

Abstract
This study examines co-located aerosol and precipitation chemistry data between 2010–2016 at Pinnacles National Monument ~65 km east of the coastline in central California. Positive matrix factorization analysis of the aerosol composition data revealed seven distinct pollutant sources: aged sea salt (25.7% of PM2.5), biomass burning (24.2% of PM2.5), fresh sea salt (15.0% of PM2.5), secondary sulfate (11.7% of PM2.5), dust (10.0% of PM2.5), vehicle emissions (8.2% of PM2.5), and secondary nitrate (5.2% of PM2.5). The influence of meteorology and transport on monthly patterns of PM2.5 composition is discussed. Only secondary sulfate exhibited a statistically significant change (a reduction) over time among the PM2.5 source factors. In contrast, PMcoarse exhibited a significant increase most likely due to dust influence. Monthly profiles of precipitation chemistry are summarized showing that the most abundant species in each month was either SO42−, NO3−, or Cl−. Intercomparisons between the precipitation and aerosol data revealed several features: (i) precipitation pH was inversely related to factors associated with more acidic aerosol constituents such as secondary sulfate and aged sea salt, in addition to being reduced by uptake of HNO3 in the liquid phase; (ii) two aerosol source factors (dust and aged sea salt) and PMcoarse exhibited a positive association with Ca2+ in precipitation, suggestive of directly emitted aerosol types with larger sizes promoting precipitation; and (iii) sulfate levels in both the aerosol and precipitation samples analyzed were significantly correlated with dust and aged sea salt PMF factors, pointing to the partitioning of secondary sulfate to dust and sea salt particles. The results of this work have implications for the region’s air quality and hydrological cycle, in addition to demonstrating that the use of co-located aerosol and precipitation chemistry data can provide insights relevant to aerosol-precipitation interactions.
Keywords: Pinnacles National Monument, Sea Salt, Biomass Burning, PMF, IMPROVE, NADP
Graphical Abstract
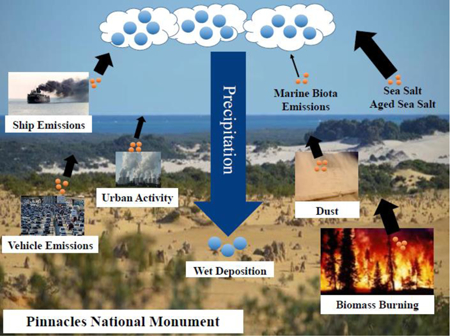
1. Introduction
The largest uncertainty in quantifying global anthropogenic radiative forcing is linked to interactions of aerosol particles with clouds (IPCC, 2013), which is partly driven by the difficulty of conducting the required measurements and separating the influence of meteorology and aerosol pollution on clouds. The impact of aerosol particles on precipitation is challenging to address but is important for reasons extending from improving model representation of clouds and precipitation to identifying the impacts of wet deposition on aquatic and terrestrial ecosystems due to inputs of nutrients and contaminants that may have originated from particles serving as cloud condensation nuclei (CCN) or ice nuclei (IN). Precipitation has important effects on aerosol particles owing to scavenging and removing them from the air, in addition to also removing gases (e.g., MacDonald et al., 2018). In this regard, monitoring the composition of wet deposition is important not just for impacts on ecosystems (e.g., Bobbink et al., 1998; Driscoll et al., 2003; Pardo et al., 2011), but for evaluating chemical tracer transport models (e.g., Rodhe et al., 1995; Liu et al., 2001). As observational studies of aerosol-cloud-precipitation interactions relying on airborne in situ measurements and remote sensing have limitations (e.g., cost, statistics, temporal resolution and coverage), alternative methods of examining this complex system can provide much needed insight. One such method is to rely on long-term records of surface-based aerosol and precipitation chemistry data, as has been demonstrated in just a few studies focused on regions such as the southwestern United States (Sorooshian et al., 2013) and Mexico City (Mora et al., 2017). Examining simultaneously collected aerosol and precipitation chemistry data can provide knowledge about topics with important implications including (i) sources of pollutants, (ii) potential interactions between gases and particles with precipitation drops including uptake processes and wet scavenging, and aerosol types impacting clouds that produce the precipitation.
Aerosol-cloud-precipitation interactions have been the subject of extensive field projects across California, including for stratiform cloud decks off the coast (e.g., Russell et al., 2013; Sorooshian et al., 2018) and for the more convective clouds inland that provide important rainfall for the state (e.g., Fan et al., 2014; Ralph et al., 2016), including intense precipitation events associated with wintertime atmospheric rivers (e.g., Ralph et al., 2004, 2013). Many of these studies have tried to provide details about sources of pollution impacting the clouds and what the sign of the precipitation response was due to an increase in aerosol concentration. This study aims to fill a gap by examining for the first time long term datasets for aerosol and precipitation chemistry at a central California site, with results that can be contrasted with findings from other works that have relied on aircraft and remote sensing.
This study reports on long-term data collected at Pinnacles National Monument, which is distinguished from most other surface monitoring areas in California because of the co-location of monitoring instruments for both aerosol and precipitation chemistry for a long-term period. Another motivation for studying this site is that it represents an area impacted by a diverse set of sources (e.g., urban, agriculture, marine, shipping, wildfires, dust) due to its geographical position. Focus is first placed on a comprehensive analysis of the aerosol composition data to characterize temporal trends, sources of the pollutants, and the impact of meteorology and transport. Subsequently, the precipitation chemistry data are summarized and then compared with the aerosol composition data.
2. Methods
2.1. Site Description
California is of interest in this study owing to its large population (~36.5 million as of 1 July 2017; US Census Bureau; https://www.census.gov/) that is impacted in various ways by aerosol pollution and precipitation. The Pinnacles National Monument (PNM) site was chosen for this analysis owing to its geographic location as an inland California site impacted by a diversity of sources (Figure 1), and because there are co-located aerosol and precipitation monitoring stations operated by the Environmental Protection Agency (EPA) Interagency Monitoring of Protected Visual Environments (IMPROVE) network and the National Atmospheric Deposition Program (NADP) National Trends Network (NTN), respectively. The PNM site is ~65 km east of the ocean and ~115 km south of San Jose (Figure 1). The California coastal range separates the marine atmosphere from the urban atmosphere over major cities such as San Jose (3rd largest in California), which when combined with Sunnyvale and Santa Clara forms a metropolitan area with an estimated population of ~1.9 million (as of 1 July 2017 according to the US Census Bureau; www.census.gov). To the north is the more populated metropolitan area including San Francisco, Oakland, and Hayward, with a combined population of 4.7 million (as of 1 July 2017; www.census.gov). Aside from anthropogenic and marine sources, the general region containing the PNM site is impacted by wildfires (Westerling et al., 2006) and long-range transport of pollution from distant regions such as Asia (VanCuren and Cahill, 2002), Mexico (Xu et al., 2006), and neighboring states (Xu et al., 2006).
Figure 1.
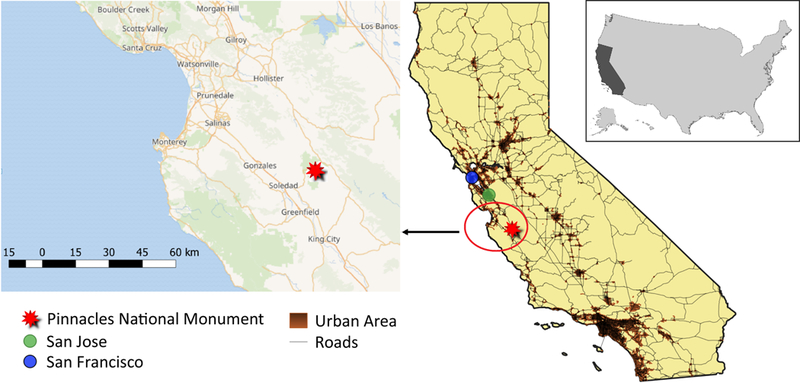
Map showing the study region, including where the collocated IMPROVE and NADP/NTN stations are at Pinnacles National Monument relative to major urban centers and the coastline.
2.2. Long-Term Surface Monitoring Stations
Data were retrieved from the NADP/NTN (36.4819°N, −121.155°W, 300 m above MSL; http://nadp.slh.wisc.edu/data/ntn/) and IMPROVE (36.4833°N, −121.1568°W, 302 m above MSL; http://views.cira.colostate.edu/fed/DataWizard/) sites between 5 January 2010 – 27 December 2016 and 2 January 2010 – 29 December 2016, respectively. At the IMPROVE site, 24 hour filter samples were collected every third day during the study period. The collection system consisted of multiple separate sampling modules to characterize different aspects of either PM2.5 or PM10. Of relevance to this study are the measurements of species mass concentrations in the PM2.5 fraction, including water soluble ions in the PM2.5 fraction measured by ion chromatography (SO4−2, NO3−, Cl−), elemental mass concentrations in the PM2.5 fraction measured by either X-ray fluorescence (Fe and heavier elements) or particle-induced X-ray emission (Na to Mn), elemental carbon (EC) and organic carbon (OC) quantified by thermal optical analysis according to IMPROVE-A protocol (Chow et al., 2007), and total PM2.5 and PM10 mass concentrations via gravimetric analysis. PMcoarse is calculated as the difference between PM10 and PM2.5 mass concentrations. Additional details of sampling protocols and measurement techniques used in IMPROVE can be found elsewhere (Solomon et al., 2014; Chow et al., 2015).
