

Abstract
Pulmonary innate immune responses involve a highly regulated multicellular network to defend the enormous surface area of the lung. Disruption of these responses renders the host susceptible to pneumonia. Alveolar epithelial cells (AEC) are a critical source of innate immune molecules such as granulocyte-macrophage colony stimulating factor (GM-CSF), that determine the functional maturation of alveolar macrophages. In many pulmonary diseases, heterogeneous ventilation leads to regional hypoxia in the lung. The effect of hypoxia on AEC innate immune function is unknown. We now report that exposure of primary murine AEC to hypoxia (1% oxygen) for 24h results in significant suppression of key innate immune molecules, including GM-CSF, CCL2, and IL-6. This exposure did not cause toxicity but did induce stabilization of hypoxia inducible factor 1α protein (HIF-1α) and shift to glycolytic metabolism. Focusing on GM-CSF, we found that hypoxia greatly decreased the rate of GM-CSF transcription. Hypoxic both decreased NF-kB signaling in AEC and induced chromosomal changes resulting in decreased accessibility in the GM-CSF proximal promoter of target sequences for NF-kB binding. In mice exposed to hypoxia in vivo (12% oxygen for 2 days), lung GM-CSF protein expression was reduced. In vivo phagocytosis of fluorescent beads by alveolar macrophages was also suppressed, but this effect was reversed by treatment with GM-CSF. These studies suggest that in critically ill patients, local hypoxia may contribute to the susceptibility of poorly ventilated lung units to infection through complementary effects on several pathways reducing AEC expression of GM-CSF and other key innate immune molecules.
Keywords: lung, innate immunity, hypoxia, NF-kB, gene transcription
INTRODUCTION
The pulmonary alveolar space forms an enormous site of interaction with the external environment. Precise control of innate immune responses in this space is critical for maintaining a balance between host defense and gas exchange in the lung. Alveolar epithelial cells (AEC2) are key participants in pulmonary host defense, producing a variety of molecules that influence the recruitment and function of inflammatory cells (1). Of particular interest for the current work, AEC secrete interleukin (IL)-6, CCL2 (also known as MCP-1) and granulocyte-macrophage colony stimulating factor (GM-CSF, or colony stimulating factor 2, csf2). GM-CSF is an important product of AEC that is necessary for normal functional maturation of alveolar macrophages (2). In its absence, alveolar macrophages revert to an immature phenotype, leading to greatly increased susceptibility to lethal pneumonia (3–5). We have shown previously that hyperoxia and other forms of oxidative stress lead to significant suppression of AEC GM-CSF expression, resulting in macrophage dysfunction and impaired defense against pneumonia (6).
In the setting of disease, the lung is frequently subject to extremely heterogeneous ventilation, resulting in regional hypoxia, with well recognized consequences for ventilation-perfusion matching and gas exchange. In a number of cell types, hypoxia has been shown to influence pathways that may have important effects on local host defense. However, the effects of hypoxia on the innate immune function of AEC have not been fully explored. In the present study, we report that hypoxia induced significant reduction of AEC expression of important innate immune molecules, in the absence of AEC toxicity. Using GM-CSF as a focal point for investigation, we have identified key mechanisms involved in this altered innate immune response and have confirmed that this effect on GM-CSF has important implications for alveolar macrophage function in vivo.
METHODS AND MATERIALS
Animals and AEC isolation and culture
Wild-type (WT) C57Bl/6 (Ly5.1; CD45.2) mice were obtained from Jackson Laboratory (Bar Harbor, ME). Male and female mice aged 6–12 weeks were used in these experiments. Mice were housed under specific pathogen-free conditions and were monitored daily by veterinary staff. The animal care committee at the Salt Lake City VA Medical Center approved these experiments.
Primary murine AEC were isolated using a modification of the method of Corti (7), as in our prior work (8–11). AEC were placed in tissue culture treated plastic dishes in DMEM + 10% FBS in an atmosphere of ambient air with 5% CO2 (normoxia). Cultures were washed with PBS after 24h. After 2 days further in culture, AEC were placed in normoxia or hypoxia (1% oxygen, 5% CO2) for up to 24 h. To establish hypoxic conditions, culture dishes were placed in a chamber (Billups- Rothenberg, Del Mar, CA) that was flushed for 10 minutes with a commercially available gas mixture of CO2 and 1% oxygen with a balance of nitrogen, adjusted to maintain a fractional concentration of oxygen of 0.01. This chamber was sealed, humidified and maintained at 370C. The volume of media in each well of a 12-well dish was 1ml and the dishes were incubated with lids. For pharmacologic stabilization of HIF proteins, cells in normoxia were exposed to dimethyloxaloylglycine (DMOG, 1 mM, Sigma) for 24h.
MLE-15 Cell line
Although primary AEC are our best model for studying cell and molecular biology of the lung epithelium in vitro, cell numbers are a significant limitation. Therefore, in an experiment assessing HIF-1α transcriptional activity, in which cell numbers are crucial, we used MLE-15 cells, an SV40-immortalized murine AEC cell line (gift of Jeffrey Whitsett, University of Cincinnati). These cells were maintained in HITES media as previously described (12).
RNA preparation and Real-time (RT)-PCR
Total cellular RNA was isolated, first strand cDNA was reverse transcribed and specific amplification in a StepOnePlus (ABI, ThermoScientific). Gene specific primers were designed using the Roche Applied Science Universal Probe Library Assay Design Center. Each biological sample was amplified in duplicate and the average of the duplicates taken as a single data point for statistical analysis. Recognizing that hypoxia may influence the expression of a number of commonly used “housekeeping” genes (13) we explored the effect of hypoxia in our model on a group of housekeeping genes. As shown in supplemental data, Figure S1, B2M did not appear to be significantly altered in hypoxia. Therefore, for analysis of mRNA expression, data were normalized to beta-2 microglobulin (B2M). A constant amount of total RNA (1 μg)was converted to cDNA and all comparative samples purified, converted to cDNA and RT-PCR performed simultaneously to reduce experimental error. In these experiments, results are expressed as fold-change over control values. The first sample of the control (normoxia) group was set to 100 and standard errors are provided for control and experimental groups. Examination of the role of the miR133 family in the effect of hypoxia on AEC was carried out as described previously (9).
Protein measurements
Expression of GM-CSF, CCL2, IL-6 and VEGF (vascular endothelial growth factor) proteins in cell-free culture supernatants was determined by ELISA (R&D Systems Bio-techne). In each experiment, media was collected, immediately centrifuged to remove cell debris and either immediately assayed for cytokines levels by ELISA, or frozen in suitable aliquots at −800C and assayed subsequently, with care to ensure only a single freeze-thaw.
Measures of oxidative stress
Reactive oxygen species (ROS) produced by AEC in hypoxia and normoxia were determined by four separate methods: the nitro-blue tetrazolium (NBT) assay was used to measure superoxide (11). The Amplex Red assay (Invitrogen, ThermoScientific) was used to measure intracellular hydrogen peroxide (14). We also measured overall intracellular ROS using relative DCF (7’-Dichlorodihydrofluorescein) fluorescence (Cell BioLabs). In addition, mitochondrial ROS levels were measured using MitoSOX (Invitrogen, ThermoScientific). The cell permeable MitoSOX red reagent is selectively targeted to the mitochondria, where the level of oxidation to a fluorescent product is proportional to mitochondrial ROS.
Cellular ATP levels and lactate production
Cellular ATP were measured using Luminescent (firefly luciferase) ATP Detection Assay kit (Abcam). Primary AEC were plated at confluence in a white 96-well plate and on day 3 of culture were exposed to room air or 1% oxygen for 24 and 48h. After incubation the cells were lysed directly in the well. This step irreversibly inactivates ATPases ensuring the luminescent signal obtained corresponds to the endogenous level of ATP. Data was expressed as relative light units (RLU).
