

The human fungal pathogen Candida albicans is the major etiological agent of vulvovaginal candidiasis (VVC). Despite this fact, other non-albicans Candida (NAC) species have frequently been reported, as well.
KEYWORDS: Candida, NAC species, VVC, immunopathogenesis, inflammasome, vaginitis, vulvovaginal
ABSTRACT
The human fungal pathogen Candida albicans is the major etiological agent of vulvovaginal candidiasis (VVC). Despite this fact, other non-albicans Candida (NAC) species have frequently been reported, as well. Despite their presence in the vaginal environment, little is known about their capacities to elicit immune responses classically associated with C. albicans-mediated immunopathology, including neutrophil recruitment and proinflammatory cytokine signaling. Therefore, using a combination of in vitro and in vivo approaches, we undertook a comparative analysis to determine whether a representative panel of NAC species could colonize, induce immunopathological markers, or cause damage at the vaginal mucosa. Using a murine model of VVC, C. albicans was found to induce robust immunopathology (neutrophils and interleukin 1β [IL-1β]) and elicit mucosal damage. However, all the NAC species tested (including C. dubliniensis, C. tropicalis, C. parapsilosis, C. krusei, C. glabrata, and C. auris) induced significantly less damage and neutrophil recruitment than C. albicans, despite achieving similar early colonization levels. These results largely correlated with a notable lack of ability by the NAC species (including C. dubliniensis and C. tropicalis) to form hyphae both in vitro and in vivo. Furthermore, both C. dubliniensis and C. tropicalis induced significantly less expression of the ECE1 gene encoding candidalysin, a key fungal virulence determinant driving VVC immunopathology. In order to determine the relative capacities of these species to elicit inflammasome-dependent IL-1β release, both wild-type and NLRP3−/− THP-1 cells were challenged in vitro. While most species tested elicited only modest amounts of IL-1β, challenge with C. albicans led to significantly elevated levels that were largely NLRP3 dependent. Collectively, our findings demonstrate that although NAC species are increasingly reported as causative agents of VVC, C. albicans appears to be exceedingly vaginopathogenic, exhibiting robust immunopathology, hypha formation, and candidalysin expression. Thus, this study provides mechanistic insight into why C. albicans is overwhelmingly the major pathogen reported during VVC.
INTRODUCTION
Vulvovaginal candidiasis (VVC) is a common mucosal infection in immunocompetent women overwhelmingly caused by the opportunistic fungus Candida albicans (1, 2). Symptomatic infection typically results in itching, burning, pain, and redness of the vaginal mucosa, often accompanied by vaginal discharge (3). VVC is the most prevalent human candidal infection, affecting ∼75% of the female population at least once in their lifetime. Moreover, 5% to 8% of all women suffer from recurrent infections (RVVC), defined as >3 episodes per year, often necessitating continuous antifungal therapy (4, 5).
A landmark live-challenge study conducted by Fidel and colleagues described VVC as an immunopathology in which the host innate immune response is dominated by an influx of polymorphonuclear leukocytes (PMNs), is nonprotective, and ultimately drives the above-mentioned vaginal symptoms (6). Recently, it was demonstrated that the NLRP3 inflammasome, a cytoplasmic protein complex involved in the release of interleukin 1β (IL-1β) and IL-18, plays an important role in recruiting PMNs to the vaginal mucosa during C. albicans challenge (7, 8). Work from our laboratory has also established that fungal colonization of the vaginal mucosa alone is insufficient to drive PMN recruitment and proinflammatory cytokine signaling (including IL-1β), as hypha-deficient mutants exhibit robust asymptomatic colonization (9). Instead, we have recently shown that both hypha formation and downstream expression of ECE1, encoding the hypha-expressed toxin candidalysin, are the crucial virulence determinants driving neutrophil recruitment and damage at the vaginal mucosa (9, 10).
While the majority (∼90%) of VVC cases are due to infection with C. albicans (1), other, non-albicans Candida (NAC) species have been implicated as causative agents of VVC, and their incidence is disproportionately high in certain geographic regions (11). Retrospective reports indicate that identification of NAC species during VVC episodes range anywhere from 5% to 50% of total Candida species isolated (12–25). Of these, C. glabrata is most frequently reported as the second leading causative agent (∼5%), and the remainder are most commonly composed of C. krusei, C. parapsilosis, and C. tropicalis (11, 20, 23). Reported vaginal symptoms caused by NAC species are often milder than those experienced during infection with C. albicans (26). However, antifungal resistance and recurrence rates are typically higher with the NAC species, necessitating prolonged and/or alternative treatment options (23, 27). Even more troubling are several reports indicating that NAC species are being increasingly identified as causative agents of VVC (28, 29). Moreover, while the virulence mechanisms driving C. albicans immunopathogenesis at the murine vaginal mucosa are well described (e.g., hypha formation, candidalysin expression, and inflammasome activation), those responsible for immunopathology caused by NAC species remain poorly defined. Since routine surveillance of NAC species in VVC is still fairly uncommon (with the exception of C. glabrata), little is known regarding the true capacities of the NAC species to elicit immunopathology at the vaginal mucosa. This is further complicated by divergent clinical views on the role of NAC species during VVC: one school of thought accredits the NAC species with a true pathogenic function, while another suggests only an incidental bystander role during infection (11, 14, 30). Thus, clarifying which species of Candida exhibit vaginopathogenicity may allow physicians to make appropriate therapeutic decisions for treatment and simultaneously impede development of azole resistance in susceptible species. Therefore, to better delineate the pathogenesis of NAC species (including C. dubliniensis, C. tropicalis, C. parapsilosis, C. krusei, C. glabrata, and C. auris) at the vaginal mucosa, a comparative study of their capacities to elicit neutrophil recruitment, damage, and NLRP3 inflammasome-dependent IL-1β was undertaken.
RESULTS
Relative immunopathogenicities of the NAC species during VVC.
In order to determine which Candida species are capable of colonizing and eliciting neutrophil recruitment, inflammatory cytokine production, and damage at the vaginal mucosa in vivo, a comparative analysis of a panel of reference isolates (i.e., those whose genomes have been sequenced) of both C. albicans and NAC species was undertaken. Using the murine model of vaginitis, we assessed these biomarkers of infection at both day 3 and day 7 postinoculation (p.i.) to determine potential differences between early and late responses. Surprisingly, colonization rates were similar for all the species tested at the early time point (Fig. 1A). However, colonization with strains capable of forming true hyphae (C. albicans, C. dubliniensis, and C. tropicalis) was generally higher and more consistent at day 7 (Fig. 1B). Despite this trend, colonization rates among species failed to reach statistical significance. C. albicans exhibited the highest level of vaginopathogenicity at both time points. The only other species that approached eliciting consistently high levels of PMNs and IL-1β was C. dubliniensis, the species most closely related to C. albicans. Conversely, C. tropicalis (the only other reported true hypha former) was among the least immunopathogenic species tested. The remaining NAC species, including those that are reported to grow as pseudohyphal or yeast forms (e.g., C. parapsilosis and C. glabrata), elicited only marginal levels of inflammatory effectors that were not significantly elevated over those of sham-treated controls (Fig. 1C to H). However, the role of the yeast-hypha switch in driving VVC immunopathology could not be completely determined without comparing fungal morphology within the vagina.
FIG 1.