NADP/NTN was initiated in 1978 to monitor the phenomena of wet deposition and its effects on terrestrial features (e.g., agriculture, forests, and water) (Lamb and Bowersox, 2000). Every NADP/NTN site utilizes two instruments for collection of precipitation. The Belfort B5–780 rain-gauge mechanically measures and records the amount of precipitation each day to the nearest 0.01 inch. The Aerochem Metrics 301 precipitation collector (ACM) automatically collects precipitation samples on a weekly basis (Tuesday to Tuesday) for chemical analysis. Weekly samples obtained via the ACM are sent to the Central Analytical Laboratory (CAL) at the Illinois State Water Survey in Champaign, Illinois to quantify the weekly values of the following parameters: pH, conductance, and water-soluble ion concentrations (NH4+, Ca2+, Cl−, Mg2+, NO3−, K+, Na+, SO42−) measured by ion chromatography. The NTN has a vigorous quality assurance program described in more detail elsewhere (See et al., 1989; NADP, 2014). Weekly values of the chemical measurements and the precipitation amounts were used for calculation of monthly/annual precipitation-weighted averages of pH, ion concentrations, and ion mass fractions. Moreover, wet deposition amounts were calculated by multiplying the weekly ion concentrations by the weekly amounts of precipitation. For all calculations, values below detection limits were substituted with half of the detection limits (DL). A summary of DL values for species in the NADP/NTN and IMPROVE datasets is provided in the supporting information (Tables S1-S2).
2.3. Meteorological Data
Meteorological data including wind speed and direction, temperature, relative humidity, and solar radiation were obtained from the EPA Air Quality System (AQS) database (https://www.epa.gov/outdoor-air-quality-data) for the PNM IMPROVE site. Precipitation accumulation data were obtained from the NADP/NTN site. Planetary boundary layer height (PBLH) data were obtained from the Modern Era-Retrospective Analysis for Research and Applications (MERRA-2) model with 0.5° × 0.625° spatial resolution. Values of near surface specific humidity and surface soil moisture (0–10 cm) were obtained from the Global Land Data Assimilation System (GLDAS) with 0.25° × 0.25° spatial resolution. Parameters obtained from both MERRA-2 and GLDAS were calculated over 0.5° × 0.625° and 0.5° × 0.5° areas, respectively, extending around the PNM site.
2.4. Calculations
2.4.1. Positive Matrix Factorization (PMF)
A PMF model (US EPA’s PMF version 5) was applied to the field data to identify sources and evaluate corresponding contributions impacting a sampling site (Paatero and Tapper, 1994; Paatero, 1997; Hopke, 2016). PMF has been widely implemented as a way to conduct source apportionment of PM2.5 (Polissar et al., 2001; Lee et al., 2006; Masiol et al., 2014). Seventeen species (SO42−, NO3−, OC, EC, Na, Al, Si, Cl−, K, Ca, V, Mn, Fe, Ni, Cu, Zn, Br) were included in the analysis and categorized as “strong” based on exhibiting a signal to noise ratio (S/N) above one. Summary statistics for PM2.5 and speciated concentrations included in PMF analysis are given in Table S2. PM2.5 was added to the model as a total variable, categorized as a “weak” species to minimize its effect in driving the model results (Kim et al., 2003). Based on Polissar et al. (1998), concentration values below the DL were substituted with a value equal to half of the DL, with an uncertainty of 5/6 of the associated DL. For missing values, median species concentrations were used, with an uncertainty of four times the median. An additional 10% uncertainty was added to account for unconsidered errors for all species.
The uncertainty associated with the model output was assessed via bootstrapping (BS), displacement (DISP), and bootstrapping with displacement (BS-DISP). For BS, 300 resamples were used with a threshold value of 0.6 for the correlation coefficient (r) to qualify as successful mapping in each run. For DISP and BS-DISP analyses, factor swaps and uncertainty in the model output were assessed for dQmax equal to four.
2.4.2. Concentration Weighted Trajectory (CWT) Analysis
As a way of tracking the trajectory of specific aerosol chemical signatures to the sample site, Concentration Weighted Trajectory (CWT) analysis was employed (e.g., Hsu et al., 2003). This method considers grid cells that are assigned a weighted concentration calculated by taking the mean of sample concentrations that have associated trajectories crossing a particular grid cell. The GIS-based software TrajStat (Wang et al., 2009) was used to obtain the CWT profiles. In order to obtain CWT profiles, 96 h back-trajectories were used from the Hybrid Single-Particle Lagrangian Integrated Trajectory (HYSPLIT) model (Stein et al., 2015; Rolph, 2016) ending 500 m above ground level at PNM. Trajectories were obtained every six hours using the National Centers for Environmental Prediction/National Center for Atmospheric Research (NCEP/NCAR) reanalysis data with the “Model vertical velocity” method. Using a domain of −180° to −100° longitude and 20° to 70° latitude with grid cell size 0.5°×0.5° resulted in an average of ~20 trajectory end points for each cell. A point filter was applied to reduce weight in the effect of the cells with only a small number of end points (Wang et al., 2009). Trajectory frequency maps from 2010 to 2016 are provided in Figure S1, showing that the majority of air masses originated over the Pacific Ocean, especially in the summer, which exhibited a more distant trajectory of air down the coastline of the western United States towards the PNM site.
3. Results and Discussion
3.1. Meteorological Profile
The monthly meteorological profile is summarized in Figure 2 for the study region. Precipitation occurred between October and June without any accumulation in the summer months between July and September. The peak of precipitation accumulation was between December and March. Soil moisture expectedly followed the same general monthly profile as precipitation, suggestive of a greater likelihood of local soil emissions in summer months, especially when coupled to higher wind speeds in those months. Specific humidity was most enhanced in the summer months (July-August) unlike relative humidity, which is sensitive to temperature and exhibited higher levels in the winter months. The PBLH expectedly followed the same general trend as ambient temperature and solar radiation with higher levels in the summer months.
Figure 2.
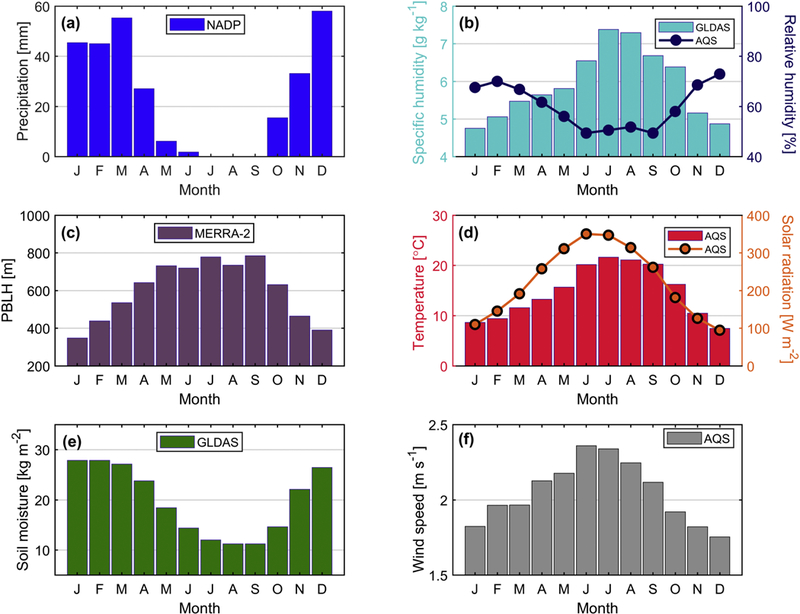
Monthly profiles of (a) precipitation accumulation, (b) specific humidity and relative humidity, (c) planetary boundary layer height (PBLH), (d) temperature and solar radiation, (e) soil moisture, and (f) wind speed based on data between 2010 and 2016. These profiles are obtained from different data sources including EPA AQS monitoring network, MERRA-2, and GLDAS.
3.2. Source Apportionment of PM2.5
PMF analysis was conducted to identify key sources, and multiple PMF solutions ranging from having three to 10 characteristic pollutant sources were analyzed. The final solution with seven sources was chosen based on the physical validity of the results (described in Supporting Information, Figures S2-S6), which also showed the proximity of the calculated ratio of Qtrue/Qexpected (1.1) to 1.0. The optimal solution also exhibited the following characteristics: i) all factors were mapped in BS runs; ii) during DISP, no factor swap was observed (Table S3) and the decrease in Q values was less than 1%; and iii) there were no factor swaps in BS-DISP (Table S3) where OC, EC, SO42−, NO3−, Cl−, K, and Na were actively displaced. In addition, there was a high coefficient of variation between measured and predicted PM2.5 (r2 = 0.91), which added confidence in relying on the PMF model for source apportionment of PM2.5.
The seven resolved source profiles of PM2.5 are shown in Figure 3, with their associated monthly profiles summarized in Figure 4, which were derived from BS resampling (n = 10000). Time series plots of PM2.5 mass concentrations from each source are shown in the supporting information (Figure S7). The smoothed results of CWT analysis associated with each factor for the full study period are shown in Figure 5, with seasonal maps shown in Figure S8.
Figure 3.
