

Abstract
Extracellular vesicles (EVs) actively participate in intercellular communication and pathological processes. Studying the molecular signatures of EVs is the key to reveal their biological functions and clinical values, which, however, is greatly hindered by their sub-100-nm dimensions, the low quantities of biomolecules each EV carries, and the large population heterogeneity. Here, we report the single-EV Flow Cytometry Analysis technique that realizes single EV counting and phenotyping in a conventional flow cytometer for the first time, enabled by Target-Initiated Engineering (TIE) of DNA nanostructures on each EV. By illuminating multiple markers on single EVs, we reveal statistically significant differences among the molecular signatures of EVs originated from several breast cancer cell lines, and successfully recognize the cancer cell-derived EVs among the heterogeneous EV populations. Thus, our approach holds great potential for various biological and biomedical applications.
Keywords: Single Extracellular vesicle analysis, Flow cytometry analysis, Heterogeneity, Molecular signature, Engineering
Graphical Abstract
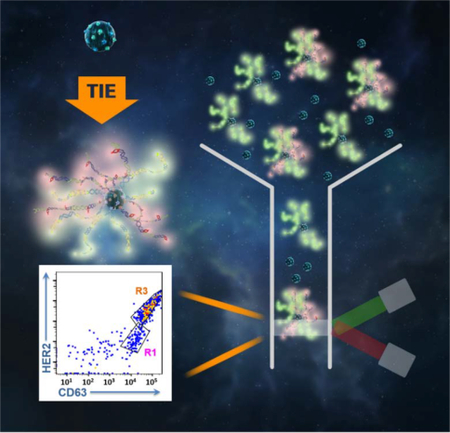
Single extracellular vesicle (EV) flow cytometry analysis enabled by target initiated engineering (TIE) of DNA nanostructures reveals unique molecular signatures of individual vesicles and differentiates EV sub-populations.
Extracellular vesicles (EVs) are membrane-encapsulated vesicles (30–100 nm) secreted by all cell types and present in various biological fluids. [1] They constitute an emerging target for liquid biopsy in cancer diagnosis because they actively participate in cellcell communication by shuttling signalling molecules between cells and modulate pathological processes like tumor initiation, progression and metastasis.[2] Study of the molecular signature of EVs is thus critical for profoundly understanding their biological functions and clinical values. [3] Existing methods for EV study are primarily focused on bulk analysis of a large number of EVs, because the small physical dimensions of EVs limit the total amounts of biomolecules to be carried in each EV, greatly enhancing the difficulty in molecular profiling. [5] However, subtle molecular differences at the single EV level may yield significant variation in EV biological functions, [6] and the highly heterogeneous nature of EV population demands the development of techniques capable of profiling individual EVs. [4] Most recently, single EV counting with a nanochip[7] and imaging single vesicles with advanced fluorescence microscopy[8] have been reported, and showed that EVs from different cell of origin can carry distinct surface markers mimicking their parent cells. Still, they require EV immobilization steps, limiting the down-stream investigations on EV functions and biogenesis.
Flow cytometry has been widely employed to distinguish different cell types in mixed populations based on the expression of cellular markers.[9] Similarly, it could be used to study the heterogeneous EVs. However, the sizes of EVs fall well below the detection limit of conventional flow cytometers, making it impossible to do single-EV analysis without significant instrumentation development.[10] Herein, we report the first single-EV Flow Cytometry Analysis (FCA) in conventional flow cytometers enabled by Target-Initiated Engineering (TIE) of DNA nanostructures on individual EVs. This technique employs conformation-switchable DNA probe to bind to the EV surface marker, which triggers the engineering of a DNA nanostructure by hybridization chain reaction (HCR).[11] The HCR products not only enlarge the overall size of the single EV to be beyond 500 nm, but also can bind to multiple fluorophores to amplify the signal from the few marker molecules locating on the limited area of EV surface, both enabling visualization of single EVs in a conventional flow cytometer, and greatly simplifying measurement of multiple markers on the same EV.