NAC species fail to elicit robust inflammation or mucosal damage at the vaginal mucosa. Groups of estrogen-treated C57BL/6 mice (n = 8) were intravaginally challenged with PBS, C. albicans, C. dubliniensis, C. tropicalis, C. parapsilosis, C. krusei, C. glabrata, or C. auris. Vaginal lavage fluid was assessed longitudinally at day 3 and day 7 for fungal burden (the horizontal lines indicate medians) by microbiological plating (A and B), for PMNs (means plus SEM) by microscopy (C and D), for the damage biomarker LDH (means plus SEM) by enzymatic assay (E and F), or for IL-1β (means plus SEM) by ELISA (G and H). Experiments in all the inoculation groups were performed in duplicate, and the data were combined. The data were tested for normality using the Shapiro-Wilks test. Statistical significance was calculated using one-way ANOVA and the Kruskal-Wallis posttest (nonnormally distributed data) or Dunnett’s posttest (normally distributed data). *, P < 0.05.
Morphogenesis among the NAC species.
In order to determine whether the lack of robust immunopathology observed among the NAC species was due to a failure in the ability to switch from yeast to hyphae, vaginal lavage fluids were stained to elucidate both neutrophils and fungi by microscopy. Intravaginal challenge with C. albicans led to robust levels of PMN recruitment (Fig. 2A, yellow arrows) and copious amounts of hyphal filaments (green arrows), indicating robust activation of the morphogenetic switch. In fact, C. albicans is found almost exclusively in the hyphal form during murine VVC. Analysis of lavage fluid from mice challenged with C. dubliniensis revealed only very sporadic filamentation and recruitment of relatively few PMNs (Fig. 2B). Interestingly, challenge with C. tropicalis revealed colonization with only yeast cells, as no hyphae or pseudohyphae could be detected throughout all the prepared slides (Fig. 2C). Unsurprisingly, this correlated with the presence of very few PMNs. Similarly, despite the capacity to form pseudohyphae, challenge with C. parapsilosis and C. krusei led to only the observation of yeast cells with few PMNs present (Fig. 2D and E). As expected, C. glabrata and C. auris (species restricted to the yeast morphology) were found in the vaginal fluid as only yeast forms among a few PMNs (Fig. 2F and G). Thus, these data are well correlated with previously observed results (Fig. 1), strongly suggesting that the capacity to form hyphae in vivo robustly drives the immunopathogenesis of VVC (9).
FIG 2.
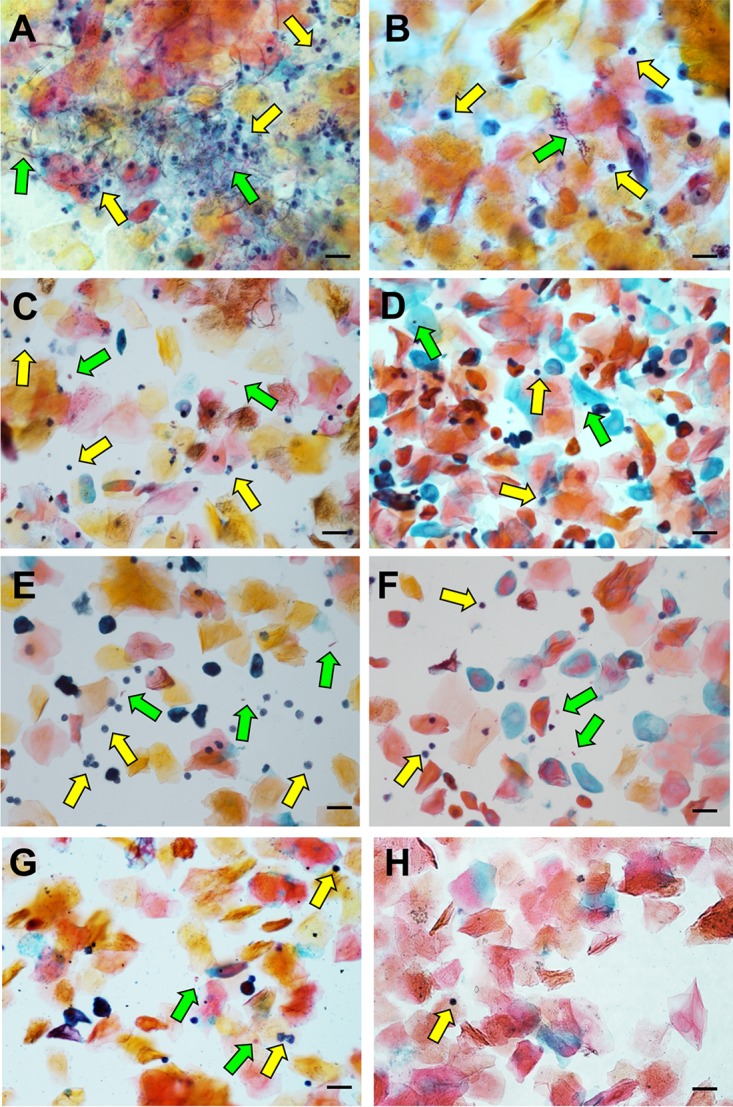
NAC species largely fail to robustly form hyphae or recruit neutrophils in vivo. Vaginal lavage fluids (10 μl) from day 3 p.i. were smeared onto glass slides, fixed, and stained by the Papanicolaou technique. Images of five nonadjacent fields were captured by standard light microscopy, and a representative of each is depicted for C. albicans (A), C. dubliniensis (B), C. tropicalis (C), C. parapsilosis (D), C. krusei (E), C. glabrata (F), C. auris (G), and mock-challenged mice (H). The green arrows indicate fungi, and the yellow arrows indicate neutrophils. Scale bars, 20 μm.
We next wanted to determine whether these morphogenetic phenotypes were restricted to colonization of the murine vagina or were more broadly conserved in vitro under hypha-inducing conditions. Planktonic growth of the isolates in RPMI medium (a tissue culture medium that robustly induces filamentation in C. albicans) led to very similar results, where C. albicans robustly formed hyphal filaments while C. dubliniensis and C. tropicalis formed only infrequent and comparatively stunted hyphae (see Fig. S1 in the supplemental material). The remaining species grew exclusively as yeast forms under these conditions. Therefore, hyphal growth observed in vivo at the vaginal mucosa is largely reflective of growth in vitro under conditions that permit filamentation, suggesting that the pathogenesis of each species/strain is under unique morphogenetic control. Moreover, these results also demonstrate that, among the strains tested, C. albicans exhibits the greatest pathogenicity (e.g., hyphal growth and mucosal damage) at the vaginal mucosa, likely explaining its dominance as the major etiological agent of VVC.
NAC species do not effectively elicit NLRP3 inflammasome-dependent IL-1β release.
Since we and others have previously demonstrated an important role for the NLRP3 inflammasome in governing early neutrophil recruitment and inflammatory cytokine signaling (including IL-1β) during VVC, we wished to determine the relative capacities of the Candida species to stimulate NLRP3-dependent IL-1β release (7, 8, 31). In order to assess comparative pathogenicity, we challenged both wild-type (WT) and NLRP3−/− differentiated THP-1-derived human macrophages with equivalent amounts of each Candida species (multiplicity of infection [MOI], 5:1). With the exception of C. dubliniensis, the NAC species elicited only negligible IL-1β release during challenge (Fig. 3). Although modest amounts of IL-1β were observed during C. dubliniensis challenge, the levels induced by all the NAC species were significantly lower than the levels induced by C. albicans. Despite these differences, at least some IL-1β release was observed for all the species tested, and it was largely NLRP3 dependent, as challenge of NLRP3−/− cells led to significantly reduced (or, in some cases, absent) levels of this cytokine. Challenge with lipopolysaccharide (LPS) and ATP (strong activators of the NLRP3 inflammasome) led to NLRP3-dependent IL-1β release equivalent to that observed during C. albicans challenge, indicating robustness of the experimental system (Fig. 3). Thus, in comparison to C. albicans, these representative NAC species are impaired in their relative capacities to activate the NLRP3 inflammasome.
