

Abstract
Effective humoral immunity requires class switch recombination (CSR) catalyzed by activation-induced deaminase (AID). In response to T cell-dependent (TD) antigens, CSR can be induced by CD40 signaling in B cells. Tumor necrosis factor receptor associated factors 2 and 3 (TRAF2/TRAF3) function as adaptors of the CD40 signaling pathway. B cell-intrinsic TRAF2 or TRAF3 knockout mice (B-TRAF2 or B-TRAF3 KO) were previously reported to have indistinguishable phenotypes in gene expression, B cell survival and development as well as enlarged peripheral lymphoid organs. However, it remains unknown whether deficiency of B-TRAF2 or B-TRAF3 differentially affects TD humoral immune responses and CD40-induced CSR. Here, we show that B-TRAF2 is essential for optimal isotype-switching induced by in vivo TD antigen immunization or by engaging CD40 in vitro. Our data clarify the controversial role of B-TRAF3 and confirm its dispensability in CD40-induced CSR. Mechanistically, CD40-induced AID expression was markedly impaired by B-TRAF2 but not B-TRAF3 deficiency. Moreover, B-TRAF2 deficiency causes defective activation of the NF-κB1 complex in a CD40-autonomous manner, and restoring CD40-induced NF-κB1 activation in TRAF2-deficient B cells rescues AID expression and CSR. We conclude that TRAF2 is essential but TRAF3 is dispensable for TD humoral immunity and CD40-induced CSR. Our studies provide significant biological bases for optimizing treatment of B cell-associated immune disorders by targeting CD40 signaling.
Keywords: class switch recombination, T cell-dependent antibody responses, activation induced deaminase, TRAF2, TRAF3
Introduction
The major function of B cells is to generate different isotypes of Abs to execute humoral immune responses. Upon activation, B cells switch from expressing or secreting IgM to IgA, IgE or IgG, which display diversified effector functions (1). Ab isotype-switching is achieved by DNA recombination events at the Igh locus, termed class switch recombination (CSR) (1, 2). CSR is initiated by activation-induced cytidine deaminase (AID) (3), and completed by non-homologous end-joining (NHEJ) repair (4, 5). Another prerequisite of CSR is the germline transcription (GLT) of the Igh constant (C) region, which allows AID to access the switch region of the Igh locus and to deaminate cytidine into uracil (6). Subsequently, AID-initiated DNA lesions are converted into DNA double-stranded breaks (DSBs) (7) that are rejoined by the NHEJ pathway (4, 5, 8, 9), leading to the replacement of the Cμ region with a downstream Cγ, Cα or Cε region and the subsequent production of IgG, IgA or IgE Abs, respectively. DNA repair proteins are constitutively expressed and recruited to the Igh locus by AID-initiated DNA lesions. AID expression and Igh GLT are induced only upon B cell activation by BCR, CD40, TLR or other receptors in the presence of proper cytokine milieu (10, 11). Therefore, signals emanating from various receptors to induce CSR are convergent at regulation of AID expression and Igh GLT.
CD40 signaling in B cells plays a pivotal role in CSR induced by immunization with T cell-dependent (TD) antigens (12, 13). CD40 on B cells interacts with CD40 ligand (CD40L) on helper T cells, which induces CD40 clustering, leading to recruitment of signaling molecules (14, 15). Among them are tumor necrosis factor receptor (TNFR) associated factor 2 and 3 (TRAF2 and TRAF3). In resting B cells, TRAF3 associates with NF-κB inducing kinase (NIK), while TRAF2 associates with cellular inhibitors of apoptosis protein 1 and 2 (cIAP1/2). Within this cytoplasmic complex, TRAF2 and TRAF3 heteromeric interaction allows cIAP1/2 to induce NIK degradation. In the membrane CD40 signalosome, TRAF2 and TRAF3 interact with CD40, which leads to degradation of TRAF3 (16, 17), thereby releasing NIK. NIK in turn phosphorylates inhibitory κB (IκB) kinase-α (IKKα). Activated IKKα phosphorylates NF-κB2 p100 and triggers p100 proteolytic cleavage into p52. Engaging CD40 also activates transforming growth factor activated kinase 1 (TAK1) (18–21). TAK1 phosphorylates IKKβ to promote IKKα/β/γ complex assembly, which phosphorylates IκBα and triggers ubiquitination-mediated IκBα degradation. Removal of IκBα promotes the cleavage of NF-κB1 precursor p105 into active p50. Both NF-κB1 and NF-κB2 are implicated in AID expression (10, 22–25), and deletion of either one dramatically impairs TD humoral immune responses (26, 27). CD40 may also employ TRAF3 to regulate AKT activation (28), which inhibits AID expression and CSR by targeting forkhead box protein O1 (FOXO1) for degradation (29, 30). However, it remains incompletely understood how TRAF2 and TRAF3 regulate CD40-induced AID expression.
Germline TRAF2 or TRAF3 deletion causes perinatal lethality (31–33). TRAF3 KO fetal liver cell-reconstituted chimeric mice exhibited defective Ab isotype-switching in response to TD Ag immunization (32). Mice carrying both germline deletion of TRAF2 and TNFRI survived and exhibited impaired isotype-switching against virus infection (31). Since TRAF2 and TRAF3 are ubiquitously expressed in T and B cells as well as APCs (34–36), these prior studies, despite suggesting an important role of TRAF2 and TRAF3 in Ab isotype-switching, could not define the role of B cell-intrinsic TRAF2 or TRAF3 (B-TRAF2 or B-TRAF3) in isotype-switching. TRAF2 deletion in the CH12 B cell line impaired CD40-induced IgM secretion (16); however, this study did not address whether and how TRAF2 deletion affects CSR. WT or mutant CD40 lacking the ability to bind TRAF2 or TRAF3 or both were employed to restore CD40 expression in CD40−/− B cells (37). This study reported that both B-TRAF2 and B-TRAF3 are required for CD40-induced Ab isotype-switching and the two proteins independently signal to induce CSR upon CD40 engagement (37). In contrast, B-TRAF3 KO mice showed no defects in isotype-switching in response to TD Ag immunization (38); moreover, engaging CD40 in vitro induced a higher level of secreted IgG1 in TRAF3-KO B cells than WT ones (39). These studies suggest that B-TRAF3 is dispensable for CD40-induced isotype-switching. Thus, the role of B-TRAF3 in TD humoral immune responses and CD40-induced CSR remains controversial. Despite of a number of previous studies investigating the role of TRAF2 in immune responses, CD40 signaling, B cell development and survival as well as lymph organ homeostasis (16, 31, 37, 40, 41), it remains unknown whether and how TRAF2 deletion in B cells affects CD40-induced CSR and TD humoral immune responses.
In the current study, we demonstrate that B-TRAF2 KO mice have impaired TD humoral immune responses and B-TRAF2 is an essential signaling element for the CD40 pathway to activate the NF-κB1 complex, and to induce AID expression and CSR. More importantly, we show that restoring CD40-induced NF-κB1 activation can rescue defective Ab isotype-switching in TRAF2-KO B cells. Lastly, we show that B-TRAF3 is dispensable for TD humoral immune responses and CD40-induced CSR, thereby settling the long-standing controversies.
Materials and methods
Mice, in vivo Ag immunization and ELISA
TRAF2flox/flox (40) and TRAF3flox/flox (38) mice were generated previously. To achieve B cell-specific deletion of traf2 and/or traf3 genes, TRAF2flox/flox and/or TRAF3flox/flox mice were bred with CD19Cre transgenic mice (38). Littermate controls (LMC) were TRAF2flox/flox, TRAF3flox/flox, or TRAF2flox/flox/TRAF3flox/flox. For some of biochemical assays, WT B6 mice were also used and purchased from Jackson Laboratory. In general, 8 to 12 week-old mice were used for all experiments. For in vivo immunization experiments, some of the mice used were up to 14 weeks old. Animal work was approved by the Institutional Animal Care and Use Committee of the University of Colorado Anschutz Medical Campus (Aurora, CO).
