 1% d-glucose. Deletion of selected regions or genes was performed by allelic exchange using the pSW7848T suicide plasmid [36, 37]. To this end, two 500-bp fragments flanking the target region or gene were amplified (see primer details in Table S4), cloned into pSW7848T as previously described [9], and transferred by conjugation from E. coli as a donor to Vibrio as a recipient. Subsequently, the first and second recombinations leading to pSW7848T integration and elimination were selected on Cm/glucose and arabinose-containing media, respectively. Gene inactivation was performed by cloning 500 bp of the target gene in pSW23T [38] and selecting on Cm the suicide plasmid integration obtained by a single recombination [8]. For complementation experiments, the R5.7 gene was cloned in Apa1/Xho1 sites of the pMRB plasmid known to be stable in Vibrio spp. [39], resulting in a constitutive expression from a Plac promoter [9]. Conjugation between E. coli and Vibrio was performed at 30 °C as described previously [36]. The BL21(DE3) strains expressing the R5.7_GFP fusion proteins were constructed using the Gibson assembly method (New England Biolabs, NEB) as previously described [40]. Briefly, the fragment encoding the green fluorescent protein (GFP) was amplified from plasmid pMRB-GFP [6] (see primer details in Table S4). R5.7F12 and R5.7crass genes, including a flexible linker were PCR-amplified from genomic DNA of strains B8 and J2-9, respectively. The pFO4 vector, that contains a His6-tag at the N terminus for affinity purification, was amplified by PCR inside-out. All constructs were confirmed by sequencing prior to electrocompetent BL21(DE3) transformation.
Purification of recombinant proteins
Recombinant R5.7F12 and R5.7crass proteins were purified as previously described [40]. Briefly, after expression of the proteins in ZYP 5052 (20 °C, 72 h) and chemical cell lysis, the lysate was clarified at 13,865 × g for 45 min at 4 °C. The supernatant was loaded on a 5-ml GE HisTrap HP column, washed twice with 20 mM Na3PO4, pH 8.0, 290 mM NaCl, and 5 mM imidazole, and eluted using a gradient of 1–100% 20 mM Na3PO4, pH 8.0, 290 mM NaCl, and 1 M imidazole. Fractions containing the protein of interest were pooled, concentrated (molecular weight cutoff of 10 kDa), and loaded onto a Superdex S200 column in HEPES that contains 20 mM NaCl at 150 mM, pH 7.4. Fractions were again pooled and concentrated. The concentration was calculated from A280 and the extinction coefficient was calculated using the ProtParam tool from ExPASy (ε0.1% of 1.04).
Experimental determination of mortality
To determine the virulence of isolates, bacteria were grown under constant agitation at 20 °C for 24 h in Zobell media. One hundred microliters of the culture (108 colony-forming units, cfu) were injected intramuscularly into oysters. Co-injections of the ΔR5.7 mutant with the recombinant purified R5.7F12 or R5.7crass proteins were performed following the same procedure, except that the tested protein (30 μg/animal) or the corresponding volume of protein solubilization buffer (as a control) were added to the bacterial suspension 1 h before injection. The bacterial concentration was confirmed by conventional dilution plating on Zobell agar. After injection, the oysters were transferred to aquaria (20 oysters per 2.5 l of aquarium) containing 1 l of aerated 5-µm-filtered seawater at 20 °C, and kept under static conditions. Experiments were performed in duplicate at least twice, and mortality was assessed after 24 h.
Results
Virulence is an ancestral trait of several ecological populations
We first investigated the frequency of virulence among 405 strains representing 15 ecological populations sampled randomly from Plum Island (MA) [15, 17] by injecting them individually into SPF oysters. This showed that nine populations contained exclusively non-virulent strains, while six of them were potentially virulent with the majority (5/6) of these being in the Splendidus clade (Fig. 1, populations belonging to the Splendidus clade are each indicated by distinct colours). All the strains of population F12, which represents a new species closely related to V. crassostreae [41], induced high levels of oyster mortality, while populations F15 (taxonomically assigned to the species V. cyclitrophicus), F17 (V. tasmaniensis), and F18 (V. splendidus) are predominantly virulent, and population F16 (V. tasmaniensis) mostly contains non-virulent strains. The only highly virulent population outside the Splendidus clade is F3, which is taxonomically assigned to V. ordalii, a species that has been associated with fish disease [42]. The observed high prevalence of virulence among populations in the Splendidus clade led us to test the hypothesis that the ability to kill oysters might be an ancestral trait for the entire clade by (i) identifying putative virulence genes by comparative genomics and genetic knockout and (ii) reconstructing the acquisition and inheritance of the virulence genes among populations.
Fig. 1.

Mortality induced in oysters by Vibrio populations. The virulence potential mapped onto the isolate phylogeny (Vibrio collection from Plum Island, MA, USA). The tree is based on the genetic marker hsp60 and comprises different genotypic clusters previously found to have a cohesive ecology and hypothesized to represent samples from natural ecological populations [15]. The closest named species to populations are as follows: F1: Enterovibrio calviensis; F2: Enterovibrio norvegicus; F3: V. ordalii; F4: V. rumoiensis; F5: V. alginolyticus; F7 Aliivibrio fischeri/logei; F8: A. fischeri; F9: V. superstes; F15: V. cyclitrophicus; F16–17: V. tasmaniensis; F18: V. splendidus. Populations F6, 10, 12, and 13 were not assigned to a species and were thus named here as Vibrio sp. nov. The node leading to the Splendidus clade in this tree is indicated by a red dot and the six populations belonging to the Splendidus clade are each indicated by distinct colors (also used in subsequent figures). Note that the presence of populations F9 and F10 within this cluster on the hsp60 tree is an artifact of a recombination event in that gene and they are not members of the Splendidus clade, as determined by a full genome analysis. Red bars indicate weighted mortality (using V. crassostreae J2-9 mortality rate as a reference to evaluate variability between experiments) induced by individual strains 24 h after injection in oysters (n = 20). The range of variability for J2-9 between experiments (50–90%, data not shown) was lower than the variability between populations. Stars indicate the available genome sequences
The same genes are necessary for full virulence across the Splendidus clade
To test the hypothesis of ancestral virulence in the Splendidus clade, we first sought to identify genes potentially responsible for virulence using comparative genomics of populations F12 (Vibrio sp. nov) and F13 (Vibrio sp. nov), since these are the most closely related populations that contain exclusively virulent and non-virulent strains, respectively (Fig. 1). Using genomes of five isolates from each population, we identified 120 genes to be specific to the virulent population F12, of which 87 genes were localized in 24 genomic regions, designated regions a through x (i.e., Ra through Rx) (Table S5). Annotation implicates many of these genes in regulation, detoxification, xenobiotic degradation, siderophore production, or acquisition, i.e., the ability to respond to transient selective pressure in the environment.