To prove the working principle, we designed the conformation-switchable probe to recognize CD63, a classic tetraspanin marker that is highly abundant in various EVs (Figure 1A). This probe contained a target recognition domain with the sequence of an anti-CD63 aptamer[12] and a trigger domain for initiation of DNA growth via HCR. These two domains were flanked by a hinge sequence to achieve conformation change triggered by aptamer-target interaction (Fig. 1A, Table S1): a part of the target recognition domain hybridized with the trigger domain (i.e. “deactivated state”) to form a hairpin structure, which could be opened upon target binding to expose the sequence (i.e. “activated state”) for hybridization with Hairpin 1 (H1). Then sequential hybridization between H1 and H2 would occur to build the long dsDNA product on EV (Fig. 1A). The design of target-initiated engineering of DNA nanostructure could effectively eliminate non-specific DNA growth without the target. In addition, the DNA nanostructures constructed upon recognition of the free, non-EV bound target would not locate on EV surface and should not reach the size range detectible by the conventional flow cytometer. Both features significantly reduce the background in FCA and render our technique ultra-high simplicity: no washing is necessary to remove the unreacted probes.
Figure 1.

A) Schematic of the Single Extracellular Vesicle Flow Cytometry Analysis technique enabled by target-initiated engineering of DNA nanostructures. B) Analysis of the long DNA products by gel electrophoresis: Lane 1 - reaction probes only; Lane 2, 3, and 4 - reaction triggered by the simple initiator, CD63 protein, and CD63+ EVs, respectively. C) TEM image of the Engineered EV (Engr. EV). D) Fluorescence microscopy image of the Engr. EV tagged with rhodamine-nanoparticles (Engr. EV-RhNPs; shown in red) or QDs-525 (Engr. EV-QDs shown in green). Circles – Engr. EV-RhNPs or Engr. EV-QDs, well separated from each other; Square – one Engr. EV-QDs located nearby another Engr. EV-RhNPs; Triangle – Engr.EV labelled with both QDs and Rh-NPs.
Successful DNA hybridization cascade initiated by CD63 or the CD63-containing EVs was verified by native agarose gel electrophoresis. We can see from the gel image (Fig. 1B) that, long DNA products were formed in the presence of CD63 protein (Lane 3), accompanied with significant consumption of H1 & H2, similar to the reaction between the hairpins and a simple initiator (SI) (Lane 2), a sequence of the trigger domain that can directly hybridize with H1 without target recognition and initiate DNA growth. With the CD63-containing EVs (referred to as “standard EVs” hereafter), most of the long DNA products were trapped inside the loading well probably by binding to the EVs (Lane 4). We employed Transmission Electron Microscopy (TEM) to confirm the size enlargement in EVs produced by TIE. As clearly illustrated in Fig. 1C, after TIE, each standard EV exhibited the “hairy” morphology on its surface, with the overall size increasing from the original tens of nanometers to hundreds of nanometers and maintaining the spherical shape (Figs. S1A&B). In contrast, the DNA hybridization products triggered by SI were in random shapes without the densely stained core (Fig. S1C). The growth effect was also viewed by Atomic Force Microscopy (AFM) (Fig. S2) and nanoparticle tracking analysis (NTA) (Fig. S1D): after TIE, DNA strands up to 250 nm long were seen surrounding the individual EVs under AFM; and the size distribution profile in NTA shifted to the larger size region with a new peak at 500 nm appeared.
We examined the enlarged EVs by a confocal fluorescence microscope. Labelled the hairpins with biotin, the long DNA products can be tagged with the streptavidin-conjugated fluorescent probes, like the Qdot™ 525 Streptavidin Conjugate (QDs, Ex 488 nm/Em 525 nm) and the streptavidin/rhodamine-conjugated iron oxide nanoparticles (RhNPs, Ex 547 nm/Em 572 nm). These two tags were used to produce two distinct EV populations in separate reaction tubes, which were mixed and inspected under microscope. Indeed, the engineered EVs labelled with either the QDs or the RhNPs were clearly discernible in the confocal mode with an objective of 40× (Fig. 1D). We even found particles that were overlapped (highlighted by a triangle), located close to each other (highlighted by a square), or completely separated (indicated by circles), proving that each fluorescent particle represented one individual vesicle. The negative controls, i.e. the QDs-labelled DNA nanostructure established by the SI, and the RhNPs or QDs by themselves, were not observable even under a higher magnification of 60× or 100× (Fig. S3), due to the few numbers of QDs on each DNA nanostructure. On contrary, each EV could carry multiple surface markers, and be labeled by multiple dsDNAs (Fig. S2) that not only enlarge the overall size, but also amplify the total fluorescence signal, making the single EV visible under the same imaging setting. This result proves that the EVs enlarged by TIE were visible by conventional optical imaging tools.