Lactate levels in AEC culture medium were measured using an EnzyChromTM L-lactate Assay Kit (BioAssay Systems, catalog number: ECLC-100 according to the manufacturer’s recommendations). Briefly, cell supernatants were harvested from AEC exposed to normoxia or hypoxia for prescribed times. After removing the media, the cells were lifted and counted. Cell supernatants and a standard curve of lactate (up to 1mM) were placed in wells of a 96-well plate and assay buffer added. Absorption (565 nm) was measured at time 0 and after 20 minutes. The delta OD was determined for the samples and the standard curve and the concentration of lactate in each sample determined from the standards. The quantity of lactate was normalized to number of AEC.
GM-CSF mRNA stability, transcription rate and accessibility of regions of the GM-CSF proximal promoter.
Turnover of GM-CSF mRNA was determined by RT-PCR with RNA extracted at increasing time following inhibition of transcription with actinomycin D (5 ng/ml), as in our previous work (9, 11). Relative GM-CSF transcription was determined using a PCR method, with primers designed to span the first intron/exon boundary in order to identify newly transcribed nuclear RNA (nRNA) (15). We assessed the relative accessibility of the GM-CSF proximal promoter region by evaluating the sensitivity of intact chromatin to nuclease digestion as in our prior work (15). Using the EpiQuik Chromatin accessibility kit (EpiGentek), accessible regions are digested with a nuclease mix and the PCR product amplified using site specific primers is reduced compared to undigested chromatin (16). Inaccessible chromatin is resistant to this digestion and therefore can by amplified by primers spanning the site. Conversely, chromatin regions in a relatively open configuration are digested and cannot be amplified by primers spanning the site. Un-denatured DNA was prepared and evaluated by PCR with or without EpiQuik nuclease digestion, using primers spanning −195 to −78 bases (encompassing NF-kB/Sp1/AP-1 sites) relative to the transcription start site (17). Accessibility of the chromatin region in question is then determined as the difference in ct for PCR of digested minus the ct for PCR of undigested samples. Thus increasing accessibility to digestion results in greater numeric value. In each experiment, primers for an accessible gene in euchromatin (GAPDH) and an inaccessible gene in heterochromatin (hemoglobin, not expressed in these cells) were used to determine if chromatin digestion by the EpiQuik nuclease was successfully and specifically achieved. A qPCR assay of euchromatin shows a large Ct shift between digested and undigested samples. A qPCR assay of heterochromatin shows an insignificant Ct shift between digested and undigested samples. These requirements were met in our experiments.
HIF-1α induction and signaling
HIF-1α protein in primary AEC cell extracts was measured by ELISA (R&D Systems Bio-techne). The lower limit of detection in this assay is 15 ng/107 cells. In order to confirm HIF-1α transcriptional activity, nuclear proteins were extracted from MLE-15 cells under appropriate conditions and assayed for specific DNA binding activity using a commercially available kit (abcam 133104). These nuclear proteins were added to 96-well plates in which a specific dsDNA sequence (5’-ACGTG-3’) containing the HIF-1α response element had been immobilized. HIF-1α bound to this target sequence was then detected using an anti-HIF-1α antibody. This experiment was carried out using MLE-15 cells because the low abundance of HIF-1α protein required a large number of cells (107/sample) for this assay.
NF-kB signaling
To assess post-translational events required for NF-kB signaling, phospho-p65 was determined by ELISA (Cell Signaling) to assess NF-kB activation. NF-kB signaling was also assessed using a reporter assay in which a Lentivirus system was used to transduce AEC to expression an NF-kB reporter construct (Cignal, Qiagen). Lentivirus at an MOI of 50 was transduced into the cells using 5μg/ml polybrene (Sigma). We typically attained transduction efficiency of 40–50% (GFP, data not shown). AEC were subject to 24h in hypoxia or room air beginning 48h after transduction. NF-kB reporter activity was measured using a Dual Luciferase Reporter Assay (Promega) and the experimental data (Firefly Luciferase) were normalized for viral transduction based on the Renilla Luciferase signal. Similar to the approach for HIF-1α above, NF-kB transcriptional activity in AEC was determined using a commercially available kit (abcam 133112) in which binding of extracted nuclear protein to immobilized NF-kB target nuclear sequences (5’-AGGAAATTCCG-3’) is detected with an anti-p65 antibody. Due to the increased abundance of NF-kB protein compared to HIF-1α, this experiment could be carried out using primary murine AEC.
In vivo exposure or mice to hypoxia and phagocytosis of fluorescent beads by alveolar macrophages
Mice were exposed to 12% oxygen or room air (21% oxygen) for 48h. In initial experiments, lungs were harvested and snap frozen in dry ice/methanol. Subsequently the lungs were homogenized in Tissue Extraction Reagent I (Invitrogen). Total protein was determined using the BAC assay (ThermoFischer). GM-CSF protein in lung homogenates was determined by ELISA. In order to determine the effects of hypoxia and treatment with GM-CSF on alveolar macrophage phagocytosis, mice were treated IN with recombinant murine GM-CSF IN (100 ng, carrier-free R&D Systems) or saline at the onset and again after 24h exposure. After 48h exposure to hypoxia or normoxia, FITC labeled polyacrylamide beads (1μm- Bangs Laboratories, Inc.) were delivered IN in 30μl of saline (2 × 107 beads/mouse). The beads were coated with BSA and sonicated immediately prior to administration to prevent non-specific binding and agglutination. After 2 hrs, mice were euthanized and alveolar macrophages were collected by bronchoalveolar fluid lavage (3 × 1 ml of saline, each passed into the lungs 3 times). The total number of macrophages was determined and 104 cells were cytospun (Shandon) and air-dried. Five fields of 100 cells were scored for FITC positive cells by an observer blinded to the treatment.
Statistical analysis
Data are presented as mean ± standard error of the mean (SEM). Statistical analysis was carried out using GraphPad Prism v4C software (GraphPad, Inc.). Two-tailed tests of significance were used. Differences between multiple groups were compared with one-way analysis of variance. Comparisons were deemed statistically significant for P values < 0.05.
RESULTS
Hypoxia suppresses AEC innate immune function
In order to investigate the impact of hypoxic conditions on the expression of key innate immune molecules by AEC, primary murine AEC were placed in an atmosphere of 1% oxygen for 24h. This hypoxic exposure resulted in very significant suppression of constitutive GM-CSF mRNA and protein expression (figure 1a and 1b). In addition to this constitutive expression of GM-CSF, we have shown previously that AEC respond to inflammatory stimulation with further increase in GM-CSF expression (8). Here we find that LPS induces increased GM-CSF expression and that hypoxia also suppressed this induction (supplemental figure S2). AEC also secrete inflammatory mediators such as CCL2 and IL-6, particularly in response to IL-1β. Exposure of AEC to 1% oxygen resulted in significantly suppressed expression of CCL2 and IL-6 by AEC both at baseline and in response to IL-1β (figure 1c and 1d). Thus hypoxia broadly suppressed expression of key innate immune molecules by AEC.
Figure 1. Effects of hypoxia expression of GM-CSF, CCL2 and IL-6 by primary murine AEC.

Primary murine AEC were cultured for 24h in normoxic (21% oxygen, black bars) or hypoxic (1% oxygen, gray bars) conditions. Constitutive expression of GM-CSF was determined in media alone (a and b). Expression of CCL2 and IL-6 was determined in the absence or presence of IL-1ß (10ng/ml; c and d). mRNA (RT-PCR) is expressed relative to control (normoxia) with the value of the first control sample set at 100. Protein was measured by ELISA in the culture media collected after 24h. Data are representative of 3 independent experiments and expressed as mean ± SEM, n=3. **p<0.01, *p<0.05.