Using a genetic knockout approach, we next investigated the importance of these loci for the virulence of population F12 strain 10N.286.48.B8 (hereafter named B8). None of the 24 deletions impaired bacterial growth in culture media (data not shown) and only the deletion of region Rg resulted in a decrease in oyster mortalities after experimental infection (Fig. 2a). This region contains two genes VB12B8_v1_40197 and VB12B8_v1_40198, each encoding an exported protein of unknown function. Notably, these genes showed 91 and 90% identity to the R5.7 and R5.8 proteins of V. crassostreae, respectively [9]. For simplicity, these genes are subsequently referred to as R5.7/8cras or R5.7/8F12 for V. crassostreae and population F12, respectively. A mutant lacking the R5.7F12 gene (ΔR5.7F12) revealed that this gene contributes to B8 virulence (Fig. 2b), as previously shown for R5.7cras. When constitutively expressed in trans from a plasmid, R5.7F12 or R5.7cras were sufficient to partially restore the virulence of the mutant ΔR5.7F12. Furthermore, a complete restoration of virulence was observed by co-injecting the ΔR5.7F12 with the recombinant purified R5.7F12 or R5.7cras proteins (Fig. 2c). On the other hand, these proteins did not induce mortality when injected alone or co-injected with the non-virulent strain J2-8, showing that R5.7 is not sufficient to induce mortality. Finally, the deletion of the R5.8 gene did not alter the virulence of the B8 strain, showing that only R5.7 is necessary for the contribution of Rg to virulence.
Fig. 2.

Oyster mortality in response to experimental infection with Vibrio wild-type strains and derivatives. a Twenty four population-specific regions were deleted in Vibrio sp. nov F12 strain B8. Virulence of the wild-type B8 strain (wt), derivatives (deletion of regions a to x), and Vibrio sp. nov F13-like strain J2-8 as a negative control was compared after injection of strains (108 cfu/animal) into 20 oysters and counting the percent mortality at 24 h (y axis). b Comparison of mortality induced by B8 wild-type strain (wt), the mutants obtained by R5.7 or R5.8 single-gene deletion (∆R5.7 or ∆R5.8), and the complementation assay in ∆R5.7 after the transfer of a plasmid that expressed R5.7F12 or R5.7cras constitutively from a Plac promoter (PlacR5.7F12 and PlacR5.7cras). c Complementation assays using recombinant purified protein R5.7, the wild-type strains (B8, J2-8) injected with buffer (wt + buffer), the mutant obtained by R5.7 single-gene deletion with buffer (∆R5.7 + buffer), the mutant ∆R5.7 or J2-8 co-injected with the recombinant purified proteins R5.7F12 or R5.7cras, the recombinant proteins, or buffer as control. All experiments were performed in duplicate and at least twice. Means with the same letter in italic are not significantly different from each other (ANOVA, p < 0.05 and Tukey HSD test)
R5.7/8 appear highly specific to the Splendidus clade, as evidenced from the distribution in 872 available genomes from 30 different species of Vibrio. First, R5.7 is always colocalized with R5.8 (Fig. S1). Second, this gene cluster is restricted to the Splendidus clade encompassing the populations F12 (Vi brio sp. nov), F15 (V. cyclitrophicus), and F18 (V. splendidus) from the Plum Island collection, as well as populations of strains previously isolated from a French oyster farming area and assigned to V. chagasii, V. splendidus, and V. crassostreae [6, 9] (Fig. 3). We note that population F17 (V. tasmaniensis) shown in Fig. 1 from the Plum Island collection was not evaluated since there are currently no genomes available. We also confirmed the importance of the R5.7 homolog for virulence in V. chagasii and V. splendidus by inactivating this gene in strains representative of these species (Fig. S2). Finally, the genomic comparison showed that the R5.7/8 gene cluster is absent in three out of eight populations within the Splendidus clade (Fig. 3), populations F13 (Vibrio sp. nov) and F16 (V. tasmaniensis) from the Plum Island collection, and also in Vibrio sp. nov F13-like isolated in France. Moreover, a single non-virulent V. splendidus strain, 1S_14, was lacking the gene (confirmed by PCR, data not shown). Altogether, these results suggest that the R5.7 gene is necessary for full virulence across the entire Splendidus clade.
Fig. 3.

Distribution of the R5.7 and R5.8 cluster and the surrounding genes in the Splendidus clade. The phylogenetic tree was reconstructed on an alignment of 707 concatenated core proteins and rooted with V. breoganii FF50. The length of the outgroup branch has been arbitrarily defined (dashed line) to increase the resolution of the tree. The eight species considered are highlighted in alternating grays. Strains isolated from Plum Island are indicated by the same color code as in Fig. 1 (outer-circle). Species assignment and correspondence to population assignment in Fig. 1 are also indicated. Strains isolated from oyster-farming areas in France, i.e., V. chagasii, V. crassostreae, and Vibrio sp. nov F13-like are indicated in gray (outer-circle). The absence and presence of the R5.7/8 cluster are represented by white and red squares, respectively. The presence of the surrounding genes is indicated by a black square if they belong to the same syntenic group of the R5.7/8 gene cluster (see also Figure S1). Note that in V. splendidus, only the strain 1S_14 lacks the R5-7/8, while strains FF_139, FF-144, and 1S-157 lack at least one flanking gene. Red and white circles on the tree indicate acquisition and loss of R5.7/8, respectively
We next tested whether the R5.7/8 gene cluster distribution in the Splendidus clade is best explained by (i) ancestral acquisition followed by multiple losses or (ii) multiple independent acquisitions. Reconstruction of the most parsimonious ancestral character state of the R5.7/8 genes suggests an acquisition in the most recent common ancestor (MRCA) of the Splendidus clade followed by three distinct losses in the MRCAs of populations F13, F16, and F13-like (Fig. 3). The R5.7/8 gene tree is highly congruent with the species tree (Fig. 4), suggesting vertical inheritance and persistence of the gene cluster through speciation events that likely involved differential ecological specialization of the populations. The main exception to this vertical inheritance pattern is population F12, which is most closely related to V. crassostreae in the species tree but is sister to V. chagasii in the R5.7/8 tree indicating horizontal acquisition. Although the R5.7/8 gene tree clusters the vast majority of strains within their previously assigned populations, four strains are an exception: one V. cyclitrophicus (out of 27) and three V. splendidus (out of 56) appear to have acquired the R5.7/8 locus horizontally (Fig. 4). This more recent acquisition most likely involved homologous recombination since annotation of genes present within 20 kb upstream and downstream of the R5.7/8 locus did not identify any potential insertion sites for mobile genetic elements. However, such gene conversions appear to be the exception and our analysis predicts that R5.7/8 was ancestrally acquired and subsequently predominantly vertically inherited in the Splendidus clade.