Compared to microscopic imaging, flow cytometry provides fast and automatic particle counting at the rate of thousands particles per second; and can sort pure particle populations defined by fluorescence patterns, much more ideal for interrogating single biological particles. Thus, we explored whether the engineered EVs could be detected in a conventional flow cytometer FACSCanto™ that is widely used in research and clinical settings. Analysis of the standard beads confirmed that this instrument could not see particles smaller than 500 nm. In this experiment, the DNA nanostructure carried by the engineered EVs were tagged by either Alexa Fluor® 488 Streptavidin Conjugates (i.e. Engr. EV-Alexa488) or the green QDs-525 (i.e. Engr. EV-QDs). Interestingly, both EV samples revealed a significant particle cluster in the flow cytometry plots of FSC (forward scatter) vs. SSC (side scatter) and FL1 (Fluorescence channel, λex = 488 nm) vs. SSC (Fig. 2A). These particles exhibited larger FSC and higher fluorescence (FL) than the background particles, proving the significant EV size enlargement induced by the CD63-initiated EV engineering; and the signal amplification from the multiple fluorescent tags bound to the long DNA chains on each EV. Gating the particle cluster by R1 (for the QD label) or R2 (for the Alexa488 label) illustrated that the enlarged EVs represented about 50% of the total particles detected for each sample (Table S2). In contrast, no distinct particle cluster was detected on the flow plots obtained from the negative control samples, including the EVs labelled by the anti-CD63-conjugated QDs without any size enlargement (i.e. EV-QDs) (Fig. 2A), and the EVs mixed with just the hairpins and QDs (Fig. S4). The background particles present in Fig. 2A and Fig. S4 with low forward scatter and fluorescence signals might be produced from non-specific adsorption of the hairpins and QDs on the EVs, as well as random aggregation of QDs in solutions. Furthermore, TIE on free CD63 did not produce observable particles, ensuring the counting was from intact EVs instead of free markers released by cells or from vesicle breakage.
Figure 2.

A) Representative scatter plots of flow cytometry analysis of the EVs before and after TIE. Top to bottom are: Standard EVs directly labelled with QDs (EV + QDs); Engineered EVs labelled with Alexa488 or QDs-525. The particle populations shown in green and purple on the light scatter plots of FSC vs. SSC were those included in R2 and R1, respectively, on the flow plots of FL1 vs. SSC. B) Histograms for the signals of FSC, SSC, and fluorescence produced by the engineered EVs labelled with Alexa488 (green) and QDs-525 (purple). All samples started with ~ 109 EV particles.
Using R1 and R2, we compared the scatter and FL signals obtained with Alexa488 and QDs (Fig. 2B). Both labels gave out comparable FSC readings (the mean FSC intensity ratio of QD/Alexa = 0.92 ± 0.07 (n = 3)), indicating the size enlargement effect was mainly resulted from the growth of the dsDNA, not the additional size of the fluorescent tag. Labelling with QDs resulted in 70.5 ± 6.2 (n = 3) times higher FL than with Alexa488. The higher FL shifted the enlarged EVs further away from the background particles, making the population more distinct – a critical characteristic for future sorting of specific EV populations. This is the first time a new population of detectible EVs appearing on the flow cytometric plots to enable clear and unambiguous recognition of pure EV population in a conventional flow cytometer, owing to TIE-enabled transformation of the “invisible” EVs to the “visible” particles.
