

Clostridium perfringens type F (formerly enterotoxigenic C. perfringens type A) strains produce an enterotoxin (CPE) to cause acute cases of food poisoning and chronic nonfoodborne human gastrointestinal diseases (NFD), e.g., antibiotic-associated diarrhea (AAD). NFD strains also produce NanI sialidase, an extracellular enzyme that releases sialic acids from sialyated host macromolecules.
KEYWORDS: Clostridium perfringens, intestinal colonization, mice, NanI sialidase
ABSTRACT
Clostridium perfringens type F (formerly enterotoxigenic C. perfringens type A) strains produce an enterotoxin (CPE) to cause acute cases of food poisoning and chronic nonfoodborne human gastrointestinal diseases (NFD), e.g., antibiotic-associated diarrhea (AAD). NFD strains also produce NanI sialidase, an extracellular enzyme that releases sialic acids from sialyated host macromolecules. Recent in vitro studies suggested that NanI may contribute to NFD strain intestinal colonization by enhancing the adherence of such strains to intestinal cells and promoting their bacterial growth using generated sialic acid as an energy source. The current study tested this hypothesis by developing a mouse intestinal colonization model involving clindamycin pretreatment to produce conditions mimicking those during AAD. In this model, the type F NFD strain F4969 persisted for at least 4 days in the small intestine, cecum, and colon. When clindamycin-pretreated mice were challenged by oral gavage with equivalent numbers of F4969 bacteria or its isogenic nanI null mutant, significantly lower numbers of the nanI mutant were recovered from all intestinal segments, and it was completely cleared from the small intestine by day 4. Complementation of the mutant to restore NanI production also promoted colonization. When the same nanI null mutant strain was coinoculated into the mouse model together with a nanI-producing strain, the numbers of this mutant were restored to wild-type F4969 levels in all intestinal segments. This result suggests that sialidases produced by other bacteria might also provide some support for C. perfringens intestinal colonization. Collectively, these in vivo findings identify NanI to be the first known significant contributor to chronic intestinal colonization by NFD strains.
INTRODUCTION
Clostridium perfringens is an important pathogen that causes intestinal and histotoxic infections in humans and animals (1, 2). The virulence of this Gram-positive, spore-forming anaerobe involves the production of a large repertoire of toxins, many of which are implicated in specific diseases (3–5). The recently updated toxin-based typing system classifies C. perfringens isolates into seven types (types A through G) on the basis of the production of six typing toxins: alpha, beta, epsilon, iota, necrotic enteritis B-like toxin, and enterotoxin (CPE) (6).
CPE is responsible for several important intestinal infections in humans and, perhaps, animals (1, 3, 7). Specifically, C. perfringens type F (formerly known as CPE-positive C. perfringens type A) strains cause nearly 1 million cases of human food poisoning (FP) per year in the United States (8). This foodborne disease is clinically characterized by acute diarrhea and abdominal cramps (8). C. perfringens type F strains also cause several nonfoodborne human gastrointestinal (GI) diseases (NFDs), including antibiotic-associated diarrhea (AAD) and sporadic diarrhea, both of which are often chronic conditions (3, 9).
Sialidases, also known as neuraminidases, release sialic acids from complex glycoconjugates, including those present on host cell surfaces and in the mucus of the gastrointestinal and respiratory tracts (10, 11). Their action also exposes, for release by other glycosylases and proteases, underlying sugars and amino acids that are subsequently metabolized by some microorganisms (11). C. perfringens produces up to three sialidases, including NanH, which has a cytoplasmic location during log-phase growth, and two secreted sialidases, named NanJ and NanI (12–15). When produced, NanI accounts for most of the exosialidase activity of this bacterium in vitro (12, 16). Several previous in vitro studies by our group suggested that NanI sialidase could be a factor favoring intestinal colonization of NanI-producing C. perfringens. For example, this sialidase facilitates the adherence of NanI-producing C. perfringens type C, D, and F strains to enterocyte-like Caco-2 cells in vitro by modifying the surface of those host cells (13, 14). NanI can also contribute to C. perfringens growth and survival in the presence of Caco-2 cells or semipurified mucin (17). Besides those possible contributions to colonization, NanI also enhances the binding and cytotoxic activity of several important enteric toxins produced by C. perfringens, including CPE (13–15, 18). Additionally, NanI production leads to the increased in vitro production of epsilon toxin by C. perfringens type D (19). Lastly, intestinal proteases were shown to activate NanI ex vivo, suggesting that the in vitro effects described above may be further enhanced during C. perfringens intestinal infections (18).
Despite its pathogenic importance, colonization of the gastrointestinal tract mucosal surface (12) by C. perfringens remains poorly understood. Since NanI sialidase has been associated with increased host cell adherence and the growth of C. perfringens in vitro, it is reasonable to hypothesize that this enzyme, when produced, contributes to intestinal colonization in vivo. However, the lack of a relevant animal model for C. perfringens intestinal colonization has precluded direct testing of this postulate. In response, the current study reports the development of a novel mouse model for studying C. perfringens intestinal colonization. This model was then used with wild-type type F strain F4969, an isogenic nanI null mutant, and a nanI-complemented strain to identify NanI sialidase as the first proven intestinal colonization factor of C. perfringens.
RESULTS
Development of a mouse model for C. perfringens intestinal colonization.
In the absence of a suitable animal model, the first goal of this study was to develop a mouse model to study C. perfringens NFD strain intestinal colonization. After numerous trials, various parameters, such as the challenge dose, the timing of antibiotic administration, and the timing and dose of C. perfringens challenge after antibiotic administration, were established to develop an optimized protocol for a C. perfringens intestinal colonization model in mice (Fig. 1).
FIG 1.

Development of a mouse model for Clostridium perfringens intestinal colonization. Male or female BALB/c mice (weight, 20 to 25 g) were treated orally for 4 days with 1.0 mg of clindamycin dissolved in 0.5 ml of 0.9% sodium chloride by using sterilized oral gavage needles. After 48 h with no treatment, groups of mice were orally challenged with TH broth only (no infection) or ∼108 CFU/ml of wild-type F4969 in TH broth. Some mice were challenged with the same number of nanI null mutant or complemented cells instead of the wild type. During the following 4 days, subgroups of mice (total = 8 mice per subgroup) were euthanized daily and mucosal scrapings from the small intestine, cecum, and colon were collected and weighed. Samples were resuspended in TH medium, and serial dilutions were plated on selective TSC agar plates for C. perfringens and anaerobically incubated overnight at 37°C to calculate the number of CFU per gram of mucosal scraping. A representative number of black colonies obtained 24 h later was screened by PCR for the cpe gene. TSC, tryptose-sulfite-cycloserine medium; compl., complemented.