Fig. 4.

Comparison of the R5.7 and R5.8 concatenated gene tree with the species tree for populations of the Splendidus clade. The species tree (right) was reconstructed based on an alignment of 707 concatenated core proteins and rooted with V. breoganii FF50. Both trees are represented without branch lengths and the outgroup is represented with a dashed line on the tree. Black points on branches indicate a support value above 80% based on the rapid bootstrap analysis implemented in RAxML. Species are linked with different colors in both trees, and population correspondence with Fig. 1 is also indicated. Supported incongruences between R5.7/8 gene cluster (left) and species trees were identified for Vibrio sp. nov F12 (pink), one V. cyclitrophicus (darker green), and three V. splendidus strains (darker turquoise)
Acquisition and loss of MARTX toxin modulates virulence
The above genomic analysis also showed that R5.7 was present in non-virulent strains belonging to populations F15 and F18 (Figs. 1 and 3), suggesting that the gene is necessary but not sufficient for virulence and that other genes may play a role that these strains might have lost or never acquired. To test this hypothesis of additional virulence genes, we first computed the correlation between gene family presence/absence and induced mortalities using phylogenetic logistic regression in population F18 (V. splendidus) (see Materials and methods).
Among 12,158 families, only six genes, which were all colocalized in a single locus, showed a high correlation factor (c = 4.01; P = 0.005) (Fig. 5a). This locus encodes a putative toxin (MARTX encoded by the gene rtxA), a putative acyltransferase (rtxC), an uncharacterized protein (rtxH), and a putative type-I secretion system (rtxBDE). Because this comparison strongly suggested rtxACHBDE as additional virulence genes, we genetically assessed the importance of this locus for V. splendidus virulence.
Fig. 5. Distribution, evolutionary history, and domain variation of the rtxACHBDE cluster in V. splendidus related to induced mortality in oysters.

a The phylogenetic tree is based on an alignment of 2290 concatenated core genes. Weighted mortalities (WM, using V. crassostreae J2-9 mortality rate as a reference to evaluate variability between experiments) after injection in oysters (n = 20) are indicated by a color gradient from white (0%) to red (150%). The range of variability for J2-9 between experiments (50–90%, data not shown) was lower than the variability between populations. Gene presence and absence is represented by red and white squares, respectively. Losses are indicated when the rtxACHBDE cluster is predicted to be absent on a node (or leaf) while being present in the parent node. b Diversity of MARTX identified in V. splendidus. The domain abbreviations are Rtx repeats in toxin, ACD actin cross-linking domain, ABH alpha/beta hydrolase, MCF makes caterpillars floppy, RRSP Ras/Rap1-specific endopeptidase, CPD cysteine protease domain [44]. A scale bar indicating protein length in amino acids is present on the top right of the figure
The deletion of the complete rtxACHBDE locus in three strains (ZS_173, ZS_185, and ZS_213) did not impair growth in culture media, but resulted in a considerable decrease in mortalities after injection of bacteria (Fig. 6), demonstrating a role of this locus in V. splendidus virulence. MARTX toxins have been defined as “multifactorial effector cargo translocation, processing and delivery machines” (reviewed by refs. [43, 44]). As in other bacteria, the MARTXs found in V. splendidus (MARTXVs) are extremely large (3646–5097 aa) and contain several domains (Fig. 5b). MARTXVs is predicted to contain the three domains found in all MARTXs, i.e., the core structure composed of two conserved regions at the amino- and carboxyl-termini and a cystein protease domain (CPD) [45]. Only one effector domain, a Ras/Rap1-specific endopeptidase (RRSP) [46, 47] is present in all MARTXVs. A homolog of the actin cross-linking domain (ACD) [48] allows distinguishing two main variants of MARTXVs but the presence of ACD is not associated with heightened virulence among strains. The α/β hydrolase (ABH) and the cysteine protease (MCF) domains [49] are absent from only two V. splendidus strains (5S-272 and ZS-117) out of 24, that are still virulent. Finally, the Rho inactivation domain (RID) [50] found in V. cholerae and V. vulnificus and associated with a “cell rounding” effect is absent from V. splendidus. Hence, none of the domains ACD and RID previously demonstrated to affect polymerized actin in target eukaryotic cells, seem to be key domains of V. splendidus MARTX activity, while the RRSP may be hypothesized as an essential domain for its activity.
Fig. 6.