For TD Ab responses, mice were immunized i.p. with 100μg of TNP(40)-BGG (Biosource Technologies) precipitated in alum on day0 and boosted on day14. Sera were collected 28 days after the first immunization. Serum titers of anti-TNP-BGG IgG1, IgG2a, and IgE were measured by ELISA. TNP-BGG was dissolved in a carbonate buffer (pH9.5) at 20μg/ml and coated on 96-well plates (Thermo Scientific) at 50μl/well for at least 12 hours at 4˚C. Coated plates were blocked with 200μl/well blocking buffer (2% BSA in PBS) for 2 hours at room temperature (RT). Sera were diluted with blocking buffer at 1:400, 1:800, 1:1600, and 1:3200 for IgG1 and IgG2a, or at 1:100; 1:200, 1:400, and 1:800 for IgE ELISA. Diluted samples, in duplicate, were loaded onto plates and incubated at RT for 2 hours. After serum incubation, plates were washed with PBST. TNP-BGG-specific IgG1, IgG2a, and IgE were detected by HRP-conjugated goat anti-mouse IgG1 (1:3000), IgG2a (1:7000), and IgE (1:2500) secondary Abs, respectively. After a 2 hour incubation at RT, plates were washed four times with tap water, and HRP substrate (1-Step Ultra TMB-ELISA, Thermo Scientific) was added. Plates were incubated for 5–60 minutes depending on the Ab isotype to allow color development. The HRP substrate reaction was stopped with 2M H2SO4. The OD value was read by Nanoquant infinite M200 at wavelength 450nm. All plates were normalized to a reference serum sample diluted in 7 serials and to blank controls. Standard curves were calculated by Graphpad Prism using the OD values of 7 serial dilutions of the reference sample. OD values for each sample were averaged between duplicates and the value from one concentration (e.g., 1:400 to fit within the linear range of the standard curve) was employed to interpolate the relative titers based on the standard curves and dilution factors. For TI Ab responses, mice were immunized i.p. with 100μg of TNP(40)-FICOLL (Biosource Technologies) precipitated in alum. Sera were collected 14 days after immunization. Serum titers of TNP-specific IgG1, IgG2a, and IgE were measured by ELISA. To analyze in vivo isotype-switched B cells, splenocytes from immunized mice were isolated and analyzed by flow cytometry using Abs against B220, CD19, IgM, and IgG1 on a BD LSRFortessa.
Antibodies and chemicals
Details of the Abs used are included in Supplemental Table I. Abs were obtained from the following companies: Cell Signaling Technology (Beverly, MA), Santa Cruz Biotechnology (Santa Cruz, CA), Jackson Immunoresearch Laboratory (West Grove, PA), Southern Biotech (Birmingham, AL), abcam (Cambridge, MA), Biolegend (San Diego, CA), and BD Biosciences (San Jose, CA). PMA was purchased from LC laboratories (Woburn, MA). 5Z-7-Oxozeaenol and LPS (E.coli 0111: B4) were purchased from Sigma (St. Louis, MO). All chemicals were dissolved in H2O or DMSO. DMSO was titrated to determine its effect on B cell function including B cell proliferation, CSR, AID expression and others. When DMSO was diluted more than 1:1000, it had no effects on B cell function. Because all chemicals were used at 1:2000 to 1:100,000 dilution from stocks, vehicle controls were not included in these assays.
Cell culture and flow cytometry
Naïve splenic B cells were isolated with mouse B cell isolation EASY kit (StemCell) as described previously (29, 42). FO and MZ B cells were purified with MACS isolation kit on auto MACS Pro Separator (Milttenyi Biotec Inc, Auburn, CA). Purified B cells (0.5×106/ml, 3 ml/well in 6-well plate) were stimulated in vitro with anti-CD40 (1μg/ml, Biolegend, clone HM40–3) plus IL-4 (10ng/ml, GenScript, Piscataway, NJ), or LPS (2μg/ml) plus IL-4 (10ng/ml) in 10% FBS RPMI lymphocyte medium for indicated days in a 5% CO2 incubator. Activated B cells were examined by flow cytometry to detect the percentage of IgG1+ or IgE+ isotype-switched B cells. Methods for intracellular IgE staining were described in details in our previous publication (29). Briefly, single cell suspension was treated for 1 min with acid buffer (0.085M NaCl, 0.005M KCl, 0.01M EDTA, and 0.05M NaAcetate [pH 4]) to remove cytophilic IgE (defined as extrinsic, CD23-bound) (43, 44), then the samples were neutralized with 3 ml of cell culture medium and washed with 2% FBS 1×PBS buffer sequentially. Cells were fixed, permeabilized and stained with BD Fixation/Permeabilization Solution Kit according to manufacturer’s instructions (BD Biosciences, San Jose, CA), using APC-anti-mouse IgG1 and PE-anti-mouse IgE. Proliferation assays were performed with the CellTrace™ CFSE Cell Proliferation Kit for flow cytometry (Thermo Fisher Scientific) according to the manufacturer’s instructions. Flow cytometry was performed on BD LSRII, BD LSRFortessa, or FACSCalibur (BD Biosciences). Data were analyzed with Flow-Jo software.
Biochemical assays and Western blotting
For AID expression, purified B cells were treated by inhibitors or untreated, and stimulated with anti-CD40/IL-4 for 3 days. Cells were then harvested and lysed with lysis buffer (50mM Tris-base pH7.5, 150mM NaCl, 2mM EDTA, 2mM Na3O4V, 4mM NaF, 1% Triton-X100, 0.1% SDS, 0.5% Sodium deoxycholate) for 30 minutes on ice. Lysates were centrifuged at 12000 RPM for 10 minutes at 4˚C. Supernatants were collected for subsequent analysis. For signaling studies, freshly purified B cells were aliquoted into each tube (10×106 cells in 0.5 ml RPMI1640), treated with inhibitors or activators, then stimulated with 5μg anti-CD40 or 10μg LPS for indicated times in a 5% CO2 incubator (37°C). After stimulation, cells were immediately cooled down with cold PBS on ice. Cell lysates were prepared as above. Protein concentrations were determined with a BCA protein assay kit (Thermo scientific). 20μg of protein per sample was separated on SDS-PAGE (Bio-Rad, Hercules, CA) and transferred onto nitrocellulose membranes (Thermo scientific). Membranes were blocked and probed with specific Abs followed by HRP-conjugated anti-mouse or anti-rabbit secondary Abs, respectively. Protein bands were read with ECL (Thermo scientific) on a G:Box Chemi-XX6 platform (Syngene, Frederick, MD).
Quantitative PCR for Cγ1 and Cε GLTs and AID transcripts
Purified B cells were stimulated as above for 2 days. Total RNA was isolated with TriPure (Roche, Indianapolis, IN) and quantified. The same amount of RNA of each sample was used for reverse transcription (RT) reactions according to manufacturer’s instructions (Promega, Madison, WI). The primers are as follows: β-actin-For: 5’-TGGAATCCTGTGGCATCCATGAAAC-3’; β-actin-Rev: 5’-TAAAACGCAGCTCAGTAACAGTCCG-3’; Cγ1 GLT (Iγ1)-For: 5’-GGCCCTTCCAGATCTTTGAG-3’. Cγ1 GLT (Cγ1 exon1)-Rev: 5’-CAGGGTCACCATGGAGTTAGTT-3’. Cε GLT (Iε)-For: 5’-GCAGAAGATGGCTTCGAATAAGAACAGT-3’; Cε GLT (Cε exon1)-Rev: 5’-TCGTTGAATGATGGAGGATGTGTCACGT-3’. AID-For: 5’-CGCGCCTCTACTTCTGTGAA-3’; AID-Rev: 5’-GTCTGGTTAGCCGGACAGAA-3’. qPCR reagent: absolute qPCR SYBR green ROX mix (Thermo Scientific). qPCR was performed on QuanStudio 7 Flex Real-Time PCR system (Applied Biosystem), with the type of experiment set-up as Comparative CT (ΔΔCT). PCR conditions were as follows (default setting): 1) hold stage: 1.6°C/s, 50°C 02:00, 1.6°C/s, 95°C 10:00; 2) PCR (40 cycles) stage: 1.6°C/s, 95°C 00:15, 1.6°C/s, 60°C 01:00; 3) melt curve stage: 1.6°C/s, 95°C 00:15, 1.6°C/s, 60°C 01:00, 0.05°C/s, 95°C 00:15 (dissociation). Dissociation curves were evaluated after each reaction to ensure the specificity of the amplification. The arbitrary unit of the relative transcripts was calculated as follows: ΔCT=CT(AID) or CT(Cε) or C(Cγ1) − CT(β-actin), respectively. All ΔCT were compared with the reference ΔCT (obtained from TRAF2-KO B cells stimulated by anti-CD40/IL-4) to acquire ΔΔCT. Arbitrary unit=2^(−ΔΔCT).