In this model, mice were treated with clindamycin for 4 days, before a brief rest and challenge with C. perfringens. This antibiotic treatment produces conditions mimicking those occurring during AAD, which is one of the most common spontaneous C. perfringens type F nonfoodborne GI diseases. Prior to challenge, no C. perfringens bacteria were recovered from these BALB/c mice after a 4-day oral treatment with clindamycin and a 2-day rest. After a 2-day rest, the clindamycin-treated mice were orally challenged with ∼108 CFU/ml of wild-type cpe-positive strain F4969, a NanI-producing type F NFD strain, in either Todd-Hewitt (TH) broth or TH broth only (no infection). A 4-day-postchallenge experimental period was used in this model since it falls within the natural duration of type F nonfoodborne GI diseases, which last from 1 to 21 days (20). Daily after challenge, animals were sacrificed and intestinal samples were collected.
One day after this challenge, the level of colonization by wild-type F4969 (per gram of mucosal scraping) was ∼105 CFU in the small intestine and ∼107 CFU in both the cecum and colon (Fig. 2). This colonization persisted for at least 4 days in all intestinal regions, gradually declining to 103 CFU in the small intestine and 106 CFU in the colon and cecum (Fig. 2). PCR for the cpe gene was always positive when performed on representative colonies recovered from the F4969-challenged mice, confirming that those colonies had derived from the initial bacterial challenge (Fig. 1). By comparison, no C. perfringens colonies were recovered from mice challenged with TH broth alone (Fig. 2), further confirming that the C. perfringens bacteria recovered from the F4969-challenged mice were the challenge strain.
FIG 2.

Intestinal colonization of F4969 and isogenic derivatives using the new mouse intestinal colonization model. Mice pretreated for 4 days with clindamycin were orally challenged with ∼108 CFU/ml of the wild-type (WT) F4969, nanI null mutant, or nanI-complemented strain in TH broth or with TH broth only. Each day thereafter up to day 4, some mice were euthanized and intestinal scrapings were collected for plating on TSC agar. Significantly lower numbers of the nanI mutant were recovered on days 3 and 4 from the small intestine (A) and cecum (B) and on all days from the colon (C) after bacterial challenge. *, P < 0.05. Each data point represents the mean value for 8 mice (from three independent experiments). Error bars show standard errors of the means.
C. perfringens localization in the intestine.
Samples of the three intestinal segments that had been challenged with wild-type F4969 or sterile TH broth alone (no infection) were collected, processed for histology, and either stained with periodic acid-Schiff (PAS) and Gram stain for covisualization of mucus and Gram-positive rods or tested by C. perfringens immunohistochemistry (IHC) using an indirect immunoperoxidase technique. One day after bacterial challenge, many Gram-positive rods were visualized in the preserved mucus covering the mucosa of all intestinal segments of the mice inoculated with F4969 (Fig. 3A). Some of these bacteria directly abutted the epithelial cells (not shown). The great majority of those Gram-positive rods were positive by C. perfringens IHC (Fig. 3B). No Gram-positive rods or C. perfringens immunostaining was observed in the intestinal segments of mice challenged with TH broth alone (no infection) (Fig. 3).
FIG 3.
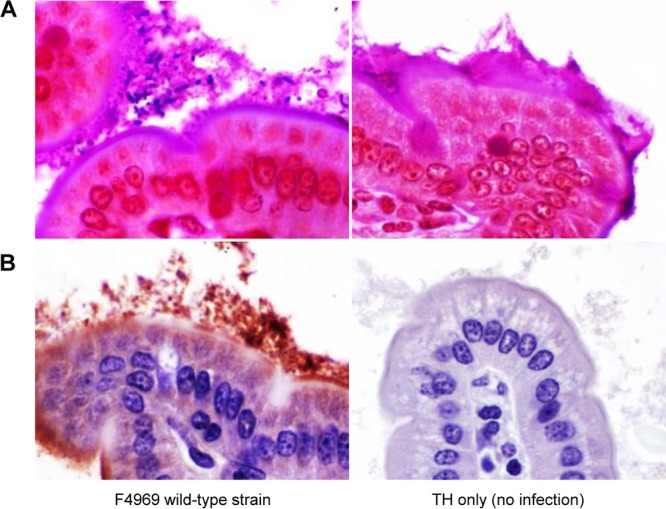
C. perfringens localization in the intestine. (A) PAS and Gram staining of the small intestine of a mouse 1 day after challenge with the F4969 wild-type strain (left) or TH broth alone (right). Numerous Gram-positive rods are visible within the mucus layer. (B) C. perfringens immunohistochemistry on the small intestine of a mouse challenged with the F4969 wild-type strain. Most of the immunopositive staining is present within the mucus layer. Note that no immunostaining was present in the absence of F4969 challenge. TH only indicates challenge with sterile medium (no infection). Magnifications, ×600.
Construction and characterization of a C. perfringens F4969 nanI null mutant.
To investigate whether NanI sialidase can play a role during intestinal colonization by NFD strains, the nanI gene in the transformable type F, human NFD strain F4969 was insertionally inactivated using the Clostridium-modified TargeTron method, as described previously (13). Prior to performing mutagenesis, PCR analyses confirmed previous reports (15) that wild-type F4969 is cpe positive and carries all three recognized C. perfringens sialidase genes (Fig. 4A), including nanH, which encodes the NanH sialidase with a cytoplasmic location in log-phase cultures, and nanI and nanJ, which encode the NanI and NanJ exosialidases, respectively. Identification of these genes was demonstrated by PCR using primers specific for the internal nanH, nanI, and nanJ open reading frame (ORF) sequences; those primers amplified the expected PCR products of 285, 467, and 306 bp, respectively (Table 1).
FIG 4.

Characterization of the C. perfringens F4969 nanI null mutant. (A) PCR analysis of the wild-type F4969 strain shows genes for all three C. perfringens sialidases (nanH, nanI, nanJ) and the cpe gene. The mutant strain carries the wild-type nanH, nanJ, and cpe genes but has a 0.9-kb intron insertion in its nanI gene. The leftmost lane contains molecular size markers (the numbers on the left are in base pairs). (B) Southern blot hybridization of an intron-specific probe with DNA from the wild-type F4969 or the nanI null mutant strain. A single intron insertion was detected using DNA only from the nanI null mutant. The leftmost lane contains DNA fragment size markers. (C) Sialidase activity analyses for the wild-type F4969, nanI null mutant, and complemented strains using 16-h TH broth culture supernatants. *, P < 0.05. The error bars indicate standard errors. OD595, optical density at 595 nm.