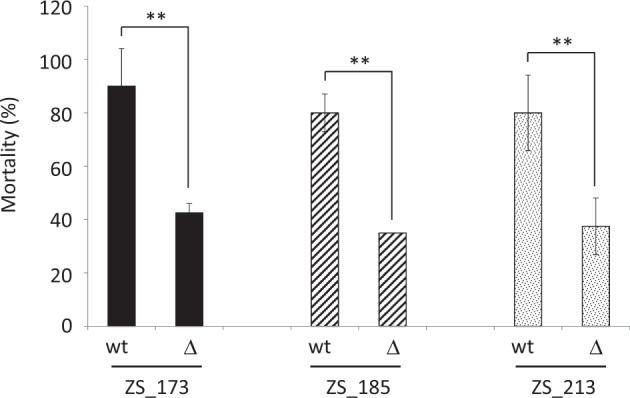
Oyster mortality in response to experimental infection with V. splendidus wild-type strains and ∆rtxACHBDE derivatives. The rtxACHBDE locus was deleted in three V. splendidus strains (ZS_173, ZS_185, and ZS_213). Virulence of the wild type (wt) and derivative (∆) was compared after injection of strains (108 cfu/animal) in 20 oysters and counting the percentage of mortalities at 24H (y axis). Experiments were performed in duplicate and repeated twice. Two stars indicate highly significant differences between treatments (Wilcoxon test, p > 0.005)
In the Splendidus clade, the rtxACHBDE cluster is found only in V. splendidus but is also present in several more distantly related Vibrio species (Fig. 7). Within V. splendidus, the locus is present in 24/35 members (Fig. 5a), is chromosomally encoded, and does not show any signs of recent acquisition by HGT involving non-homologous mechanisms. Across all 872 genomes of Vibrionaceae (Fig. 7), this cluster is identified in seven additional species that have all been associated with disease in humans or animals [43, 44]. The frequency of the locus (strains carrying the rtxACHBDE cluster/total) appears different between species. In V. cholerae, V. vulnificus, and V. ordalii, the cluster is present in the vast majority of the strains, suggesting strong selection for its maintenance, irrespective of their source of isolation. In V. cholerae and V. vulnificus, 153 of 167 and 15 of 16 clinical isolates, and 37 of 43 and 16 of 16 environmental isolates contained the rtxACHBDE cluster, respectively. Conversely, in the shrimp pathogen V. nigripulchritudo, the cluster is restricted to six strains that belong to the same pathogen-containing lineage and is carried by a plasmid [51, 52]. Similarly, in V. harveyi and V. aestuarianus, the locus is detected in only 3 of 23 and 1 of 12 strains, respectively, and annotation of flanking regions indicates a recent acquisition by HGT (plasmid genes, transposase, and syntenic rupture). Hence rtxACHBDE, although widespread among Vibrio spp., appears to be present at variable frequencies within species, indicating potentially different selective pressures on the gene cluster.
Fig. 7.

Distribution of the rtxACHBDE cluster in the genomes of 30 Vibrio species. The phylogenetic tree was reconstructed using the RctB protein sequence. The inner circle indicates the presence (red square) or absence (white square) of the six genes in the rtxACHBDE cluster. The outer circle indicates the isolation site of the V. splendidus strains with a turquoise for Plum Island and gray for oyster-farming areas in France. The 30 different Vibrio species and two Vibrionaceae genus outgroups are represented using alternating grays with the following letters: (a) Photobacterium spp.; (b) Enterovibrio spp.; (c) V. breoganii; (d) V. nigripulchritudo; (e) V. aestuarianus; (f) V. ordalii; (g) V. scophthalmi; (h) V. coralliilyticus; (i) V. furnissii; (j) V. fluvialis; (k) V. metoecus; (l) V. mimicus; (m) V. cholerae; (n) V. navarrensis; (o) V. cidicii; (p) V. vulnificus; (q) V. natriegens; (r) V. parahaemolyticus; (s) V. alginolyticus; (t) V. rotiferianus; (u) V. harveyi; (v) V. campbellii; (w) V. owensii; (x) V. jasicida; (y) V. mediterranei; (F15) V. cyclitrophicus; (aa) V. crassostreae; (ab) V. chagasii; (ac) Vibrio sp. nov F13-like; (F12) Vibrio sp. nov; (F13) Vibrio sp. nov; (F16) V. tasmaniensis; (F18) V. splendidus. The tree was rooted with Photobacterium spp. and Enterovibrio spp. Populations of the Splendidus clade are grouped between the two arrows and population correspondence with Fig. 1 is indicated by the same color code
Discussion
Ecological and evolutionary dynamics of virulence genes have been poorly documented for Vibrionaceae in the wild, and most knowledge stems from a handful of human pathogens isolated from clinical cases where the focus has been on pathogen emergence by gene acquisition and expansion of virulent clones [53–56]. Here we show that virulence toward juvenile oysters appears to be an ancestral trait in the Splendidus clade triggered by the acquisition and subsequent vertical inheritance of the R5.7/8 gene cluster. Populations F12 (closely related to V. crassostreae), F15 (V. cyclitrophicus), and F18 (V. splendidus) have all previously been implicated in mortality of a wide range of animals [8, 57], and our results show that the ability to be virulent is characteristic of the entire populations, rather than recently emerged clones. Consequently, non-virulent populations and strains have evolved by gene loss and expansion. However, we also show that increased virulence likely involves additional gene acquisitions, as demonstrated for the rtxACHBDE gene cluster, which in our collection was specifically acquired by V. splendidus. Because our results indicate that the R5.7 gene is necessary but not sufficient for virulence, we hypothesize that the other virulent populations within the Splendidus clade (e.g., F15, V. cyclitrophicus) may contain additional yet-to-be-determined virulence genes.
The only virulent population outside the Splendidus clade in our collection is F3, V. ordalii, which has been associated with fish disease [42] and belongs to the Anguillarum clade that contains the fish pathogen V. anguillarum and the oyster pathogen V. aestuarianus [26]. This widespread distribution of pathogenicity among closely related species suggests that similar to the Splendidus clade, virulence traits might have been acquired by the MRCA of the Anguillarum clade. One candidate gene is a zinc metalloprotease, which has been demonstrated in V. aestuarianus to be sufficient to cause exotoxicity [58] and to be necessary for virulence in V. anguillarum [59]. Moreover, we also found this gene in all V. ordalii genomes (not shown). Hence, as is the case in the Splendidus clade, this zinc metalloprotease may be an ancestral virulence determinant gene that has been preserved through speciation events and that plays an important role in the biology of this clade.
Within the Splendidus clade, all virulent populations share the R5.7/8 gene cluster for which we show this to be ancestrally acquired. Only R5.7 seems necessary for full virulence and complementation assays using the purified R5.7F12 or R5.7cras proteins strongly suggest that these conserved proteins interact with the external surface of Vibrio and/or its cellular target in oysters. The injection of the R5.7 protein alone or in the presence of non-virulent Vibrio strains did not affect oyster viability, showing that the protein alone does not have a lethal effect. Finally, a strong genetic linkage between R5.7/8 suggests functional interaction between these exported proteins of unknown function, in a mechanism that remains to be determined.