FIG 3.
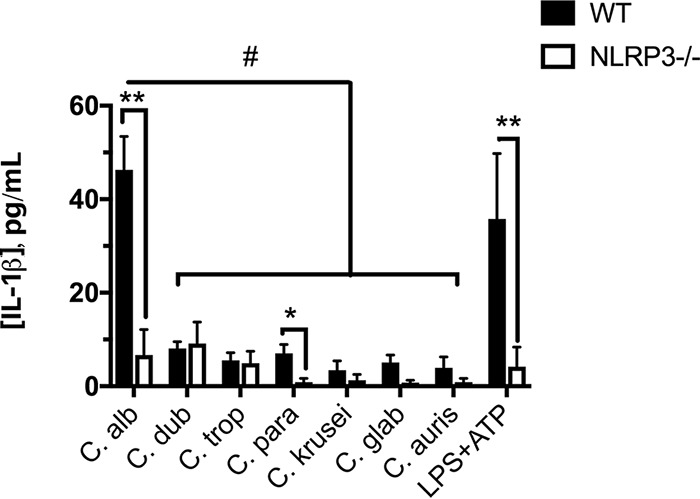
NAC species fail to robustly activate the NLRP3 inflammasome. C. albicans and NAC species (C. dubliniensis, C. tropicalis, C. parapsilosis, C. krusei, C. glabrata, and C. auris) were added to differentiated WT and NLRP3−/− THP-1 cells at an MOI of 5:1 for 4 h. Cells were also challenged with LPS plus ATP as a positive control for inflammasome activation. Cell-free culture supernatants were analyzed for IL-1β release. The solid bars represent WT cells, and the open bars depict NLRP3−/− cells. The numbers of technical replicates (n = 4) per experiment were averaged. The data are depicted as the average of 3 independent biological repeats (means plus SEM). Statistical significance was calculated using one-way ANOVA and Dunnett’s posttest. Comparison between WT and NLRP3−/− cells, *, P < 0.05, and **, P < 0.01; comparison between species, #, P < 0.05.
ECE1 gene expression is significantly reduced in the NAC species.
We have previously demonstrated that expression of the ECE1 gene encoding candidalysin is required for robust immunopathology at the vaginal mucosa during C. albicans infection. It is also highly inducible under conditions that promote the yeast-to-hypha switch (even at time points before hyphae are visually observable) (32). C. dubliniensis and C. tropicalis are the only Candida species with known ECE1 orthologs, so we used a quantitative real-time PCR (qRT-PCR) approach with species-specific primers (Table 1) to quantify the relative expression of ECE1 at 4 h after the switch to RPMI medium. Importantly, the primers were designed to yield equivalent PCR efficiencies and were confirmed prior to qRT-PCR analysis (data not shown). As expected, C. albicans robustly upregulated ECE1 expression (∼5,000-fold) (Fig. 4A). However, C. dubliniensis only modestly expressed ECE1 (∼50-fold), while only exceedingly little induction was detected for C. tropicalis (Fig. 4A). In vivo ECE1 expression was also determined following intravaginal challenge, with C. albicans demonstrating robustly elevated levels (Fig. 4B). Consistent with significantly reduced expression levels in vitro, quantifiable ECE1 transcripts could not be reliably detected for C. dubliniensis and C. tropicalis. Expression levels of ACT1 for these species were similar to that of C. albicans, indicating robustness of the extraction method (data not shown). These results largely correlate with the relative inflammasome activation observed in THP-1 cells and the capacity to switch from yeast to hypha.
TABLE 1.
Primer names and sequences used to determine ACT1 and ECE1 expression in C. albicans, C. dubliniensis, and C. tropicalis
| Primer namea | Sequence (5′→3′) |
|---|---|
| CaACT1qPCR-F | TTGGATTCTGGTGATGGTGTTA |
| CaACT1qPCR-R | TCAAGTCTCTACCAGCCAAATC |
| CaECE1qPCR-F | TTGCTAATGCCGTCGTCAGA |
| CaECE1qPCR-R | GAACGACCATCTCTCTTGGCAT |
| CdACT1qPCR-F | GGATTCTGGTGATGGTGTTAC |
| CdACT1qPCR-R | GTAAGTCTCTACCAGCCAAATC |
| CdECE1qPCR-F | GTTACTAATGCCATCGTCAGA |
| CdECE1qPCR-R | CAACACCGTCTCTCTTGG |
| CtACT1qPCR-F | CTTGGATTCTGGTGATGGTGTTA |
| CtACT1qPCR-R | TCAAGTCTCTACCAGCCAAGTC |
| CtECE1qPCR-F | GCACTCAAATTCTTGGCTCA |
| CtECE1qPCR-R | GCTGAAGGTATCAGAATTGGC |
Ca, C. albicans; Cd, C. dubliniensis; Ct, C. tropicalis.
FIG 4.
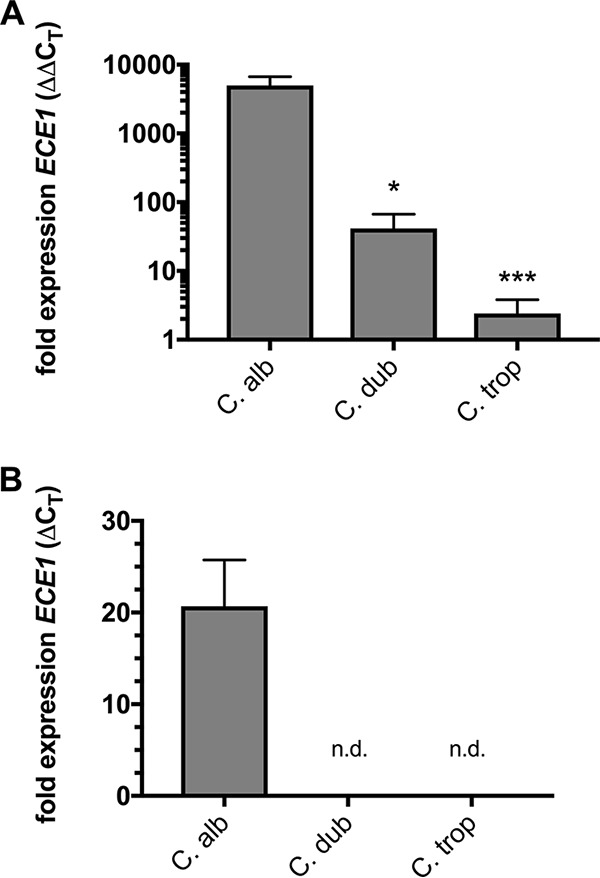
C. dubliniensis and C. tropicalis fail to strongly upregulate ECE1 expression in vitro or in vivo. (A) Overnight YPD cultures of C. albicans, C. dubliniensis, and C. tropicalis were transferred to either fresh YPD or RPMI medium for 4 h. RNA was extracted, normalized, and reverse transcribed, and the expression of ECE1 and the housekeeping gene ACT1 was monitored. The normalized fold expression of ECE1 (mean plus SEM) was calculated from the average independent biological repeats (n = 3) using the ΔΔCT method. Statistical significance was calculated using one-way ANOVA and Dunnett’s posttest. *, P < 0.05; ***, P < 0.001. (B) C. albicans, C. dubliniensis, and C. tropicalis were intravaginally inoculated in estrogen-treated mice as described in the text. At day 3 p.i., the mice underwent two rounds of vaginal lavage with PBS to recover fungal cells, and RNA was immediately prepared as described above. The normalized fold expression of ECE1 (mean plus SEM) was calculated from the average of independently inoculated animals (n = 4) and compared to ACT1 expression using the ΔCT method. n.d., not detected.