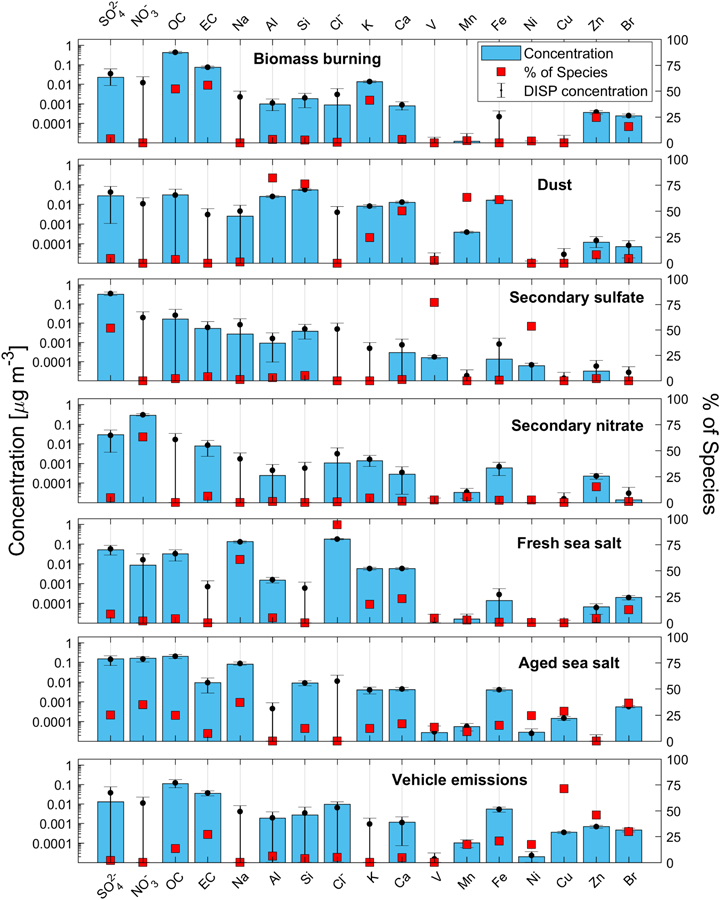
Seven source factor profiles from PMF analysis using chemically-speciated PM2.5 data from PNM IMPROVE station. Blue bars represent the mass concentration of the species while red filled markers signify the percent contribution of a particular source factor to the mass sum of a species’ overall concentration. Black circles and error bars represent mean and range, respectively, of DISP values of the species mass concentration.
Figure 4.
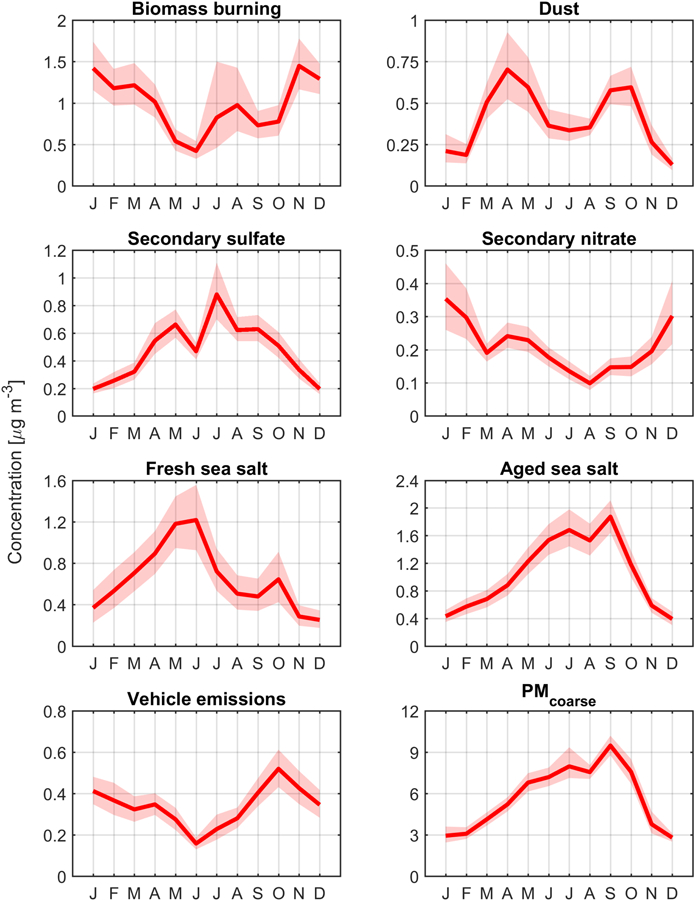
Monthly mean statistics of source factor mass concentrations. Solid lines and shadings represent the mean and 95th confidence intervals, respectively, calculated using bootstrapping (n = 10000).
Figure 5.
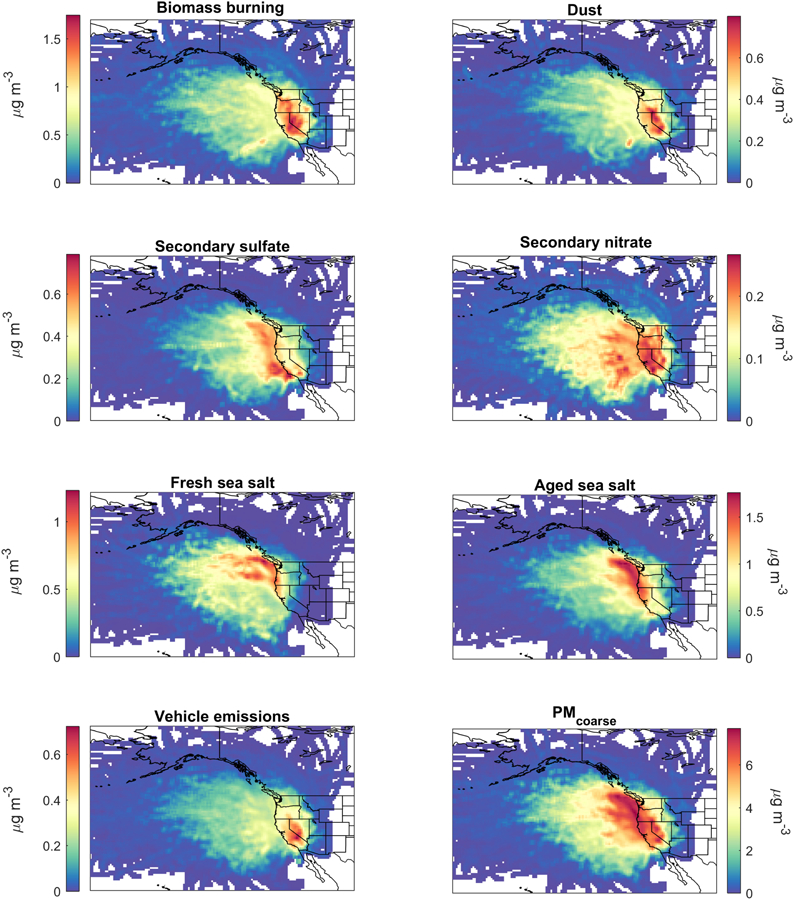
Concentration Weighted Trajectory (CWT) maps of PMF source factors in addition to PMcoarse.
3.2.1. Biomass Burning
The mass concentration associated with the biomass burning source factor accounted for 24.2% of total PM2.5. As compared to other sources, biomass burning contributed the most to the overall mass concentrations of EC (55.7%), OC (52.0%), and K (41.1%) (Figure 3), the cluster of which is known to be associated with biomass burning in the western United States (Cahill et al., 2008; Schlosser et al., 2017) owing mainly to wildfires and wood burning for domestic heating (Wang et al., 2012; Masiol et al., 2017). The temporal profile of biomass burning differs from the other six source factors in that it exhibits its highest concentrations (> 1 µg m−3) for an extended period of the year (November – April), and with a minor concentration peak in the summer months of July-August (Figure 4). The more pronounced and longer concentration peak stems most likely from domestic heating, while the smaller peak in the summertime, coupled with the highest variability in mass concentration, is linked to wildfire activity (Westerling et al., 2006; Dennison et al., 2014) (Figure 4). The CWT plot presented in Figure 5 shows that the strongest sources of this pollutant type are expectedly over the western United States, with strong influence from the neighboring states of Oregon and Nevada.
3.2.2. Dust
The dust source factor accounted for 10.0% of total PM2.5. This factor is linked to dust, supported by it contributing the most to the mass concentrations of the following crustal elements (e.g., Malm et al., 1994; Chow et al., 2003; Prabhakar et al., 2014a): Al (81.8%), Si (76.3%), Mn (63.3%), Fe (61.1%), and Ca (50.2%). Dust exhibited a bimodal temporal profile with minimum concentrations between November and February owing to low wind speeds and high soil moisture. Maximum mass concentrations of dust occurred between the spring months of March and May, likely related to dust transport from distant sources including Asia (Tratt et al., 2001; VanCuren and Cahill, 2002; Wells et al., 2007; Kavouras et al., 2009; Creamean et al., 2014; Lopez et al., 2016) and neighboring states such as Nevada (Figure 5). Locally generated dust likely did not contribute significantly in those months as PMcoarse did not exhibit its highest levels (Figure 4). Ideal conditions for local dust emissions (low soil moisture, high wind speed) occurred later in the summer months. In contrast to March-May, the second concentration peak for dust between September-October was most likely driven by local sources, supported by PMcoarse exhibiting maximum mass concentrations (Figure 4) and its highest correlation with dust during these months (Table 1); furthermore, soil moisture is near its lowest values in these months too.
Table 1.
