

Abstract
ABSTRACT: Overlap syndrome is often defined as an entity that satisfies the classification criteria for at least two distinct connective tissue disease. We present the case of a female patient, 39 years old, hospitalized in the Dermatology clinic of Emergency County Hospital in september 2014, for ulcerative lesions on the left hallux and the second and third left toes, associated with pain and difficulties in walking. After performing the clinical exam and paraclinical tests, we decided for both intravenous synthetic analogue of prostacyclin PGI2-iloprost and local therapeutic measures, with a favourable outcome of the ulcerations. Due to the fact that overlap syndrome is an entity with several visceral involvement and unpredictable evolution, we must not disregard the skin manifestation, so that we can prevent a severe evolution and improve the outcome. The case represents a permanent challenge for the multidisciplinary team that examine, survey and periodically adjust the treatment, based on the biological status and predominant symptoms.
Keywords: overlap, systemic lupus erythematosus, systemic sclerosis
Introduction
Overlap syndrome (OS) is often defined as an entity that satisfies the classification criteria for at least two connective tissue diseases (CTDs) occurring at the same time or at different times in the same patient [1]. CTDs include systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), systemic sclerosis (SSc), polymyositis/dermatomyositis (PDM), and Sjögren syndrome (SS). Any combinations of coexisting rheumatic diseases are reported in the literature. Cardinal signs of one specific connective tissue disease may also be part of the normal clinical spectrum of another connective tissue disease, since many clinical findings and laboratory markers lack specificity [2, 3]. The accurate identification of OS is useful in order to clarify the prognosis and apply the proper therapeutic measures.
Two approaches have been suggested in categorizing such conditions. We can approach this conditions either by detecting a specific antibody combined with peculiar clinical findings, or by identyfing a certain pattern of clinical features, without a specific serologic marker [1, 4].
The pharmacological therapeutic options are mainly based on corticosteroids, disease modifying agents (DMARDs), synthetic or biological, and immunosuppressive drugs. Biologic drugs, like anti-TNFα or anti-CD20 monoclonal antibodies, have been used as alternative treatments in refractory cases. Moreover, there are some concerns with the use of anti-TNF agents in patients with systemic autoimmune diseases, due to the risk of triggering disease exacerbations [1, 2]. The genetic and immunological profile could therefore be useful in predicting a certain therapeutic response.
Case report
We present the case of a 39 years old female patient, hospitalized in the Dermatology clinic of Emergency County Hospital in september 2014, for ulcerative lesions on the left hallux and the second and third left toes, associated with pain and difficulties in walking. Moreover, the patient presented inflammatory pain of the radio-carpal joint, asthenia, dyspneea and myalgia, especially in upper limbs. The patient was diagnosed in 2007 with overlap syndrome-systemic lupus erythematosus, antiphospholipid syndrome, systemic sclerosis-diffuse form and dermatomyositis, based on laboratory findings-presence of antinuclear antibodies-ANA, anti-dsDNA antibodies, anti-Scl70 and anti-U1-RNP antibodies, increased levels of creatine kinase-CK, positive biopsy for systemic sclerosis and dermatomyositis. The histopathological exam, for skin biopsy, revealed atrophic epidermis with hyperpigmentation of the basal layer, with straight lines of the basal membrane and papilloma areas; underlying dense collagen sclerosis, with hyaline areas, partial atrophy of skin and dermal appendages, and for muscle biopsy: degeneration of skeletal muscle fibers and interstitial inflammatory infiltrate.
From the personal history, we note multiple spontaneous abortion in the first 3-4 weeks of pregnancy. The pathologic history reveals chronic kidney disease, an ischemic stroke in 2011 (objectivized by computed tomography and magnetic resonance), hypertension, coronary cardiac disease and cardiac insufficiency treated and monitored periodically by the cardiologist.