The kinetics of TIE was studied by changing the HCR time from 0 hrs to 24 hrs. A gate was applied on the flow plots to identify the particle cluster displaying higher FL and larger FSC than background, which were considered as the “detectible events”. We found that, a reaction time of 2 hrs produced only very few numbers of the detectible events, which dramatically increased at 4 and 8 hrs (Fig. S5). NTA measurement also confirmed that with the reaction time increasing to 4 and 8 hrs, more engineered EVs larger than 250 nm were produced (Fig. S1D). The “mean fluorescence intensity (MFI)” gradually increased with the reaction time going from 0 to 8 hrs (Fig. S5&S6). But extending the reaction to 12 hrs did not induce significant changes in the number of “detectible events” and the MFI value detected in the flow cytometer, neither big change was observed in the size distribution profile obtained with NTA. This result indicates that, recognition of the CD63-positive EVs may have reached a steady state and no new engineered EVs could be produced with longer reaction time. Still, the length of the DNA product continued to increase with longer reaction duration, which generated the engineered EVs larger than 500 nm at 24 hrs observed by NTA (Fig. S1D). However, space hindrance and winding of the long DNA strand may have prevented more QDs from binding to the DNA; and after being kept at 37 °C for 24 hrs, the EVs may no longer be stable, both leading to the decrease in MFI and “detectible events” measured by flow cytometry (Fig. S6). While detailed study of the kinetics of engineering DNA nanostructures on EVs needs to be conducted in future works, the present work employed overnight reaction to obtain stable signals in flow cytometric analysis of single EVs.
Under the optimized TIE conditions, we confirmed that the number of detectible events was linearly (R2 = 0.9974) proportional to the number of EVs in the sample within the range of 20 μg/mL to 500 μg/mL (Fig. S7), proving the capability of our method in EV quantification. Furthermore, the robustness of our method was verified by the intraday and interday replicates (Table S3). The Student’s t test obtained a p value smaller than 10E-4 when evaluating the MFI or mean forward scatter (MFS) among all these replicates, proving that EV engineering is highly reproducible. More importantly, we showed that our method can directly engineer the EVs in biofluids like cell culture medium (Fig. S8) and serum (Fig. S9) by simply treating the matrix with sodium citrate to inhibit exonuclease activity.[13] Direct EV detection in biological samples with minimum sample pretreatment and without EV enrichment or immobilization is a big advancement compared to existing technologies and makes our method highly suitable for studying the heterogeneity of EV populations and examining their clinical values.
Differentiation of heterogeneous EVs requires recognition of multiple markers. The TIE system can be easily adjusted to target different surface biomarkers by simply switching the aptamer sequence in the conformation-switchable DNA probe. To demonstrate this, we designed another TIE system to target human epidermal growth factor receptor 2 (HER2), a typical breast cancer marker overexpressed in ~20% of breast cancer patients and present on a subset of EVs derived from breast cancer cell lines.[14] We substituted the anti-CD63 aptamer with an anti-HER2 aptamer[15] in the target-recognition domain, and kept the trigger domain intact. The hinge domain was adjusted slightly to achieve effective conformational switching upon HER2 recognition (Table S1). Since the HCR triggered by HER2 remains the same as that by CD63, we define it as the “single hybridization cascade system”, and applied it to analyze HER2 and CD63 expression separately on single EVs secreted by three different cell lines: the breast cancer cells of SKBR3 (highly metastatic) and MCF-7 (poorly metastatic) and the non-tumour epithelial cell MCF-10A. We found that, the anti-HER2 system produced more detectable events from the SKBR3-derived EVs than those from MCF-7 and MCF-10A (Fig. S11a). In addition, 2 times more events were detected by the anti-HER2 system on the SKBR3 EVs than by the anti-CD63 system. On the other hand, the EVs originated from MCF-10A and MCF-7 resulted in comparable numbers of detectable events for detection of CD63 or HER2. These results agree well with the bulk analysis by ELISA (Fig. S10), which confirmed the overexpression of HER2 in SKBR3 cells as reported in literature,[16] and found 2 times more HER2 than CD63 in the SKBR3-derived EVs, but comparable amounts of HER2 and CD63 in the EVs from MCF-7 and MCF-10A cells.