Correlation coefficients (r) between PMcoarse and seven PMF factors corresponding to the PM2.5 fraction of sampled aerosol. Values are only shown when statistically significant based on p < 0.05.
| PMcoarse |
|||||
|---|---|---|---|---|---|
| Annual | DJF | MAM | JJA | SON | |
| Biomass burning | − | − | − | 0.22 | − |
| Dust | 0.55 | 0.79 | 0.46 | 0.58 | 0.83 |
| Secondary sulfate | 0.38 | − | 0.34 | 0.36 | 0.21 |
| Secondary nitrate | − | − | 0.47 | 0.41 | − |
| Fresh sea salt | 0.24 | − | 0.39 | 0.26 | − |
| Vehicle emissions | 0.18 | 0.23 | 0.33 | 0.33 | 0.29 |
| Aged sea salt | 0.65 | 0.36 | 0.55 | 0.68 | 0.49 |
The CWT map of dust is somewhat similar to that of biomass burning in that major sources are California, Nevada, and Oregon. Dust can be co-emitted with biomass burning emissions due to the buoyancy and turbulence associated with fires near the surface (e.g., Kavouras et al., 2012; Popovicheva et al., 2014; Maudlin et al., 2015). The impact of Asian dust is recognizable from seasonal CWT maps (Figure S8) where spring months showed the greatest dust concentrations for trajectories transported over the Pacific Ocean. As Asian dust has previously been linked to California orographic precipitation (Ault et al., 2011), the PMF and CWT results emphasize the importance of considering also locally generated dust and plumes from neighboring states with regard to impacts on clouds and precipitation in the study region. At high altitudes, dust has been shown to be an important source of IN (e.g., Heintzenberg et al., 1996; Twohy and Gandrud, 1998; DeMott et al., 2003).
3.2.3. Secondary Sulfate
The secondary sulfate source factor (11.7% of total PM2.5) contributed the most of any factor to the mass concentrations of sulfate (51.5%), V (77.0%) and Ni (53.5%), the group of which is linked to fuel oil combustion such as with shipping (Moldanova et al., 2009; Coggon et al., 2012). Aside from secondary production of sulfate stemming from SO2 emitted by ships and potentially other regional combustion sources, another significant source of sulfate in the region is secondary production from dimethlysulfide (DMS) emissions over the ocean. Sulfur emissions from ships were previously found to be of the same order of magnitude as that of DMS fluxes in a marine environment (Corbett and Fischbeck, 1997). The peak in mass concentration for secondary sulfate in July is consistent with many past studies showing a summertime peak owing to enhanced photochemical activity and reactions facilitated in conditions of high moisture (e.g., Hidy et al., 1978; Baumgardner et al., 1999; Tai et al., 2010; Hand et al., 2012).
The CWT map shows that strong contributions come from marine areas, especially along the southern coast where both the Port of Los Angeles and the Port of Long Beach are situated. These two ports are among the busiest ones in the United States by container volume (US Department of Transportation, 2017). The high sulfate levels stretch from between those ports to farther north by Oakland and San Francisco where there is major port activity as well.
Analysis using the non-parametric Mann-Kendall test at the 95% significance level revealed that secondary sulfate is the only PM2.5 factor showing a significant trend versus time in its mean annual concentration. In agreement with previous studies showing sulfate reductions (e.g., Malm et al., 2002; Sorooshian et al., 2011; Chan et al., 2018), this factor exhibited a decreasing trend (−0.06 μg m−3 yr−1) over the study period most likely due to the reduction in SO2 emissions (Hand et al., 2012). This is an important result with regard to impacts on regional aerosol properties as sulfate is a hygroscopic component of ambient particles and its reduced levels can alter overall water-uptake properties; also, less sulfate in ambient particles can allow for more ammonium nitrate production as formation of this latter salt is thermodynamically less favorable than ammonium sulfate (e.g., Seinfeld and Pandis, 2016). Less particulate sulfate also has the important effect of limiting the ability of this species to reduce precipitation acidity, where H2SO4 has been shown to be the more significant acidifying agent in wet deposition in contrast to the other major inorganic acid, HNO3, for some regions (e.g., Tuncer et al., 2001; Naimabadi et al., 2018).
3.2.4. Secondary Nitrate
The secondary nitrate source factor accounted for 5.2% of total PM2.5, the lowest of any factor. As expected, this source was the dominant contributor to nitrate (63.2%), which is a result of secondary formation from NOx, sources of which are diverse in the region including ship exhaust, biomass burning, agricultural activity, and urban emissions (Prabhakar et al., 2014b). The monthly profile of secondary nitrate mimics what would be expected for a species favoring conditions associated with colder conditions in winter months (December – February), coincident with more wood burning and a shallower PBLH. Thermodynamically, nitrate favors the aerosol phase at lower temperatures (e.g., Johnson et al., 1994). In contrast to the other source factors, the CWT map for secondary nitrate exhibits strong sources both over the ocean and over the western United States spanning the states of California, Nevada, Oregon, and Washington, consistent with precursor sources being over land and the ocean.
3.2.5. Fresh and Aged Sea Salt
The fresh sea salt source factor accounted for 15.0% of total PM2.5. This factor contributes the most to the overall mass concentrations of Na (60.6%) and Cl− (93.9%). The monthly profile reveals that the highest PM2.5 concentration of fresh sea salt was in May and June. The CWT map expectedly shows greater mass concentrations associated with trajectories coming from the northwest over the Pacific Ocean.
The aged sea salt factor contributed 25.7% to total PM2.5. In contrast to fresh sea salt, this factor contributed more to a wider range of species Na (37.1%), Br (36.3%), nitrate (34.9%), Cu (28.6%), OC (24.7%), sulfate (25.1%), and Ni (24.4%), in addition to having a negligible contribution to Cl−. The high concentration of sulfate, nitrate, and OC can be attributed to sea salt aging processes. Sea salt aging includes the reaction of sea salt aerosol with acidic gases that replace Cl− (chloride depletion) by other anions and release hydrochloric acid (Meinert and Winchester, 1977; Eldering et al., 1991). Sulfuric acid and nitric acids are among most abundant inorganic acids causing chloride depletion (Sievering et al., 1991; ten Brink, 1998; Yao et al., 2003). Moreover, organic acids emitted from various sources (e.g., wildfires) will alter sea salt composition. For instance, in a study at a nearby coastal site, Braun et al. (2017) showed that Cl−exhibited substantially diminished concentrations during the Soberanes Fire.
The temporal profile of aged sea salt differs from that of fresh sea salt in that the peak concentrations were between June and September rather than May-June. This may be explained by more efficient production of secondary aerosol species in the summertime owing to higher incident solar radiation and photochemical reactivity (Lack et al., 2004; Tai et al., 2010). More specifically, there are enhanced biogenic volatile organic compound emissions during the summer in the study region (Sorooshian et al., 2015), which, when coupled to high concentrations of oxidants (e.g., hydroxyl radicals), can produce high levels of secondary organic aerosol (SOA) species such as organic acids that have been shown to partition to coarse sea salt particles (Laskin et al., 2012; Braun et al., 2017). Also notable during the summertime in the region is efficient aqueous chemistry to produce SOA, especially organic acids, and secondary sulfate (Sorooshian et al., 2007) that can subsequently interact with sea salt particles. Similar to fresh sea salt, the CWT map for aged sea salt shows that the major sources reside over the Pacific Ocean, with the difference being that there is more influence from sources, including ship exhaust, closer to the coastline extending from central California to British Columbia. The spatial pattern of highest concentrations is expectedly close to that of secondary sulfate. The findings associated with sea salt emphasize the importance of distinguishing between aged and fresh sea salt owing to different chemical profiles, and thus varying hygroscopic and radiative properties.
3.2.6. Vehicle Emissions
The vehicle emission source factor contributed 8.2% to total PM2.5. This factor contributed the most to EC (27.0%) and OC (13.6%), in addition to metals including Cu (71.4%), Zn (46.0%), Fe (20.6%), and Mn (17.3%). OC and EC are established markers of both diesel and gasoline vehicular exhaust (El Haddad et al., 2009; Zhu et al., 2010; Pant and Harrison, 2013). The presence of metal tracers in this factor is due not only to vehicular exhaust, but also non-exhaust emissions such as brake wear, tire wear, and re-suspension of dust (Wahlin et al., 2006; Fabretti et al., 2009; Pant and Harrison, 2013). There is also a notable contribution from this factor to Br (30%), which has been observed in vehicle exhaust in previous studies (Lim et al., 2010; Peltier et al., 2011). In fact, Br is common additive element used in gasoline and lubricating oil (Huang et al., 1994).
The monthly profile of the vehicle emissions factor differs from all other sources in that it exhibited a dominant peak in October, which is coincidentally the month where dust exhibited a secondary peak and linked earlier to locally generated sources rather than long-range transport. Dust can be emitted via vehicular activity (i.e., road dust), which is the connection between vehicular emissions tracers and dust. The lowest concentrations of this source factor in the summer months may be due to more dilution owing to the highest year-round PBLHs (Figure 2). The CWT map shows that the main source regions coincide with areas containing busy roadways including the Central Valley, southern California, and western Nevada.
3.3. PMcoarse
Although not a PMF source factor, PMcoarse is relevant to the discussion as it provides insight into the nature of larger aerosol particles, specifically dust and sea salt. Its monthly profile most closely resembles that of aged sea salt rather than dust or fresh sea salt owing to the proximity to the ocean and because the aerosol has been aged by the time it reaches the PNM site, respectively. Consistent with that result, PMcoarse concentrations are best correlated with those of aged sea salt (r = 0.65) followed by dust (r = 0.55) on annual time scales (Table 1). PMcoarse is best correlated with aged sea salt for half of the year (MAM, JJA), while it was best correlated with dust for the other half of the year (DJF, SON). The CWT profiles of PMcoarse expectedly represent a hybrid of the two sea salt factors and dust.
PMcoarse exhibited a statistically significant upward trend in its mean annual concentration during the study period (0.17 µg m−3 yr−1), which, as will be shown subsequently, may have been driven to at least some extent by increasing levels of coarse dust particles. This speculation is based on a significant relationship between PMcoarse and a dust tracer in wet deposition (Ca2+), whereas PMcoarse exhibited insignificant relationships with both major sea salt constituents (Na+ and Cl−) (Table 2).