On admission, the clinical exam revealed a patient with a facies with lack of frontal folds, pinched nose, retraction of the lips, with limited oral aperture, Cushing conformation, telangiectasia on the cheeks and livedo reticularis, with poikilodermic aspect (1), pale chest with sharply demarcated and erythematous papulo-squamous lesions (Fig.1), left hallux ulceration, of 1.5/1 cm, with a haematic scall, ulcerations of 0.5cm on the second and third toe, dry eyes, diffuse alopecia, muscular hypotonia and hypokinesia, muscle atrophy of the shoulder and pelvic girdle. We found inflammatory signs in radio-carpal, metacarpophalangeal (MCP) joints and proximal interphalangeal (PIP) joints. The pulmonary and cardiac clinical exam showed no pathological findings. The muscular testing, using MMT-8 scale, that included evaluation of axial, bilateral proximal and distal muscles for upper and lower limbs, showed a score of 91, of 150 maximum.
Figure 1.
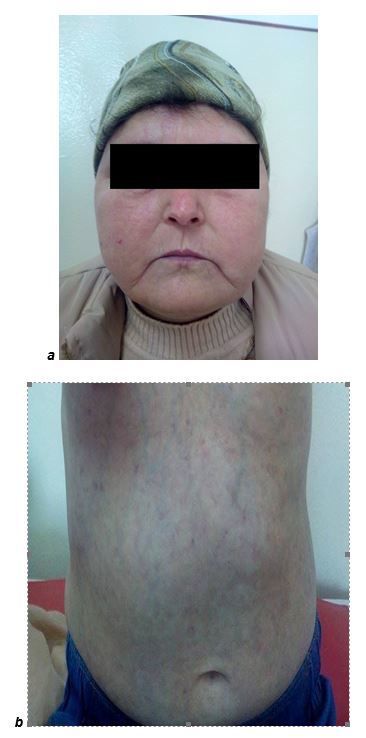
a.Fool moon facies/ b.poikilodermic aspect
The biological analyses show mild normochromic, normocytic anemia, inflammatory syndrome (ESR 38/60mm and CRP 6mg/dl), creatinine of 1.92mg/dl, urea 69mg/dl, normal aminotransferases, CK 250U/l; urinalysis: presence of albumin, a few leucocytes and red blood cells; proteinuria/24 hours-650mg/24h. The immunological tests show a low complement: C4 8.3mg/dl and C3 80mg/dl; anti-dsDNA antibodies 425UI/ml.
The chest X-ray shows interstitial pulmonary fibrosis, mostly in medium and inferior areas. The cardiac ultrasonography, performed with 5 days before the admission in our department, showed a mild systolic and diastolic dysfunction, pulmonary hypertension and small pericardial effusion.
In order to assess the peripheric microcirculation, we performed nailfold capillaroscopy, that showed avascular areas (1), dilated capillaries, frequent haemorrhages (2; 3), bushy capillaries, multiple capillaries with increased tortuosity (3) and giant capillaries (4), findings specific to a late scleroderma pattern.
Figure 2.

a.Avascular area, dilated capillaries, relative recent haemorrhage/ b.bushy capillaries, increased tortuosity
Figure 3.

Frequent dilated capillaries, haemorrhage
Figure 4.

Giant capillaries
The treatment included intravenous administration of synthetic analogue of prostacyclin PGI2-Iloprost for 3 days and local measures, like antiseptic solutions and epithelization agents.
The patient also continued the chronic treatment prescribed by the rheumatologist: corticotherapy-Methylprednisolone 32mg/day, immunosuppressive drugs-Azathioprine 100mg/day, peripheric vasodilators-Pentoxiretard 800mg/day, and cardiologist-antihypertensive drugs-Atacand 32mg/day, Norvasc 5mg/day, platelets antiaggregant-Aspenter 75mg/say and antithrombotic-Vessel due F 2 capsules/day.