The R5.7/8 cluster can be present in non-virulent strains demonstrating that this cluster is necessary but not sufficient for virulence. Indeed, we show that an additional locus, the rtxACHBDE cluster, is necessary for full virulence towards oysters in V. splendidus. In other Vibrio species, the toxin MARTX has been demonstrated to work in concert with a cytolysin (in V. vulnificus) and a hemolysin (in V. cholerae), and it has been associated with the severity of infection by contributing to the evasion of innate immune defense [44]. An accessory role of MARTX in virulence might be expected in the shrimp pathogen V. nigripulchritudo [52] since it has been recently demonstrated that a lethal toxin, nigritoxin, is the major virulence factor of this pathogen [40, 51]. Among V. splendidus strains, MARTX contains diverse effector domains and only RRSP is shared by all strains. This domain encoding an endopeptidase has been implicated in other species in the cleavage of Ras and Rap1 proteins [46, 47]. Importantly these small GTPases play a role as regulatory nodes of intracellular signaling and membrane trafficking, including trafficking of Toll-like receptor 4 (TLR4) that is involved in innate immune response in both vertebrates and invertebrates [60]. Hence the presence and conservation of a RRSP domain suggest that in V. splendidus, MARTX might be involved in virulence by impairing the host innate immune responses.
The R5.7/8 and rtxACHBDE loci showed differential evolutionary dynamics that indicate distinct selective regimes. The high frequency within virulent populations and mostly vertical transmission of R5.7/8 suggest that it is maintained by positive selection. That three non-virulent populations within the Splendidus clade have lost the gene cluster further suggests that it is involved in niche differentiation at the species level. On the other hand, the rtxACHBDE cluster has been independently acquired by V. splendidus and distantly related other species, and this gene cluster varies in frequency across these species. Moreover, within V. splendidus, and similarly in V. vulnificus [61, 62], the composition of the effector domains appears heterogeneous, ranging from 4 to 6 effectors per toxin with only one, RRSP, found in all MARTXvs. In V. vulnificus, such compositional variation has been shown to result from homologous recombination, shuffling effector domains such that different strains within the same species can express variants of the toxins that are likely to have distinct roles in the niche [44, 61–63]. Among the diverse MARTXvs, 15 and 22 of 24 share an ACD and ABH domain, respectively. Moreover, 22 of 24 MARTXvs share at least one MCF domain with the MARTX type III of V. vulnificus. Although the functional consequences of this domain variation are not fully understood, the MARTX type III of V. vulnificus appears to play a dual role in eel infection as well as in protection from predation by amoebae by causing the lysis of this grazer [64]. In both V. splendidus and V. vulnificus, the intermittent presence and domain diversity of the MARTX cluster are potential indicators of frequency-dependent selection [65]. Because MARTX is a very large secreted protein and hence costly to express, we hypothesize that public good dynamics may arise where producers are favored only when low in frequency within the population [66]. Additionally, variation in toxin types may arise as a result of the evolutionary arms race between bacteria and predators or host immune system and, hence, is similar to frequency-dependent selection imposed on O-antigen structure or outer membrane proteins [67–70].
Our analysis shows that a number of environmental Vibrio populations present a risk of virulence independent of intensive aquaculture settings. Because virulence represents a function encoded in the core genome of these populations, specific markers can be used for risk assessment rather than overall Vibrio abundance, as is frequently the practice. At a minimum, the R5.7/8 gene cluster may be used to indicate overall risk. However, population specific markers will facilitate a more detailed evaluation of the epidemiology of oysters. An important component of such surveys is the determination of population dynamics outside the oyster host to specifically recognize ecological conditions that may trigger population increases and hence risk of infection. Importantly, recent analysis of microbial dynamics in the coastal ocean has shown rapid, near weekly, turnover of communities that are characterized by short-lived blooms of different bacteria and eukaryotes [71]. Exploration to what extent such transient communities allow for specific blooms of different Vibrio populations may elucidate whether there are periods of increased risk from multiple co-occurring populations or whether risk is similar across extended periods due to sequential blooms of populations. In support of the former, we previously showed that several virulent populations of Vibrio can co-occur in diseased oysters, e.g., V. crassostreae with V. splendidus or V. chagasii [6]. We identified a plasmid that is present specifically in virulent strains of V. crasssotreae and necessary for full virulence [6]. Here we show that the MARTX gene cluster is involved in V. splendidus virulence and absent from the other virulent populations within the Splendidus clade. An interesting hypothesis to explore in the future is that other virulent populations such as V. chagasii or V. cyclitrophicus carry specific virulent traits. In a context of oyster infection by multiple co-occurring populations, these diverse virulence traits could act synergistically, increasing the vulnerability of the host. Indeed, experimental infections have demonstrated that some strains, belonging to the same [72] or different species [73], are moderately virulent when injected into oyster individually, and display heightened virulence in mixed experimental infections. Hence oyster disease may result from microbial interaction within and between populations and should be evaluated as potentially polymicrobial.
Disclaimer
The material represents an original result and has not been submitted for publication elsewhere.
Electronic supplementary material
Acknowledgements
We thank the staff of the station Ifremer Argenton and Bouin, the ABIMS and CRBM (Roscoff) and LABGeM (Evry) plateforms, Astrid Lemire, David Goudenège, Remy Van Geersdaële, Julien Le Blanc and Christiane Le Roux for technical support. We are grateful to Jesse Shapiro (University of Montréal, Canada) for his thoughtful comments, which improved the manuscript. This work was supported by grants from the Agence Nationale de la Recherche (ANR-13-ADAP-0007-01 « OPOPOP» and ANR-16-CE32-0008-01 « REVENGE ») to FLR, the U.S. National Science Foundation (OCE-1441943) to MFP, Ifremer to MB, AJ, and DP, and the Region Bretagne to AJ and DP.
Author contributions
YL, AJ, DP, SC, BP, and FLR performed experiments. MB performed the in silico analyses. MB, FLR, and MFP designed the experiments, interpreted results, and wrote the paper.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest. The material represents an original result and has not been submitted for publication elsewhere.
Contributor Information
Martin F. Polz, Phone: +1 617 253 7128, Email: mpolz@mit.edu
Frédérique Le Roux, Phone: +33 2 98 29 56 47, Email: frederique.le-roux@sb-roscoff.fr.
Electronic supplementary material
The online version of this article (10.1038/s41396-018-0245-3) contains supplementary material, which is available to authorized users.