Interestingly, using the MFI and MFS values obtained from both markers as the four variables to conduct principal component analysis (PCA), the EVs from each cell line can be clearly differentiated by their cells of origin (Fig. S11b), hinting that, the size and FL increase resulted from TIE can help to differentiate EV sub-populations. If the signals from both markers can be acquired simultaneously, the flow plots should directly show the presence of EV sub-populations in a heterogeneous EV mixture without the help of statistical tools, which is necessary for single vesicle analysis using microscopic methods. [8] Herein, we designed a “dual hybridization cascade system” to simultaneously amplify signals from HER2 and CD63 on a single EV by two separate hybridization cascade reactions (Table S1). In this system, CD63 signal was still derived from the streptavidin-conjugated QDs, but HER2 signal was from the Alexa660 labelled DNA tag hybridized with the overhang in H3 on the HCR product (Fig. 3B and Table S1). The fluorescence of QDs and Alexa660 were detected in the FACSCanto’s fluorescence channel of FL1 and FL4, respectively. As expected, the detectible events from the EVs produced by SKBR3, MCF-7 and MCF-10A located at distinct positions on the fluorescence flow cytometric plots, each EV sub-population exhibiting characteristic distribution patterns (Fig. 3A and Fig. S12). Viewing CD63 as the internal standard, we calculated the MFI ratio of FL4/FL1 to represent the relative content of HER2 and CD63 on the same EV. Significant difference in the MFI ratios between EVs from the MCF-10A and SKBR3 cells (p = 0.009), or between those from the MCF-7 and SKBR3 cells (p = 0.02) was confirmed with the Student’s t test (Fig. 3C), proving the effectiveness of dual labelling on the same EV for robust differentiation of the SKBR3-derived EVs from those produced by the MCF cells. The EVs from MCF-7 and MCF-10A were not differentiable using these two markers, because the expression levels of both markers were comparable in these two EVs (Fig. S10).
Figure 3.

A) Representative flow cytometry plots of the particle cluster determined by the relative expression levels of HER2 and CD63 for the EVs from different cell lines. B) Scheme of the dual hybridization cascade system for recognition of two markers on the same EV. C) Box chart of the mean fluorescence intensity (MFI) ratio between FL4 and FL1 of the EVs from three cell lines. *p < 0.05, and **p < 0.01, n = 5. IQR - interquartile range. D) Flow cytometry scatter plots and fluorescence histograms for analysis of EV mixtures. R1 and R3 are defined in Fig. 3A using the EVs from the corresponding cell lines. The particle clusters were coloured on the scatter plot of FSC vs. SSC and the histograms based on the gates defined in the HER2 vs. CD63 plots.
The significant difference between the MFI values from different EV sub- population indicates that our technique has the power to reveal the presence of EV sub-populations among heterogeneous mixtures. To confirm this, we tested the mixture of EVs from MCF10A and SKBR3 prepared at a 1:1 ratio. Using the distribution patterns identified with the pure EV populations, we could clearly see the presence of two EV populations in the mixture (Fig. 3D). The particle count ratio for those locating within R1 and R3 was equal to 0.98 ± 0.07 (n = 3), agreeing with the mixing ratio of the two EV sub-populations. In contrast, bulk analysis of the same EV mixture using ELISA only reflected a slight increase in the HER2 content, but failed to recognize the presence of two distinct EV sub-populations (Fig. S10). More strikingly, varying the mixing ratios of these two EV sub-populations, the ratio of the particle counts in the gated regions of R1 and R3 showed a strongly linear relationship (R2 = 0.9581) with the ratio of the added EV numbers between two cell lines (Fig. S13). These results demonstrate the great potential of our method in recognizing EV heterogeneity and differentiating EV sub-populations.
In summary, we have developed a single-EV FCA technique to visualize individual EV in a conventional flow cytometer for the first time, which can help gain in-depth insights into the molecular signatures of EV sub-populations under regular lab settings. While our work demonstrates simultaneous recognition of dual surface markers on the same vesicle, more markers can be targeted by simply revising the conformation-switchable probes to improve more effective differentiation of more EV sub-populations. Furthermore, our technique opens the opportunity for EV sorting and collection based on surface marker profiles under typical clinical lab settings. The pure EV sub-populations obtained will definitely benefit clear correlation between EV composition and their biogenesis and functions. Overall, we believe this single-EV FCA technique is a valuable tool for gaining more understanding on the roles of EVs in cell-cell communication and pathological development, and its high simplicity and good adaptivity to clinical labs will be highly beneficial for screening for EV markers for liquid biopsy applications.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grants R01- CA188991 to W. Z., R01 CA218140 and R01 CA206911 to S.E.W.. G. B. A. was supported by the Research Training Grant in Environmental Toxicology from the National Institute of Environmental Health Sciences (T32ES018827); and Q. S. J. was supported by the Environmental Toxicology Program fund.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201xxxxxx.