TABLE 1.
Primers used in this study
| Primer name | Sequence (5′–3′) | Target gene | Product size (bp) |
|---|---|---|---|
| nanHKOF | AATTGGATGGCTAGGTGGAGTT | nanH | 285 |
| nanHKOR | CAGGTGCTTCCTAAATCGTGAG | ||
| nanIKOF | CAAGAGTTGGTTTTGAGC | nanI | 467 (wild type), 1,367 (null mutant) |
| nanIKOR | AAATAAGGCTGGTATTCTG | ||
| nanJKOF | CTGCAATTCAAGGTGTTGGTG | nanJ | 306 |
| nanJKOR | CTTGTCTTCTAAGCTCATATCC | ||
| CPEnteroF | GGAGATGGTTGGATATTAGG | cpe | 233 |
| CPEnteroR | GGACCAGCAGTTGTAGATA |
However, consistent with the insertion of an ∼900-bp intron into the targeted nanI ORF, the same nanI primers amplified a PCR product of ∼1,400 bp from the isogenic nanI null mutant strain generated by TargeTron mutagenesis (Fig. 4A). As expected, PCR also showed that the nanI mutant still carried the nanH, nanJ, and cpe genes (Fig. 4A). A nanI-complemented strain of F4969 was then prepared, as described previously (13), by transforming the nanI mutant with a pJIR750 shuttle plasmid carrying the nanI gene expressed under the control of its own promoter.
To assess whether only a single intron had inserted into the nanI null mutant, DNA from the F4969 wild-type strain or the nanI mutant was subjected to Southern blot analysis using an intron-specific probe (13). The presence of only a single intron insertion was detected on this Southern blot using DNA from the nanI null mutant (Fig. 4B). In contrast, no probe hybridization to wild-type DNA was detected.
Western blots then compared sialidase expression between F4969 and its derivative strains. This analysis demonstrated that sonicated 8-h TH broth cultures of both wild-type F4969 and the nanI-complemented strains contained the 127-kDa NanJ, the 77-kDa NanI, and the 43-kDa NanH sialidases; in contrast, the F4969 nanI null mutant produced only the NanH and NanJ sialidases. For phenotypic characterization, exosialidase activity assays were carried out using 16-h TH broth culture supernatants of the F4969 wild-type, nanI null mutant, and nanI-complemented strains. The results confirmed that exosialidase activity was significantly reduced in the supernatants from the nanI null mutant strain compared with that in the supernatants from wild-type F4969 (Fig. 4C). This result is consistent with previous reports (15, 17) that NanI is the predominant exosialidase of this strain. Furthermore, complementation of the mutant significantly increased exosialidase activity (Fig. 4C).
Inactivation of the nanI sialidase gene impairs C. perfringens F4969 intestinal colonization in mice.
Our new mouse model (Fig. 1) was used to evaluate whether NanI can contribute to F4969 intestinal colonization. Clindamycin-pretreated mice were orally challenged with equivalent numbers of bacteria of the F4969 wild-type, the isogenic nanI null mutant, or the nanI-complemented strain. By day 4 postchallenge, significantly lower numbers of the nanI mutant were recovered from the small intestine, cecum, and colon of these mice than from the same organs of mice receiving a similar challenge using the F4969 wild-type or nanI-complemented strain (Fig. 2). After day 2 postchallenge, the clearance rate for the nanI mutant from the colon paralleled the clearance rate for the NanI-producing strains (Fig. 2C), but the clearance rate was higher from the small intestine and cecum (Fig. 2A and B). Of note, the nanI mutant was completely cleared from the small intestine by day 4 postchallenge, while the wild-type and nanI-complemented strains still persisted in that intestinal region until the end of the experiments at day 4 (Fig. 2A).
C. perfringens IHC was also performed on intestinal samples from mice 4 days after they had been orally challenged (Fig. 5), and the results mirrored the results presented in Fig. 2. Specifically, the abundant presence of C. perfringens was detected in the small intestine, cecum, and colon from mice orally challenged with wild-type F4969. In samples from mice orally challenged with the F4969 nanI mutant, fewer C. perfringens bacteria were present in the cecum and colon and none were present in the small intestine. As expected on the basis of the results presented in Fig. 2, no C. perfringens bacteria were evident after immunohistochemical staining of these intestinal regions from mice challenged with sterile TH broth, confirming that the C. perfringens bacteria recovered from the mice challenged with F4969 or the F4969 nanI mutant were from the inoculum.
FIG 5.

Microscopy comparison of wild-type and nanI mutant strain abundance in mouse intestine. Note that inactivation of the nanI sialidase gene decreases the amount of C. perfringens F4969 in the intestine of mice. Representative C. perfringens IHC images of the intestines of mice on day 4 postchallenge are shown. Consistent with the findings presented in Fig. 2, lower numbers of the nanI mutant than the wild-type strain were visualized on day 4 in the small intestine, cecum, and colon after bacterial challenge in clindamycin-pretreated mice.
Coinfection with a nanI-producing strain enhances intestinal colonization by an isogenic nanI null mutant.
In Fig. 2, much lower numbers of C. perfringens bacteria were recovered from all intestinal segments of mice inoculated with the isogenic nanI null mutant than from mice challenged with wild-type F4969 or the complemented strain. Therefore, an experiment was performed to distinguish whether coinfection with both an isogenic nanI mutant and a nanI-producing strain would either (i) allow the nanI producer to outcompete the mutant for intestinal colonization or, instead, (ii) enhance the colonization of the mutant. For this study, the mouse model for intestinal colonization described earlier was employed with a few modifications: (i) clindamycin-pretreated mice were orally and simultaneously challenged with equivalent numbers of both the F4969 nanI null mutant and the nanI-complemented strain (the nanI-complemented strain was used in this experiment to allow the rapid quantitative discrimination of the nanI-complemented chloramphenicol [CM]-resistant strain from the CM-sensitive nanI mutant by plating intestinal tissue samples on both CM-free and CM-containing tryptose-sulfite-cycloserine [TSC] agar), (ii) serial dilutions from homogenized mucosal scrapings obtained at days 1 and 3 were plated in duplicate on TSC agar plates with or without CM (since the nanI-complemented strain carries a gene for resistance to this antibiotic), and (iii) representative numbers of the recovered colonies from the CM-free TSC plates were tested by nanI PCR using primers nanIKOF and nanIKOR (Table 1), to confirm the proportions of mutant versus complemented strains indicated by plate count results.