Results
B cell-intrinsic TRAF2 is essential but TRAF3 is dispensable for TD humoral immune responses
Previous studies showed that B-TRAF2 and B-TRAF3 KO mice shared similar phenotypes in gene expression, B cell survival and development as well as enlarged peripheral lymph organs (38, 40, 41). While B-TRAF3 deletion did not impair TD humoral immune responses, it remains unknown whether B-TRAF2 deletion affects TD humoral immune responses. To address this question, B-TRAF2-KO (CD19Cre+/TRAF2fl/fl), B-TRAF3-KO (CD19Cre+/TRAF3fl/fl) and correspondent littermate control (LMC, TRAF2fl/fl or TRAF3fl/fl, respectively) mice were immunized with the TD Ag, 2,4,6-trinitrophenol-conjugated bovine γ-globulin (TNP-BGG). Splenocytes were isolated from immunized mice and analyzed by flow cytometry. We found that the percentage of switched IgG1+ B cells (gated on B220+CD19+) was significantly reduced in B-TRAF2-KO mice compared with LMC mice (Figure 1A, B). In contrast, the percentage of IgG1+ B cells was not significantly different between B-TRAF3-KO and LMC mice (Figure 1A, B). We measured the serum level of TNP-BGG-specific isotype-switched Abs by ELISA. Consistent with FACS data, B-TRAF2-KO mice produced significantly less TNP-BGG specific IgG1, IgG2a and IgE Abs than LMC mice (Figure 1C). However, the level of TNP-BGG specific IgG1, IgG2a, or IgE Abs was not significantly altered between B-TRAF3-KO and LMC mice (Figure 1C), consistent with a previous report (38). Taken together, our data demonstrate that B cell-intrinsic TRAF2 is required for TD Ag-induced humoral immune responses, whereas TRAF3 is dispensable. Intriguingly, when immunized with a T cell-independent (TI) Ag, TNP-FICOLL, both B-TRAF2-KO and B-TRAF3-KO mice produced a higher level of isotype-switched Abs than LMC mice (Supplemental Figure 1A). TD Ag-induced isotype-switching depends on CD40, while TI Ag-induced one does not (45, 46). Hence, our data suggest that TRAF2 deletion in B cells specifically impairs CD40 autonomous signaling to reduce isotype-switching.
Figure 1. Effects of B cell-intrinsic TRAF2 or TRAF3 deficiency on TD Ag-induced Ab isotype-switching.
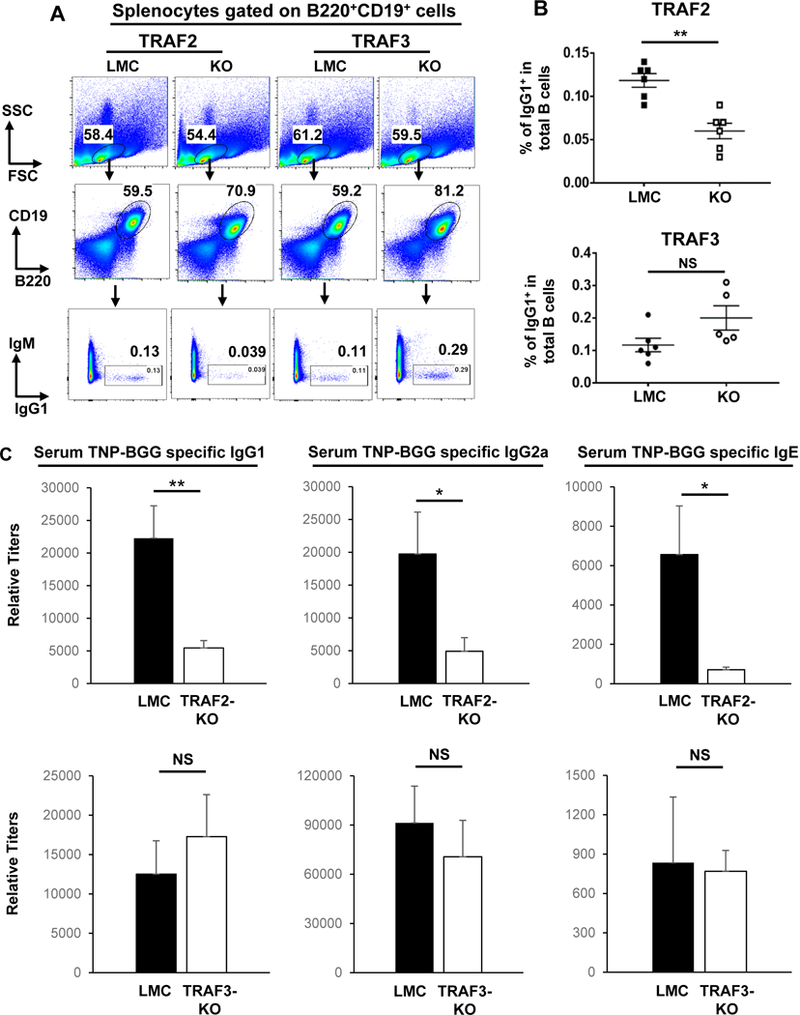
(A) Representative FACS data showing the gating strategy and the percentage of IgG1+ B cells. 28 days post first immunization, splenocytes were isolated from mice immunized with TNP-BGG Ag and analyzed by flow cytometry. Numbers indicate the percentage of IgG1+ B cells in the B220+CD19+ population. (B) Quantification of FACS data. The percentage of IgG1+ isotype-switched B cells were significantly reduced in B-TRAF2-KO but not in B-TRAF3-KO mice (n=6 per group). (C) Representative ELISA data of serum IgG1, IgG2a and IgE Abs against TNP-BGG. Data are presented as mean±s.e.m (n=5 or 6 per group). All experiments were repeated three times independently and representative data from one experiment are shown. P values were calculated by a Student’s t-test, *p≤0.05, **p≤0.01, which was applied to graphic data in all figures. LMC: littermate control (TRAF2fl/fl or TRAF3fl/fl, respectively). KO: knockout. TNP-BGG: 2,4,6-trinitrophenol-conjugated bovine γ-globulin.
CD40-induced CSR is markedly reduced in TRAF2 but not TRAF3 deficient B cells ex vivo
To further dissect the role of B cell-intrinsic TRAF2 or TRAF3 in CD40-driven CSR, we investigate how B-TRAF2 and B-TRAF3 deficiency affect Ab isotype-switching induced by engaging CD40 ex vivo. We isolated B cells from B-TRAF2-KO, B-TRAF3-KO and LMC mice and stimulated them with an agonistic Ab against CD40 or with LPS, a ligand for TLR4, in the presence of IL-4. IL-4 alone does not induce IgG1 and IgE CSR in LMC, B-TRAF2-KO or B-TRAF3-KO B cells (data not shown). IL-4 is included in all CSR assays to support B cell survival and is not mentioned further for CD40/IL-4 or LPS/IL-4 induced CSR. Consistent with our in vivo data, TRAF2 deficiency in B cells almost abrogated CD40-induced CSR to IgG1 or IgE but enhanced TLR4-induced CSR (Figure 2A, B). These results indicate that CSR machinery in B-TRAF2-KO B cells is intact for IgG1 or IgE; however, it cannot be fully activated by engaging CD40. In contrast, TRAF3 deficiency in B cells did not affect CD40-induced CSR but significantly increased TLR4-induced CSR compared with LMC (Figure 2A, B), consistent with a previous report (39). Therefore, we conclude that TRAF2 is essential while TRAF3 is dispensable for CD40 signaling to induce CSR, and both B cell-intrinsic TRAF2 and TRAF3 are not required for TLR4-induced CSR in the presence of IL-4.
Figure 2. B cell-intrinsic TRAF2 but not TRAF3 deficiency impairs CD40-induced CSR ex vivo.