Tolerance and metabolic consequences of hypoxic exposure for primary murine AEC
Primary murine AEC were tolerant of culture in an atmosphere with 1% oxygen for 24h. Exposure to hypoxic conditions did not result in increased release of LDH from the cells and did not impede cellular metabolic activity as measured in the MTT assay (supplemental figure S3). Thus hypoxia suppressed expression of key innate immune molecules by AEC without causing toxicity. However, hypoxia did result in a significant increase in lactate production (figure 2a) and decrease in AEC ATP content, as shown in figure 2b, consistent with the proposition that these cells responded to the stress of hypoxia by shifting metabolism to glycolytic pathways so as to decrease oxygen consumption, while preserving essential cellular processes to maintain key functions and cellular integrity (18).
Figure 2. Effect of hypoxia on measures of AEC lactate production and ATP stores.
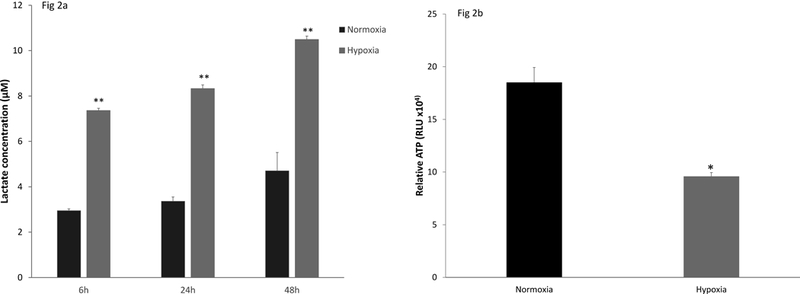
Primary murine AEC were cultured for 24h in normoxic (21% oxygen, black bars) or hypoxic (1% oxygen, gray bars) conditions. Lactate was measured in the cell supernatants over 48h. The data are expressed as mean ± SEM, n= 4. * p<0.001 and are representative of 2 independent experiments (Figure 2a). Cellular ATP stores (Figure 2b) were determined after 24h in hypoxia using a luminescent assay and are expressed as relative light units (RLU). Data are representative of 3 independent experiments and expressed as mean ± SEM, n= 4. * p<0.001.
Induction of HIF protein expression in AEC by hypoxia
The prototypical consequence of hypoxia exposure in most cells is stabilization of HIF protein and induction of HIF-responsive genes. As anticipated, HIF-1α protein expression was induced during hypoxia (figure 3a). We also confirmed translocation of transcriptionally active HIF-1α to the nucleus of MLE-15 cells exposed to hypoxia, as shown by an assay for binding of nuclear protein to the HIF-1α target sequences (figure 3b). HIF-1α was active in hypoxia-exposed AEC, based on induction of iNOS, HO-1, and VEGF, a group of HIF-responsive genes (19), in AEC cultured under hypoxic conditions (figure 3c). These data demonstrate that AEC responded to hypoxia under these culture conditions by activation of a HIF-1α pathway.
Figure 3. AEC expression of HIF-1α and of HIF-target genes in hypoxia.
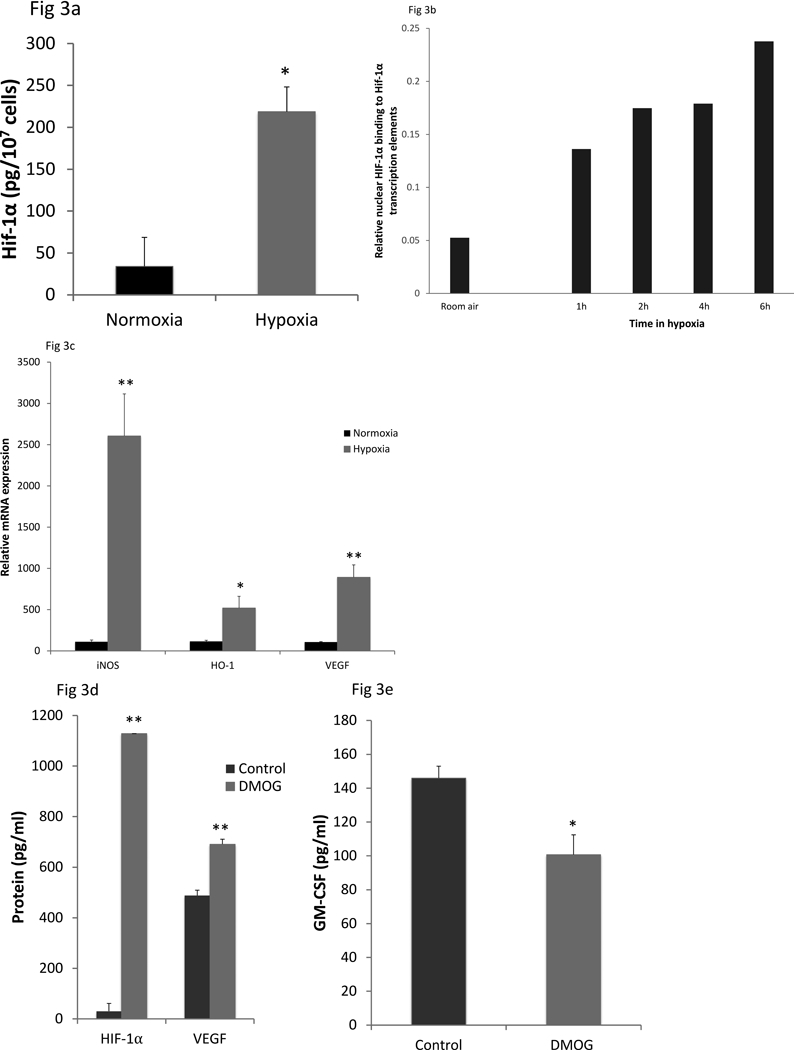
Whole cell HIF-1α protein levels were measured by ELISA on cell lysates in primary murine AEC after exposure to normoxia (21% oxygen) or hypoxia (1% oxygen) for 24h (Figure 3a). Data shown are mean ± SEM of three independent AEC isolations, with the cells from each isolation were pooled. Translocation of active HIF-1α to the nucleus in MLE15 cells exposed to hypoxia (1% oxygen) was measured as binding of nuclear protein to HIF-1α transcription elements, detected by HFI1α antibody (Figure 3b). Data presented are the mean of two and are representative of two independent experiments. To confirm functional HIF induction in primary murine AEC, mRNA expression of three classic HIF responsive genes (iNOS, HO-1, VEGF) was determined after 24h in normoxia or hypoxia (Figure 3c). The data are expressed relative to normoxic levels with the value of the first control sample set at 100. The experiment shown is representative of 2 independent experiments. AEC were exposed to the HIF stabilizing agent, DMOG (1mM), for 24h and intracellular HIF-1α (Figure 3d pg protein/107 cells), secreted VEGF (Figure 3d, pg/ml) and secreted GM-CSF (Figure 3e) protein levels measured. The experiment shown is representative of 2 independent experiments. **p<0.01, *p<0.05.
HIFs are transcription factors whose direct effects typically are to induce gene transcription. In order to determine whether HIF-stabilization might contribute indirectly to suppression in AEC innate immune function, we evaluated the effect of the HIF stabilizer, DMOG, on AEC. Treatment with DMOG greatly increased HIF-1α protein and suppressed AEC GM-CSF expression (figure 3d and 3e), although the effect on GM-CSF expression of DMOG was not as great as in hypoxia (figure 1). These data suggest that downstream effects of HIF proteins contribute to the suppression of GM-CSF expression in hypoxia. They also suggest that HIF alone cannot account for the full effect of hypoxia on GM-CSF expression.