Table 2.
Correlation coefficients (r) of both weekly mass cocentrations (values before “/”) and mass fractions (values after “/”) of precipitation species versus the weekly averaged aerosol mass concetrations for seven PMF factors and also total PM2.5 and PMcoarse. Values are only shown when statistically significant based on p < 0.05.
| Ca2+:Rain | Mg2+:Rain | K+:Rain | Na+:Rain | NH4+:Rain | NO3−:Rain | Cl−:Rain | SO42−:Rain | pH:Rain | |
|---|---|---|---|---|---|---|---|---|---|
| Biomass burning | − / − | − / − | − / − | − / − | − / − | − / − | − / − | − / − | - |
| Dust | 0.36 / 0.34 | − / − | − / − | − / −0.25 | 0.31 / − | 0.31 / − | − / −0.24 | 0.30 / 0.37 | - |
| Secondary sulfate | −/ 0.27 | − / − | − / − | − / − | − / − | − / − | − / − | − / 0.45 | −0.23 |
| Secondary nitrate | − / − | − / − | − / − | − / − | − / − | − / − | − / − | − / − | - |
| Fresh sea salt | − / − | − / − | − / − | − / − | − / − | − / −0.20 | − / − | − / − | - |
| Vehicle emissions | − / − | − / − | − / −0.19 | − / − | − / − | − / 0.25 | − / − | − / − | −0.31 |
| Aged sea salt | 0.26 / 0.25 | − / − | − / − | − / − | − / − | 0.23 / − | − / − | − / 0.24 | −0.24 |
| PM2.5 | 0.24 / − | − / − | − / − | − / − | − / − | 0.19 / − | − / − | 0.20 / 0.21 | −0.23 |
| PMcoarse | 0.28 / 0.22 | − / − | − / − | − / − | 0.19 / − | 0.24 / − | − / − | 0.23 / 0.34 | - |
3.4. Cumulative Precipitation Chemistry Profile
The monthly profiles and time series of species concentrations and pH are summarized in Figure 6a and Figure S9, respectively. The non-parametric Kruskal-Wallis test was applied (Table S4) to the NADP/NTN data to determine whether the species concentrations and pH values exhibited significant (p < 0.05) monthly differences. Except for Na+, Cl−, and Mg2+, all other species and pH showed significant differences in averaged monthly values. Monthly averaged values of pH ranged from 5.33 to 5.51. Of note are the higher concentrations for all species and lowest pH in May, owing largely to very low precipitation in that month and presumably less dilution in solution as compared to other months. This would have led to higher concentrations of acidic species. For analagous reasons, the month with the highest precipitation accumulation, December, exhibited among the lowest ion concentrations and the highest pH. The non-parametric Mann-Kendall test was applied to the precipitation species concentrations and pH to see if any significant trends were evident during the study period, and none appeared.
Figure 6.
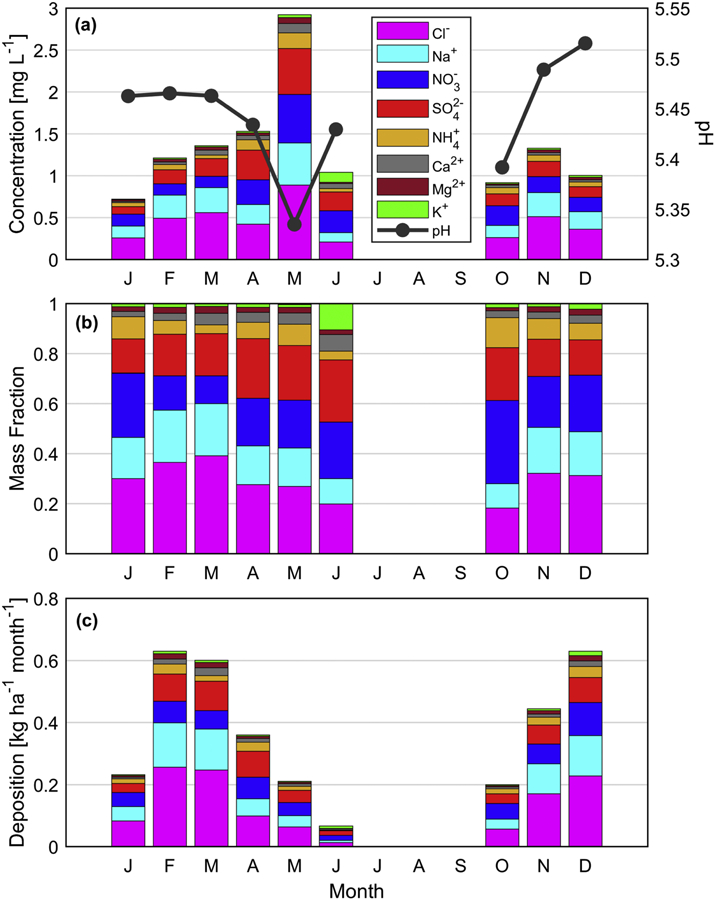
Monthly (a) precipitation-weighted average of weekly species concentrations, (b) precipitation-weighted average of weekly species mass fractions, and (c) wet deposition flux of species from the PNM site between 2010 and 2016. The number of samples in each month ranged from 6–23, except for June (n = 3) and July-September (n = 0).
The monthly profile of mass fractions reveals that the most abundant species was either Cl− (0.18 – 0.39), SO42− (0.14 – 0.25), or NO3− (0.11 – 0.33). The least abundant species was usually K+ with the exception of June when it mass fraction reached 0.11 in contrast to being < 0.02 in other months. It is unclear though as to what the source was as the biomass burning factor (a major source of K+) exhibited its lowest concentration in that month.
Another way to examine the precipitation data is by examining deposition fluxes (Figure 6c), which has similarity to the monthly profile of precipitation accumulation. Among months with precipitation, deposition fluxes of the species examined peaked in February, March, and December, with the lowest fluxes being in June. In most months with precipitation, the major sea salt components (Na+ and Cl−) accounted for most of the deposition by mass among the species examined. Sulfate and NO3− were the next highest in terms of overall deposition flux among the nine months with recorded data, with especially high levels in June and October, as evident by their high combined mass fractions in those months.
There were strong interrelationships between the various ions measured (Table S5), with the strongest being between sea salt constituents: Na+ and Cl− (r = 1.00), Mg2+ and Cl− (r = 0.99), Mg2+ and Na+ (r = 0.99). The significant relationships between species typically associated with secondary production mechanisms (SO42−, NO3−, NH4+) and the rest which are commonly associated with sea salt and/or crustal particles, is consistent with other regions (Hutchings et al., 2009; Satyanarayana et al., 2010; Sorooshian et al., 2013; Mora et al., 2017). This can be explained by the partitioning of secondarily produced species to the coarser aerosol particle types (sea salt, dust) or by scavenging of the former by hydrometeors (Granat et al., 2002; van der Swaluw et al., 2011). The pH of wet deposition exhibited a significant relationship with only NO3− (r = −0.35) indicative of how that species, at the minimum, is associated with more acidic precipitation. Liquid water in the form of drops readily takes up HNO3, which is a likely explanation for the reduction in pH.
3.5. Aerosol-Precipitation Chemical Interrelationships
Mass fractions of the precipitation species were compared to concentrations of the PMF factors, PM2.5, and PMcoarse (Table 2), with the results essentially resembling those when comparing concentrations of the precipitation species with those of PMF factors. The pH of precipitation was inversely related to factors associated with more acidic elements such as secondary sulfate and aged sea salt. The fact that pH was only correlated with NO3− among the precipitation species and not secondary nitrate in the aerosol phase is further supportive of the speculation that there was uptake of HNO3 in the liquid phase, which would not manifest itself in the surface aerosol measurements.
The mass fraction of Ca2+ in precipitation exhibited its highest r value (0.34) with the dust source factor, suggestive of dust seeding precipitation leading to the higher Ca2+ levels in precipitation samples. As described in previous studies, dust aerosol can play a role as IN and increase preciptation in mixed-phase clouds (e.g., Muhlbauer and Lohmann, 2009; Rosenfeld et al., 2011). A similar link between dust and precipitation has been observed in nearby regions such as Arizona and New Mexico (Hutchings et al., 2009; Sorooshian et al., 2013), Texas (Ponette-González et al., 2018), and the Sierra Nevada in California (Ault et al., 2011; Creamean et al., 2013). In contrast to fresh sea salt, the aged sea salt source factor was also correlated with Ca2+ in precipitation (r = 0.25), suggesting that marine emissions are another contributor to Ca2+ in the region’s wet deposition.
For the three most abundant components in precipitation based on mass fractions, Cl− was only correlated with dust, SO42− was correlated with dust, secondary sulfate, and aged sea salt, while NO3− was correlated with fresh sea salt and vehicle emissions (Table 2). It is noteworthy that there was a significant relationship between SO42− in precipitation with both the dust and aged sea salt factors of PM2.5, in addition to PMcoarse, owing most likely to the partitioning of secondary sulfate to coarse aerosol surfaces (Usher et al., 2002; Manktelow et al., 2010; Wang et al., 2016). In the aerosol phase, the secondary sulfate factor was positively correlated with the dust factor (r = 0.32, p < 0.05), aged sea salt factor (r = 0.30, p < 0.05), and PMcoarse (r = 0.38, p 0.05). Size-resolved aerosol measurements closer to the coast in central California show that SO42−, NO3−, and organic acids reside in the same size range as where sea salt and dust aerosol exhibit their highest mass concentrations (Maudlin et al., 2015). Lastly, it was of note that the biomass burning source factor did not exhibit a significant relationship with the mass fraction of any precipitation species.