Consistent with the results presented in Fig. 2, plate counting indicated and colony PCR confirmed that the nanI-complemented strain persisted at high levels in all intestinal segments even 3 days after the coinfection challenge (Fig. 6 and 7). Furthermore, both of those approaches demonstrated that the nanI null mutant persisted in all intestinal segments at similar numbers as the nanI-producing strain throughout the 3-day coinfection challenge (Fig. 6 and 7). The number of nanI null mutant colonies recovered in all intestinal segments from mice coinfected with both strains (Fig. 6 and 7) was significantly higher than the number recovered from mice receiving the F4969 nanI null mutant strain alone (Fig. 2).
FIG 6.

Coinfection with a C. perfringens nanI-producing strain enhances intestinal colonization by an isogenic nanI null mutant. Clindamycin-pretreated mice were orally challenged with equivalent numbers of the F4969 nanI null mutant and the nanI-complemented strains. On days 1 and 3 thereafter, groups of mice were euthanized and intestinal scrapings were collected for plating on TSC agar (with or without chloramphenicol). The nanI null mutant persisted at similar numbers as the nanI-producing strain throughout the 3-day challenge in the small intestine (A), cecum (B), and colon (C). ns, not significantly different. Each data point represents the mean value for 5 mice. Error bars show standard errors of the means.
FIG 7.

PCR of C. perfringens colonies isolated from the coinfection experiment. Representative PCR electrophoresis images of recovered colonies tested for nanI by PCR using the primers nanIKOF and nanIKOR to distinguish the nanI null mutant (1,367 bp) from the nanI-complemented (467 bp) strain are shown. Clindamycin-pretreated mice were orally challenged with equivalent numbers of the F4969 nanI null mutant and the nanI-complemented strains. Representative numbers of C. perfringens colonies from intestinal mucosal scrapings were tested by PCR on days 1 and 3. The numbers on the left are molecular size markers (in base pairs). S.I., small intestine.
A control experiment was performed using a similar challenge with the complemented strain alone to assess whether the high colonization rate of the mutant is an artifact of in vivo nanI plasmid loss from the complemented strain, which would cause those strains to appear by plate counting and PCR as the mutant input strain. This control experiment detected by PCR the in vivo loss of the nanI plasmid from only 5% to 10% of the complemented strains by day 3 postchallenge. The results in Fig. 6 were therefore corrected to account for the 10% in vivo conversion of complemented cells to the mutant that was due to spontaneous nanI plasmid loss during the experiment.
DISCUSSION
Mucosal surfaces are frontline barriers that limit invasion by both commensal and pathogenic bacteria into deeper tissues. However, commensals and some pathogens (particularly those causing chronic diseases) colonize and persist on mucosal surfaces (21). An important mucosal colonization strategy used by some of those bacteria is to produce one or more sialidases (22). The paradigm bacterial pathogen for understanding sialidase contributions to mucosal surface colonization is Streptococcus pneumoniae, which produces up to three sialidases (NanA, NanB, and NanC) that promote respiratory tract colonization (23).
Sialidases can contribute to mucosal colonization in several ways. First, they generate free sialic acid, which can then be metabolized for growth or survival. Several commensal and pathogenic intestinal bacteria are known to use sialic acid as a source of carbon and energy, e.g., C. difficile (24), C. perfringens (25, 26), Escherichia coli (27), Vibrio cholerae (28), and Vibrio vulnificus (29). Some of those enteric pathogens, e.g., C. difficile (24), use the sialic acid generated by sialidases produced by other bacteria. However, many intestinal bacteria, including some pathogens (e.g., V. cholerae and C. perfringens), produce their own sialidase. In addition to generating sialic acid for growth, sialidases may expose other underlying sugars or amino acids on intestinal glycoconjugates for release by other degradative enzymes, e.g., C. perfringens hyaluronidase or proteases, followed by the uptake and metabolism of those nutrients. Last, producing their own sialidase may help these intestinal pathogens to adhere to the mucosal surface of the intestines by modifying the mucosal surface to reduce charge repulsions and/or expose occluded adhesin receptors (12).
Despite their potential importance, sialidase contributions to colonization by intestinal pathogens have received relatively limited study. However, recent in vitro studies suggested a potential intestinal colonization role for NanI sialidase by showing that two C. perfringens wild-type intestinal disease strains, i.e., CN3718 and F4969, adhered more efficiently to enterocyte-like Caco-2 cells than their isogenic mutants that did not produce the NanI sialidase (13, 15). Restoring NanI production by complementation boosted adherence, confirming an important role for NanI in the attachment of those C. perfringens strains to Caco-2 cells (13, 15). In addition, NanI sialidase was shown to contribute to the in vitro growth and survival of F4969 using two intestinally relevant nutrient sources, i.e., mucin or enterocyte-like cells (17).
Given the limited direct information available regarding the role of sialidases in intestinal colonization, the present study sought to extend our previous in vitro observations using a newly developed clindamycin-treated mouse model of C. perfringens intestinal colonization that produces conditions that mimic those during AAD, a major type F nonfoodborne GI disease. Using this model, we first showed that F4969 can persistently colonize the small intestine, cecum, and colon. When F4969 wild-type, isogenic nanI mutant, and complemented strains were tested in this mouse model, NanI sialidase production was shown to increase significantly the persistence of the pathogenic C. perfringens type F F4969 strain in all intestinal segments for at least 4 days after intragastric bacterial inoculation. Specifically, inactivation of the nanI gene significantly reduced the ability of F4969 to colonize the mouse gut, while complementation restored colonization, ruling out the possibility that nonspecific secondary mutations caused the reduced colonizing ability of the nanI mutant. To our knowledge, this is the first clear demonstration of a bacterial pathogen producing its own sialidase to enhance intestinal colonization in vivo. On the basis of the previously presented results of in vitro studies discussed in the preceding paragraph, the contribution of NanI to NFD strain intestinal colonization is likely complex, involving (at a minimum) both an enhancement of in vivo adherence and the growth/survival of these strains.