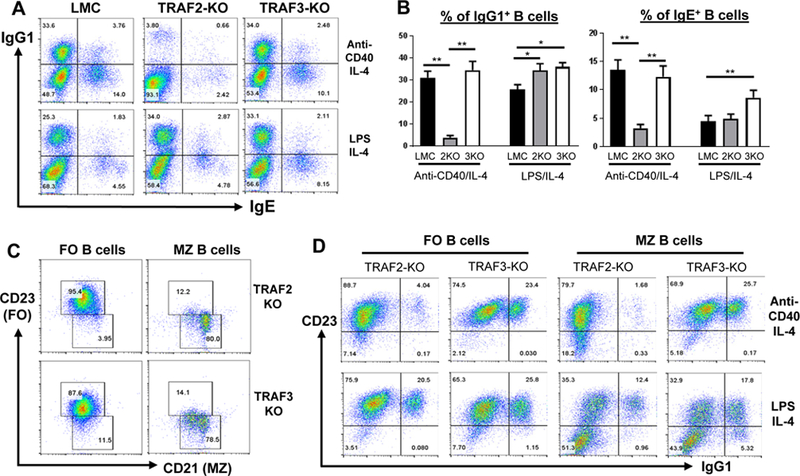
(A) Representative FACS data of IgG1 and IgE CSR. Purified B cells from various genotypes were stimulated with anti-CD40/IL-4 or LPS/IL-4 for 4 days and analyzed by flow cytometry. (B) Quantification of FACS data. The percentage of IgG1+ or IgE+ isotype-switched B cells was calculated from triplicates of each indicated genotype in one experiment. LMC: littermate control (TRAF2fl/fl or TRAF3fl/fl); 2KO: B-TRAF2-KO; 3KO: B-TRAF3-KO. Statistical analysis was performed as described in Figure 1. For panel A and B, all experiments were repeated five times independently and representative data from one experiment were shown. (C) Representative FACS data of purified MZ and FO B cells. FO B cells are CD23highCD21low, whereas MZ B cells are CD21highCD23low. (D) Representative FACS data of IgG1 CSR of FO and MZ B cells. Purified FO vs. MZ B cells from B-TRAF2-KO or B-TRAF3-KO mice were stimulated with anti-CD40/IL-4 or LPS/IL-4 for 4 days. For panel C and D, all experiments were repeated three times independently and representative data from one experiment are shown.
Consistent with previous studies (38, 41), we found that both follicular (FO) and marginal zone (MZ) B cells were significantly expanded in B-TRAF2 or B-TRAF3 KO mice (Supplemental Figure 1Band C). We thus investigate whether CD40-induced CSR is differentially affected by TRAF2 or TRAF3 deficiency in MZ vs. FO B cells. To do so, we purified splenic MZ and FO B cells from B-TRAF2-KO and B-TRAF3-KO mice (Figure 2C) and subjected them to CSR assay. Intriguingly, we found that almost all isotype-switched IgG1+ B cells acquired the phenotype of FO B cells (CD23+)(Figure 2D, right panel). As expected, both B-TRAF2-KO FO and MZ B cells failed to undergo CD40-induced CSR (Figure 2D, top panel). However, both FO and MZ B cells from B-TRAF2-KO or B-TRAF3-KO mice robustly underwent TLR4-induced CSR (Figure 2D, bottom panel). These data demonstrate that B-TRAF2-KO or B-TRAF3-KO B cells are capable of undergoing LPS-induced CSR regardless of B cell subsets, and suggest that defective CD40-induced CSR in B-TRAF2-KO B cells is probably not due to altered B cell development. Taken together, we conclude that B-TRAF2 deficiency specifically impairs CD40 autonomous signaling to reduce Ab isotype-switching.
CD40-induced AID expression and GLT of Igh C regions are significantly reduced by TRAF2 but not TRAF3 deficiency
Upon stimulation, B cells express AID and activate Igh C region GLT, both of which are required for CSR (2, 3, 47). To address the mechanism by which TRAF2 deficiency in B cells impairs CD40-induced CSR, we measured AID expression and Igh GLT induced by engaging CD40 or TLR4. B cell-specific TRAF2 deficiency led to a decreased level of Cγ1 and Cε GLT induced by CD40 (Figure 3A, B), in line with the reduced level of CSR to IgG1 and IgE in TRAF2 deficient B cells (Figure 2A, B). In contrast, TRAF3 deficiency did not reduce Cγ1 and Cε GLT (Figure 3A, B). B-TRAF2-KO B cells expressed much fewer AID transcripts induced by CD40 compared with LMC B cells (Figure 3C). Consistently, CD40-induced AID protein level was also drastically reduced in B-TRAF2-KO B cells (Figure 3D). Contrary to B-TRAF2-KO B cells, B-TRAF3-KO B cells expressed a comparable level of AID transcripts with LMC B cells (Figure 3C), and showed no defects in the expression of AID protein (Figure 3D). However, TLR4-induced AID transcription and protein expression were not significantly affected by TRAF2 or TRAF3 deficiency (Figure 3C, D). Thus, TRAF2 deficiency specifically impairs CD40-induced AID transcription. Taken together, we conclude that CD40-induced AID expression and Igh GLT require B-cell intrinsic TRAF2, but not TRAF3. These data lay a mechanistic foundation to explain why B cell-intrinsic TRAF2 deletion significantly reduces CD40-induced Ab isotype-switching.
Figure 3. Effects of B cell-intrinsic TRAF2 or TRAF3 deficiency on AID expression and GLT of Cγ1 or Cε induced by CD40.
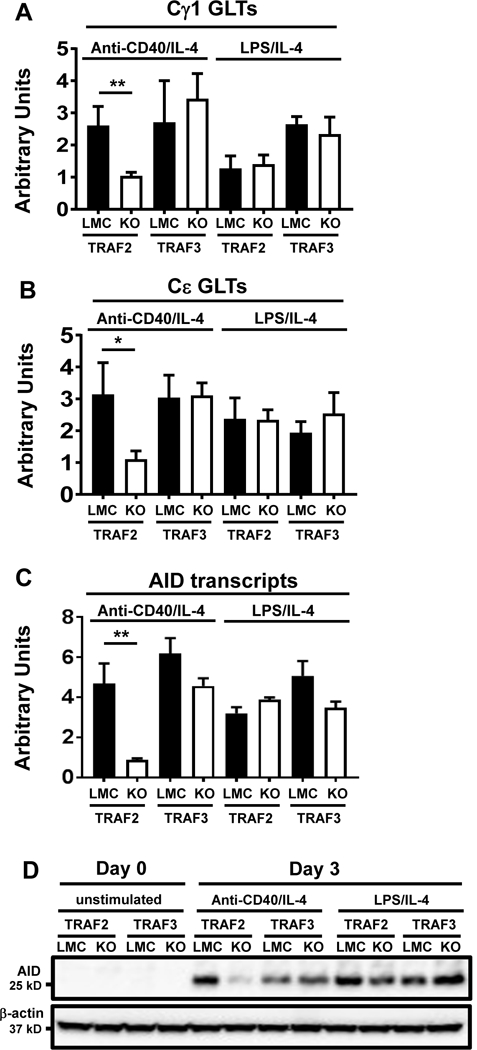
Quantitative RT-PCR (qPCR) data of relative transcripts of Cγ1 GLT (A), Cμ GLT (B) and AID (C). Purified B cells of indicated genotypes were stimulated with anti-CD40/IL-4 or LPS/IL-4 for 2 days. qPCR was conducted as described in Materials and Methods. Statistical significance analysis was performed by student t test, **p<0.01, *p<0.05. (D) Western blot of AID protein expression in the indicated B cells that were either unstimulated (day 0) or stimulated with anti-CD40/IL-4 or LPS/IL-4 for 3 days. β-actin is the loading control. LMC: littermate control (TRAF2fl/fl or TRAF3fl/fl). 2KO: B-TRAF2-KO. 3KO: B-TRAF3-KO. All experiments were repeated three times independently and representative data from one experiment are shown.
TRAF2-mediated activation of NF-κB1 complex is critical for CD40-induced CSR
AID transcription was significantly reduced in anti-CD40/IL-4-stimulated B-TRAF2-KO B cells. Previous studies implicated the critical role of NF-κB in activating AID transcription (10, 22–25); thus, we next investigated the status of NF-κB activation in B cells. Consistent with previous reports (38, 41), we found that active NF-κB2 (p52) was elevated in both resting B-TRAF2-KO and B-TRAF3-KO B cells compared with LMC B cells (Supplemental Figure 1D), possibly due to constitutive NIK accumulation. These data suggested that reduced AID transcription probably was not due to altered NF-κB2 activation in B-TRAF2-KO B cells; thus, we focused on examining activation of the NF-κB1 complex.
While the protein level of RelA (p65), which forms a complex with active NF-κB1 (p50), was similar in resting (0 min) or anti-CD40-activated (5 or 30 min) B cells of different genotypes, we found that B-TRAF2-KO B cells exhibited a significantly lower level of phospho-RelA upon 5 min stimulation, compared with LMC or B-TRAF3-KO B cells (Figure 4A). Consistently, the degradation level of IκBα, a NF-κB1 inhibitor, was much lower in B-TRAF2-KO B cells than LMC B cells stimulated with anti-CD40 (Figure 4A). Taken together, these data demonstrated that B-TRAF2 KO B cells reduced the activation of the NF-κB1complex upon CD40 stimulation. On the other hand, B-TRAF2-KO B cells appeared to have no intrinsic defects in activating NF-κB1 since LPS-induced activation of the NF-κB1 complex was comparable among B cells of different genotypes (Figure 4B).