Suppression of AEC GM-CSF expression by hypoxia is not a consequence of oxidative stress and accelerated mRNA turnover.
In some cell systems hypoxia has been found to induce expression of ROS and oxidative stress (20, 21). GM-CSF is an oxidant sensitive gene whose expression in AEC is reduced in response to oxidative stress associated with hyperoxia or H2O2 exposure, through effects on mRNA turnover (9, 11). Therefore, we evaluated the potential role of oxidative stress in hypoxia-induced suppression of GM-CSF. AEC did not exhibit measureable oxidative stress during hypoxia, as determined in the NBT and Amplex red assays for ROS or by the rate of formation of fluorescent DCF and MitoSOX (supplemental figure S4). Furthermore, PEG-catalase, which we previously found reversed oxidant-induced GM-CSF suppression, did not preserve GM-CSF expression during hypoxia (data not shown), also indicating that GM-CSF suppression in hypoxic AEC is not mediated by ROS.
In previous work we found that AEC GM-CSF expression in the setting of oxidative stress due to hyperoxic is controlled at the level of mRNA turnover (9, 10). We measured turnover of GM-CSF mRNA in hypoxia compared to normoxia to determine whether suppression in this environment involved a similar molecular mechanism to that seen in the response to hyperoxia. Interestingly, mRNA decay after inhibition of transcription with actinomycin D was not accelerated in hypoxia compared to normoxia (figure 4a). In additional experiments, we found that hypoxia did not induce expression of the specific family of microRNA (miR133) responsible for GM-CSF mRNA turnover during oxidative stress (9) (data not shown). Together, these data confirmed that reduced expression of GM-CSF in AEC in hypoxic environments is attributable to mechanisms distinct from those at play in hyperoxia and is not a direct consequence of oxidative stress.
Figure 4. Effects of hypoxia on AEC GM-CSF mRNA stability and gene transcription.
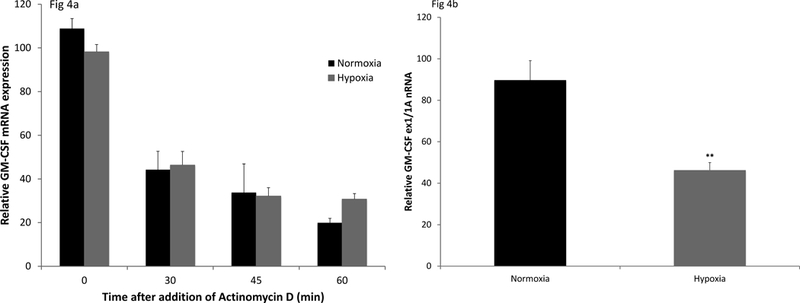
AEC were placed in culture in normoxic (21%) or hypoxic (1%) conditions for 24h. Actinomycin D (5 μg/ml) was then added to stop transcription. Relative mRNA for GM-CSF was determined (RT-PCR) at time points from 0 to 60 min. Black bars represent cells in normoxia; gray bars represent cells in hypoxia (Figure 4a). Data are presented as mean ± SEM, n=4. The experiment is representative of 2 independent experiments. Figure 4b; relative GM-CSF transcription was determined after exposure of AEC to normoxia or hypoxia (24h) by RT-PCR, using primers designed to span and amplify the junction of Exon 1/ Intron A of murine Csf2 (GM-CSF) gene. Values are normalized to expression of the small nuclear RNA, RNu6. Data are representative of more than 3 independent experiments and are expressed as mean ± SEM, n=3. **p<0.01.
Hypoxia inhibits GM-CSF gene transcription in AEC
Because hypoxia did not accelerate GM-CSF mRNA turnover, we next examined the effect of hypoxia on relative gene transcription measured by RT-PCR, with primers spanning GM-CSF exon 1/intron A (ex1/IA) to assess the relative amount of unspliced exon/intron junction nuclear RNA. We found that hypoxia significantly decreased GM-CSF transcription (figure 4b). Once again, this result is in distinct contrast to that in hyperoxia, in which GM-CSF transcription is not reduced compared to normoxia (15).
Effect of hypoxia on NF-kB signaling in AEC
The AEC innate immune molecules of interest, GM-CSF, CCL2 and IL-6, all are suppressed during hypoxia and share NF-kB binding targets in their proximal upstream promoters. We have found previously that pharmacologic inhibition of NF-kB (with BAY 11–7082) results in complete suppression of constitutive GM-CSF expression. We first evaluated the effect of hypoxia on NF-kB signaling in AEC using a lentiviral system, in which AEC were transduced to express an NF-kB reporter construct, prior to exposure to hypoxia or normoxia. Compared to cells in normoxia, AEC in hypoxia demonstrated significantly decreased NF-kB reporter activity (figure 5a). This change was confirmed in experiments demonstrating hypoxia-induced suppression of AEC nuclear protein binding to NF-kB target sequences (figure 5b), showing that the relative nuclear accumulation of active p65 was significantly decreased compared to cells under normoxic conditions. We also evaluated the effect of hypoxia on expression of phosphorylated p65 (RelA). This phosphorylation event is a prerequisite for the movement of NF-kB from the cytoplasm to the nucleus and is an indication of NF-kB activation. AEC in hypoxia expressed decreased phospho-p65 and compared to cells in normoxia (figure 5c). Together, these data strongly suggest that diminished NF-kB signaling during hypoxia contributes to suppression of AEC innate immune responses.
Figure 5. Effect of hypoxia on NF-kB signaling in AEC.

AEC were exposed to normoxia (21% oxygen) or hypoxia (1% oxygen) for 24h. NF-kB-regulated signal transduction was measured using a Lentivirus NF-kB reporter (Figure 6a). The reporter expresses the firefly luciferase upon activation of NF-kB. Data are expressed as the average luciferase activity with the first normoxia sample assigned a value of 100% ± SEM, n=3; *p<0.02. The experiment shown is representative of 4 independent experiments. Translocation of active NF-kB to the nucleus was measured by determining the amount of p65 protein (in nuclear protein extracts from primary murine AEC exposed to normoxia or hypoxia) that bound to the NF-kB transcription element, detected using a specific p65 antibody. Data are presented as mean ± SEM, n=6. **p<0.01 (Figure 5b)., Phosphorylated p65 (at Ser536) and total p65 were measured whole cell extracts from primary murine AEC exposed to normoxia or hypoxia, using a cell-based ELISA, and activation of NF-kB assessed as the ratio of phospho-p65 to total p65 (Figure 5c). The first normoxic control was assigned the value 100. Data are representative of 2 independent experiments, mean ± SEM, n=4; **p<0.001.To determine the effect of hypoxia on accessibility of the GM-CSF proximal promoter chromatin, un-denatured DNA was isolated from primary AEC and digested with nuclease or left untreated. RT-PCR of the NF-kB region of murine GM-CSF proximal promoter was performed and the data analyzed as the ct differential between digested sample and undigested samples as described in Methods (Figure 5d). Data are expressed as mean ± SEM, n=4; **p<0.01. The experiment shown is representative of 2 independent experiments.
The experiment using the lentiviral reporter system does not incorporate epigenetic effects on NF-kB signaling. In order to assess the contribution of such changes to suppression of GM-CSF transcription, we assessed the impact of hypoxia on relative accessibility of the NF-kB binding site in the GM-CSF proximal promoter. Compared to AEC in normoxia, hypoxia resulted in decreased accessibility of this binding region in the promoter GM-CSF proximal promoter (figure 5d). Interestingly, this region also includes binding sites for AP-1 and Sp1, transcription factors that have been found to cooperate with NF-kB for promotion of GM-CSF transcription in other systems (17). Together, these data suggest that hypoxia may decrease activation of NF-kB while also decreasing accessibility of the promoter in a region targeted by multiple transcription factors, thereby reducing GM-CSF gene transcription in AEC.