DISCUSSION
A plethora of large-scale retrospective and prospective clinical studies examining thousands of isolates have demonstrated that C. albicans is clearly the most frequent cause of VVC; commonly reported incidence rates suggest 70 to 90% of the total cases observed per study (18, 20, 23). The gold standard for determining the species of recovered vaginal isolates remains plating on chromogenic agar, which distinguishes Candida species based on colony color (33). While distinct color phenotypes do exist for several of the species (including C. albicans, C. tropicalis, and C. krusei), differences between the remaining species can be difficult to distinguish, even for the trained laboratory technician (34). Moreover, C. dubliniensis is almost indistinguishable from C. albicans using this methodology, possibly leading to low reported rates of C. dubliniensis as an etiological agent of VVC (35). That said, molecular diagnostics (e.g., PCR) to distinguish Candida species largely report findings similar to those of culture-based identification (12, 24, 36). Thus, imperfect or infrequently used diagnostic tools, self-diagnosis, and availability of over-the-counter treatment options may contribute to the potentially underreported rate of NAC vaginitis.
Interestingly, a number of small-scale (hundreds of isolates) clinical studies have revealed quite a different scenario, in which NAC species make up as much as 50% of the total isolates recovered (13, 15, 37). In some situations, NAC species isolated even outnumber C. albicans (17, 38, 39). These somewhat surprising results could be explained as follows. In many cases, the findings arise from assessing symptomatic infection in African or Asian populations, suggesting either ancestral host genetic susceptibility to NAC isolates or geographic dominance of a given species or strain. Also, many of the studies examined patient populations seeking treatment at tertiary-care vaginitis clinics, which are sought only after routine treatment therapy has failed or when chronic recurrence is an issue. Given the association between recurrence and treatment failure with the NAC species, results from these studies may be biased by an already susceptible patient population (22). Nonetheless, our data strongly support the broad clinical finding that C. albicans is more adept at causing symptomatic VVC, thus providing a rationale for its increased incidence compared to NAC species.
Our results largely mirror the reduced pathogenicity of the NAC species compared to C. albicans reported during infection of oral epithelium (40). Findings by Moyes et al. revealed that of all the pathogenic Candida species tested, only C. albicans and C. dubliniensis were capable of activating mitogen-activated protein kinase (MAPK) signaling in oral epithelial cells—a process strongly correlated with hypha formation (41). However, mucosal damage and downstream cytokine signaling induced by C. dubliniensis were significantly impaired in comparison to C. albicans, which robustly activated these responses. Furthermore, the remaining fungi tested (C. tropicalis, C. parapsilosis, C. krusei, C. glabrata, and Saccharomyces cerevisiae) failed to approach the magnitude of responses observed during C. albicans challenge (40). Similar to these findings, other studies have demonstrated that NAC species inefficiently activate the NLRP3 inflammasome compared to C. albicans (Fig. 3), requiring a much higher MOI and/or greatly expanded kinetic time courses (42–44). However, our study represents the first systematic in vivo comparison of the pathogenicities of the major candidal causative agents of VVC.
Despite the strong disparity between C. albicans and NAC species pathogenicities delineated in this study, we recognize that these findings should be taken with caution. Genome-sequenced reference isolates represent our greatest understanding of an organism’s biology and virulence, as significant amounts of published work regarding pathogenicity and fitness exist for these strains. Thus, in this study, we chose to use common reference isolates (or those with whole-genome sequences available) to comparatively assess vaginopathogenicity. However, these data are representative of a single isolate of each species, and the phenotypes should not be broadly conferred across all clinical isolates. For example, Asmundsdottir and colleagues demonstrated that murine systemic infection with different C. dubliniensis clinical isolates (n = 9) led to lethal outcomes resembling that of C. albicans infection in 30% of the cases (45). The remaining isolates were largely avirulent, highlighting dramatic interstrain variability. A similar outcome may likely be true for the other species and/or mucosal disease, including that caused by C. albicans. Differences in biofilm formation and hemolysin, protease, or phospholipase secretion likely impact strain-to-strain pathogenicities (46–48).
This possibility raises some intriguing questions. Given that vaginal colonization is not dependent on invasiveness (as evidenced by lack of robust lactate dehydrogenase [LDH] release elicited by C. dubliniensis), why has C. albicans evolved to be so highly vaginopathogenic? For a commensal organism that inhabits humans as its primary niche, alarming the immune system seems a counterintuitive long-term colonization strategy. However, several plausible hypotheses may explain this. Humans, like mice, are more susceptible to VVC during periods of estrogenic activity (49). Indeed, estrogen induces keratinization of the vaginal epithelium, causing the cornified cells to slough away from the vaginal wall (50). Perhaps C. albicans robustly forms hyphae under these conditions to remain anchored to the mucosa to prevent clearance. This hypothesis may be supported by colonization data (Fig. 1), in which species incapable of forming hyphae were largely cleared or exhibited erratic colonization at day 7. However, given that C. albicans can covalently bind to epithelial cells (e.g., via Hwp1p-mediated interactions), this hypothesis may provide a less feasible explanation (51). Estrogen also significantly increases the glycogen content of the vaginal epithelium. Dennerstein and Ellis tested a panel of Candida species for the capacity to catabolize glycogen in vitro. Interestingly, only C. albicans was able to utilize this complex carbohydrate source to support growth (52). Thus, it is possible that C. albicans has a metabolic advantage at the vaginal mucosa compared to the NAC species. It would be intriguing to investigate genetic disruption of this pathway and its effect on C. albicans vaginopathogenesis. Also, fine-tuning of ECE1 expression or evolved allelic diversity among Candida species may help explain their relative pathogenicities (Fig. 4). Lastly, it is possible that C. albicans is better equipped to deal with stressors and host defense mechanisms encountered in the vaginal environment (53). Whatever the explanation, it is clear that the capacity to form hyphae and cause damage seemingly confers selective fitness at the vaginal mucosa, given the high isolation rate of C. albicans during VVC.
Despite the relative frequency with which they are isolated, the role of NAC species during VVC remains highly contested. Some clinicians feel that NAC species are simply colonizers that are present during an idiopathic bout of vaginal symptoms (11, 14). In support of this, a clinical study by Dennerstein et al. demonstrated that of 44 women presenting with vaginitis-like symptoms and harboring a NAC yeast, 86% reported spontaneous improvement without requiring antifungal intervention (30). On the other hand, the relatively large number (∼5% to 10%) of VVC cases universally caused by C. glabrata cannot be denied, suggesting that NAC species do actively contribute to symptomatic VVC (20, 23, 24, 28). C. glabrata remains a formidable clinical challenge, as these infections are largely unresponsive to standard fluconazole therapy, given the intrinsic resistance of C. glabrata to the azole class (54). Often, prolonged treatment regimens (weeks to months) with alternative topical azole (e.g., miconazole and terconazole) or antifungal (e.g., flucytosine and amphotericin B) drugs are required, in conjunction with boric acid vaginal suppositories (55, 56). However, rates of recurrence and relapse remain unacceptably high.