Figure 4. TRAF2 is required for CD40 signaling to activate the NF-κB1 complex and induce isotype-switching.
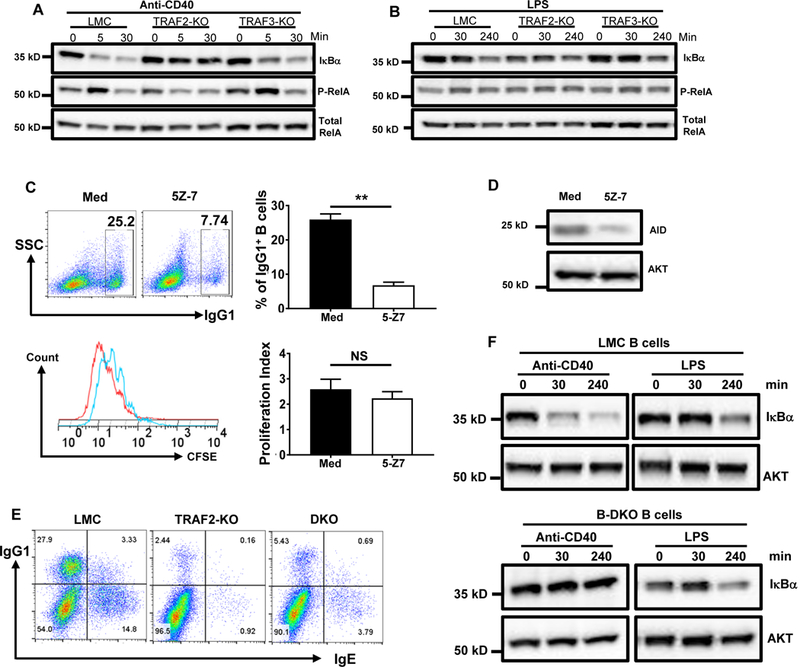
(A) Western blot of IκBα degradation and RelA phosphorylation in CD40-stimulated B cells. Purified B cells of indicated genotypes were stimulated with anti-CD40 for 0, 5 or 30 mins. CD40 stimulation did not induce IκBα degradation or RelA phosphorylation in B-TRAF2-KO B cells. (B) Western blot of IκBα degradation and RelA phosphorylation in LPS-stimulated B cells. Purified B cells of indicated genotypes were stimulated with LPS for 0, 30 or 240 mins that exhibited comparable levels of IκBα degradation and RelA phosphorylation. (C) Representative FACS data of IgG1 CSR (top panel) and CFSE dilution (bottom panel). LMC B cells were labeled with CFSE, then either treated with 5Z-7 (0.5μM) or untreated (Med), and stimulated with anti-CD40/IL-4 for 3 days. Top panel: representative of FACS data for IgG1 CSR and averaged percentage of IgG1+ B cells from triplicates. Bottom panel: representative FACS data for CFSE dilution (Med: red; 5Z-7 treated: blue) and the averaged proliferation index from triplicates. Proliferation index was calculated using Flowjo proliferation analysis program. P values were calculated by a Student’s t-test, **p<0.01, NS: not significant. (D) Western blot data show that 5Z-7 also significantly reduced AID expression. (E) Representative FACS data of IgG1 and IgE CSR. B cells of different genotypes were stimulated by anti-CD40/IL-4 for 4 days and analyzed by flow cytometry. TRAF3 deficiency in B-TRAF2-KO B cells did not rescue CD40-induced CSR. (F) Western blot of IκBα degradation in CD40- or LPS-stimulated LMC B cells (top panel) and B-DKO B cells (bottom panel). B cells were stimulated by anti-CD40 or LPS for 0, 30 or 240 mins and subjected to western blotting. TRAF3 deficiency in B-TRAF2-KO B cells did not rescue the defective CD40-induced IκBα degradation. B-DKO: double knockout of B-TRAF2 and B-TRAF3; LMC: littermate control (CD19Cre−/TRAF2fl/fl/TRAF3fl/fl). Med: culture medium with anti-CD40 plus IL-4. All experiments were repeated independently three or five times and representative data from one experiment are shown.
We hypothesize that the defective NF-κB1 activation in B-TRAF2-KO B cells is responsible for impaired CD40-induced CSR. If so, inhibiting NF-κB1 activation in LMC B cells should significantly reduce CD40-induced CSR. As such, we employed an inhibitor, (5Z)-7-Oxozeaenol (5Z-7), which covalently binds and inhibits TAK1. TAK1 is a kinase that phosphorylates IKKβ to activate the IKK complex (18). 5Z-7 markedly reduced CD40-induced CSR in LMC B cells (Figure 4C, top panel), while this concentration of 5Z-7 treatment did not significantly affect B cell proliferation (Figure 4C, bottom panel). 5Z-7 treatment also inhibited AID protein expression (Figure 4D) by blocking NF-κB1 activation.
Previous studies indicate that TRAF3 functions as an inhibitor of NF-κB1 activation induced by TNFRs (34, 48). CD40 is a member of the TNFR family. Hence, we asked whether TRAF3 deficiency can rescue the defective CD40-induced NF-κB1 activation and CSR in B-TRAF2-KO B cells. To test this possibility, we generated B-TRAF2 and B-TRAF3 double KO (B-DKO) mice. While the B-DKO B cells had a slightly increased CSR level compared with B-TRAF2-KO B cells, it was still much lower than that of LMC B cells (Figure 4E). These data indicate that, without TRAF2, CD40 cannot fully activate the NF-κB1 pathway. Consistently, B-DKO B cells exhibited little IκBα degradation upon engaging CD40 compared with LMC ones (Figure 4F). In contrast, we found no intrinsic defects in NF-κB1 activation in B-DKO B cells since TLR4-mediated activation of the NF-κB1 complex occurred normally (Figure 4F). Taken together, we conclude that the TRAF2 deficiency-induced CSR defect is caused by impaired NF-κB1 activation induced by CD40, which could not be rescued by TRAF3 deficiency.
Restoring NF-κB1 activation rescues CD40-induced CSR in TRAF2 deficient B cells
If NF-κB1 pathway activation is critical, restoring NF-κB1 activation should rescue CD40-induced AID expression and CSR in B-TRAF2-KO B cells. To test this notion, we employed phorbol 12-myristate 13-acetate (PMA), a NF-κB1 activator (49). We found that PMA stimulation alone induced IκBα degradation in LMC and B-TRAF2-KO B cells comparably (Figure 5A), further indicating that B-TRAF2-KO B cells had no intrinsic defects in NF-κB1 activation. Of note, PMA alone did not promote RelA phosphorylation (Figure 5A). While anti-CD40 stimulation failed to induce IκBα degradation and RelA phosphorylation, stimulating B-TRAF2-KO B cells with both anti-CD40 Ab and PMA rescued such defects by inducing IκBα degradation and RelA phosphorylation to the same extent as anti-CD40-stimulated LMC B cells (Figure 5B). Thus, these data suggest that PMA rescues the defective CD40-induced activation of the NF-κB1 complex in B-TRAF2-KO B cells.
Figure 5. PMA rescues the defective CD40-induced NF-κB1 complex activation and CSR in B-TRAF2-KO B cells.
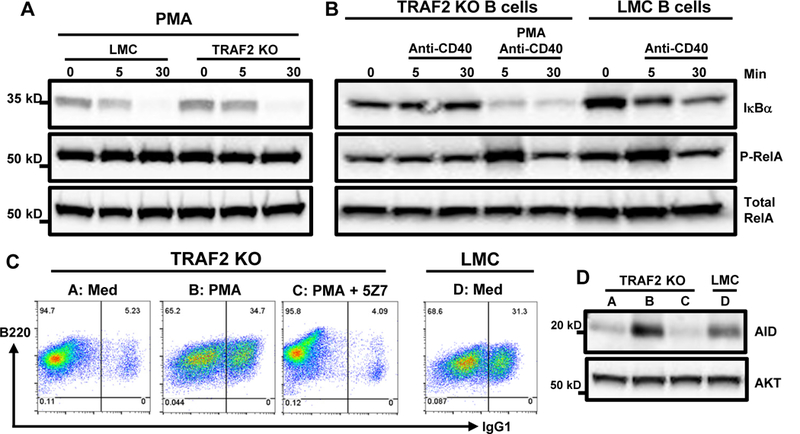
(A) Western blot of IκBα degradation induced by PMA stimulation alone. LMC and B-TRAF2-KO B cells were stimulated with PMA (1μM) for 0, 5 or 30 min and comparable IκBα degradation was observed in both groups. (B) Western blot of IκBα degradation and RelA phosphorylation in anti-CD40- vs. anti-CD40/PMA-stimulated B cells. LMC and B-TRAF2-KO B cells were stimulated as indicated in (A). (C) Representative FACS data of IgG1 CSR. Med: culture medium with anti-CD40/IL-4. B-TRAF2-KO (group A-C) or LMC (group D) B cells were stimulated in medium with anti-CD40/IL-4 and stimulator or inhibitor as indicated for 4 days. PMA (2nM) rescued CD40-induced CSR in B-TRAF2-KO B cells (A vs. B). However, the rescue effect of PMA was completely abrogated by 5Z-7 treatment (B vs. C). (D) Western blot of AID expression in different groups (A-D). B cells were stimulated as indicated in panel (C) and subjected to western blot analysis. P-RelA: phospho-RelA. All experiments were repeated independently three or five times and representative data from one experiment are shown.