Hypoxic exposure in vivo suppresses lung GM-CSF protein and mRNA expression and AM phagocytic activity
In order to extend these observations to the in vivo circumstance, mice were exposed to a hypoxic atmosphere of 12% oxygen for 2 days. Compared to mice in normoxia, this exposure induces very modest acute lung injury, as demonstrated by the increased protein concentration in BAL fluid (860±40 μg protein/ml vs. 534±41 μg protein/ml, p<0.01). This increase is far less than the approximately 10-fold increase found in sublethal hyperoxia. GM-CSF protein in lung homogenates was significantly reduced in hypoxia compared to normoxia (figure 6a). In order to explore the consequences of this effect on lung innate immune function, we assessed AM phagocytosis in vivo of fluorescent beads, introduced IN to mice that had been in normoxia or hypoxia for 24 hrs. AM phagocytic activity was significantly suppressed in the hypoxic mice compared to normoxic controls (figure 6b). Furthermore, treatment with recombinant GM-CSF administered IN thoroughly reversed this effect.
Figure 6. Effect of hypoxia exposure in vivo on lung GM-CSF expression and AM phagocytic activity.
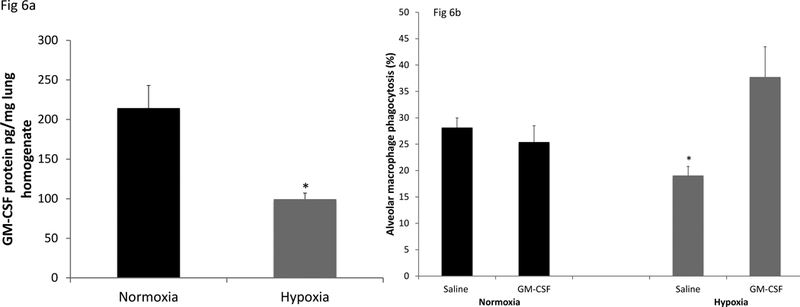
C57BL/6 mice were exposed to room air (21% oxygen) or hypoxia (12% oxygen) for 48h. GM-CSF protein levels in whole lung homogenates were determined (ELISA), normalized to total protein content (Figure 6a). The data are the mean ± SEM; n=5 *p<0.01.Alveolar macrophages were obtained by lung lavage after 48h exposure in 21% or 12% oxygen from mice given saline or GM-CSF (100ng in 40μl saline, IN) at 0 and 24h time of exposure (Figure 6b). Two hours prior to collection of the alveolar macrophages, fluorescent beads (2 ×107 in 40μl saline) were given intra-nasally. The results are expressed as the % macrophages that had taken up beads. 500 macrophages from each mouse were scored and the data are the mean ± SEM; n=5 *p<0.01.
DISCUSSION
Alveolar epithelial cells play key roles in pulmonary innate immunity, recognizing invading pathogens and secreting molecules that recruit and support the host defense function of inflammatory cells. In murine models of pneumonia, the loss of inflammatory responses from AEC and other parenchymal cells significantly impairs pulmonary host defense and increases susceptibility to pneumonia (22–24). In particular, expression of GM-CSF by AEC is critically important for innate immune responses and for pulmonary homeostasis (2, 5). Its absence has far reaching consequences for host defense, leading to greatly increased susceptibility to lethal pneumonia from bacterial, viral or fungal pathogens (5, 25–28). Suppression of GM-CSF for even short periods, alveolar macrophages fail to maintain their mature state, rapidly reverting to an immature phenotype with important consequences for host defense (3, 6). Similarly, in the absence of CCL2 (29) or IL-6 (30, 31) host defense against bacterial pneumonia is greatly impaired. Thus, AEC derived factors determine the context of innate immune response in the alveolar space. Although an inflammatory response may be mounted to invading pathogens in the absence of these AEC-derived factors, this response is dysregulated, disturbing the balance between antimicrobial defense and preservation of gas exchange.
In the setting of pulmonary disease, regions of the lung may be subjected to a very broad range of oxygen concentrations. We have shown that hyperoxia may have important adverse implications for pulmonary host defense (6, 11). Because a variety of pulmonary disease states result in areas of hypoxia in the lung, we have examined the effects of hypoxia on expression of GM-CSF and other key innate immune molecules by AEC. We now report that hypoxia induces significant suppression of GM-CSF, CCL-2 and IL-6 expression in primary murine AEC, without evidence of toxicity or oxidative stress. Furthermore, exposure of mice to a hypoxic environment in vivo leads to suppression of lung GM-CSF expression. As might be anticipated, in the setting of hypoxia-induced reduction in GM-CSF expression, alveolar macrophage phagocytic activity, assessed using fluorescent beads, was also reduced, an effect that was reversed by intrapulmonary treatment with recombinant GM-CSF.
Decreased oxygen tension globally across the alveolar space may occur at altitude, during profound alveolar hypoventilation or in very severe cases of acute lung injury, among other causes. However, regional hypoxia due to localized limitation of ventilation attributable to airway obstruction or alveolar filling with fluid or inflammatory exudate is a hallmark of conditions as varied as pneumonia, COPD, asthma and the acute respiratory distress syndrome (ARDS). In the modern intensive unit, individuals with respiratory failure who are receiving supplemental oxygen (with or without mechanical ventilation) most often have extensive heterogeneity of local oxygen exposure within the lung, with a mixture of hypoxic regions and regions exposed to normal or elevated oxygen tension. Infection and local hypoxia often coexist in these disease states.
Alveolar epithelial cells appear to be generally quite tolerant of hypoxic environments, in part due to adaptations that result in both increased glycolysis and decreased ATP consumption (21, 32). Prior studies have investigated the impact of hypoxia on sodium channel and Na,K-ATPase activity, and demonstrated consequences for alveolar fluid clearance (33). However, there is less known concerning the effects of regional hypoxia on AEC host defense functions.
There is a complex interaction between inflammation and hypoxia that differs based on the organ and cell lineage (34). Individuals who develop mountain sickness at high altitude have increased levels of circulating inflammatory cytokines and develop pulmonary edema. Recent work by Pugliese et al. investigated the impact of 4–14 days exposure to hypoxic conditions on gene expression by interstitial and alveolar macrophages, using RNA sequencing and bioinformatics analysis (35). Using principal component analysis, they identified changes in the alveolar macrophage transcriptome suggesting both mitochondrial dysfunction and increased proinflammatory gene activation. Many other organs show evidence of acute inflammation when exposed to hypoxia. Conversely, in the hypoxic microenvironment within many solid tumors, macrophages and T cells have been found to be reprogrammed to suppress local immune and inflammatory responses, thereby limiting anti-tumor defenses (36, 37).
The suppression of AEC expression of GM-CSF, CCL2 and IL-6 by hypoxia, in concert with disruption in transcription of GM-CSF, suggested that the NF-kB signaling pathway might be involved in this response. We found that NF-kB is active in AEC under basal conditions. Although this might seem counterintuitive, it is of note that Ward et al. have found that pharmacologic inhibition of NF-kB activity results in loss of tight junction protein localization and impaired barrier function in primary rat AEC. Recognizing the limitations of pharmacologic approaches to NF-kB inhibition, their work suggests that constitutive activity of NF-kB is important for maintenance of tight junctions and normal alveolar epithelial barrier function (38). We found high levels of expression of phosphorylated p65 and activity of the NF-kB reporter construct in unstimulated cells, similarly suggesting that basal NF-kB activation is a feature of AEC, even in the absence of inflammation. Because alveolar macrophage recycling of pulmonary surfactant is dependent upon AEC expression of GM-CSF, which may in turn be dependent upon AEC NF-kB activation, it would be anticipated that the latter would continue in the absence of inflammation. Otherwise, pulmonary alveolar proteinosis would be common (39). The potential contribution of other transcription factors to constitutive expression of GM-CSF in these cells is the subject of ongoing investigation.