References
- [1].(a) Antonyak MA, Cerione RA, Proc. Natl. Acad. Sci. USA 2015, 112, 3589–3590. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Brown L, Wolf JM, Prados-Rosales R, Casadevall A, Nat. Rev. Microbiol. 2015, 13, 620–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].(a) Sullivan LB, Nat. Chem. Bio. 2017, 13, 924–925. [DOI] [PubMed] [Google Scholar]; (b) Thompson AG, Gray E, Heman-Ackah SM, Mäger I, Talbot K, Andaloussi SE, Wood MJ, Turner MR, Nat. Rev. Neurol. 2016, 12, 346–357. [DOI] [PubMed] [Google Scholar]
- [3].(a) Ruivo CF, Adem B, Silva M, Melo SA, Cancer Res. 2017, 77, 6480–6488. [DOI] [PubMed] [Google Scholar]; (b) Han L, Xu J, Xu Q, Zhang B, Lam EW, Sun Y, Med. Res. Rev. 2017, 37, 1318–1349. [DOI] [PubMed] [Google Scholar]
- [4].Tkach M, Thery C, Cell 2016, 164, 1226–1232. [DOI] [PubMed] [Google Scholar]
- [5].(a) Jiang Y, Shi M, Liu Y, Wan S, Cui C, Zhang L, Tan W, Angew. Chem. Int. Ed. 2017, 56, 11916–11920. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Greening DW, Xu R, Ji H, Tauro BJ, Simpson RJ, Methods Mol. Biol. 2015, 1295, 179–209. [DOI] [PubMed] [Google Scholar]; (c) Pocsfalvi G, Stanly C, Fiume I, Vekey K, Chromatogr JA 2016, 1439, 26–41. [DOI] [PubMed] [Google Scholar]; (d) Zhao Z, Yang Y, Zeng Y, He M, Lab Chip 2016, 16, 489–496. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Rider MA, Hurwitz SN, Meckes DG, Sci. Rep. 2016, 6, 23978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Leslie M, Science 2011. 331, 24–26. [DOI] [PubMed] [Google Scholar]
- [7].(a) Friedrich R, Block S, Alizadehheidari M, Heider S, Fritzsche J, Esbjörner EK, Westerlund F, Bally M, Lab Chip 2017, 17, 830–841. [DOI] [PubMed] [Google Scholar]; (b) Liang K, Liu F, Fan J, Sun D, Liu C, Lyon CJ, Bernard DW, Li Y, Yokoi K, Katz MH, Koay EJ, Zhao Z, Hu Y, Nat. Biomed. Eng. 2017, 1, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lee K, Fraser K, Ghaddar B, Yang K, Kim E, Balaj L, Chiocca EA, Breakefield XO, Lee H, Weissleder R, ACS Nano 2018, 12, 494–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Brown M, Wittwer C, Clin. Chem. 2000, 46, 1221–1229. [PubMed] [Google Scholar]
- [10].Tian Y, Ma L, Gong M, Su G, Zhu S, Zhang W, Wang S, Li Z, Chen C, Li L, Wu L, Yan X, ACS Nano 2018, 12, 671–680. [DOI] [PubMed] [Google Scholar]
- [11].(a) Dirks RM, Pierce NA, Proc. Natl. Acad. Sci. USA 2004, 101, 15275–15278. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cangialosi A, Yoon C, Liu J, Huang Q, Guo J, Nguyen TD, Gracias DH, Schulman R, Science 2017, 357, 1126–1130. [DOI] [PubMed] [Google Scholar]
- [12].Zhou Q, Rahimian A, Son K, Shin DS, Patel T, Revzin A, Methods 2016, 97, 88–93. [DOI] [PubMed] [Google Scholar]
- [13].Kolarevic A, Yanchev D, Kocic G, Smelcerovic A, Eur. J. Med. Chem. 2014, 88, 101–111. [DOI] [PubMed] [Google Scholar]
- [14].Wang SE, Lin R-J, MicroRNA 2013, 2, 137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].(a) Mahlknecht G, Maron R, Mancini M, Schechter B, Sela M, Yarden Y, Proc. Natl. Acad. Sci. USA 2013, 110, 8170–8175. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Squadrito ML, Cianciaruso C, Hansen SK, De Palma M, Nat. Methods 2018, 15, 183–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Subik K, Lee JF, Baxter L, Strzepek T, Costello D, Crowley P, Xing L, Hung MC, Bonfiglio T, Hicks DG, Tang P, Breast Cancer: Basic and Clinical Research 2010, 4, 35–41. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.