C. glabrata is unable to form hyphae, has no candidalysin ortholog, and exhibits virtually no immunopathogenicity in the murine model of VVC, yet it clearly does so in women. How is this so? The simplest explanation is that clear differences in human immunity result in exacerbated symptomatology compared to murine challenge. It is also possible that specific isolates of C. glabrata are vaginopathogenic while others only asymptomatically colonize. Although much less common than C. glabrata, the highly antifungal-resistant emerging pathogen C. auris has also been implicated in causing VVC (57). In other cases, even the benign food grade yeast S. cerevisiae has been isolated as a causative agent of vaginitis (58). Like C. glabrata, these species are restricted to growth in the yeast form. Thus, it is possible that mechanisms unrelated to the fungal pathogen drive vaginal symptoms (e.g., itching and burning) in a subset of women. Perhaps allergic or hypersensitivity responses mediated by population level immunogenetics contribute to heightened responses to fungal antigens or virulence determinants. Given that VVC is largely diagnosed based on symptoms (even in the absence of microbiological evidence), it may be better regarded as a syndrome, in which multifactorial root causes result in similar clinical presentations. This classification would help unify pathogenicity mechanisms for which there is solid foundational biological evidence with otherwise inexplicable clinical findings.
Overall, our results demonstrate that, with the exception of C. dubliniensis, the representative NAC species are incapable of forming hyphae in the murine vagina and consequently do not elicit robust immunopathology characteristic of C. albicans-mediated disease. Consistent with these observations, these NAC species also fail to robustly activate the NLRP3 inflammasome, contributing to reduced immunopathogenicity. These findings provide a supportive rationale for C. albicans being the primary causative agent of VVC.
MATERIALS AND METHODS
Ethics statement.
The animals used in this study were housed in AAALAC-approved facilities located in the Regional Biocontainment Laboratory (RBL) at the University of Tennessee Health Sciences Center (UTHSC). The UTHSC Animal Care and Use Committee approved all animals and protocols. The mice were given standard rodent chow and water ad libitum. The mice were monitored for signs of distress, including noticeable weight loss and lethargy.
Microorganism growth.
When possible, genome-sequenced reference isolates were used. C. albicans SC5314, C. dubliniensis CD36, C. tropicalis MYA3404, C. parapsilosis CDC317, C. krusei 81-B-5 (59), C. glabrata CBS138, and C. auris [429]0382 were maintained as glycerol stocks stored at −80°C. A small amount of stock was spread onto yeast extract-peptone-dextrose (YPD) agar and incubated at 30°C for 48 h to obtain isolated colonies. A single colony was transferred to 10 ml of YPD and incubated at 30°C with shaking at 200 rpm for 18 h prior to vaginal or THP-1 challenge.
Murine model of vulvovaginal candidiasis.
The well-established murine model of vulvovaginal candidiasis was performed as described previously (60, 61). Briefly, female 6- to 8-week-old C57BL/6 mice were purchased from Charles River Laboratories and housed in isolator cages mounted on ventilated racks. The mice were administered 0.1 mg of estrogen (β-estradiol 17-valerate; Sigma) dissolved in 0.1 ml of sesame oil subcutaneously 72 h prior to intravaginal inoculation with Candida species. Stationary-phase cultures of Candida isolates were prepared as described above. Cell suspensions were counted and adjusted to 5 × 108 CFU/ml in sterile endotoxin-free phosphate-buffered saline (PBS). The estrogen-treated mice were intravaginally inoculated with 10 μl of the standardized blastoconidial cell suspension, generating an inoculum size of 5 × 106 blastoconidia. The mice underwent vaginal lavage with 100 μl of PBS at days 3 and 7 p.i. The resultant lavage fluids were briefly centrifuged (2,500 rpm; 1 min) to remove debris and spiked with 1 μl of 100× EDTA-free protease inhibitors (Complete; Roche) and kept on ice or stored at −80°C until they were processed for immunopathological markers. All animal experiments (n = 4 per group) were conducted in duplicate, and the resulting data were combined.
Assessment of fungal burden and vaginitis immunopathology.
Immunopathological markers were assessed as described previously (62). Lavage fluid was serially diluted 10-fold by the drop plate method and plated onto YPD agar containing 50 μg/ml chloramphenicol. The plates were incubated for 24 h at 37°C, colonies were enumerated, and numbers of CFU/ml were reported as the median. Lavage fluid (10 μl) was smeared onto glass slides, fixed with CytoFix spray, and stained by the Papanicolaou technique to assess PMN recruitment (small blue cells with multilobed nuclei). PMNs were counted in 5 nonadjacent fields by standard light microscopy, using a 40× objective, and values were reported as means plus standard errors of the mean (SEM). Murine IL-1β was assessed in clarified, diluted vaginal lavage fluid using a murine Ready-Set-Go enzyme-linked immunosorbent assay (ELISA) kit (eBioscience) according to the manufacturer's protocol, and the results were reported as means plus SEM. LDH activity was measured in diluted lavage fluid using the commercially available CytoTox 96 nonradioactive cytotoxicity assay (Promega), and the results were reported as means plus SEM.
Hyphal growth assay.
Candida species were grown overnight in YPD, washed in PBS as described above, diluted to 5 × 105 cells/ml in phenol red-free RPMI 1640 containing 25 mM HEPES (Corning), and shaken in a 37°C incubator. Aliquots were removed at several time points, and wet mounts were prepared and imaged by standard light microscopy.
Cultivation of cell lines.
WT (THP1 null; InvivoGen) and NLRP3−/− (THP1-defNLRP3; InvivoGen) THP-1 cells were cultured according to the manufacturer’s protocol in RPMI 1640 with 25 mM HEPES supplemented with 10% heat-inactivated fetal bovine serum (FBS), 100 U/ml Pen-Strep, and normocin. THP-1 cells were counted on a Countess II FL (Life Technologies) and frozen as aliquots of ∼5 × 106 cells in liquid nitrogen.
Macrophage challenge assay.
Upon recovery from cryopreservation, cells were incubated for 3 days at 37°C, 5% CO2, and 90% humidity in a T25 flask in RPMI 1640 medium (10% heat-inactivated FBS, 100 U/ml Pen-Strep). After 3 days, THP-1 cells were counted on the Countess II, assessed for high viability by exclusionary trypan blue staining, and diluted to 5.56 × 105 cells/ml in RPMI 1640 (25 mM HEPES, 10% heat-inactivated FBS, 100 U/ml Pen-Strep), and 180-μl aliquots were seeded at a final density of 1 × 105 cells/well in 96-well polystyrene plates. Phorbol 12-myristate 13-acetate (PMA) (InvivoGen) was added at a final concentration of 100 nM to differentiate cells to a macrophage phenotype, and the cells were incubated at 37°C with 5% CO2 for 24 h. Following incubation, the spent culture medium was replaced with 180 μl fresh phenol red-free RPMI 1640 containing 25 mM HEPES. Overnight cultures of Candida species were prepared as described above, washed 3 times in sterile PBS, and adjusted to 2.5 × 107 CFU/ml, and 20 μl of the suspension was added to wells containing THP-1 cells, generating a 5:1 MOI. Mock-infected controls using medium alone were also included. A positive control for inflammasome activation was also prepared by challenging cells with 1 μg/ml lipopolysaccharide (Escherichia coli 0111:B4; InvivoGen) for an equivalent time, followed by addition of 5 mM ATP (InvivoGen) 30 min prior to the endpoint. The cells were incubated for 4 h and gently centrifuged at 200 × g for 2 min to settle the cells/Candida, and 100 μl of culture supernatant was transferred to a polystyrene plate containing 100 µl of prediluted 1× ELISA/enzyme-linked immunosorbent spot (ELISPOT) assay buffer (eBioscience) and stored at −20°C. Culture supernatants were assessed for IL-1β using the Human Ready-Set-Go ELISA kit (eBioScience). ELISA optical density values from mock-infected controls were subtracted from those of Candida-challenged samples. Experiments were conducted in technical replicates (n = 4) and repeated independently in triplicate. Data are reported as means plus SEM.
Expression of ECE1 by qRT-PCR.