Identifying NanI as an important C. perfringens intestinal colonization factor has implications for understanding disease outcomes. For example, it helps to explain our previous observations (15) that nanI is (i) present in C. perfringens enteric disease strains that persistently colonize the intestine, such as type F strains causing CPE-associated chronic NFD, but (ii) absent from C. perfringens type F FP strains, which cause only an acute infection. Coupling the results of the current study with those epidemiologic association patterns suggests that NanI is an important contributor to the more chronic nature of NFD, where prolonged colonization plays a critical role during repetitive cycles of intestinal vegetative growth and sporulation with associated CPE production. Further supporting a role for NanI in intestinal colonization is the observation that commensal type A strains of C. perfringens isolated from the intestines of healthy humans also produce NanI (15). In contrast, limited colonization due to an inability to produce NanI may help to explain the acute nature of C. perfringens type F FP.
Interestingly, intestinal colonization by the F4969 NanI null mutant strain was restored to wild-type levels if the mutant was coinoculated with a nanI-complemented strain into mice. The ability of the NanI exosialidase produced by this complemented strain to sustain colonization of both the mutant and the complemented strains could, on the basis of in vitro results (13, 15, 17), be attributable to NanI enhancing F4969 adherence and the growth/survival of the mutant. If NanI similarly affects C. perfringens in vivo growth, that would resemble the effect previously observed for the C. difficile 630 strain (24), which encodes the nan operon but lacks a sialidase (30). Consequently, it is proposed that C. difficile acquires and metabolizes free sialic acids generated by other sialidase-producing microorganisms in the intestine (11, 24, 31). It is possible that sialidases of commensal bacteria might also provide some nutrients to NanI-negative type F strains during FP. However, that effect would be unlikely to achieve intestinal sialidase levels comparable to those present during infections by NanI-positive strains during NFD, where up to ∼109 C. perfringens bacteria per gram of stool can occur (32). In addition, sialidases differ in their substrate specificity and other properties, so the sialidases of commensal bacteria may not provide levels of nutrients equivalent to those generated by high levels of NanI during NFD. Similarly, low levels of other sialidases many not cause the same mucosal surface modifications as NanI, which may be important because (i) those surface modifications can facilitate C. perfringens NFD strain adherence to Caco-2 cells (13, 15) and (ii) the absence of NanI production by FP strains suggests an explanation for their relatively rapid clearance from the intestine during FP.
NanI sialidase is likely to be particularly important for C. perfringens growth in nutrient-limited environments like those that can occur in the intestine. For example, the levels of glucose in the small intestine are highly variable (33), which is significant, since abundant glucose reduces the production of NanI (26). When produced, NanI sialidase has been shown to generate sialic acids from sialyated host macromolecules, like mucins (17). Mucins are major components of the mucus that covers the surface of cells lining the digestive tract (34). The presence of many C. perfringens cells on the mucosal surface, as observed in this study, would provide these bacteria with easy access to mucin. Mucin glycoproteins are synthesized by goblet cells present in all intestinal segments (35). However, the proportion of goblet cells along the intestinal mucosa is not uniform; i.e., it increases caudally from the duodenum (4%) to the distal colon (16%) (35, 36). This correlates with the increasing number of microorganism present along the intestine, with the distal colon having the highest numbers (36). The lower numbers of C. perfringens bacteria, particularly those lacking NanI, recovered from the small intestine than from the cecum or colon in the current study may be due, at least in part, to this uneven distribution of goblet cells and the smaller amount of mucin available in this intestinal segment.
Previous studies have provided evidence that NanI sialidase also increases the cytotoxic effects of several C. perfringens toxins involved in diseases originating in the GI tract, e.g., beta and epsilon toxins and CPE (13, 18). This NanI-induced increase in cytotoxicity involves the enhanced binding of those toxins to host cells. The mechanism by which NanI improves toxin binding is not completely understood, but it may involve physical changes on the surface of host cells by removing sialic acids, thus reducing charge repulsion effects and facilitating interaction of the toxins with their receptors (12, 18). While the C. perfringens F4969 strain used in this study can produce CPE, the present animal model of C. perfringens colonization is not suitable to reproduce the intestinal pathology caused by CPE since it does not induce significant in vivo sporulation of C. perfringens, which is needed for CPE production. Therefore, further studies will be needed to develop another model to evaluate the role of NanI sialidase in CPE-associated intestinal disease in vivo. This is particularly important considering the potential use of sialidase inhibitors as an alternative method to treat intestinal infections caused by C. perfringens (12).
MATERIALS AND METHODS
Bacterial strain, plasmids, media, and chemicals.
F4969, a C. perfringens type F (formerly CPE-positive type A) nonfoodborne human GI disease strain (37), was used in this study. This strain carries the cpe gene encoding CPE and all three sialidase genes (nanJ, nanI, and nanH). The plasmids used in this study to prepare a F4969 nanI null mutant strain and nanI-complemented strain were created as described previously (13).
The following media were used for culturing C. perfringens: cooked meat medium (CMM; Difco Laboratories), fluid thioglycolate (FTG) medium (Sigma-Aldrich), TH medium (Bacto Todd-Hewitt broth [Becton, Dickinson] with 0.1% sodium thioglycolate [Sigma-Aldrich]), TGY medium (3% tryptic soy broth [Becton, Dickinson], 2% glucose [Fisher Scientific], 1% yeast extract [Becton, Dickinson], 0.1% sodium thioglycolate [Sigma-Aldrich]), brain heart infusion (BHI) agar plates (Becton, Dickinson), and tryptose-sulfite-cycloserine (TSC) agar plates made of SFP agar base (Becton, Dickinson) with 0.04% d-cycloserine (Sigma-Aldrich). Mice were treated with clindamycin hydrochloride (MP Biomedicals) dissolved in 0.9% sodium chloride (Baxter Healthcare Corporation).
DNA extraction and PCR analysis.
DNA was extracted from the C. perfringens strains using a MasterPure Gram-positive DNA purification kit (Epicentre) following the instructions of the manufacturer. The primers used in this study are listed in Table 1. For all gene targets, PCR amplification conditions were the following: cycle 1 was 95°C for 5 min; cycles 2 through 35 were 95°C for 30 s, 55°C for 40 s, and 68°C for 90 s; and the final extension step was for 5 min at 68°C. An aliquot (20 µl) of each PCR sample was visualized after electrophoresis on a 1.5% agarose gel stained with ethidium bromide.
Preparation of isogenic nanI null mutant and complemented strains.
The Clostridium-modified TargeTron system was utilized to inactivate the nanI gene in F4969 via the targeted insertion of a group II intron (13). As described previously, a TargeTron plasmid that targets insertion of a sense-oriented intron (∼900 bp) between nucleotides 730 and 731 of the nanI open reading frame (ORF) was prepared (13). Primers nanIKOF and nanIKOR were used to screen for the intron insertion into the nanI gene by PCR. This PCR amplifies a 467-bp product from the wild-type nanI gene but a 1,367-bp product from a nanI null mutant strain. As also described previously (13), a pJIR750 shuttle plasmid carrying the nanI gene expressed from its own promoter was prepared and introduced by electroporation into the F4969 nanI null mutant strain to create the F4969 nanI-complemented strain.