Discussion
The etiopathogeny of immune mediated connective tissue diseases is not completely understood and their classification constitutes a permanent challenge, directly related to different patterns of clinical and immunological expression [1, 4]. Different intricate pathological features, clinical and biological, define a certain evolution and prognosis for the patients. The major diagnosis, and mostly therapeutic difficulty, derives from this fusion of features, like SLE or SSc. Moreover, the association of inflammatory myopathies, was reported in a significant percentage in OS [1, 5]. OS-systemic sclerosis/myositis were described both in adults and teenagers. A study performed by Troyanov et al, that included a Canadian cohort of 100 patients with inflammatory myophaties, showed a percentage of 42%, from the 24 patients with overlap syndromes that presented criteria for systemic sclerosis [5]. Another study, published in Clinical Rheumatology, in 2014, by Aguila et al, that had as an objective the comparative analysis of 31 patients with poly/dermatomyositis associated with SSc, SLE or AR, described the association of SSc in 48% of the cases, LES in 29% and AR in 22.6%. Pulmonary manifestations (fibrosis) and cutaneous involvement (Raynaud phenomena, digital ulcerations) were reported with a high incidence in those with findings of SSc [6]. In a large cohort, of 332 patients with SSc overlap syndromes, Pakozdi et al found that patients with SSc/poly or dermatomyosists carried anti-PM/Scl antibodies in 33.1% of cases; however, this antibody is not disease specific. Other reported autoantibodies were anti-Scl70 in 7.9% and anti U1-RNP in 15.7% of the patients [7]. In our case anti-PM/Scl antibodies were not present in either one of the analysis performed during monitoring visits, but we found the presence of anti-Scl70 and anti-U1RNP antibodies. One possible explanation could be the fact that anti-PM/Scl antibodies seem to be associated with limited SSc and myosistis, and a low prevalence of gastrointestinal features, our patient having diffuse SSc and gastrointestinal involvement from the beginning of disease [8].
The overlap of SLE/SSc was described in different studies, with a low percentage, a report that included all the cases communicated form the nineties to 2013, showed a number of 101 patients with this subtype of OS [1, 7, 8, 9, 10, 11]. The prognosis of these patients is unpredictable and the therapeutic approach difficult to handle, polyserositis, pulmonary hypertension, aseptic femoral head necrosis and leucoencephalopathy being described in the course of disease [2]. There was no specific serological marker identified yet, but a high incidence of anti-dsDNA and anti-Scl70 antibodies has been reported, antibodies that are also present in our case [1]. In SLE patients, the presence of pulmonary hypertension imposes a fast initiation of immunosuppression among with glucocorticoids and cytotoxic drugs. Both causes of pulmonary hypertension, SSc and SLE, have benefits after administering vasodilators, as antagonists for endothelin receptors, analogues of prostacyclin and calcium channel blockers [2, 12]. In our case, was instituted a constant therapy with vasodilators-calcium channel blockers and Iloprost every 6 months, with benefits for the pulmonary hypertension, Raynaud phenomena and for the treatment or prevention of ulcerations. The necessity of high doses of glucocorticoids in SLE, and also in inflammatory myopathies, may be limited by the concomitant SSc-diffuse form, because of the potential of a renal crisis, and may require early use of cytotoxic drugs, like the case of our patient. In addition, in patients with SLE/SS overlap, it is important to distinguish lupus nephritis and renal crisis, in order to establish the proper management of the patient [1, 2, 6, 12]. The particular finding in our case, is the chronic renal disease, with a complex mechanism, by contribution of lupus nephritis and hypertensive nephropathy, our patient having high blood pressure values from the age of 18 years.
Conclusion
OS are often difficult to treat, given the multi-systemic character of the entity, that imposes the physicians to institute a symptomatic therapeutic approach, step by step.
The case presented is a permanent challenge for the multidisciplinary team that examine and survey periodically the evolution, in order to adjust the treatment, depending on the biologic and immunologic status.
The presence of several complications, mentioning the cardiac and the chronic renal disease, the multi-systemic dysfunction, limit our treatment and induce the necessity of a permanent revision of the doses and therapy, in order to decrease the risk of side effects and control the disease activity.