4. Conclusions
This study leveraged a long-term dataset of co-located aerosol and precipitation composition measurements at a central California site (Pinnacles National Monument) that is impacted by diverse pollutant types including marine emissions, dust, wildfires, and urban emissions. The goal was to critically examine the data to understand pollutant sources and to additionally use interrelationships between aerosol and precipitation species to gain insight into both aerosol types impacting clouds that produce the precipitation and interactions between gases and particles with precipitation drops (e.g., uptake processes and wet scavenging).
A thorough characterization of pollution sources was presented including their relative contributions to total PM2.5 and the influence of transport, meteorology, and time of year. Aged sea salt was the most influential source on a mass basis to PM2.5, which was shown to exhibit a distinct chemical profile relative to fresh salt in that there was a likely contribution from inorganic and organic acids stemming from precursors emissions from both natural and anthropogenic sources. Interstate transport of pollution was shown to be important with regard to aerosol types linked to dust, wildfire emissions, secondary nitrate, and vehicular emissions, while Asian dust was shown to be impactful in the study region in spring months. Of note was that secondary sulfate represented the only PM2.5 pollutant source exhibiting a significant trend during the study period as its levels have decreased, with implications of this phenomenon discussed. In contrast, PMcoarse exhibited a significant increase owing most likely to the influence of dust.
Intercomparison of aerosol and precipitation data revealed that the two aerosol source factors (dust and aged sea salt) and PMcoarse exhibited a positive association with Ca2+ in precipitation, suggestive of directly emitted aerosol types with larger sizes impacting precipitation. Furthermore, sulfate levels in both the aerosol and precipitation samples analyzed were significantly correlated with dust and aged sea salt PMF factors, pointing to the partitioning of secondary sulfate to dust and sea salt particles. Presumably other acids may partition to these coarse aerosol types as well and impact precipitation, such as organic acids; however, speciated organics were unavailable in the precipitation dataset. Intercomparisons of the aerosol and precipitation datasets reveal that there likely was efficient uptake of HNO3 to drops, which served to reduce precipitation pH values.
The results of this analysis revealed that simultaneously collected aerosol and precipitation chemistry data at a single site can indeed provide useful insight into processes associated with aerosol effects on clouds (via CCN and IN sources) and also precipitation effects on aerosol (via scavenging and uptake of gases). Further research is warrranted to validate some of the explanations provided in this intercomparison as the current datasets cannot provide unambiguous proof of cause-and-effect relationships. However, a major motivation of the intercomparison is to infer about processes to subsequently validate with more focused measurements and modeling studies.
Supplementary Material
Highlights.
Characterization of aerosol and precipitation chemistry at a central California site
Co-located aerosol and wet deposition data examined between 2010 and 2016
Positive matrix factorization and concentration weighted trajectory maps were used
Seven sources were identified with temporal profiles dependent on meteorology
Insights were gained into aerosol-precipitation interactions
Acknowledgements
All data used can be obtained from websites and references provided in Section 2. This work was funded by Grant 2 P42 ES04940 from the National Institute of Environmental Health Sciences (NIEHS) Superfund Research Program, NIH and ONR grants N00014–16-1–2567 and N00014–10-1–0811. IMPROVE is a collaborative association of state, tribal, and federal agencies, and international partners. US Environmental Protection Agency is the primary funding source, with contracting and research support from the National Park Service. The Air Quality Group at the University of California, Davis is the central analytical laboratory, with ion analysis provided by Research Triangle Institute, and carbon analysis provided by Desert Research Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ault AP, Williams CR, White AB, Neiman PJ, Creamean JM, Gaston CJ, et al. Detection of Asian dust in California orographic precipitation. Journal of Geophysical Research-Atmospheres 2011; 116. [Google Scholar]
- Baumgardner RE, Isil SS, Bowser JJ, and Fitzgerald KM. Measurements of rural sulfur dioxide and particle sulfate: Analysis of CASTNet data, 1987 through 1996. Journal of Air Waste Management 1999; 49, 1266–1279. [DOI] [PubMed] [Google Scholar]
- Bobbink R, Hornung M, Roelofs JGM. The effects of air-borne nitrogen pollutants on species diversity in natural and semi-natural European vegetation. Journal of Ecology 1998; 86: 717–738. [Google Scholar]
- Braun RA, Dadashazar H, MacDonald AB, Aldhaif AM, Maudlin LC, Crosbie E, et al. Impact of Wildfire Emissions on Chloride and Bromide Depletion in Marine Aerosol Particles. Environmental Science & Technology 2017; 51: 9013–9021. [DOI] [PubMed] [Google Scholar]
- Cahill CF, Cahill TA, Perry KD. The size- and time-resolved composition of aerosols from a sub-Arctic boreal forest prescribed burn. Atmospheric Environment 2008; 42: 7553–7559. [Google Scholar]
- Chan EAW, Gantt B, and Mcdow S. The reduction of summer sulfate and switch from summertime to wintertime PM2.5 concentration maxima in the United States. Atmospheric Environment 2018; 175: 25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow JC, Watson JG, Ashbaugh LL, Magliano KL. Similarities and differences in PM10 chemical source profiles for geological dust from the San Joaquin Valley, California. Atmospheric Environment 2003; 37: 1317–1340. [Google Scholar]
- Chow JC, Watson JG, Chen LWA, Chang MCO, Robinson NF, Trimble D, and Kohl S. The IMPROVE-A temperature protocol for thermal/optical carbon analysis: maintaining consistency with a long-term database, Journal of the Air Waste Management Association 2007; 57: 1014–1023. [DOI] [PubMed] [Google Scholar]
- Chow JC, Lowenthal DH, Chen LWA, Wang XL, Watson JG. Mass reconstruction methods for PM2.5: a review. Air Quality Atmosphere and Health 2015; 8: 243–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coggon MM, Sorooshian A, Wang Z, Metcalf AR, Frossard AA, Lin JJ, et al. Ship impacts on the marine atmosphere: insights into the contribution of shipping emissions to the properties of marine aerosol and clouds. Atmospheric Chemistry and Physics 2012; 12: 8439–8458. [Google Scholar]
- Corbett JJ, Fischbeck P. Emissions from ships. Science 1997; 278: 823–824. [Google Scholar]
- Creamean JM, Suski KJ, Rosenfeld D, Cazorla A, DeMott PJ, Sullivan RC, et al. Dust and Biological Aerosols from the Sahara and Asia Influence Precipitation in the Western U.S. Science 2013; 339: 1572–1578. [DOI] [PubMed] [Google Scholar]
- Creamean JM, Spackman JR, Davis SM, White AB. Climatology of long-range transported Asian dust along the West Coast of the United States. Journal of Geophysical Research-Atmospheres 2014; 119: 12171–12185. [Google Scholar]
- DeMott PJ, Sassen K, Poellot MR, Baumgardner D, Rogers DC, Brooks SD, et al. African dust aerosols as atmospheric ice nuclei. Geophysical Research Letters 2003; 30. [Google Scholar]
- Dennison PE, Brewer SC, Arnold JD, and Moritz MA: Large wildfire trends in the western United States, 1984–2011, Geophysical Research Letter 2014; 41, 2928–2933. [Google Scholar]
- Driscoll CT, Whitall D, Aber J, Boyer E, Castro M, Cronan C, et al. Nitrogen pollution in the northeastern United States: Sources, effects, and management options. Bioscience 2003; 53: 357–374. [Google Scholar]
- El Haddad I, Marchand N, Dron J, Temime-Roussel B, Quivet E, Wortham H, et al. Comprehensive primary particulate organic characterization of vehicular exhaust emissions in France. Atmospheric Environment 2009; 43: 6190–6198. [Google Scholar]
- Eldering A, Solomon PA, Salmon LG, Fall T, Cass GR. Hydrochloric-Acid - a Regional Perspective on Concentrations and Formation in the Atmosphere of Southern California. Atmospheric Environment Part a-General Topics 1991; 25: 2091–2102. [Google Scholar]
- Fabretti JF, Sauret N, Gal JF, Maria PC, Scharer U. Elemental characterization and source identification of PM2.5 using Positive Matrix Factorization: The Malraux road tunnel, Nice, France. Atmospheric Research 2009; 94: 320–329. [Google Scholar]
- Fan J, Leung LR, DeMott PJ, Comstock JM, Singh B, Rosenfeld D, et al. Aerosol impacts on California winter clouds and precipitation during CalWater 2011: local pollution versus long-range transported dust. Atmospheric Chemistry and Physics 2014; 14: 81–101. [Google Scholar]
- Granat L, Norman M, Leck C, Kulshrestha UC, Rodhe H. Wet scavenging of sulfur compounds and other constituents during the Indian Ocean Experiment (INDOEX). Journal of Geophysical Research-Atmospheres 2002; 107. [Google Scholar]
- Hand JL, Schichtel BA, Malm WC, and Pitchford ML. Particulate sulfate ion concentration and SO2 emission trends in the United States from the early 1990s through 2010. Atmospheric Chemistry & Physics 2012; 12: 10353–10365. [Google Scholar]
- Heintzenberg J, Okada K, Strom J. On the composition of non-volatile material in upper tropospheric aerosols and cirrus crystals. Atmospheric Research 1996; 41: 81–88. [Google Scholar]
- Hidy GM, Mueller PK, Tong EY. Spatial and Temporal Distributions of Airborne Sulfate in Parts of United-States. Atmospheric Environment 1978; 12: 735–752. [Google Scholar]
- Hopke PK. Review of receptor modeling methods for source apportionment. Journal of the Air & Waste Management Association 2016; 66: 237–259. [DOI] [PubMed] [Google Scholar]