NF-kB has been found to play an important role in the response to hypoxia in other organs and cells (40–42). HIF pathways and NF-kB signaling are intimately related (34, 43). NF-kB is often required for HIF gene expression and for inflammatory mediator expression in response to hypoxia (34, 40, 44). Conversely, the hypoxic environment has been found to lead to inhibition of NF-kB signaling, possibly due to post-translational changes in NF-kB constituents that alter binding to target DNA sequences within the promoter regions of key genes (34, 41). Our data identify two points at which NF-kB signaling in AEC might be altered with effects on GM-CSF transcription. Decreased phospho-p65 might contribute to decreased transcription of a variety of NF-kB target genes (45–48). Moreover, we found that the NF-kB binding site in the GM-CSF 5’ promoter region became less accessible in the context of hypoxia. Together, these observations suggest that inhibition of NF-kB signaling in response to hypoxia is a key step in the suppression of GM-CSF expression when AEC are exposed to hypoxic conditions.
Chromatin remodeling is an energy requiring process that is influenced by availability of ATP (49, 50). Diminished AEC ATP stores during hypoxic exposure is potentially an important mechanism by which hypoxia may alter accessibility of the GM-CSF proximal promoter. In fact, pharmacologic inhibition of the electron transport chain with agents such as sodium azide or rotenone induces a similar effect on AEC GM-CSF expression (data not shown). The same region in the GM-CSF proximal promoter that contains a binding site for NF-kB includes a close by binding site for the transcription factor, AP-1. The approach we have taken to evaluating accessibility of promoter regions does not distinguish between effects on NF-kB and AP-1. However, these transcription factors are often found to work in concert. For instance, NF-kB and AP-1 sites are found within 38 bases of each other in the proximal promoter of the CCL2 gene. Induction of CCL2 expression by IL-1 was shown to depend on cooperative interaction of these two sites (51). Thus alterations in chromatin structure influencing accessibility of this region may influence GM-CSF transcription through effects on binding of more than one transcription factor.
The culture conditions used in these experiments induced a prototypical hypoxic response in AEC, with induction of HIF protein expression and of a number of HIF target genes. Although there is a wide array of HIF target genes involved in pathways influencing metabolism, apoptosis, inflammation and more, GM-CSF has not been identified as a HIF-responsive gene in large screens (52, 53). Nor does GM-CSF contain likely HIF binding sequences within the 5’ proximal promoter. However, it is likely that HIF has indirect effects that contribute to the inhibition of inflammatory pathways, including GM-CSF expression, by hypoxia. In fact, pharmacologic stabilization of HIF by DMOG results in suppression of GM-CSF expression. However, this suppression of GM-CSF expression was more modest than that induced by 24 hrs in hypoxia despite the fact that the increase in HIF protein was greater in DMOG compared to hypoxia, suggesting that additional mechanisms beyond HIF pathways are involved. Overall, it is apparent that all three mechanisms contribute to the suppression of AEC activity for pulmonary innate immunity following hypoxic exposure. Neither HIF effects, nor decreased activity of NF-kB, nor changes in promoter accessibility are sufficient to induce the extent of suppression observed. Rather, it appears that these mechanisms are in fact complementary.
An important feature of these studies is their use of primary murine AEC in the great majority of experiments. The response to hypoxia is cell-type dependent across a variety of organs. Furthermore, lung cell lines often are poorly representative of AEC in vivo or in vitro (11, 54). This discrepancy has been particularly important with respect to GM-CSF expression in AEC. Although primary AEC constitutively express GM-CSF and demonstrate suppression in the face of hyperoxia, commonly used cell lines exhibit a markedly different pattern, expressing GM-CSF in the setting of cytokine stimulation and demonstrating induction with hyperoxia (10). Of note, our in vivo experiments in hypoxia confirm that primary AEC recapitulate changes in the intact lung and emphasize the significance of short term changes in GM-CSF expression for lung macrophage function.
These studies have focused on mechanisms influencing the contribution of the alveolar epithelium to alterations in pulmonary innate immune responses during hypoxia. Clearly multiple different types of cells contribute to these responses, including alveolar macrophages. In vivo studies of hypoxic mice confirmed our findings of decreased lung GM-CSF expression and demonstrate impaired alveolar macrophage phagocytic activity. Because alveolar macrophages function in a microenvironment heavily influenced by the alveolar epithelium, we believe this approach provides the most accurate reflection of the broader effects of hypoxia. Future studies will explore the influence of hypoxia on macrophage antimicrobial activity within the alveolar setting. Furthermore, pulmonary innate immune responses are finely tuned to support antimicrobial activity while limiting tissue injury from dysregulated activity of macrophages or circulating leukocytes. Loss of expression of key molecules such as GM-CSF by AEC may result in both impaired pathogen clearance and lung injury due to poorly calibrated inflammatory responses, including increased activation of inflammatory pathways in alveolar macrophages in the absence of pathogens (35).
There are several limitations in this work to consider. We have been generally very successful in using primary cells for the conduct of studies investigating molecular pathways, despite well-known limitations in the numbers of cells available. However, in order to confirm nuclear translocation of active HIF-1α it was necessary to use a model AEC cell line (MLE-15 cells) due to the number of cells required to detect this low abundance protein. Because the result of this experiment aligns well with experiments in primary AEC showing activation of HIF-target genes in the setting of hypoxia, we are confident that the experiment directly assessing HIF-1α activity is indeed a reflection of changes in AEC. Similarly, we found luciferase activity of our lentivirally induced NF-kB reporter to be decreased in the setting of hypoxia. It has been reported that hypoxia itself can induce luciferase activity in some cell systems using reporter constructs for a variety of genes, independent of effects on the gene of interest (55). This direct effect would suggest that our finding in fact underestimates the extent of suppression of NF-kB activation by hypoxia. Our in vivo phagocytosis experiment does not assess the direct effect of hypoxia on macrophage phagocytic function outside of the alveolar environment. It is likely that there are additional contributions to altered macrophage phagocytic activity, beyond the changes induced by suppression of AEC GM-CSF expression. However, this experiment, including the reversal of this hypoxia effect by treatment with recombinant GM-CSF, clearly supports a major role for GM-CSF in the local environment determining macrophage phagocytic function during hypoxic stress in vivo.
Although we demonstrated significant impact of hypoxia on AEC expression of IL-6 and CCL2, we have focused our mechanistic studies on GM-CSF. It is likely that the effects of hypoxia on NF-kB signaling for induction of gene transcription will at least partially apply to each of these inflammatory molecules, although we have not yet investigated effects on accessibility of the proximal promoters for IL-6 and CCL2. Similarly, it is possible that hypoxia may induce additional effects on chromatin accessibility and transcription factor binding further upstream from the transcription start site for GM-CSF.
In summary, we have found that exposure of primary murine AEC to a hypoxic environment results in impaired expression of the important innate immune molecules, GM-CSF, IL-6 and CCL2. In particular, hypoxia leads to suppression of GM-CSF transcription in association with effects on accessibility of the NF-kB/AP-1 binding sites in the GM-CSF 5’ proximal promoter region, and decreased NF-kB signaling. These findings provide insights into mechanisms by which regional hypoxia found in a wide variety of pulmonary disease states might increase susceptibility to pneumonia and offer potential targets for therapeutic intervention to prevent infection or enhance antimicrobial defenses in this context.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Dr. Jeffrey Whitsett of the University of Cincinnati for the generous gift of MLE-15 immortalized alveolar epithelial cells.