References
- 1.Baker-Austin C, Trinanes JA, Taylor NGH, Hartnell R, Siitonen A, Martinez-Urtaza J. Emerging Vibrio risk at high latitudes in response to ocean warming. Nat Clim Change. 2013;3:73–7. doi: 10.1038/nclimate1628. [DOI] [Google Scholar]
- 2.Le Roux F, Wegner KM, Baker-Austin C, Vezzulli L, Osorio CR, Amaro C, et al. The emergence of Vibrio pathogens in Europe: ecology, evolution, and pathogenesis (Paris, 11-12th March 2015) Front Microbiol. 2015;6:830. doi: 10.3389/fmicb.2015.00830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vezzulli L, Grande C, Reid PC, Helaouet P, Edwards M, Hofle MG, et al. Climate influence on Vibrio and associated human diseases during the past half-century in the coastal North Atlantic. Proc Natl Acad Sci USA. 2016;113:E5062–71. doi: 10.1073/pnas.1609157113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barbosa Solomieu V, Renault T, Travers MA. Mass mortality in bivalves and the intricate case of the Pacific oyster, Crassostrea gigas. J Invertebr Pathol. 2015;131:2–10. doi: 10.1016/j.jip.2015.07.011. [DOI] [PubMed] [Google Scholar]
- 5.Travers MA, Boettcher Miller K, Roque A, Friedman CS. Bacterial diseases in marine bivalves. J Invertebr Pathol. 2015;131:11–31. doi: 10.1016/j.jip.2015.07.010. [DOI] [PubMed] [Google Scholar]
- 6.Bruto M, James A, Petton B, Labreuche Y, Chenivesse S, Alunno-Bruscia M, et al. Vibrio crassostreae, a benign oyster colonizer turned into a pathogen after plasmid acquisition. ISME J. 2017;11:1043–52. doi: 10.1038/ismej.2016.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gay M, Berthe FC, Le Roux F. Screening of Vibrio isolates to develop an experimental infection model in the Pacific oyster Crassostrea gigas. Dis Aquat Organ. 2004;59:49–56. doi: 10.3354/dao059049. [DOI] [PubMed] [Google Scholar]
- 8.Le Roux F, Zouine M, Chakroun N, Binesse J, Saulnier D, Bouchier C, et al. Genome sequence of Vibrio splendidus: an abundant planctonic marine species with a large genotypic diversity. Environ Microbiol. 2009;11:1959–70. doi: 10.1111/j.1462-2920.2009.01918.x. [DOI] [PubMed] [Google Scholar]
- 9.Lemire A, Goudenege D, Versigny T, Petton B, Calteau A, Labreuche Y, et al. Populations, not clones, are the unit of Vibrio pathogenesis in naturally infected oysters. ISME J. 2014;9:1523–31. doi: 10.1038/ismej.2014.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Petton B, Bruto M, James A, Labreuche Y, Alunno-Bruscia M, Le Roux F. Crassostrea gigas mortality in France: the usual suspect, a herpes virus, may not be the killer in this polymicrobial opportunistic disease. Front Microbiol. 2015;6:686. doi: 10.3389/fmicb.2015.00686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perez-Cataluna A, Lucena T, Tarazona E, Arahal DR, Macian MC, Pujalte MJ. An MLSA approach for the taxonomic update of the Splendidus clade, a lineage containing several fish and shellfish pathogenic Vibrio spp. Syst Appl Microbiol. 2016;39:361–9. doi: 10.1016/j.syapm.2016.03.010. [DOI] [PubMed] [Google Scholar]
- 12.Sawabe T, Koizumi S, Fukui Y, Nakagawa S, Ivanova EP, Kita-Tsukamoto K, et al. Mutation is the main driving force in the diversification of the Vibrio splendidus clade. Microbes Environ. 2009;24:281–5. doi: 10.1264/jsme2.ME09128. [DOI] [PubMed] [Google Scholar]
- 13.Spratt BG, Maiden MC. Bacterial population genetics, evolution and epidemiology. Philos Trans R Soc Lond B Biol Sci. 1999;354:701–10. doi: 10.1098/rstb.1999.0423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sundberg LR, Ketola T, Laanto E, Kinnula H, Bamford JK, Penttinen R, et al. Intensive aquaculture selects for increased virulence and interference competition in bacteria. Proc Biol Sci. 2016;283:20153069. doi: 10.1098/rspb.2015.3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hunt DE, David LA, Gevers D, Preheim SP, Alm EJ, Polz MF. Resource partitioning and sympatric differentiation among closely related bacterioplankton. Science. 2008;320:1081–5. doi: 10.1126/science.1157890. [DOI] [PubMed] [Google Scholar]
- 16.Preheim SP, Boucher Y, Wildschutte H, David LA, Veneziano D, Alm EJ, et al. Metapopulation structure of Vibrionaceae among coastal marine invertebrates. Environ Microbiol. 2011;13:265–75. doi: 10.1111/j.1462-2920.2010.02328.x. [DOI] [PubMed] [Google Scholar]
- 17.Preheim SP, Timberlake S, Polz MF. Merging taxonomy with ecological population prediction in a case study of Vibrionaceae. Appl Environ Microbiol. 2011;77:7195–206. doi: 10.1128/AEM.00665-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Szabo G, Preheim SP, Kauffman KM, David LA, Shapiro J, Alm EJ, et al. Reproducibility of Vibrionaceae population structure in coastal bacterioplankton. ISME J. 2012;7:509–19. doi: 10.1038/ismej.2012.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hehemann JH, Arevalo P, Datta MS, Yu X, Corzett CH, Henschel A, et al. Adaptive radiation by waves of gene transfer leads to fine-scale resource partitioning in marine microbes. Nat Commun. 2016;7:12860. doi: 10.1038/ncomms12860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shapiro BJ, Friedman J, Cordero OX, Preheim SP, Timberlake SC, Szabo G, et al. Population genomics of early events in the ecological differentiation of bacteria. Science. 2012;336:48–51. doi: 10.1126/science.1218198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cordero OX, Ventouras LA, Delong EF, Polz MF. Public good dynamics drive evolution of iron acquisition strategies in natural bacterioplankton populations. Proc Natl Acad Sci USA. 2012;109:20059–64. doi: 10.1073/pnas.1213344109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cordero OX, Wildschutte H, Kirkup B, Proehl S, Ngo L, Hussain F, et al. Ecological populations of bacteria act as socially cohesive units of antibiotic production and resistance. Science. 2012;337:1228–31. doi: 10.1126/science.1219385. [DOI] [PubMed] [Google Scholar]