C. albicans, C. dubliniensis, and C. tropicalis were grown overnight in YPD medium at 30°C, washed in PBS, and added to 5 ml of fresh YPD or phenol red-free RPMI 1640 containing 25 mM HEPES and incubated with shaking (200 rpm) at 30°C or 37°C, respectively, for 4 h. RNA was extracted by the acid phenol-chloroform procedure as previously described, followed by precipitation with 3 M sodium acetate-ethanol, washing in 70% ethanol, and air drying (63). The pellets were resuspended in sterile water, and RNA integrity (1 μl) was assessed by morpholinepropanesulfonic acid (MOPS) gel electrophoresis and visualization of intact 18S and 28S bands. RNA concentrations were measured using a NanoDrop spectrophotometer to assess A260/A280. RNA concentrations were equalized among samples, and 200-ng aliquots were treated with RNase-free DNase (Thermo Fisher) according to the manufacturer’s instructions. RNA was reverse transcribed using random hexamers and the RevertAid kit (Thermo Fisher) according to the manufacturer’s protocol. Candida species-specific forward and reverse primers (final concentration, 0.5 μM) for ECE1 or ACT1 open reading frames (ORFs) were used in conjunction with 2× Maxima SYBR green mix (Thermo Fisher) according to the manufacturer’s instructions to amplify approximately 100-bp fragments from 20 ng of cDNA. Primers were designed to have equivalent melting temperatures. Primer sets were also validated by PCR, amplifying genomic DNA extracted from each species, and gel electrophoresis was used to confirm amplification of target sequences. qRT-PCRs were monitored and analyzed with an Applied Biosystems 7500 platform and software. Expression levels of ECE1 were compared to those of a reference gene (ACT1) and growth in YPD or RPMI medium using the ΔΔCT method (64). RNA obtained from vaginal lavage fluids of mice 3 days following challenge with Candida species was similarly prepared and analyzed for ECE1 expression, except that levels were reported as the ratio of ECE1 to ACT1 using the ΔCT method.
Imaging.
Standard light microscopy images of Papanicolaou-stained vaginal lavage fluids or cells from the hyphal growth assay were captured using a Nikon Ni-U microscope with the NIS Elements software package. Images were processed (cropped or resized) using Adobe Photoshop CS3, and any adjustments (e.g., brightness or contrast) were applied uniformly across the entire image using ImageJ (NIH). Images of five nonadjacent fields were taken and are representative.
Statistical analyses.
All experiments were conducted using groups of mice (n = 4) and repeated in duplicate as determined by power analyses. All data were plotted and analyzed for statistical significance using GraphPad Prism software. Data sets were tested for normality using the Shapiro-Wilks test. Data were compared by one-way analysis of variance (ANOVA) and Dunnett’s (parametric) or Kruskal-Wallis (nonparametric) posttest. Graphs in figures are annotated to indicate significance levels.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dave Rogers (University of Tennessee Health Sciences Center) for C. tropicalis, C. parapsilosis, and C. glabrata isolates. We thank Mary Ann Jabra-Rizk (University of Maryland—Baltimore) for C. dubliniensis isolate CD36. We also thank Anja Forche (Bowdoin College) for C. krusei isolate 81-B-5. Lastly, we thank the Division of Healthcare Quality Promotion, Centers for Disease Control and Prevention (CDC), Clinical and Environmental Microbiology Branch for providing the Candida auris panel (to G.E.P.), including C. auris isolate [429]0382.
Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under awards K22AI110541 (B.M.P.), R21AI127942 (B.M.P.), R01AI134796 (B.M.P.), and R01AI099080 (G.E.P.).
The content is solely our responsibility and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00527-18.
REFERENCES
- 1.Achkar JM, Fries BC. 2010. Candida infections of the genitourinary tract. Clin Microbiol Rev 23:253–273. doi: 10.1128/CMR.00076-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sobel JD. 2007. Vulvovaginal candidosis. Lancet 369:1961–1971. doi: 10.1016/S0140-6736(07)60917-9. [DOI] [PubMed] [Google Scholar]
- 3.Carr PL, Felsenstein D, Friedman RH. 1998. Evaluation and management of vaginitis. J Gen Intern Med 13:335–346. doi: 10.1046/j.1525-1497.1998.00101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Foxman B, Barlow R, D’Arcy H, Gillespie B, Sobel JD. 2000. Candida vaginitis: self-reported incidence and associated costs. Sex Transm Dis 27:230–235. doi: 10.1097/00007435-200004000-00009. [DOI] [PubMed] [Google Scholar]
- 5.Sobel JD. 1997. Vaginitis. N Engl J Med 337:1896–1903. doi: 10.1056/NEJM199712253372607. [DOI] [PubMed] [Google Scholar]
- 6.Fidel PL Jr, Barousse M, Espinosa T, Ficarra M, Sturtevant J, Martin DH, Quayle AJ, Dunlap K. 2004. An intravaginal live Candida challenge in humans leads to new hypotheses for the immunopathogenesis of vulvovaginal candidiasis. Infect Immun 72:2939–2946. doi: 10.1128/IAI.72.5.2939-2946.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Borghi M, De Luca A, Puccetti M, Jaeger M, Mencacci A, Oikonomou V, Pariano M, Garlanda C, Moretti S, Bartoli A, Sobel J, van de Veerdonk FL, Dinarello CA, Netea MG, Romani L. 2015. Pathogenic NLRP3 inflammasome activity during Candida infection is negatively regulated by IL-22 via activation of NLRC4 and IL-1Ra. Cell Host Microbe 18:198–209. doi: 10.1016/j.chom.2015.07.004. [DOI] [PubMed] [Google Scholar]
- 8.Bruno VM, Shetty AC, Yano J, Fidel PL Jr, Noverr MC, Peters BM. 2015. Transcriptomic analysis of vulvovaginal candidiasis identifies a role for the NLRP3 inflammasome. mBio 6:e00182-15. doi: 10.1128/mBio.00182-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peters BM, Palmer GE, Nash AK, Lilly EA, Fidel PL Jr, Noverr MC. 2014. Fungal morphogenetic pathways are required for the hallmark inflammatory response during Candida albicans vaginitis. Infect Immun 82:532–543. doi: 10.1128/IAI.01417-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Richardson JP, Willems HME, Moyes DL, Shoaie S, Barker KS, Tan SL, Palmer GE, Hube B, Naglik JR, Peters BM. 2017. Candidalysin drives epithelial signaling, neutrophil recruitment, and immunopathology at the vaginal mucosa. Infect Immun doi: 10.1128/IAI.00645-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kennedy MA, Sobel JD. 2010. Vulvovaginal candidiasis caused by non-albicans Candida species: new insights. Curr Infect Dis Rep 12:465–470. doi: 10.1007/s11908-010-0137-9. [DOI] [PubMed] [Google Scholar]