Southern blot analysis.
DNA was isolated from the C. perfringens F4969 wild-type and nanI null mutant strains using the MasterPure Gram-positive DNA purification kit (Epicentre). Each DNA sample was then digested with EcoRI overnight at 37°C and run on a 1% agarose gel. After alkali transfer to a nylon membrane (Roche), the blot was hybridized with a digoxigenin-labeled, intron-specific probe as previously described (38). This intron-specific probe was prepared using the primers KO-IBS and KO-EBS1d (38) and a PCR digoxigenin labeling kit (Roche Applied Science) following the instructions of the manufacturer.
Western blot analysis.
A 200-μl aliquot of an overnight FTG culture of the C. perfringens F4969 wild-type, nanI null mutant, or nanI-complemented strain was inoculated into 10 ml of TH medium. Samples were collected, and each supernatant or the sonicated whole culture was mixed with SDS loading buffer and boiled for 5 min. Those mixtures were electrophoresed on an 8% polyacrylamide gel containing SDS for analyzing sialidase proteins. The gels were then subjected to Western blotting using appropriate antibodies, as described previously (13).
Measurement of sialidase activity in culture supernatant.
The NanI sialidase activity in the supernatants of overnight cultures of F4969 was reported to represent most of the total sialidase activity associated with this strain (15). This was confirmed, as previously described (13). Briefly, a 0.2-ml aliquot of an FTG overnight (∼16-h) culture of the F4969 wild-type, the nanI null mutant, or the nanI-complemented strain was transferred to 10 ml of fresh TH medium and cultured overnight at 37°C. The next day, a 0.2-ml aliquot was transferred to a new 10 ml of fresh TH medium and cultured for 16 h at 37°C. A 20-µl aliquot of supernatant from that TH broth culture was added to 60 µl of 0.05 M Tris-HCl buffer (pH 7.2) in a microtiter plate. A 20-µl aliquot of substrate (4 mM 5-bromo-4-chloro-3-indolyl-α-d-N-acetylneuraminic acid [Sigma-Aldrich]) was added, and the mixture was incubated at 37°C for 30 min. The absorbance at 595 nm was then measured using a Bio-Rad microplate reader.
Mouse model for C. perfringens F4969 intestinal colonization.
All procedures involving animals were reviewed and approved by the University of California, Davis, Committee for Animal Care and Use (permit 19186). The experimental protocol is summarized in Fig. 1. Roughly equal numbers of male and female BALB/c mice (weight, 20 to 25 g) were treated orally daily for 4 days with 1 mg of clindamycin dissolved in 0.5 ml of 0.9% sodium chloride by using sterilized oral gavage needles (22 gauge, 50 mm long). The animals were kept in sterilized cages with sterile bedding and offered feed and water (both of which were sterile) ad libitum. Each day, mice were placed into new sterilized cages. This was followed by a 48-h period with no treatment. Then, three groups of mice were challenged with 0.5 ml of overnight TH broth cultures containing ∼108 CFU/ml of the F4969 wild-type (n = 32), F4969 nanI null mutant (n = 32), or F4969 nanI-complemented (n = 32) strain by using sterilized oral gavage needles. A fourth group (n = 32) was challenged with 0.5 ml of sterile TH broth. During the following 4 days, groups of mice were euthanized daily and samples were collected from the small intestine, cecum, and colon. Those samples were fixed by immersion in Carnoy’s fixative for 2 h and then transferred to 10% buffered formalin, pH 7.2, for 24 to 72 h, dehydrated through alcohols to xylene, and embedded in paraffin wax. Approximately 4-cm-long sections of these segments were collected aseptically, opened longitudinally, and gently washed in sterile 0.9% sodium chloride, and mucosal scrapings were then collected using scalpel blades. The mucosal scrapings were weighed and resuspended in TH medium. Serial dilutions (10−1 to 10−8) in TH medium were plated on selective TSC agar plates for C. perfringens and anaerobically incubated overnight at 37°C. Twenty-four hours later, black colonies, indicative of C. perfringens (39), were counted for calculation of the number of CFU per gram. A representative number of those colonies was screened by PCR for the cpe gene to confirm that they originated from the inoculated F4969 strain.
C. perfringens microscopy.
Four-micrometer sections were cut from the paraffin blocks of all intestinal segments of mice inoculated with C. perfringens and TH broth alone, and they were costained with periodic acid-Schiff (PAS) and Gram stain for covisualization of mucus and Gram-positive rods. Four-micrometer-thick paraffin-embedded sections of small intestine, cecum, and colon were also processed by an indirect immunoperoxidase technique for C. perfringens as previously described (40), using a Dako EnVision kit (Dako, Carpenteria, CA) according to the instructions of the manufacturer. The primary antibody was rabbit polyclonal anti-C. perfringens (GenWay Bio, San Diego, CA). Intestinal samples from mice receiving clindamycin but no C. perfringens inoculation were used as negative controls. Additional negative controls consisted of serial tissue sections of the test tissue incubated with normal rabbit serum instead of the specific antibodies. The colon of a goat from which C. perfringens had been isolated was used as a positive control.
Mouse model for coinfection with the F4969 nanI null mutant and nanI-complemented strains.
For the mouse model of coinfection with the F4969 nanI null mutant and nanI-complemented strains, after 4 days of clindamycin treatment and 48 h of rest, mice (n = 10) were inoculated with 0.5 ml of overnight TH broth cultures containing ∼108 CFU/ml of the F4969 nanI null mutant and ∼108 CFU/ml of the F4969 nanI-complemented strains by using sterilized oral gavage needles. On the first and third days after inoculation, groups of mice were euthanized and mucosal scrapings of small intestine, cecum, and colon were collected as described above and resuspended in TH medium. Serial dilutions (10−1 to 10−8) in TH medium were plated in duplicate on selective TSC agar plates for C. perfringens with or without chloramphenicol (CM) and incubated anaerobically overnight at 37°C. The rationale behind this differential plating is that the nanI-complemented strain carries a gene encoding resistance to this antibiotic, so both the nanI mutant and complemented strains can grow on TSC agar without CM but only the complemented strain can grow on TSC agar with CM. Twenty-four hours later, black colonies were counted for calculation of the number of CFU per gram. A representative number of those colonies (about 20 per intestinal segment) was screened by PCR for nanI using the primers nanHKOF and nanHKOR (Table 1) to evaluate the proportion of nanI null mutant and nanI-complemented strains recovered on each day and, thus, confirm the plate count results. A control experiment was performed using a challenge with the complemented strain alone to examine for spontaneous plasmid loss, which would cause those bacteria to appear as the mutant strain when plated on TSC plates with CM or by PCR on TSC plates without CM. This control experiment detected the loss of the nanI plasmid by ∼5 to 10% of the complemented strains by day 3 postchallenge. The number of colonies counted on CM plates was then increased by 10% to account for this spontaneous nanI plasmid loss. With this information, the final number of nanI null mutant colonies in the coinfection assay was calculated as follows: the total number of colonies of the nanI null mutant and nanI-complemented strains (colonies counted on TSC plates with no CM) minus the number of nanI-complemented strain colonies (colonies counted on TSC plates with CM).