This work was supported by a VA Merit Review grant from the Department of Veterans Affairs and by the Kenneth P. Burbidge Presidential Endowed Chair for Pulmonary Medicine and Lung Transplantation (both to RP) and by NHLBI training grant support (for AF).
Abbreviations:
- AEC
alveolar epithelial cell
- DMOG
(dimethyloxaloylglycine)
- HIF
hypoxia inducible factor
- VEGF
vascular endothelial cell growth factor
REFERENCES
- 1.Whitsett JA, and Alenghat T. 2015. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat Immunol 16: 27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Trapnell BC, and Whitsett JA. 2002. Gm-CSF regulates pulmonary surfactant homeostasis and alveolar macrophage-mediated innate host defense. Annual review of physiology 64: 775–802. [DOI] [PubMed] [Google Scholar]
- 3.Bozinovski S, Jones J, Beavitt SJ, Cook AD, Hamilton JA, and Anderson GP. 2004. Innate immune responses to LPS in mouse lung are suppressed and reversed by neutralization of GM-CSF via repression of TLR-4. American journal of physiology. Lung cellular and molecular physiology 286: L877–885. Epub 2003 November 2014. [DOI] [PubMed] [Google Scholar]
- 4.Paine R 3rd, Morris SB, Jin H, Wilcoxen SE, Phare SM, Moore BB, Coffey MJ, and Toews GB. 2001. Impaired functional activity of alveolar macrophages from GM-CSF-deficient mice. American journal of physiology. Lung cellular and molecular physiology 281: L1210–1218. [DOI] [PubMed] [Google Scholar]
- 5.Shibata Y, Berclaz PY, Chroneos ZC, Yoshida M, Whitsett JA, and Trapnell BC. 2001. GM-CSF Regulates Alveolar Macrophage Differentiation and Innate Immunity in the Lung through PU.1. Immunity 15: 557–567. [DOI] [PubMed] [Google Scholar]
- 6.Baleeiro CE, Christensen PJ, Morris SB, Mendez MP, Wilcoxen SE, and Paine R 3rd. 2006. GM-CSF and the impaired pulmonary innate immune response following hyperoxic stress. American journal of physiology. Lung cellular and molecular physiology 291: L1246–1255. [DOI] [PubMed] [Google Scholar]
- 7.Corti M, Brody AR, and Harrison JH. 1996. Isolation and primary culture of murine alveolar type II cells. American Journal of Respiratory Cell & Molecular Biology 14: 309–315. [DOI] [PubMed] [Google Scholar]
- 8.Mir-Kasimov M, Sturrock A, McManus M, and Paine R 3rd. 2012. Effect of alveolar epithelial cell plasticity on the regulation of GM-CSF expression. American journal of physiology. Lung cellular and molecular physiology 302: L504–511. [DOI] [PubMed] [Google Scholar]
- 9.Sturrock A, Mir-Kasimov M, Baker J, Rowley J, and Paine R 3rd. 2014. Key role of microRNA in the regulation of granulocyte macrophage colony-stimulating factor expression in murine alveolar epithelial cells during oxidative stress. The Journal of biological chemistry 289: 4095–4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sturrock A, Seedahmed E, Mir-Kasimov M, Boltax J, McManus ML, and Paine R 3rd. 2012. GM-CSF provides autocrine protection for murine alveolar epithelial cells from oxidant-induced mitochondrial injury. American journal of physiology. Lung cellular and molecular physiology 302: L343–351. [DOI] [PubMed] [Google Scholar]
- 11.Sturrock A, Vollbrecht T, Mir-Kasimov M, McManus M, Wilcoxen SE, and Paine R 3rd. 2010. Mechanisms of suppression of alveolar epithelial cell GM-CSF expression in the setting of hyperoxic stress. American journal of physiology. Lung cellular and molecular physiology 298: L446–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wikenheiser KA, Vorbroker DK, Rice WR, Clark JC, Bachurski CJ, Oie HK, and Whitsett JA. 1993. Production of immortalized distal respiratory epithelial cell lines from surfactant protein C/simian virus 40 large tumor antigen transgenic mice. Proc Natl Acad Sci U S A 90: 11029–11033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Caradec J, Sirab N, Keumeugni C, Moutereau S, Chimingqi M, Matar C, Revaud D, Bah M, Manivet P, Conti M, and Loric S. 2010. ‘Desperate house genes’: the dramatic example of hypoxia. British journal of cancer 102: 1037–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ballinger MN, Hubbard LL, McMillan TR, Toews GB, Peters-Golden M, Paine R 3rd, and Moore BB. 2008. Paradoxical role of alveolar macrophage-derived granulocyte-macrophage colony-stimulating factor in pulmonary host defense post-bone marrow transplantation. American journal of physiology. Lung cellular and molecular physiology 295: L114–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sturrock A, Baker JA, Mir-Kasimov M, and Paine R 3rd. 2015. Contrasting effects of hyperoxia on GM-CSF gene transcription in alveolar epithelial cells and T cells. Physiological reports 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holloway AF, Rao S, Chen X, and Shannon MF. 2003. Changes in chromatin accessibility across the GM-CSF promoter upon T cell activation are dependent on nuclear factor kappaB proteins. The Journal of experimental medicine 197: 413–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brettingham-Moore KH, Rao S, Juelich T, Shannon MF, and Holloway AF. 2005. GM-CSF promoter chromatin remodelling and gene transcription display distinct signal and transcription factor requirements. Nucleic Acids Res 33: 225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hochachka PW, Buck LT, Doll CJ, and Land SC. 1996. Unifying theory of hypoxia tolerance: molecular/metabolic defense and rescue mechanisms for surviving oxygen lack. Proceedings of the National Academy of Sciences of the United States of America 93: 9493–9498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wenger RH 2002. Cellular adaptation to hypoxia: O2-sensing protein hydroxylases, hypoxia-inducible transcription factors, and O2-regulated gene expression. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 16: 1151–1162. [DOI] [PubMed] [Google Scholar]
- 20.Araneda OF, and Tuesta M. 2012. Lung oxidative damage by hypoxia. Oxid Med Cell Longev 2012: 856918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clerici C, and Planes C. 2009. Gene regulation in the adaptive process to hypoxia in lung epithelial cells. American journal of physiology. Lung cellular and molecular physiology 296: L267–274. [DOI] [PubMed] [Google Scholar]
- 22.Quinton LJ, Jones MR, Robson BE, Simms BT, Whitsett JA, and Mizgerd JP. 2008. Alveolar epithelial STAT3, IL-6 family cytokines, and host defense during Escherichia coli pneumonia. Am J Respir Cell Mol Biol 38: 699–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Quinton LJ, Jones MR, Simms BT, Kogan MS, Robson BE, Skerrett SJ, and Mizgerd JP. 2007. Functions and regulation of NF-kappaB RelA during pneumococcal pneumonia. J Immunol 178: 1896–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yamamoto K, Ahyi AN, Pepper-Cunningham ZA, Ferrari JD, Wilson AA, Jones MR, Quinton LJ, and Mizgerd JP. 2014. Roles of lung epithelium in neutrophil recruitment during pneumococcal pneumonia. Am J Respir Cell Mol Biol 50: 253–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.LeVine AM, Reed JA, Kurak KE, Cianciolo E, and Whitsett JA. 1999. GM-CSF-deficient mice are susceptible to pulmonary group B streptococcal infection. The Journal of clinical investigation 103: 563–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ballinger MN, Paine R 3rd, Serezani CH, Aronoff DM, Choi ES, Standiford TJ, Toews GB, and Moore BB. 2006. Role of granulocyte macrophage colony-stimulating factor during gram-negative lung infection with Pseudomonas aeruginosa. American journal of respiratory cell and molecular biology 34: 766–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen GH, Olszewski MA, McDonald RA, Wells JC, Paine R 3rd, Huffnagle GB, and Toews GB. 2007. Role of granulocyte macrophage colony-stimulating factor in host defense against pulmonary Cryptococcus neoformans infection during murine allergic bronchopulmonary mycosis. The American journal of pathology 170: 1028–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paine R 3rd, Preston AM, Wilcoxen S, Jin H, Siu BB, Morris SB, Reed JA, Ross G, Whitsett JA, and Beck JM. 2000. Granulocyte-macrophage colony-stimulating factor in the innate immune response to Pneumocystis carinii pneumonia in mice. Journal of immunology 164: 2602–2609. [DOI] [PubMed] [Google Scholar]