Stimulation of B-TRAF2-KO B cells with both anti-CD40 Ab and PMA completely rescued CSR defects to a level comparable with LMC B cells stimulated by anti-CD40 (Figure 5C). To ascertain that the rescue of CD40-induced CSR in B-TRAF2-KO B cells is due to the restored NF-κB1 complex activation by PMA, we pretreated B-TRAF2-KO B cells with 5Z-7, the specific inhibitor of the NF-κB1 complex, then stimulated the pretreated B cells with anti-CD40 plus PMA. Our results showed that the NF-κB1-specific inhibitor completely abrogated the PMA-induced restoration of CSR (Figure 5C). In line with CSR phenotypes, PMA rescued AID expression induced by CD40 in B-TRAF2-KO B cells (Figure 5D). Thus, we conclude that TRAF2-mediated activation of the NF-κB1 complex is critical for CD40 signaling to induce AID expression and Ab isotype-switching.
Discussion
In the current study, we made several important discoveries: (1) B-TRAF2 is essential for optimal isotype-switching induced by in vivo TD Ag immunization or by engaging CD40 in vitro; (2) B-TRAF3 is dispensable for CD40-induced CSR, thereby clarifying its controversial role; (3) CD40-induced AID expression was markedly impaired by B-TRAF2 but not B-TRAF3 deficiency; and (4) B-TRAF2 deficiency causes defective activation of the NF-κB1 pathway in a CD40-autonomous manner, and restoring CD40-induced NF-κB1 activation in B-TRAF2-KO B cells rescues CD40-induced AID expression and CSR. TRAF2 and TRAF3 are critical signaling adaptors for various types of receptor families including TNFRs, TLRs, and cytokine receptors. Our study provides the basis to distinguish the effects of B cell-intrinsic TRAF2 from TRAF3 on TD humoral immunity and CD40-induced AID expression and CSR. Hence, our study generates novel insights into treating immunodeficiency caused by defects in the CD40 signaling pathway. In addition, if uncontrolled, AID may target non-Ig loci in the B cell genome to induce DNA damage, contributing to lymphomagenesis (8, 50, 51). Given that mutations in TRAF2 and TRAF3 are observed in human B cell lymphomas (52, 53), our study may also provide new insights into AID regulation during lymphomagenesis.
Our results appear to be contrary to a previous study reporting that B-TRAF3 is required for TD humoral immune responses and that TRAF3 independently transduces CD40 signaling from TRAF2 to induce Ab isotype-switching (37). This discrepancy is due to different model systems used. In the previous study, WT or mutant CD40 genes were used to reconstitute CD40 expression in B cells of CD40−/− mice (37). It was shown that the CD40 mutant that cannot bind TRAF3 resulted in defective Ab isotype-switching (37). Therefore, the conclusion was based on CD40’s binding ability to TRAF3 instead of the genetic deletion of traf3 per se. Of note, upon its binding with CD40, TRAF3 is degraded by cIAP1/2, then NIK can be accumulated to activate NF-κB2 pathway (17). NF-κB2 activation is required for germinal center formation and Ab isotype-switching (26). Thus, mechanistically, B-TRAF3 is a negative regulator and its degradation is probably required for optimal CD40-induced CSR and TD humoral immune responses. Presumably, the CD40 mutant that loses its binding ability to TRAF3 would not mediate TRAF3 degradation. As such, this mutant cannot induce NF-κB2 activation upon CD40 engagement by CD40L. In summary, the results from the CD40 reconstitution model (37) and our B-TRAF3 KO model can be reconciled at the mechanistic level of TRAF3 and CD40 interaction.
Another prior study showed that TRAF3 deletion in all hematopoietic lineages impaired TD humoral immune responses (32). While this study indicates the importance of TRAF3 in hematopoietic cells, it cannot define the role of TRAF3 in B cells because TRAF3 is also expressed in T cells and APCs. Given that B-TRAF3 is dispensable for TD humoral immune responses, we suggest that the presence of TRAF3 in other cell types may be required for mediating TD humoral immunity. Indeed, T cell-intrinsic TRAF3 deletion impairs TCR signaling (54). However, it remains to be addressed whether such an impairment affects TD humoral immune responses.
Germline deletion of TRAF2 causes perinatal lethality (33). Because TRAF2 KO fetal liver cells are too sensitive to TNF-induced cell death (33), it is impossible to generate chimera mice with TRAF2-KO hematopoietic cells. Germline deletion of both TRAF2 and TNFRI allows mice to survive (31); these double KO mice showed a defect in immunity against virus infection and a defect in CD40-induced NF-κB1 activation and proliferation of splenocytes (31). However, it remains to be addressed whether these double KO splenocytes have CSR defects and whether the defect in CD40-induced NF-κB1 activation is attributed to B cells or other cell types. Due to germline deletion of TRAF2, this model cannot define the role of B-TRAF2 in CSR and TD humoral immunity, which is also complicated by TNFRI deletion given that TNF is an important mediator of immune and inflammatory responses. Later, the traf2 gene was deleted in the CH12 cell line (16), a model cell line for studying CSR. However, this study did not report whether TRAF2 deletion affects CD40-induced CSR, while it was reported to impair CD40-induced IgM secretion (16). In contrast to TRAF2/TNFRI double KO splenocytes, TRAF2 KO CH12 cells did not show defects in CD40-induced NF-κB1 activation (16), probably due to the compensation of endogenous TRAF6 for TRAF2 loss. While CD40’s binding ability to TRAF2 was shown to be required for TD humoral immunity and CD40-induced CSR (14, 37), the limitation of such a CD40 reconstitution approach has been discussed above. Finally, studies using B-TRAF2 KO mice reported that CD40-induced NF-κB1 activation was defective in B cells; however, they did not investigate CD40-induced CSR ex vivo or TD humoral immune responses in vivo (41). In the end, the precise role of B-TRAF2 remained unclear in CD40-induced CSR and TD humoral immunity. Our current studies definitively demonstrate a B cell-intrinsic role of TRAF2 in mediating TD but not TI humoral immune responses. B-TRAF2 KO B cells exhibited defects in CD40-induced but not TLR4-induced CSR, and NF-κB1 pathway activation was only impaired when these B cells were activated by CD40 engagement but not TLR4 engagement (Figure 4). Taken together, these studies show that TRAF2 in B cells is an essential signaling element for the CD40 pathway to activate the NF-κB1 complex and to induce AID expression and CSR. This data highlights a clear difference in the function of TRAF2 and TRAF3 in B cells, and also highlights important differences in how B cells get activated using the standard in vitro “stimulator” LPS vs. anti-CD40. CSR also depends on GLT and chromatin looping anchored at Eμ and 3’ Igh enhancer, and these processes are also dependent on NF-κBs (55, 56). Consistently, we found that, in the absence of TRAF2, the level of GLT was reduced compared with LMC controls. Thus, it remains possible that TRAF2 loss may also affect Igh locus looping, thereby impairing GLT level, which warrants further investigation.