Statistical analyses.
All statistical analyses were performed using R (v3.3.1). For comparison of sialidase activity in culture supernatants, one-way analysis of variance (ANOVA) was applied with post hoc analysis using Tukey’s multiple-comparison test. Bacterial counts were compared by negative binomial regression analysis. Differences were considered significant when the P value was less than 0.05.
ACKNOWLEDGMENTS
This work was generously supported by grant R21 AI125796-2 (to B.A.M., J.L., and F.A.U.) from the National Institute of Allergy and Infectious Diseases. M.A.N. is supported by Becas Chile, CONICYT, Gobierno de Chile.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
REFERENCES
- 1.Uzal FA, Freedman JC, Shrestha A, Theoret JR, Garcia J, Awad MM, Adams V, Moore RJ, Rood JI, McClane BA. 2014. Towards an understanding of the role of Clostridium perfringens toxins in human and animal disease. Future Microbiol 9:361–377. doi: 10.2217/fmb.13.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Theoret JR, McClane BA. 2016. Toxins of Clostridium perfringens, p 45–69. In Uzal FA, Songer JG, Prescott JF, Popoff MR (ed), Clostridial diseases of animals. Wiley Blackwell, Ames, IA. [Google Scholar]
- 3.McClane BA, Robertson SL, Li J. 2013. Clostridium perfringens, p 465–489. In Doyle MP, Buchanan RL (ed), Food microbiology: fundamentals and frontiers, 4th ed ASM Press, Washington, DC. [Google Scholar]
- 4.Li J, Adams V, Bannam TL, Miyamoto K, Garcia JP, Uzal FA, Rood JI, McClane BA. 2013. Toxin plasmids of Clostridium perfringens. Microbiol Mol Biol Rev 77:208–233. doi: 10.1128/MMBR.00062-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Navarro M, McClane B, Uzal F. 2018. Mechanisms of action and cell death associated with Clostridium perfringens toxins. Toxins 10:212. doi: 10.3390/toxins10050212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rood JI, Adams V, Lacey J, Lyras D, McClane BA, Melville SB, Moore RJ, Popoff MR, Sarker MR, Songer JG, Uzal FA, Van Immerseel F. 2018. Expansion of the Clostridium perfringens toxin-based typing scheme. Anaerobe, in press. doi: 10.1016/j.anaerobe.2018.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Freedman JC, Shrestha A, McClane BA. 2016. Clostridium perfringens enterotoxin: action, genetics, and translational applications. Toxins 8:73. doi: 10.3390/toxins8030073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Centers for Disease Control and Prevention (CDC). 2017. CDC estimates of foodborne illness in the United States: Clostridium perfringens. Centers for Disease Control and Prevention, Atlanta, GA. Accessed 31 August 2018 https://www.cdc.gov/foodsafety/diseases/clostridium-perfringens.html.
- 9.Carman RJ. 1997. Clostridium perfringens in spontaneous and antibiotic associated diarrhoea of man and other animals. Rev Med Microbiol 8:S43–S45. [Google Scholar]
- 10.Tailford LE, Crost EH, Kavanaugh D, Juge N. 2015. Mucin glycan foraging in the human gut microbiome. Front Genet 6:81. doi: 10.3389/fgene.2015.00081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Juge N, Tailford L, Owen CD. 2016. Sialidases from gut bacteria: a mini-review. Biochem Soc Trans 44:166–175. doi: 10.1042/BST20150226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li J, Uzal FA, McClane BA. 2016. Clostridium perfringens sialidases: potential contributors to intestinal pathogenesis and therapeutic targets. Toxins 8:341. doi: 10.3390/toxins8110341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li J, Sayeed S, Robertson S, Chen J, McClane BA. 2011. Sialidases affect the host cell adherence and epsilon toxin-induced cytotoxicity of Clostridium perfringens type D strain CN3718. PLoS Pathog 7:e1002429. doi: 10.1371/journal.ppat.1002429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li J, McClane BA. 2014. The sialidases of Clostridium perfringens type D strain CN3718 differ in their properties and sensitivities to inhibitors. Appl Environ Microbiol 80:1701–1709. doi: 10.1128/AEM.03440-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li J, McClane BA. 2014. Contributions of NanI sialidase to Caco-2 cell adherence by Clostridium perfringens type A and C strains causing human intestinal disease. Infect Immun 82:4620–4630. doi: 10.1128/IAI.02322-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chiarezza M, Lyras D, Pidot SJ, Flores-Díaz M, Awad MM, Kennedy CL, Cordner LM, Phumoonna T, Poon R, Hughes ML, Emmins JJ, Alape-Girón A, Rood JI. 2009. The NanI and NanJ sialidases of Clostridium perfringens are not essential for virulence. Infect Immun 77:4421–4428. doi: 10.1128/IAI.00548-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li J, McClane BA. 2018. NanI sialidase can support the growth and survival of Clostridium perfringens strain F4969 in the presence of sialyated host macromolecules (mucin) or Caco-2 cells. Infect Immun 86:e00547-17. doi: 10.1128/IAI.00398-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Theoret JR, Li J, Navarro MA, Garcia JP, Uzal FA, McClane BA. 2018. Native or proteolytically activated NanI sialidase enhances the binding and cytotoxic activity of Clostridium perfringens enterotoxin and beta toxin. Infect Immun 86:e00730-17. doi: 10.1128/IAI.00730-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li J, Freedman JC, McClane BA. 2015. NanI sialidase, CcpA, and CodY work together to regulate epsilon toxin production by Clostridium perfringens type D strain CN3718. J Bacteriol 197:3339–3353. doi: 10.1128/JB.00349-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brett MM, Rodhouse JC, Donovan TJ, Tebbutt GM, Hutchinson DN. 1992. Detection of Clostridium perfringens and its enterotoxin in cases of sporadic diarrhoea. J Clin Pathol 45:609–611. doi: 10.1136/jcp.45.7.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ribet D, Cossart P. 2015. How bacterial pathogens colonize their hosts and invade deeper tissues. Microbes Infect 17:173–183. doi: 10.1016/j.micinf.2015.01.004. [DOI] [PubMed] [Google Scholar]