- 29.Winter C, Herbold W, Maus R, Langer F, Briles DE, Paton JC, Welte T, and Maus UA. 2009. Important role for CC chemokine ligand 2-dependent lung mononuclear phagocyte recruitment to inhibit sepsis in mice infected with Streptococcus pneumoniae. Journal of immunology 182: 4931–4937. [DOI] [PubMed] [Google Scholar]
- 30.Jones MR, Quinton LJ, Simms BT, Lupa MM, Kogan MS, and Mizgerd JP. 2006. Roles of interleukin-6 in activation of STAT proteins and recruitment of neutrophils during Escherichia coli pneumonia. J Infect Dis 193: 360–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van der Poll T, Keogh CV, Guirao X, Buurman WA, Kopf M, and Lowry SF. 1997. Interleukin-6 gene-deficient mice show impaired defense against pneumococcal pneumonia. J Infect Dis 176: 439–444. [DOI] [PubMed] [Google Scholar]
- 32.Lottes RG, Newton DA, Spyropoulos DD, and Baatz JE. 2014. Alveolar type II cells maintain bioenergetic homeostasis in hypoxia through metabolic and molecular adaptation. American journal of physiology. Lung cellular and molecular physiology 306: L947–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jain M, and Sznajder JI. 2005. Effects of hypoxia on the alveolar epithelium. Proceedings of the American Thoracic Society 2: 202–205. [DOI] [PubMed] [Google Scholar]
- 34.Eltzschig HK, and Carmeliet P. 2011. Hypoxia and inflammation. The New England journal of medicine 364: 656–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pugliese SC, Kumar S, Janssen WJ, Graham BB, Frid MG, Riddle SR, El Kasmi KC, and Stenmark KR. 2017. A Time- and Compartment-Specific Activation of Lung Macrophages in Hypoxic Pulmonary Hypertension. J Immunol 198: 4802–4812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leblond MM, Gerault AN, Corroyer-Dulmont A, MacKenzie ET, Petit E, Bernaudin M, and Valable S. 2016. Hypoxia induces macrophage polarization and re-education toward an M2 phenotype in U87 and U251 glioblastoma models. Oncoimmunology 5: e1056442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Noman MZ, Hasmim M, Messai Y, Terry S, Kieda C, Janji B, and Chouaib S. 2015. Hypoxia: a key player in antitumor immune response. A Review in the Theme: Cellular Responses to Hypoxia. American journal of physiology. Cell physiology 309: C569–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ward C, Schlingmann B, Stecenko AA, Guidot DM, and Koval M. 2015. NF-kappaB inhibitors impair lung epithelial tight junctions in the absence of inflammation. Tissue Barriers 3: e982424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trapnell BC, Carey BC, Uchida K, and Suzuki T. 2009. Pulmonary alveolar proteinosis, a primary immunodeficiency of impaired GM-CSF stimulation of macrophages. Current opinion in immunology 21: 514–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fitzpatrick SF, Tambuwala MM, Bruning U, Schaible B, Scholz CC, Byrne A, O’Connor A, Gallagher WM, Lenihan CR, Garvey JF, Howell K, Fallon PG, Cummins EP, and Taylor CT. 2011. An intact canonical NF-kappaB pathway is required for inflammatory gene expression in response to hypoxia. Journal of immunology 186: 1091–1096. [DOI] [PubMed] [Google Scholar]
- 41.Loftis LL, Johanns CA, Lechner AJ, and Matuschak GM. 2000. Brief hypoxic stress suppresses postbacteremic NF-kappaB activation and TNF-alpha bioactivity in perfused liver. Am J Physiol Regul Integr Comp Physiol 279: R99–R108. [DOI] [PubMed] [Google Scholar]
- 42.Matsushita H, Morishita R, Nata T, Aoki M, Nakagami H, Taniyama Y, Yamamoto K, Higaki J, Yasufumi K, and Ogihara T. 2000. Hypoxia-induced endothelial apoptosis through nuclear factor-kappaB (NF-kappaB)-mediated bcl-2 suppression: in vivo evidence of the importance of NF-kappaB in endothelial cell regulation. Circ Res 86: 974–981. [DOI] [PubMed] [Google Scholar]
- 43.Morgan MJ, and Liu ZG. 2011. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell research 21: 103–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bruning U, Fitzpatrick SF, Frank T, Birtwistle M, Taylor CT, and Cheong A. 2012. NFkappaB and HIF display synergistic behaviour during hypoxic inflammation. Cell Mol Life Sci 69: 1319–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kelleher ZT, Matsumoto A, Stamler JS, and Marshall HE. 2007. NOS2 regulation of NF-kappaB by S-nitrosylation of p65. The Journal of biological chemistry 282: 30667–30672. [DOI] [PubMed] [Google Scholar]
- 46.Kelleher ZT, Potts EN, Brahmajothi MV, Foster MW, Auten RL, Foster WM, and Marshall HE. 2011. NOS2 regulation of LPS-induced airway inflammation via S-nitrosylation of NF-{kappa}B p65. American journal of physiology. Lung cellular and molecular physiology 301: L327–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marshall HE, and Stamler JS. 2001. Inhibition of NF-kappa B by S-nitrosylation. Biochemistry 40: 1688–1693. [DOI] [PubMed] [Google Scholar]
- 48.Ckless K, van der Vliet A, and Janssen-Heininger Y. 2007. Oxidative-nitrosative stress and post-translational protein modifications: implications to lung structure-function relations. Arginase modulates NF-kappaB activity via a nitric oxide-dependent mechanism. American journal of respiratory cell and molecular biology 36: 645–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hargreaves DC, and Crabtree GR. 2011. ATP-dependent chromatin remodeling: genetics, genomics and mechanisms. Cell research 21: 396–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vignali M, Hassan AH, Neely KE, and Workman JL. 2000. ATP-dependent chromatin-remodeling complexes. Mol Cell Biol 20: 1899–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cockerill PN 2016. Receptor Signaling Directs Global Recruitment of Pre-existing Transcription Factors to Inducible Elements. Yale J Biol Med 89: 591–596. [PMC free article] [PubMed] [Google Scholar]
- 52.Benita Y, Kikuchi H, Smith AD, Zhang MQ, Chung DC, and Xavier RJ. 2009. An integrative genomics approach identifies Hypoxia Inducible Factor-1 (HIF-1)-target genes that form the core response to hypoxia. Nucleic Acids Res 37: 4587–4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ortiz-Barahona A, Villar D, Pescador N, Amigo J, and del Peso L. 2010. Genome-wide identification of hypoxia-inducible factor binding sites and target genes by a probabilistic model integrating transcription-profiling data and in silico binding site prediction. Nucleic Acids Res 38: 2332–2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Paine R 3rd, and Simon RH. 1996. Expanding the frontiers of lung biology through the creative use of alveolar epithelial cells in culture. The American journal of physiology 270: L484–486. [DOI] [PubMed] [Google Scholar]
- 55.Doran DM, Kulkarni-Datar K, Cool DR, and Brown TL. 2011. Hypoxia activates constitutive luciferase reporter constructs. Biochimie 93: 361–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.