It was suggested that CD19cre-mediated TRAF3 deletion might affect B cell development, which in turns affects AID expression and CSR (37). In this regard, B-TRAF2 and B-TRAF3 KO mice share the similar phenotypes of having increased numbers of FO and MZ B cells and enlarged peripheral lymph organs (41), suggesting that the deletion of TRAF2 or TRAF3 in B cells results in similar effects on B cell development. Consistent with previous reports (38, 41), we did not find any developmental defects in B-TRAF2 KO MZ or FO B cells compared to their counterparts in B-TRAF3 KO mice. Thus, we suggest that the differential effects of B-TRAF2 and B-TRAF3 deletion on TD humoral immunity and CD40-induced CSR are not attributed to their potential effects on B cell development. Instead, B-TRAF2 is specifically required for the CD40 signaling pathway to induce AID expression and CSR. Consistent with the scenario of TRAF2’s role in CSR, we previously showed that the deletion of phosphatase and tensin homolog (PTEN) led to defective CD40-induced CSR ex vivo (29); furthermore, this phenotype is mature B cell-intrinsic and not influenced by the effects of PTEN deletion on B cell development (29). Of note, our data show that FO B cells undergo CSR more robustly, and MZ B cells appear to acquire FO phenotypes (CD23+) before they can undergo CSR (Figure 2D), an interesting finding that warrants future studies. Given that therapy targeting CD40/CD40L is emerging in cancer immunotherapy and primary immunodeficiency (57, 58), our studies lay a scientific foundation for properly treating these disorders by targeting the CD40 signaling pathway.
Supplementary Material
Acknowledgments
We thank Dr. Douglas Mann (Washington University, St. Louis, MO) for generously providing TRAF2flox/flox mice. We thank Nicole Koochi, Stephanie Cung, Brittany C. Waschke, Amanda M Perras, and Erin Kitten for technical help. We apologize to those whose work was not cited due to length restrictions.
This work was supported by University of Colorado School of Medicine and Cancer Center startup funds to Z.C. and J.H.W., R21-CA184707, R21-AI110777, R01-CA166325 and R21-AI133110 to J.H.W., a fund from the Cancer League of Colorado and American Cancer Society (IRG 57–001-53) to Z.C. R.A.W. is supported by a NIH F31 fellowship (F31DE027854). X.W. is supported by an AAI Careers in Immunology Fellowship.
References
- 1.Stavnezer J, Guikema JE, and Schrader CE 2008. Mechanism and regulation of class switch recombination. Annu Rev Immunol 26: 261–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stavnezer J, and Schrader CE 2014. IgH chain class switch recombination: mechanism and regulation. J Immunol 193: 5370–5378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, and Honjo T 2000. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 102: 553–563. [DOI] [PubMed] [Google Scholar]
- 4.Boboila C, Alt FW, and Schwer B 2012. Classical and alternative end-joining pathways for repair of lymphocyte-specific and general DNA double-strand breaks. Adv Immunol 116: 1–49. [DOI] [PubMed] [Google Scholar]
- 5.Yan CT, Boboila C, Souza EK, Franco S, Hickernell TR, Murphy M, Gumaste S, Geyer M, Zarrin AA, Manis JP, Rajewsky K, and Alt FW 2007. IgH class switching and translocations use a robust non-classical end-joining pathway. Nature 449: 478–482. [DOI] [PubMed] [Google Scholar]
- 6.Chaudhuri J, and Alt FW 2004. Class-switch recombination: interplay of transcription, DNA deamination and DNA repair. Nat Rev Immunol 4: 541–552. [DOI] [PubMed] [Google Scholar]
- 7.Di Noia JM, and Neuberger MS 2007. Molecular mechanisms of antibody somatic hypermutation. Annu Rev Biochem 76: 1–22. [DOI] [PubMed] [Google Scholar]
- 8.Chen Z, and Wang JH 2014. Generation and repair of AID-initiated DNA lesions in B lymphocytes. Frontiers of medicine 8: 201–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boboila C, Yan C, Wesemann DR, Jankovic M, Wang JH, Manis J, Nussenzweig A, Nussenzweig M, and Alt FW 2010. Alternative end-joining catalyzes class switch recombination in the absence of both Ku70 and DNA ligase 4. J Exp Med 207: 417–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zan H, and Casali P 2013. Regulation of Aicda expression and AID activity. Autoimmunity 46: 83–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu Z, Zan H, Pone EJ, Mai T, and Casali P 2012. Immunoglobulin class-switch DNA recombination: induction, targeting and beyond. Nat Rev Immunol 12: 517–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oxenius A, Campbell KA, Maliszewski CR, Kishimoto T, Kikutani H, Hengartner H, Zinkernagel RM, and Bachmann MF 1996. CD40-CD40 ligand interactions are critical in T-B cooperation but not for other anti-viral CD4+ T cell functions. J Exp Med 183: 2209–2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Essen D, Kikutani H, and Gray D 1995. CD40 ligand-transduced co-stimulation of T cells in the development of helper function. Nature 378: 620–623. [DOI] [PubMed] [Google Scholar]
- 14.Jabara H, Laouini D, Tsitsikov E, Mizoguchi E, Bhan A, Castigli E, Dedeoglu F, Pivniouk V, Brodeur S, and Geha R 2002. The binding site for TRAF2 and TRAF3 but not for TRAF6 is essential for CD40-mediated immunoglobulin class switching. Immunity 17: 265–276. [DOI] [PubMed] [Google Scholar]
- 15.Quezada SA, Jarvinen LZ, Lind EF, and Noelle RJ 2004. CD40/CD154 interactions at the interface of tolerance and immunity. Annu Rev Immunol 22: 307–328. [DOI] [PubMed] [Google Scholar]
- 16.Hostager BS, Haxhinasto SA, Rowland SL, and Bishop GA 2003. Tumor necrosis factor receptor-associated factor 2 (TRAF2)-deficient B lymphocytes reveal novel roles for TRAF2 in CD40 signaling. J Biol Chem 278: 45382–45390. [DOI] [PubMed] [Google Scholar]
- 17.Liao G, Zhang M, Harhaj EW, and Sun SC 2004. Regulation of the NF-kappaB-inducing kinase by tumor necrosis factor receptor-associated factor 3-induced degradation. J Biol Chem 279: 26243–26250. [DOI] [PubMed] [Google Scholar]
- 18.Arcipowski KM, and Bishop GA 2012. Roles of the kinase TAK1 in TRAF6-dependent signaling by CD40 and its oncogenic viral mimic, LMP1. PLoS One 7: e42478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sato S, Sanjo H, Takeda K, Ninomiya-Tsuji J, Yamamoto M, Kawai T, Matsumoto K, Takeuchi O, and Akira S 2005. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat Immunol 6: 1087–1095. [DOI] [PubMed] [Google Scholar]
- 20.Song Z, Zhu X, Jin R, Wang C, Yan J, Zheng Q, Nanda A, Granger DN, and Li G 2014. Roles of the kinase TAK1 in CD40-mediated effects on vascular oxidative stress and neointima formation after vascular injury. PLoS One 9: e101671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Y, Huang G, Vogel P, Neale G, Reizis B, and Chi H 2012. Transforming growth factor beta-activated kinase 1 (TAK1)-dependent checkpoint in the survival of dendritic cells promotes immune homeostasis and function. Proc Natl Acad Sci U S A 109: E343–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gourzi P, Leonova T, and Papavasiliou FN 2007. Viral induction of AID is independent of the interferon and the Toll-like receptor signaling pathways but requires NF-kappaB. J Exp Med 204: 259–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park SR, Zan H, Pal Z, Zhang J, Al-Qahtani A, Pone EJ, Xu Z, Mai T, and Casali P 2009. HoxC4 binds to the promoter of the cytidine deaminase AID gene to induce AID expression, class-switch DNA recombination and somatic hypermutation. Nat Immunol 10: 540–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pone EJ, Zhang J, Mai T, White CA, Li G, Sakakura JK, Patel PJ, Al-Qahtani A, Zan H, Xu Z, and Casali P 2012. BCR-signalling synergizes with TLR-signalling for induction of AID and immunoglobulin class-switching through the non-canonical NF-kappaB pathway. Nature communications 3: 767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tran TH, Nakata M, Suzuki K, Begum NA, Shinkura R, Fagarasan S, Honjo T, and Nagaoka H 2010. B cell-specific and stimulation-responsive enhancers derepress Aicda by overcoming the effects of silencers. Nat Immunol 11: 148–154. [DOI] [PubMed] [Google Scholar]