Overall, OS must be carefully evaluated, both at onset and during the follow-up visits, for the possible complications that can appear and for a better management of the patient.
OS have complex, heterogeneous, clinical features, and the precise immunologic pattern is essential for the clinician, helping him to anticipate certain complications, to evaluate the outcome and influence the prognosis. Further multi-center analysis is necessary to better clarify the long term prognosis, often unpredictable, and improve patients’ management.
Acknowledgments
This paper received financial support through the "Program of Excellence in doctoral and postdoctoral research in multidisciplinary chronic diseases", contract no. POSDRU / 159 / 1.5 / S / 133377, financed from the European Social Fund through Sectoral Operational Programme Human Resources Development 2007-2013.
References
- 1.Laccarino L, Gatto M, Bettio S, et al. Overlap connective tissue disease syndromes. Autoimmunity Reviews. 2013;12:363–373. doi: 10.1016/j.autrev.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 2.Balbir-Gurman A, Braun-Moscovici Y. Scleroderma Overlap Syndrome. IMAJ. 2011;13:14–20. [PubMed] [Google Scholar]
- 3.Doria A, Putterman C, Sarzi-Puttini P, Szekanecz Z, et al. Controversies in reheumatism and autoimmunity. Autoimmunity Reviews. 2011;11:555–557. doi: 10.1016/j.autrev.2011.10.023. [DOI] [PubMed] [Google Scholar]
- 4.Rodriguez R, Segovia A. Overlap syndromes in the context of shared autoimmunity. Autoimmunity. 2005;38:219–223. doi: 10.1080/08916930500050145. [DOI] [PubMed] [Google Scholar]
- 5.Troyanov Y, Targoff IN, Tremblay JL, et al. Novel classification of idiopathic inflammatory myopathies based on overlap syndrome features and autoantibodies: analysis of 100 French Canadian patients. Medicine (Baltimore) 2005;84(4):231–249. doi: 10.1097/01.md.0000173991.74008.b0. [DOI] [PubMed] [Google Scholar]
- 6.Aguila L.A., Lopes M.R.U., Pretti F.Z. Clinical and laboratory features of overlap syndromes of idiopathic inflammatory myopathies associated with systemic lupus erythematosus, systemic sclerosis, or rheumatoid arthritis. Clinical Rheumatology. 2014;33(8):1093–1098. doi: 10.1007/s10067-014-2730-z. [DOI] [PubMed] [Google Scholar]
- 7.Pakozdi A, Nihtyanova S, Moinzadeh P, et al. Clinical and serological hallmarks of systemic sclerosis overlap syndromes. Journal of Rheumatology. 2011;38(11):2406–2409. doi: 10.3899/jrheum.101248. [DOI] [PubMed] [Google Scholar]
- 8.Kkoschik RW, Fertig N, Tremblay JL, et al. Anti-PM-Scl antibody in patients with systemic sclerosis. Clin Exp Rheumatol. 2012;30(2 Suppl 71):S12–S16. [PubMed] [Google Scholar]
- 9.Caramanschi P, Biasi D, Volpe A, et al. Coexistence of systemic scleosis with other autoimmun disease. Rheumatol Int. 2007;27:407–410. doi: 10.1007/s00296-006-0207-3. [DOI] [PubMed] [Google Scholar]
- 10.Hunzelmann N, Genth E, Krieg T, et al. The registry of German Network for systemic scleroderma: frequency of disease subsets and patterns of organ involvment. Rheumatology (Oxford) 2008;47:1185–1192. doi: 10.1093/rheumatology/ken179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hhudson M, Rojas A, Coral P. Polyautoimmunity and familial autoimmunity in systemic sclerosis. J Autoimmunity. 2008;31:156–159. doi: 10.1016/j.jaut.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 12.Horn H, Ottosen P, Junker P. Renal crisis in a scleroderma-lupus overlap syndrome. Lupus. 2001;10:886–888. doi: 10.1191/096120301701548382. [DOI] [PubMed] [Google Scholar]