- 23.Yawata Y, Cordero OX, Menolascina F, Hehemann JH, Polz MF, Stocker R. Competition-dispersal tradeoff ecologically differentiates recently speciated marine bacterioplankton populations. Proc Natl Acad Sci USA. 2014;111:5622–7. doi: 10.1073/pnas.1318943111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Petton B, Boudry P, Alunno-Bruscia M, Pernet F. Factors influencing disease-induced mortality of Pacific oysters Crassostreae gigas. Aquac Environ Interact. 2015;6:205–22. doi: 10.3354/aei00125. [DOI] [Google Scholar]
- 25.Petton B, Pernet F, Robert R, Boudry P. Temperature influence on pathogen transmission and subsequent mortalities in juvenile Pacific oysters Crassostrea gigas. Aquac Environ Interact. 2013;3:257–73. doi: 10.3354/aei00070. [DOI] [Google Scholar]
- 26.Goudenege D, Travers MA, Lemire A, Petton B, Haffner P, Labreuche Y, et al. A single regulatory gene is sufficient to alter Vibrio aestuarianus pathogenicity in oysters. Environ Microbiol. 2015;17:4189–99. doi: 10.1111/1462-2920.12699. [DOI] [PubMed] [Google Scholar]
- 27.Le Roux F, Wegner KM, Polz MF. Oysters and Vibrios as a model for disease dynamics in wild animals. Trends Microbiol. 2016;24:568–580. [DOI] [PubMed]
- 28.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–9. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vallenet D, Belda E, Calteau A, Cruveiller S, Engelen S, Lajus A, et al. MicroScope--an integrated microbial resource for the curation and comparative analysis of genomic and metabolic data. Nucleic Acids Res. 2013;41:D636–47. doi: 10.1093/nar/gks1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miele V, Penel S, Duret L. Ultra-fast sequence clustering from similarity networks with SiLiX. BMC Bioinform. 2011;12:116. doi: 10.1186/1471-2105-12-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17:540–52. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- 32.Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–90. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 33.Ho L, Ane C. A linear-time algorithm for Gaussian and non-Gaussian trait evolution models. Syst Biol. 2014;63:397–408. doi: 10.1093/sysbio/syu005. [DOI] [PubMed] [Google Scholar]
- 34.Ives AR, Garland T., Jr. Phylogenetic logistic regression for binary dependent variables. Syst Biol. 2010;59:9–26. doi: 10.1093/sysbio/syp074. [DOI] [PubMed] [Google Scholar]
- 35.Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif. 2005;41:207–34. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 36.Le Roux F, Binesse J, Saulnier D, Mazel D. Construction of a Vibrio splendidus mutant lacking the metalloprotease gene vsm by use of a novel counterselectable suicide vector. Appl Environ Microbiol. 2007;73:777–84. doi: 10.1128/AEM.02147-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Val ME, Skovgaard O, Ducos-Galand M, Bland MJ, Mazel D. Genome engineering in Vibrio cholerae: a feasible approach to address biological issues. PLoS Genet. 2012;8:e1002472. doi: 10.1371/journal.pgen.1002472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Demarre G, Guerout AM, Matsumoto-Mashimo C, Rowe-Magnus DA, Marliere P, Mazel D. A new family of mobilizable suicide plasmids based on broad host range R388 plasmid (IncW) and RP4 plasmid (IncPalpha) conjugative machineries and their cognate Escherichia coli host strains. Res Microbiol. 2005;156:245–55. doi: 10.1016/j.resmic.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 39.Le Roux F, Davis BM, Waldor MK. Conserved small RNAs govern replication and incompatibility of a diverse new plasmid family from marine bacteria. Nucleic Acids Res. 2011;39:1004–13. doi: 10.1093/nar/gkq852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Labreuche Y, Chenivesse S, Jeudy A, Le Panse S, Boulo V, Ansquer D, et al. Nigritoxin is a bacterial toxin for crustaceans and insects. Nat Commun. 2017;8:1248. doi: 10.1038/s41467-017-01445-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Faury N, Saulnier D, Thompson FL, Gay M, Swings J, Le Roux F. Vibrio crassostreae sp. nov., isolated from the haemolymph of oysters (Crassostrea gigas) Int J Syst Evol Microbiol. 2004;54:2137–40. doi: 10.1099/ijs.0.63232-0. [DOI] [PubMed] [Google Scholar]
- 42.Austin B. Taxonomy of bacterial fish pathogens. Vet Res. 2011;42:20. doi: 10.1186/1297-9716-42-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gavin HE, Satchell KJ. MARTX toxins as effector delivery platforms. Pathog Dis. 2015;73:ftv092. doi: 10.1093/femspd/ftv092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Satchell KJ. Multifunctional-autoprocessing repeats-in-toxin (MARTX) toxins of Vibrios. Microbiol Spectr. 2015 Jun;3. 10.1128/microbiolspec.VE-0002-2014. [DOI] [PMC free article] [PubMed]
- 45.Satchell KJ. MARTX, multifunctional autoprocessing repeats-in-toxin toxins. Infect Immun. 2007;75:5079–84. doi: 10.1128/IAI.00525-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Antic I, Biancucci M, Zhu Y, Gius DR, Satchell KJ. Site-specific processing of Ras and Rap1 Switch I by a MARTX toxin effector domain. Nat Commun. 2015;6:7396. doi: 10.1038/ncomms8396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Biancucci M, Rabideau AE, Lu Z, Loftis AR, Pentelute BL, Satchell KJF. Substrate recognition of MARTX Ras/Rap1-specific endopeptidase. Biochemistry. 2017;56:2747–57. doi: 10.1021/acs.biochem.7b00246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cordero CL, Kudryashov DS, Reisler E, Satchell KJ. The actin cross-linking domain of the Vibrio cholerae RTX toxin directly catalyzes the covalent cross-linking of actin. J Biol Chem. 2006;281:32366–74. doi: 10.1074/jbc.M605275200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Agarwal S, Zhu Y, Gius DR, Satchell KJ. The Makes Caterpillars Floppy (MCF)-like domain of Vibrio vulnificus induces mitochondrion-mediated apoptosis. Infect Immun. 2015;83:4392–403. doi: 10.1128/IAI.00570-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ahrens S, Geissler B, Satchell KJ. Identification of a His-Asp-Cys catalytic triad essential for function of the Rho inactivation domain (RID) of Vibrio cholerae MARTX toxin. J Biol Chem. 2013;288:1397–408. doi: 10.1074/jbc.M112.396309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goudenege D, Labreuche Y, Krin E, Ansquer D, Mangenot S, Calteau A, et al. Comparative genomics of pathogenic lineages of Vibrio nigripulchritudo identifies virulence-associated traits. ISME J. 2013;7:1985–96. doi: 10.1038/ismej.2013.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Le Roux F, Labreuche Y, Davis BM, Iqbal N, Mangenot S, Goarant C, et al. Virulence of an emerging pathogenic lineage of Vibrio nigripulchritudo is dependent on two plasmids. Environ Microbiol. 2011;13:296–306. doi: 10.1111/j.1462-2920.2010.02329.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Boucher Y. Sustained local diversity of Vibrio cholerae O1 biotypes in a previously cholera-free country. MBio. 2016 May 3;7. pii: e00570-16. 10.1128/mBio.00570-16. [DOI] [PMC free article] [PubMed]