- 26.Caamano JH, Rizzo CA, Durham SK, Barton DS, Raventos-Suarez C, Snapper CM, and Bravo R 1998. Nuclear factor (NF)-kappa B2 (p100/p52) is required for normal splenic microarchitecture and B cell-mediated immune responses. J Exp Med 187: 185–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sha WC, Liou HC, Tuomanen EI, and Baltimore D 1995. Targeted disruption of the p50 subunit of NF-kappa B leads to multifocal defects in immune responses. Cell 80: 321–330. [DOI] [PubMed] [Google Scholar]
- 28.Fang DF, He K, Wang N, Sang ZH, Qiu X, Xu G, Jian Z, Liang B, Li T, Li HY, Li AL, Zhou T, Gong WL, Yang B, Karin M, Zhang XM, and Li WH 2014. NEDD4 ubiquitinates TRAF3 to promote CD40-mediated AKT activation. Nature communications 5: 4513. [DOI] [PubMed] [Google Scholar]
- 29.Chen Z, Getahun A, Chen X, Dollin Y, Cambier JC, and Wang JH 2015. Imbalanced PTEN and PI3K Signaling Impairs Class Switch Recombination. J Immunol 195: 5461–5471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Omori SA, Cato MH, Anzelon-Mills A, Puri KD, Shapiro-Shelef M, Calame K, and Rickert RC 2006. Regulation of class-switch recombination and plasma cell differentiation by phosphatidylinositol 3-kinase signaling. Immunity 25: 545–557. [DOI] [PubMed] [Google Scholar]
- 31.Nguyen LT, Duncan GS, Mirtsos C, Ng M, Speiser DE, Shahinian A, Marino MW, Mak TW, Ohashi PS, and Yeh WC 1999. TRAF2 deficiency results in hyperactivity of certain TNFR1 signals and impairment of CD40-mediated responses. Immunity 11: 379–389. [DOI] [PubMed] [Google Scholar]
- 32.Xu Y, Cheng G, and Baltimore D 1996. Targeted disruption of TRAF3 leads to postnatal lethality and defective T-dependent immune responses. Immunity 5: 407–415. [DOI] [PubMed] [Google Scholar]
- 33.Yeh WC, Shahinian A, Speiser D, Kraunus J, Billia F, Wakeham A, de la Pompa JL, Ferrick D, Hum B, Iscove N, Ohashi P, Rothe M, Goeddel DV, and Mak TW 1997. Early lethality, functional NF-kappaB activation, and increased sensitivity to TNF-induced cell death in TRAF2-deficient mice. Immunity 7: 715–725. [DOI] [PubMed] [Google Scholar]
- 34.Bishop GA 2016. TRAF3 as a powerful and multitalented regulator of lymphocyte functions. Journal of leukocyte biology 100: 919–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bradley JR, and Pober JS 2001. Tumor necrosis factor receptor-associated factors (TRAFs). Oncogene 20: 6482–6491. [DOI] [PubMed] [Google Scholar]
- 36.Chung JY, Park YC, Ye H, and Wu H 2002. All TRAFs are not created equal: common and distinct molecular mechanisms of TRAF-mediated signal transduction. J Cell Sci 115: 679–688. [DOI] [PubMed] [Google Scholar]
- 37.Jabara HH, Weng Y, Sannikova T, and Geha RS 2009. TRAF2 and TRAF3 independently mediate Ig class switching driven by CD40. Int Immunol 21: 477–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xie P, Stunz LL, Larison KD, Yang B, and Bishop GA 2007. Tumor necrosis factor receptor-associated factor 3 is a critical regulator of B cell homeostasis in secondary lymphoid organs. Immunity 27: 253–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xie P, Poovassery J, Stunz LL, Smith SM, Schultz ML, Carlin LE, and Bishop GA 2011. Enhanced Toll-like receptor (TLR) responses of TNFR-associated factor 3 (TRAF3)-deficient B lymphocytes. Journal of leukocyte biology 90: 1149–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grech AP, Amesbury M, Chan T, Gardam S, Basten A, and Brink R 2004. TRAF2 differentially regulates the canonical and noncanonical pathways of NF-kappaB activation in mature B cells. Immunity 21: 629–642. [DOI] [PubMed] [Google Scholar]
- 41.Gardam S, Sierro F, Basten A, Mackay F, and Brink R 2008. TRAF2 and TRAF3 signal adapters act cooperatively to control the maturation and survival signals delivered to B cells by the BAFF receptor. Immunity 28: 391–401. [DOI] [PubMed] [Google Scholar]
- 42.Chen Z, Ranganath S, Viboolsittiseri SS, Eder MD, Chen X, Elos MT, Yuan S, Hansen E, and Wang JH 2014. AID-initiated DNA lesions are differentially processed in distinct B cell populations. J Immunol 193: 5545–5556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Katona IM, Urban JF Jr., and Finkelman FD 1985. B cells that simultaneously express surface IgM and IgE in Nippostrongylus brasiliensis-infected SJA/9 mice do not provide evidence for isotype switching without gene deletion. Proc Natl Acad Sci U S A 82: 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Katona IM, Urban JF Jr., Scher I, Kanellopoulos-Langevin C, and Finkelman FD 1983. Induction of an IgE response in mice by Nippostrongylus brasiliensis: characterization of lymphoid cells with intracytoplasmic or surface IgE. J Immunol 130: 350–356. [PubMed] [Google Scholar]
- 45.Allen RC, Armitage RJ, Conley ME, Rosenblatt H, Jenkins NA, Copeland NG, Bedell MA, Edelhoff S, Disteche CM, Simoneaux DK, and et al. 1993. CD40 ligand gene defects responsible for X-linked hyper-IgM syndrome. Science 259: 990–993. [DOI] [PubMed] [Google Scholar]
- 46.Bhushan A, and Covey LR 2001. CD40:CD40L interactions in X-linked and non-X-linked hyper-IgM syndromes. Immunologic research 24: 311–324. [DOI] [PubMed] [Google Scholar]
- 47.Revy P, Muto T, Levy Y, Geissmann F, Plebani A, Sanal O, Catalan N, Forveille M, Dufourcq-Labelouse R, Gennery A, Tezcan I, Ersoy F, Kayserili H, Ugazio AG, Brousse N, Muramatsu M, Notarangelo LD, Kinoshita K, Honjo T, Fischer A, and Durandy A 2000. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2). Cell 102: 565–575. [DOI] [PubMed] [Google Scholar]
- 48.Hacker H, Tseng PH, and Karin M 2011. Expanding TRAF function: TRAF3 as a tri-faced immune regulator. Nat Rev Immunol 11: 457–468. [DOI] [PubMed] [Google Scholar]
- 49.Wang Y, Ding M, Chaudhari S, Ding Y, Yuan J, Stankowska D, He S, Krishnamoorthy R, Cunningham JT, and Ma R 2013. Nuclear factor kappaB mediates suppression of canonical transient receptor potential 6 expression by reactive oxygen species and protein kinase C in kidney cells. J Biol Chem 288: 12852–12865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pavri R, and Nussenzweig MC 2011. AID targeting in antibody diversity. Adv Immunol 110: 1–26. [DOI] [PubMed] [Google Scholar]
- 51.Wang JH 2013. The role of activation-induced deaminase in antibody diversification and genomic instability. Immunologic research 55: 287–297. [DOI] [PubMed] [Google Scholar]
- 52.Moore CR, Edwards SK, and Xie P 2015. Targeting TRAF3 Downstream Signaling Pathways in B cell Neoplasms. J Cancer Sci Ther 7: 67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rahal R, Frick M, Romero R, Korn JM, Kridel R, Chan FC, Meissner B, Bhang HE, Ruddy D, Kauffmann A, Farsidjani A, Derti A, Rakiec D, Naylor T, Pfister E, Kovats S, Kim S, Dietze K, Dorken B, Steidl C, Tzankov A, Hummel M, Monahan J, Morrissey MP, Fritsch C, Sellers WR, Cooke VG, Gascoyne RD, Lenz G, and Stegmeier F 2014. Pharmacological and genomic profiling identifies NF-kappaB-targeted treatment strategies for mantle cell lymphoma. Nature medicine 20: 87–92. [DOI] [PubMed] [Google Scholar]
- 54.Xie P, Kraus ZJ, Stunz LL, Liu Y, and Bishop GA 2011. TNF receptor-associated factor 3 is required for T cell-mediated immunity and TCR/CD28 signaling. J Immunol 186: 143–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Feldman S, Achour I, Wuerffel R, Kumar S, Gerasimova T, Sen R, and Kenter AL 2015. Constraints contributed by chromatin looping limit recombination targeting during Ig class switch recombination. J Immunol 194: 2380–2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kenter AL, Feldman S, Wuerffel R, Achour I, Wang L, and Kumar S 2012. Three-dimensional architecture of the IgH locus facilitates class switch recombination. Ann N Y Acad Sci 1267: 86–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Beatty GL, Li Y, and Long KB 2017. Cancer immunotherapy: activating innate and adaptive immunity through CD40 agonists. Expert Rev Anticancer Ther 17: 175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Meng X, Yang B, and Suen WC 2018. Prospects for modulating the CD40/CD40L pathway in the therapy of the hyper-IgM syndrome. Innate Immun 24: 4–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.