- 22.Lewis AL, Lewis WG. 2012. Host sialoglycans and bacterial sialidases: a mucosal perspective. Cell Microbiol 14:1174–1182. doi: 10.1111/j.1462-5822.2012.01807.x. [DOI] [PubMed] [Google Scholar]
- 23.Brittan JL, Buckeridge TJ, Finn A, Kadioglu A, Jenkinson HF. 2012. Pneumococcal neuraminidase A: an essential upper airway colonization factor for Streptococcus pneumoniae. Mol Oral Microbiol 27:270–283. doi: 10.1111/j.2041-1014.2012.00658.x. [DOI] [PubMed] [Google Scholar]
- 24.Ng KM, Ferreyra JA, Higginbottom SK, Lynch JB, Kashyap PC, Gopinath S, Naidu N, Choudhury B, Weimer BC, Monack DM, Sonnenburg JL. 2013. Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens. Nature 502:96–99. doi: 10.1038/nature12503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Therit B, Cheung JK, Rood JI, Melville SB. 2015. NanR, a transcriptional regulator that binds to the promoters of genes involved in sialic acid metabolism in the anaerobic pathogen Clostridium perfringens. PLoS One 10:e0133217. doi: 10.1371/journal.pone.0133217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li J, Evans DR, Freedman JC, McClane BA. 2017. NanR regulates nanI sialidase expression by Clostridium perfringens F4969, a human enteropathogenic strain. Infect Immun 85:e00241-17. doi: 10.1128/IAI.00241-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chang D-E, Smalley DJ, Tucker DL, Leatham MP, Norris WE, Stevenson SJ, Anderson AB, Grissom JE, Laux DC, Cohen PS, Conway T. 2004. Carbon nutrition of Escherichia coli in the mouse intestine. Proc Natl Acad Sci U S A 101:7427–7432. doi: 10.1073/pnas.0307888101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Almagro-Moreno S, Boyd EF. 2009. Sialic acid catabolism confers a competitive advantage to pathogenic Vibrio cholerae in the mouse intestine. Infect Immun 77:3807–3816. doi: 10.1128/IAI.00279-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jeong HG, Oh MH, Kim BS, Lee MY, Han HJ, Choi SH. 2009. The capability of catabolic utilization of N-acetylneuraminic acid, a sialic acid, is essential for Vibrio vulnificus pathogenesis. Infect Immun 77:3209–3217. doi: 10.1128/IAI.00109-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sebaihia M, Wren BW, Mullany P, Fairweather NF, Minton N, Stabler R, Thomson NR, Roberts AP, Cerdeño-Tárraga AM, Wang H, Holden MTG, Wright A, Churcher C, Quail MA, Baker S, Bason N, Brooks K, Chillingworth T, Cronin A, Davis P, Dowd L, Fraser A, Feltwell T, Hance Z, Holroyd S, Jagels K, Moule S, Mungall K, Price C, Rabbinowitsch E, Sharp S, Simmonds M, Stevens K, Unwin L, Whithead S, Dupuy B, Dougan G, Barrell B, Parkhill J. 2006. The multidrug-resistant human pathogen Clostridium difficile has a highly mobile, mosaic genome. Nat Genet 38:779–786. doi: 10.1038/ng1830. [DOI] [PubMed] [Google Scholar]
- 31.Vimr ER, Kalivoda KA, Deszo EL, Steenbergen SM. 2004. Diversity of microbial sialic acid metabolism. Microbiol Mol Biol Rev 68:132–153. doi: 10.1128/MMBR.68.1.132-153.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Borriello SP, Barclay FE, Welch AR, Stringer MF, Watson GN, Williams RK, Seal DV, Sullens K. 1985. Epidemiology of diarrhoea caused by enterotoxigenic Clostridium perfringens. J Med Microbiol 20:363–372. doi: 10.1099/00222615-20-3-363. [DOI] [PubMed] [Google Scholar]
- 33.Ferraris RP, Yasharpour S, Lloyd KC, Mirzayan R, Diamond JM. 1990. Luminal glucose concentrations in the gut under normal conditions. Am J Physiol 259:G822–G837. doi: 10.1152/ajpgi.1990.259.5.G822. [DOI] [PubMed] [Google Scholar]
- 34.Perez-Vilar J, Hill RL. 1999. The structure and assembly of secreted mucins. J Biol Chem 274:31751–31754. doi: 10.1074/jbc.274.45.31751. [DOI] [PubMed] [Google Scholar]
- 35.Karam SM. 1999. Lineage commitment and maturation of epithelial cells in the gut. Front Biosci 4:D286–D298. doi: 10.2741/A426. [DOI] [PubMed] [Google Scholar]
- 36.Kim YS, Ho SB. 2010. Intestinal goblet cells and mucins in health and disease: recent insights and progress. Curr Gastroenterol Rep 12:319–330. doi: 10.1007/s11894-010-0131-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Collie RE, McClane BA. 1998. Evidence that the enterotoxin gene can be episomal in Clostridium perfringens isolates associated with non-food-borne human gastrointestinal diseases. J Clin Microbiol 36:30–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sayeed S, Uzal FA, Fisher DJ, Saputo J, Vidal JE, Chen Y, Gupta P, Rood JI, McClane BA. 2007. Beta toxin is essential for the intestinal virulence of Clostridium perfringens type C disease isolate CN3685 in a rabbit ileal loop model. Mol Microbiol 67:15–30. doi: 10.1111/j.1365-2958.2007.06007.x. [DOI] [PubMed] [Google Scholar]
- 39.Harmon SM, Kautter DA, Peeler JT. 1971. Improved medium for enumeration of Clostridium perfringens. Appl Microbiol 22:688–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Diab SS, Kinde H, Moore J, Shahriar MF, Odani J, Anthenill L, Songer G, Uzal FA. 2012. Pathology of Clostridium perfringens type C enterotoxemia in horses. Vet Pathol 49:255–263. doi: 10.1177/0300985811404710. [DOI] [PubMed] [Google Scholar]