- 54.Martinez-Urtaza J, van Aerle R, Abanto M, Haendiges J, Myers RA, Trinanes J, et al. Genomic variation and evolution of Vibrio parahaemolyticus ST36 over the course of a transcontinental epidemic expansion. MBio. 2017 Nov 14;8. pii: e01425-17. 10.1128/mBio.01425-17. [DOI] [PMC free article] [PubMed]
- 55.Raz N, Danin-Poleg Y, Hayman RB, Bar-On Y, Linetsky A, Shmoish M, et al. Genome-wide SNP-genotyping array to study the evolution of the human pathogen Vibrio vulnificus biotype 3. PLoS ONE. 2014;9:e114576. doi: 10.1371/journal.pone.0114576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shapiro BJ, Levade I, Kovacikova G, Taylor RK, Almagro-Moreno S. Origins of pandemic Vibrio cholerae from environmental gene pools. Nat Microbiol. 2016;2:16240. doi: 10.1038/nmicrobiol.2016.240. [DOI] [PubMed] [Google Scholar]
- 57.Roux F, Austin B. The Vibrios. Washington, DC: ASM Press; 2006. pp. 285–296. [Google Scholar]
- 58.Labreuche Y, Le Roux F, Henry J, Zatylny C, Huvet A, Lambert C, et al. Vibrio aestuarianus zinc metalloprotease causes lethality in the Pacific oyster Crassostrea gigas and impairs the host cellular immune defenses. Fish Shellfish Immunol. 2010;29:753–8. doi: 10.1016/j.fsi.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 59.Milton DL, Norqvist A, Wolf-Watz H. Cloning of a metalloprotease gene involved in the virulence mechanism of Vibrio anguillarum. J Bacteriol. 1992;174:7235–44. doi: 10.1128/jb.174.22.7235-7244.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Luo L, Wall AA, Yeo JC, Condon ND, Norwood SJ, Schoenwaelder S, et al. Rab8a interacts directly with PI3Kgamma to modulate TLR4-driven PI3K and mTOR signalling. Nat Commun. 2014;5:4407. doi: 10.1038/ncomms5407. [DOI] [PubMed] [Google Scholar]
- 61.Kwak JS, Jeong HG, Satchell KJ. Vibrio vulnificus rtxA1 gene recombination generates toxin variants with altered potency during intestinal infection. Proc Natl Acad Sci USA. 2011;108:1645–50. doi: 10.1073/pnas.1014339108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Roig FJ, Gonzalez-Candelas F, Amaro C. Domain organization and evolution of multifunctional autoprocessing repeats-in-toxin (MARTX) toxin in Vibrio vulnificus. Appl Environ Microbiol. 2011;77:657–68. doi: 10.1128/AEM.01806-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dolores J, Satchell KJ. Analysis of Vibrio cholerae genome sequences reveals unique rtxA variants in environmental strains and an rtxA-null mutation in recent altered El Tor isolates. MBio. 2013;4:e00624. doi: 10.1128/mBio.00624-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee CT, Amaro C, Wu KM, Valiente E, Chang YF, Tsai SF, et al. A common virulence plasmid in biotype 2 Vibrio vulnificus and its dissemination aided by a conjugal plasmid. J Bacteriol. 2008;190:1638–48. doi: 10.1128/JB.01484-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cordero OX, Polz MF. Explaining microbial genomic diversity in light of evolutionary ecology. Nat Rev Microbiol. 2014;12:263–73. doi: 10.1038/nrmicro3218. [DOI] [PubMed] [Google Scholar]
- 66.Jousset A, Rochat L, Pechy-Tarr M, Keel C, Scheu S, Bonkowski M. Predators promote defence of rhizosphere bacterial populations by selective feeding on non-toxic cheaters. ISME J. 2009;3:666–74. doi: 10.1038/ismej.2009.26. [DOI] [PubMed] [Google Scholar]
- 67.Rodriguez-Valera F, Martin-Cuadrado AB, Rodriguez-Brito B, Pasic L, Thingstad TF, Rohwer F, et al. Explaining microbial population genomics through phage predation. Nat Rev Microbiol. 2009;7:828–36. doi: 10.1038/nrmicro2235. [DOI] [PubMed] [Google Scholar]
- 68.Seed KD, Yen M, Shapiro BJ, Hilaire IJ, Charles RC, Teng JE, et al. Evolutionary consequences of intra-patient phage predation on microbial populations. Elife. 2014;3:e03497. doi: 10.7554/eLife.03497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wildschutte H, Preheim SP, Hernandez Y, Polz MF. O-antigen diversity and lateral transfer of the wbe region among Vibrio splendidus isolates. Environ Microbiol. 2010;12:2977–87. doi: 10.1111/j.1462-2920.2010.02274.x. [DOI] [PubMed] [Google Scholar]
- 70.Seed KD, Faruque SM, Mekalanos JJ, Calderwood SB, Qadri F, Camilli A. Phase variable O antigen biosynthetic genes control expression of the major protective antigen and bacteriophage receptor in Vibrio cholerae O1. PLoS Pathog. 2012;8:e1002917. doi: 10.1371/journal.ppat.1002917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Martin-Platero AM, Cleary B, Kauffman K, Preheim SP, McGillicuddy DJ, Alm EJ, et al. High resolution time series reveals cohesive but short-lived communities in coastal plankton. Nat Commun. 2018;9:266. doi: 10.1038/s41467-017-02571-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gay M, Renault T, Pons AM, Le Roux F. Two Vibrio splendidus related strains collaborate to kill Crassostrea gigas: taxonomy and host alterations. Dis Aquat Org. 2004;62:65–74. doi: 10.3354/dao062065. [DOI] [PubMed] [Google Scholar]
- 73.Saulnier D, De Decker S, Haffner P, Cobret L, Robert M, Garcia C. A large-scale epidemiological study to identify bacteria pathogenic to Pacific oyster Crassostrea gigas and correlation between virulence and metalloprotease-like activity. Microb Ecol. 2010;59:787–98. doi: 10.1007/s00248-009-9620-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.