- 12.Abbasi Nejat Z, Farahyar S, Falahati M, Ashrafi Khozani M, Hosseini AF, Faiazy A, Ekhtiari M, Hashemi-Hafshenjani S. 2017. Molecular identification and antifungal susceptibility pattern of non-albicans Candida species isolated from vulvovaginal candidiasis. Iran Biomed J 22:33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bitew A, Abebaw Y. 2018. Vulvovaginal candidiasis: species distribution of Candida and their antifungal susceptibility pattern. BMC Womens Health 18:94–104. doi: 10.1186/s12905-018-0607-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brandolt TM, Klafke GB, Goncalves CV, Bitencourt LR, Martinez AM, Mendes JF, Meireles MC, Xavier MO. 2017. Prevalence of Candida spp. in cervical-vaginal samples and the in vitro susceptibility of isolates. Braz J Microbiol 48:145–150. doi: 10.1016/j.bjm.2016.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Choukri F, Benderdouche M, Sednaoui P. 2014. In vitro susceptibility profile of 200 recent clinical isolates of Candida spp. to topical antifungal treatments of vulvovaginal candidiasis, the imidazoles and nystatin agents. J Mycol Med 24:303–307. doi: 10.1016/j.mycmed.2014.05.001. [DOI] [PubMed] [Google Scholar]
- 16.De Vos MM, Cuenca-Estrella M, Boekhout T, Theelen B, Matthijs N, Bauters T, Nailis H, Dhont MA, Rodriguez-Tudela JL, Nelis HJ. 2005. Vulvovaginal candidiasis in a Flemish patient population. Clin Microbiol Infect 11:1005–1011. doi: 10.1111/j.1469-0691.2005.01281.x. [DOI] [PubMed] [Google Scholar]
- 17.Kalaiarasan K, Singh R, Chaturvedula L. 2017. Fungal profile of vulvovaginal candidiasis in a tertiary care hospital. J Clin Diagn Res 11:DC06–DC09. doi: 10.7860/JCDR/2017/23578.9475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu XP, Fan SR, Peng YT, Zhang HP. 2014. Species distribution and susceptibility of Candida isolates from patient with vulvovaginal candidiasis in southern China from 2003 to 2012. J Mycol Med 24:106–111. doi: 10.1016/j.mycmed.2014.01.060. [DOI] [PubMed] [Google Scholar]
- 19.Mukasa KJ, Herbert I, Daniel A, Sserunkuma KL, Joel B, Frederick B. 2015. Antifungal susceptibility patterns of vulvovaginal Candida species among women attending antenatal clinic at Mbarara Regional Referral Hospital, South Western Uganda. Br Microbiol Res J 5:322–331. doi: 10.9734/BMRJ/2015/13804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parazzini F, Di Cintio E, Chiantera V, Guaschino S. 2000. Determinants of different Candida species infections of the genital tract in women. Sporachrom Study Group. Eur J Obstet Gynecol Reprod Biol 93:141–145. doi: 10.1016/S0301-2115(00)00289-X. [DOI] [PubMed] [Google Scholar]
- 21.Paulitsch A, Weger W, Ginter-Hanselmayer G, Marth E, Buzina W. 2006. A 5-year (2000-2004) epidemiological survey of Candida and non-Candida yeast species causing vulvovaginal candidiasis in Graz, Austria. Mycoses 49:471–475. doi: 10.1111/j.1439-0507.2006.01284.x. [DOI] [PubMed] [Google Scholar]
- 22.Powell AM, Gracely E, Nyirjesy P. 2016. Non-albicans Candida vulvovaginitis: treatment experience at a tertiary care vaginitis center. J Low Genit Tract Dis 20:85–89. doi: 10.1097/LGT.0000000000000126. [DOI] [PubMed] [Google Scholar]
- 23.Richter SS, Galask RP, Messer SA, Hollis RJ, Diekema DJ, Pfaller MA. 2005. Antifungal susceptibilities of Candida species causing vulvovaginitis and epidemiology of recurrent cases. J Clin Microbiol 43:2155–2162. doi: 10.1128/JCM.43.5.2155-2162.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi XY, Yang YP, Zhang Y, Li W, Wang JD, Huang WM, Fan YM. 2015. Molecular identification and antifungal susceptibility of 186 Candida isolates from vulvovaginal candidiasis in southern China. J Med Microbiol 64:390–393. doi: 10.1099/jmm.0.000024. [DOI] [PubMed] [Google Scholar]
- 25.Zhang JY, Liu JH, Liu FD, Xia YH, Wang J, Liu X, Zhang ZQ, Zhu N, Yan Y, Ying Y, Huang XT. 2014. Vulvovaginal candidiasis: species distribution, fluconazole resistance and drug efflux pump gene overexpression. Mycoses 57:584–591. doi: 10.1111/myc.12204. [DOI] [PubMed] [Google Scholar]
- 26.Dan M, Poch F, Levin D. 2002. High rate of vaginal infections caused by non-C. albicans Candida species among asymptomatic women. Med Mycol 40:383–386. doi: 10.1080/mmy.40.4.383.386. [DOI] [PubMed] [Google Scholar]
- 27.Ventolini G, Baggish MS, Walsh PM. 2006. Vulvovaginal candidiasis from non-albicans species: retrospective study of recurrence rate after fluconazole therapy. J Reprod Med 51:475–478. [PubMed] [Google Scholar]
- 28.Hamad M, Kazandji N, Awadallah S, Allam H. 2014. Prevalence and epidemiological characteristics of vaginal candidiasis in the UAE. Mycoses 57:184–190. doi: 10.1111/myc.12141. [DOI] [PubMed] [Google Scholar]
- 29.Spinillo A, Capuzzo E, Gulminetti R, Marone P, Colonna L, Piazzi G. 1997. Prevalence of and risk factors for fungal vaginitis caused by non-albicans species. Am J Obstet Gynecol 176:138–141. doi: 10.1016/S0002-9378(97)80026-9. [DOI] [PubMed] [Google Scholar]
- 30.Dennerstein GJ, Ellis DH, Reed CS, Bennett CM. 2011. Pathogenicity of non-albicans yeasts in the vagina. J Low Genit Tract Dis 15:33–36. doi: 10.1097/LGT.0b013e3181d94f39. [DOI] [PubMed] [Google Scholar]
- 31.Roselletti E, Perito S, Gabrielli E, Mencacci A, Pericolini E, Sabbatini S, Cassone A, Vecchiarelli A. 2017. NLRP3 inflammasome is a key player in human vulvovaginal disease caused by Candida albicans. Sci Rep 7:17877. doi: 10.1038/s41598-017-17649-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moyes DL, Wilson D, Richardson JP, Mogavero S, Tang SX, Wernecke J, Hofs S, Gratacap RL, Robbins J, Runglall M, Murciano C, Blagojevic M, Thavaraj S, Forster TM, Hebecker B, Kasper L, Vizcay G, Iancu SI, Kichik N, Hader A, Kurzai O, Luo T, Kruger T, Kniemeyer O, Cota E, Bader O, Wheeler RT, Gutsmann T, Hube B, Naglik JR. 2016. Candidalysin is a fungal peptide toxin critical for mucosal infection. Nature 532:64–68. doi: 10.1038/nature17625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Houang ET, Chu KC, Koehler AP, Cheng AF. 1997. Use of CHROMagar Candida for genital specimens in the diagnostic laboratory. J Clin Pathol 50:563–565. doi: 10.1136/jcp.50.7.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sivakumar VG, Shankar P, Nalina K, Menon T. 2009. Use of CHROMagar in the differentiation of common species of Candida. Mycopathologia 167:47–49. doi: 10.1007/s11046-008-9149-5. [DOI] [PubMed] [Google Scholar]
- 35.Eraso E, Sahand IH, Villar-Vidal M, Marcos C, Dolores Moragues M, Madariaga L, Pontón J, Quindós G. 2006. Usefulness of Candida ID2 agar for the presumptive identification of Candida dubliniensis. Med Mycol 44:611–615. doi: 10.1080/13693780600830691. [DOI] [PubMed] [Google Scholar]
- 36.Vermitsky JP, Self MJ, Chadwick SG, Trama JP, Adelson ME, Mordechai E, Gygax SE. 2008. Survey of vaginal-flora Candida species isolates from women of different age groups by use of species-specific PCR detection. J Clin Microbiol 46:1501–1503. doi: 10.1128/JCM.02485-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hedayati MT, Taheri Z, Galinimoghadam T, Aghili SR, Yazdani Cherati J, Mosayebi E. 2015. Isolation of different species of Candida in patients with vulvovaginal candidiasis from Sari, Iran. Jundishapur J Microbiol 8:e15992. doi: 10.5812/jjm.8(4)2015.15992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cetin M, Ocak S, Gungoren A, Hakverdi AU. 2007. Distribution of Candida species in women with vulvovaginal symptoms and their association with different ages and contraceptive methods. Scand J Infect Dis 39:584–588. doi: 10.1080/00365540601148491. [DOI] [PubMed] [Google Scholar]