- Hsu YK, Holsen TM, Hopke PK. Comparison of hybrid receptor models to locate PCB sources in Chicago. Atmospheric Environment 2003; 37: 545–562. [Google Scholar]
- Huang XD, Olmez I, Aras NK, Gordon GE. Emissions of Trace-Elements from Motor-Vehicles - Potential Marker Elements and Source Composition Profile. Atmospheric Environment 1994; 28: 1385–1391. [Google Scholar]
- Hutchings JW, Robinson MS, McIlwraith H, Kingston JT, Herckes P. The Chemistry of Intercepted Clouds in Northern Arizona during the North American Monsoon Season. Water Air and Soil Pollution 2009; 199: 191–202. [Google Scholar]
- IPCC: Summary for Policymakers. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change (eds Stocker TF, Qin D, Plattner GK, Tignor M, Allen SK, Boschung J, Nauels A, Xia Y, Bex V & Midgley PM) 2013; Cambridge University Press: Cambridge, United Kingdom and New York, NY, USA. [Google Scholar]
- Johnson BJ, Huang SC, Lecave M, Porterfield M. Seasonal Trends of Nitric-Acid, Particulate Nitrate, and Particulate Sulfate Concentrations at a Southwestern United-States Mountain Site. Atmospheric Environment 1994; 28: 1175–1179. [Google Scholar]
- Kavouras IG, Etyemezian V, DuBois DW, Xu J, Pitchford M. Source reconciliation of atmospheric dust causing visibility impairment in Class I areas of the western United States. Journal of Geophysical Research-Atmospheres 2009; 114. [Google Scholar]
- Kavouras IG, Nikolich G, Etyemezian V, DuBois DW, King J, Shafer D. In situ observations of soil minerals and organic matter in the early phases of prescribed fires. Journal of Geophysical Research-Atmospheres 2012; 117. [Google Scholar]
- Kim E, Hopke PK, Edgerton ES. Source identification of Atlanta aerosol by positive matrix factorization. Journal of the Air & Waste Management Association 2003; 53: 731–739. [DOI] [PubMed] [Google Scholar]
- Lack DA, Tie XX, Bofinger ND, Wiegand AN, Madronich S. Seasonal variability of secondary organic aerosol: A global modeling study. Journal of Geophysical Research-Atmospheres 2004; 109. [Google Scholar]
- Lamb D, Bowersox V. The national atmospheric deposition program: an overview. Atmospheric Environment 2000; 34: 1661–1663. [Google Scholar]
- Laskin A, Moffet RC, Gilles MK, Fast JD, Zaveri RA, Wang BB, et al. Tropospheric chemistry of internally mixed sea salt and organic particles: Surprising reactivity of NaCl with weak organic acids. Journal of Geophysical Research-Atmospheres 2012; 117. [Google Scholar]
- Lee JH, Hopke PK, Turner JR. Source identification of airborne PM2.5 at the St. Louis-Midwest Supersite. Journal of Geophysical Research-Atmospheres 2006; 111. [Google Scholar]
- Lim JM, Lee JH, Moon JH, Chung YS, Kim KH. Source apportionment of PM10 at a small industrial area using Positive Matrix Factorization. Atmospheric Research 2010; 95: 88–100. [Google Scholar]
- Liu HY, Jacob DJ, Bey I, Yantosca RM. Constraints from Pb-210 and Be-7 on wet deposition and transport in a global three-dimensional chemical tracer model driven by assimilated meteorological fields. Journal of Geophysical Research-Atmospheres 2001; 106: 12109–12128. [Google Scholar]
- Lopez DH, Rabbani MR, Crosbie E, Raman A, Arellano AF, Sorooshian A. Frequency and Character of Extreme Aerosol Events in the Southwestern United States: A Case Study Analysis in Arizona. Atmosphere 2016; 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald AB, Dadashazar H, Chuang PY, Crosbie E, Wang HL, Wang Z, et al. Characteristic Vertical Profiles of Cloud Water Composition in Marine Stratocumulus Clouds and Relationships With Precipitation. Journal of Geophysical Research-Atmospheres 2018; 123: 3704–3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malm WC, Sisler JF, Huffman D, Eldred RA, Cahill TA. Spatial and Seasonal Trends in Particle Concentration and Optical Extinction in the United-States. Journal of Geophysical Research-Atmospheres 1994; 99: 1347–1370. [Google Scholar]
- Malm WC, Schichtel BA, Ames RB, and Gebhart KA. A 10-year spatial and temporal trend of sulfate across the United States, Journal of Geophysical Research-Atmosphere 2002; 107: 4627. [Google Scholar]
- Manktelow PT, Carslaw KS, Mann GW, Spracklen DV. The impact of dust on sulfate aerosol, CN and CCN during an East Asian dust storm. Atmospheric Chemistry and Physics 2010; 10: 365–382. [Google Scholar]
- Masiol M, Squizzato S, Rampazzo G, Pavoni B. Source apportionment of PM2.5 at multiple sites in Venice (Italy): Spatial variability and the role of weather. Atmospheric Environment 2014; 98: 78–88. [Google Scholar]
- Masiol M, Hopke PK, Felton HD, Frank BP, Rattigan OV, Wurth MJ, et al. Source apportionment of PM2.5 chemically speciated mass and particle number concentrations in New York City. Atmospheric Environment 2017; 148: 215–229. [Google Scholar]
- Maudlin LC, Wang Z, Jonsson HH, Sorooshian A. Impact of wildfires on size-resolved aerosol composition at a coastal California site. Atmospheric Environment 2015; 119: 59–68. [Google Scholar]
- Meinert DL, Winchester JW. Chemical Relationships in North-Atlantic Marine Aerosol. Journal of Geophysical Research-Oceans and Atmospheres 1977; 82: 1778–1782. [Google Scholar]
- Moldanova J, Fridell E, Popovicheva O, Demirdjian B, Tishkova V, Faccinetto A, et al. Characterisation of particulate matter and gaseous emissions from a large ship diesel engine. Atmospheric Environment 2009; 43: 2632–2641. [Google Scholar]
- Mora M, Braun RA, Shingler T, Sorooshian A. Analysis of remotely sensed and surface data of aerosols and meteorology for the Mexico Megalopolis Area between 2003 and 2015. Journal of Geophysical Research-Atmospheres 2017; 122: 8705–8723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhlbauer A, Lohmann U. Sensitivity Studies of Aerosol-Cloud Interactions in Mixed-Phase Orographic Precipitation. Journal of the Atmospheric Sciences 2009; 66: 2517–2538. [Google Scholar]
- NADP, 2014. Quality Assurance Plan - Central Analytical Laboratory, NADP QA Plan 2014–01, Version 7: http://nadp.sws.uiuc.edu/lib/qaPlans/qapCal2014.pdf.
- Naimabadi A, Shirmardi M, Maleki H, Teymouri P, Goudarzi G, Shahsavani A, et al. On the chemical nature of precipitation in a populated Middle Eastern Region (Ahvaz, Iran) with diverse sources. Ecotoxicology and Environmental Safety 2018; 163: 558–566. [DOI] [PubMed] [Google Scholar]
- Paatero P, Tapper U. Positive Matrix Factorization - a Nonnegative Factor Model with Optimal Utilization of Error-Estimates of Data Values. Environmetrics 1994; 5: 111–126. [Google Scholar]
- Paatero P Least squares formulation of robust non-negative factor analysis. Chemometrics and Intelligent Laboratory Systems 1997; 37: 23–35. [Google Scholar]
- Pant P, Harrison RM. Estimation of the contribution of road traffic emissions to particulate matter concentrations from field measurements: A review. Atmospheric Environment 2013; 77: 78–97. [Google Scholar]
- Pardo LH, Fenn ME, Goodale CL, Geiser LH, Driscoll CT, Allen EB, et al. Effects of nitrogen deposition and empirical nitrogen critical loads for ecoregions of the United States. Ecological Applications 2011; 21: 3049–3082. [Google Scholar]
- Peltier RE, Lippmann M. Spatial and seasonal distribution of aerosol chemical components in New York City: (1) Incineration, coal combustion, and biomass burning. Journal of Exposure Science and Environmental Epidemiology 2011; 21: 473–483. [DOI] [PubMed] [Google Scholar]
- Polissar AV, Hopke PK, Paatero P. Atmospheric aerosol over Alaska - 2. Elemental composition and sources. Journal of Geophysical Research-Atmospheres 1998; 103: 19045–19057. [Google Scholar]
- Polissar AV, Hopke PK, Poirot RL. Atmospheric aerosol over Vermont: Chemical composition and sources. Environmental Science & Technology 2001; 35: 4604–4621. [DOI] [PubMed] [Google Scholar]
- Ponette-González AG, Collins JD, Manuel JE, Byers TA, Glass GA, Weathers KC, Gill Wet dust deposition during the 2012 US drought: An overlooked pathway for elemental flux to ecosystems. Journal of Geophysical Research-Atmospheres 2018, 123: 8238–8254. [Google Scholar]
- Popovicheva O, Kistler M, Kireeva E, Persiantseva N, Timofeev M, Kopeikin V, et al. Physicochemical characterization of smoke aerosol during large-scale wildfires: Extreme event of August 2010 in Moscow. Atmospheric Environment 2014; 96: 405–414. [Google Scholar]
- Prabhakar G, Sorooshian A, Toffol E, Arellano AF, Betterton EA. Spatiotemporal distribution of airborne particulate metals and metalloids in a populated arid region. Atmospheric Environment 2014a; 92: 339–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhakar G, Ervens B, Wang Z, Maudlin LC, Coggon MM, Jonsson HH, et al. Sources of nitrate in stratocumulus cloud water: Airborne measurements during the 2011 E-PEACE and 2013 NiCE studies. Atmospheric Environment 2014b; 97: 166–173. [Google Scholar]
- Ralph FM, Neiman PJ, Wick GA. Satellite and CALJET aircraft observations of atmospheric rivers over the eastern north pacific ocean during the winter of 1997/98. Monthly Weather Review 2004; 132: 1721–1745. [Google Scholar]
- Ralph FM, Coleman T, Neiman PJ, Zamora RJ, Dettinger MD. Observed Impacts of Duration and Seasonality of Atmospheric-River Landfalls on Soil Moisture and Runoff in Coastal Northern California. Journal of Hydrometeorology 2013; 14: 443–459. [Google Scholar]
- Ralph FM, Prather KA, Cayan D, Spackman JR, DeMott P, Dettinger M, et al. Calwater Field Studies Designed to Quantify the Roles of Atmospheric Rivers and Aerosols in Modulating Us West Coast Precipitation in a Changing Climate. Bulletin of the American Meteorological Society 2016; 97: 1209–1228. [Google Scholar]
- Rodhe H, Langner J, Gallardo L, Kjellstrom E. Global scale transport of acidifying pollutants. Water Air and Soil Pollution 1995; 85: 37–50. [Google Scholar]
- Rolph GD Real-time environmental applications and display system (ready) 2016. [Available at http://ready.arl.noaa.gov.]