- 39.Deorukhkar SC, Saini S, Mathew S. 2014. Non-albicans Candida infection: an emerging threat. Interdiscip Perspect Infect Dis 2014:615958. doi: 10.1155/2014/615958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moyes DL, Murciano C, Runglall M, Kohli A, Islam A, Naglik JR. 2012. Activation of MAPK/c-Fos induced responses in oral epithelial cells is specific to Candida albicans and Candida dubliniensis hyphae. Med Microbiol Immunol 201:93–101. doi: 10.1007/s00430-011-0209-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moyes DL, Murciano C, Runglall M, Islam A, Thavaraj S, Naglik JR. 2011. Candida albicans yeast and hyphae are discriminated by MAPK signaling in vaginal epithelial cells. PLoS One 6:e26580. doi: 10.1371/journal.pone.0026580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Joly S, Ma N, Sadler JJ, Soll DR, Cassel SL, Sutterwala FS. 2009. Cutting edge: Candida albicans hyphae formation triggers activation of the Nlrp3 inflammasome. J Immunol 183:3578–3581. doi: 10.4049/jimmunol.0901323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Toth A, Zajta E, Csonka K, Vagvolgyi C, Netea MG, Gacser A. 2017. Specific pathways mediating inflammasome activation by Candida parapsilosis. Sci Rep 7:43129. doi: 10.1038/srep43129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wellington M, Koselny K, Sutterwala FS, Krysan DJ. 2014. Candida albicans triggers NLRP3-mediated pyroptosis in macrophages. Eukaryot Cell 13:329–340. doi: 10.1128/EC.00336-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Asmundsdottir LR, Erlendsdottir H, Agnarsson BA, Gottfredsson M. 2009. The importance of strain variation in virulence of Candida dubliniensis and Candida albicans: results of a blinded histopathological study of invasive candidiasis. Clin Microbiol Infect 15:576–585. doi: 10.1111/j.1469-0691.2009.02840.x. [DOI] [PubMed] [Google Scholar]
- 46.Harriott MM, Lilly EA, Rodriguez TE, Fidel PL, Noverr MC. 2010. Candida albicans forms biofilms on the vaginal mucosa. Microbiology 156:3635–3644. doi: 10.1099/mic.0.039354-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Naglik JR, Rodgers CA, Shirlaw PJ, Dobbie JL, Fernandes-Naglik LL, Greenspan D, Agabian N, Challacombe SJ. 2003. Differential expression of Candida albicans secreted aspartyl proteinase and phospholipase B genes in humans correlates with active oral and vaginal infections. J Infect Dis 188:469–479. doi: 10.1086/376536. [DOI] [PubMed] [Google Scholar]
- 48.Udayalaxmi JS, D’Souza D. 2014. Comparison between virulence factors of Candida albicans and non-albicans species of Candida isolated from genitourinary tract. J Clin Diagn Res 8:DC15–DC17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fidel PL Jr, Cutright J, Steele C. 2000. Effects of reproductive hormones on experimental vaginal candidiasis. Infect Immun 68:651–657. doi: 10.1128/IAI.68.2.651-657.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miyagawa S, Iguchi T. 2015. Epithelial estrogen receptor 1 intrinsically mediates squamous differentiation in the mouse vagina. Proc Natl Acad Sci U S A 112:12986–12991. doi: 10.1073/pnas.1513550112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Staab JF, Bradway SD, Fidel PL, Sundstrom P. 1999. Adhesive and mammalian transglutaminase substrate properties of Candida albicans Hwp1. Science 283:1535–1538. doi: 10.1126/science.283.5407.1535. [DOI] [PubMed] [Google Scholar]
- 52.Dennerstein GJ, Ellis DH. 2001. Oestrogen, glycogen and vaginal candidiasis. Aust N Z J Obstet Gynaecol 41:326–328. doi: 10.1111/j.1479-828X.2001.tb01238.x. [DOI] [PubMed] [Google Scholar]
- 53.Vilela MM, Kamei K, Sano A, Tanaka R, Uno J, Takahashi I, Ito J, Yarita K, Miyaji M. 2002. Pathogenicity and virulence of Candida dubliniensis: comparison with C. albicans. Med Mycol 40:249–257. doi: 10.1080/mmy.40.3.249.257. [DOI] [PubMed] [Google Scholar]
- 54.Fidel PL Jr, Vazquez JA, Sobel JD. 1999. Candida glabrata: review of epidemiology, pathogenesis, and clinical disease with comparison to C. albicans. Clin Microbiol Rev 12:80–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pappas PG, Kauffman CA, Andes D, Benjamin DK Jr, Calandra TF, Edwards JE Jr, Filler SG, Fisher JF, Kullberg BJ, Ostrosky-Zeichner L, Reboli AC, Rex JH, Walsh TJ, Sobel JD. 2009. Clinical practice guidelines for the management of candidiasis: 2009 update by the Infectious Diseases Society of America. Clin Infect Dis 48:T1–T535. doi: 10.1086/598961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sobel JD, Chaim W, Nagappan V, Leaman D. 2003. Treatment of vaginitis caused by Candida glabrata: use of topical boric acid and flucytosine. Am J Obstet Gynecol 189:1297–1300. doi: 10.1067/S0002-9378(03)00726-9. [DOI] [PubMed] [Google Scholar]
- 57.Kumar D, Banerjee T, Pratap CB, Tilak R. 2015. Itraconazole-resistant Candida auris with phospholipase, proteinase and hemolysin activity from a case of vulvovaginitis. J Infect Dev Ctries 9:435–437. doi: 10.3855/jidc.4582. [DOI] [PubMed] [Google Scholar]
- 58.Sobel JD, Vazquez J, Lynch M, Meriwether C, Zervos MJ. 1993. Vaginitis due to Saccharomyces cerevisiae: epidemiology, clinical aspects, and therapy. Clin Infect Dis 16:93–99. doi: 10.1093/clinids/16.1.93. [DOI] [PubMed] [Google Scholar]
- 59.Cuomo CA, Shea T, Yang B, Rao R, Forche A. 2017. Whole genome sequence of the heterozygous clinical isolate Candida krusei 81-B-5. G3 (Bethesda) 7:2883–2889. doi: 10.1534/g3.117.043547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yano J, Fidel PL Jr. 2011. Protocols for vaginal inoculation and sample collection in the experimental mouse model of Candida vaginitis. J Vis Exp doi: 10.3791/3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yano J, Lilly E, Barousse M, Fidel PL Jr. 2010. Epithelial cell-derived S100 calcium-binding proteins as key mediators in the hallmark acute neutrophil response during Candida vaginitis. Infect Immun 78:5126–5137. doi: 10.1128/IAI.00388-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Willems HME, Bruner WS, Barker KS, Liu J, Palmer GE, Peters BM. 2017. Overexpression of Candida albicans secreted aspartyl proteinases 2 or 5 is not sufficient for exacerbation of immunopathology in a murine model of vaginitis. Infect Immun 85:e00248-17. doi: 10.1128/IAI.00248-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Collart MA, Oliviero S. 2001. Preparation of yeast RNA. Curr Protoc Mol Biol Chapter 13 Unit 13.12 doi: 10.1002/0471142727.mb1312s23. [DOI] [PubMed] [Google Scholar]
- 64.Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.