- Rosenfeld D, Yu X, Liu GH, Xu XH, Zhu YN, Yue ZG, et al. Glaciation temperatures of convective clouds ingesting desert dust, air pollution and smoke from forest fires. Geophysical Research Letters 2011; 38. [Google Scholar]
- Russell LM, Sorooshian A, Seinfeld JH, Albrecht BA, Nenes A, Ahlm L, et al. Eastern Pacific Emitted Aerosol Cloud Experiment. Bulletin of the American Meteorological Society 2013; 94: 709–729. [Google Scholar]
- Satyanarayana J, Reddy LAK, Kulshrestha MJ, Rao RN, Kulshrestha UC. Chemical composition of rain water and influence of airmass trajectories at a rural site in an ecological sensitive area of Western Ghats (India). Journal of Atmospheric Chemistry 2010; 66: 101–116. [Google Scholar]
- Schlosser JS, Braun RA, Bradley T, Dadashazar H, MacDonald AB, Aldhaif AA, et al. Analysis of aerosol composition data for western United States wildfires between 2005 and 2015: Dust emissions, chloride depletion, and most enhanced aerosol constituents. Journal of Geophysical Research-Atmospheres 2017; 122: 8951–8966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See RB, Schroder LJ, Willoughby TC. A Quality-Assurance Assessment for Constituents Reported by the National Atmospheric Deposition Program and the National Trends Network. Atmospheric Environment 1989; 23: 1801–1806. [Google Scholar]
- Seinfeld JH, Pandis SN. Atmospheric chemistry and physics (3rd ed.) 2016. New York: Wiley-Interscience. [Google Scholar]
- Sievering H, Boatman J, Galloway J, Keene W, Kim Y, Luria M, and Ray J. Heterogeneous Sulfur Conversion in Sea-Salt Aerosol-Particles - the Role of Aerosol Water-Content and Size Distribution. Atmospheric Environment. Part A. Generic Topics 1991; 25: 1479–1487. [Google Scholar]
- Solomon PA, Crumpler D, Flanagan JB, Jayanty RKM, Rickman EE, McDade CE. US National PM2.5 Chemical Speciation Monitoring Networks-CSN and IMPROVE: Description of networks. Journal of the Air & Waste Management Association 2014; 64: 1410–1438. [DOI] [PubMed] [Google Scholar]
- Sorooshian A, Lu ML, Brechtel FJ, Jonsson H, Feingold G, Flagan RC, et al. On the source of organic acid aerosol layers above clouds. Environmental Science & Technology 2007; 41: 4647–4654. [DOI] [PubMed] [Google Scholar]
- Sorooshian A, Wonaschutz A, Jarjour EG, Hashimoto BI, Schichtel BA, Betterton EA. An aerosol climatology for a rapidly growing arid region (southern Arizona): Major aerosol species and remotely sensed aerosol properties. Journal of Geophysical Research-Atmospheres 2011; 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorooshian A, Shingler T, Harpold A, Feagles CW, Meixner T, Brooks PD. Aerosol and precipitation chemistry in the southwestern United States: spatiotemporal trends and interrelationships. Atmospheric Chemistry and Physics 2013; 13: 7361–7379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorooshian A, Crosbie E, Maudlin LC, Youn JS, Wang Z, Shingler T, et al. Surface and airborne measurements of organosulfur and methanesulfonate over the western United States and coastal areas. Journal of Geophysical Research-Atmospheres 2015; 120: 8535–8548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorooshian A, MacDonald AB, Dadashazar H, Bates KH, Coggon MM, Craven JS, et al. A multi-year data set on aerosol-cloud-precipitation-meteorology interactions for marine stratocumulus clouds. Scientific Data 2018; 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein AF, Draxler RR, Rolph GD, Stunder BJB, Cohen MD, Ngan F. Noaa’s Hysplit Atmospheric Transport and Dispersion Modeling System. Bulletin of the American Meteorological Society 2015; 96: 2059–2077. [Google Scholar]
- Tai APK, Mickley LJ, Jacob DJ. Correlations between fine particulate matter (PM2.5) and meteorological variables in the United States: Implications for the sensitivity of PM2.5 to climate change. Atmospheric Environment 2010; 44: 3976–3984. [Google Scholar]
- ten Brink HM. Reactive uptake of HNO3 and H2SO4 in sea-salt (NaCl) particles. Journal of Aerosol Science 1998; 29: 57–64. [Google Scholar]
- Tratt DM, Frouin RJ, Westphal DL. April 1998 Asian dust event: A southern California perspective. Journal of Geophysical Research-Atmospheres 2001; 106: 18371–18379. [Google Scholar]
- Tuncer B, Bayar B, Yesilyurt C, Tuncel G. Ionic composition of precipitation at the Central Anatolia (Turkey). Atmospheric Environment 2001; 35: 5989–6002. [Google Scholar]
- Twohy CH, Gandrud BW. Electron microscope analysis of residual particles from aircraft contrails. Geophysical Research Letters 1998; 25: 1359–1362. [Google Scholar]
- US Department of Transportation, Port Performance Freight Statistics Program, 2017. <https:www.bts.gov>
- Usher CR, Al-Hosney H, Carlos-Cuellar S, Grassian VH. A laboratory study of the heterogeneous uptake and oxidation of sulfur dioxide on mineral dust particles. Journal of Geophysical Research-Atmospheres 2002; 107. [Google Scholar]
- VanCuren RA, Cahill TA. Asian aerosols in North America: Frequency and concentration of fine dust. Journal of Geophysical Research-Atmospheres 2002; 107. [Google Scholar]
- van der Swaluw E, Asman WAH, van Jaarsveld H, Hoogerbrugge R. Wet deposition of ammonium, nitrate and sulfate in the Netherlands over the period 1992–2008. Atmospheric Environment 2011; 45: 3819–3826. [Google Scholar]
- Wahlin P, Berkowicz R, Palmgren F. Characterisation of traffic-generated particulate matter in Copenhagen. Atmospheric Environment 2006; 40: 2151–2159. [Google Scholar]
- Wang YQ, Zhang XY, Draxler RR. TrajStat: GIS-based software that uses various trajectory statistical analysis methods to identify potential sources from long-term air pollution measurement data. Environmental Modelling & Software 2009; 24: 938–939. [Google Scholar]
- Wang YG, Hopke PK, Rattigan OV, Chalupa DC, Utell MJ. Multiple-year black carbon measurements and source apportionment using Delta-C in Rochester, New York. Journal of the Air & Waste Management Association 2012; 62: 880–887. [DOI] [PubMed] [Google Scholar]
- Wang QZ, Zhuang GS, Huang K, Liu TN, Lin YF, Deng CR, et al. Evolution of particulate sulfate and nitrate along the Asian dust pathway: Secondary transformation and primary pollutants via long-range transport. Atmospheric Research 2016; 169: 86–95. [Google Scholar]
- Wells KC, Witek M, Flatau P, Kreidenwei SM, Westphal DL. An analysis of seasonal surface dust aerosol concentrations in the western US (2001–2004): Observations and model predictions. Atmospheric Environment 2007; 41: 6585–6597. [Google Scholar]
- Westerling AL, Hidalgo HG, Cayan DR, and Swetnam TW. Warming and earlier spring increase western US forest wildfire activity. Science 2006: 313, 940–943. [DOI] [PubMed] [Google Scholar]
- Yao XH, Fang M, Chan CK. The size dependence of chloride depletion in fine and coarse sea-salt particles. Atmospheric Environment 2003; 37: 743–751. [Google Scholar]
- Xu J, DuBois D, Pitchford M, Green M, and Etyemezian V. Attribution of sulfate aerosols in Federal Class I areas of the western United States based on trajectory regression analysis. Atmospheric Environment 2006; 40, 3433–3447. [Google Scholar]
- Zhu CS, Chen CC, Cao JJ, Tsai CJ, Chou CCK, Liu SC, et al. Characterization of carbon fractions for atmospheric fine particles and nanoparticles in a highway tunnel. Atmospheric Environment 2010; 44: 2668–2673. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.