

Gcn5 is a histone acetyltransferase that was previously shown to regulate phototropic and starvation-induced autophagy in the rice blast fungus Magnaporthe oryzae, likely via modification on autophagy protein Atg7. In this study, we identified more potential substrates of Gcn5-mediated acetylation by quantitative and comparative acetylome analyses. By epifluorescence microscopy and biochemistry experiments, we verified that Gcn5 may regulate autophagy induction at both the epigenetic and posttranslational levels and regulate autophagic degradation of a critical metabolic enzyme pyruvate kinase (Pk) likely via acetylation. Overall, our findings reveal comprehensive posttranslational modification executed by Gcn5, in response to various external stimuli, to synergistically promote cellular differentiation in a fungal pathogen.
KEYWORDS: comparative acetylome, Gcn5, histone acetyltransferase (HAT), Magnaporthe oryzae
ABSTRACT
The rice blast fungus Magnaporthe oryzae poses a great threat to global food security. During its conidiation (asexual spore formation) and appressorium (infecting structure) formation, autophagy is induced, serving glycogen breakdown or programmed cell death function, both essential for M. oryzae pathogenicity. Recently, we identified an M. oryzae histone acetyltransferase (HAT) Gcn5 as a key regulator in phototropic induction of autophagy and asexual spore formation while serving a cellular function other than autophagy induction during M. oryzae infection. To further understand the regulatory mechanism of Gcn5 on M. oryzae pathogenicity, we set out to identify more Gcn5 substrates by comparative acetylome between the wild-type (WT) and GCN5 overexpression (OX) mutant and between OX mutant and GCN5 deletion (knockout [KO]) mutant. Our results showed that Gcn5 regulates autophagy induction and other important aspects of fungal pathogenicity, including energy metabolism, stress response, cell toxicity and death, likely via both epigenetic regulation (histone acetylation) and posttranslational modification (nonhistone protein acetylation).
IMPORTANCE Gcn5 is a histone acetyltransferase that was previously shown to regulate phototropic and starvation-induced autophagy in the rice blast fungus Magnaporthe oryzae, likely via modification on autophagy protein Atg7. In this study, we identified more potential substrates of Gcn5-mediated acetylation by quantitative and comparative acetylome analyses. By epifluorescence microscopy and biochemistry experiments, we verified that Gcn5 may regulate autophagy induction at both the epigenetic and posttranslational levels and regulate autophagic degradation of a critical metabolic enzyme pyruvate kinase (Pk) likely via acetylation. Overall, our findings reveal comprehensive posttranslational modification executed by Gcn5, in response to various external stimuli, to synergistically promote cellular differentiation in a fungal pathogen.
INTRODUCTION
Histone acetyltransferases (HATs) and histone deacetylation complexes (HDACs) are known to catalyze acetylation/deacetylation of histones on specific lysine residues and thus, epigenetically regulate global gene transcription, stress reaction, and metabolic flux in plants and yeasts (1–3). In addition, evidence is accumulating that HATs and HDACs are also able to directly acetylate or deacetylate cytosolic proteins besides histone proteins (4–6). Recent studies revealed that fungal HATs are involved in fungal virulence and pathogenic morphogenesis (7–14). However, our knowledge on the underlying mechanism, including the HAT or HDAC substrates and the relevant cellular events, is still limited (15, 16).
Magnaporthe oryzae is an important fungal pathogen that causes the devastating blast disease in rice and several other crops (17). M. oryzae initiates its pathogenic life cycle by forming asexual spores, conidia, upon exposure to light (18). Our recent studies showed that carbohydrate catabolism and homeostasis are spatially and temporally regulated via autophagy to ensure successful conidiation in M. oryzae (19, 20). Phototropic regulation of autophagy was recently found to be mediated by Gcn5, a histone acetyltransferase that could acetylate autophagy protein Atg7 to repress autophagy, in M. oryzae (21). Gcn5 was also shown to be an important regulator of M. oryzae pathogenicity (21), but the exact substrates, other than Atg7, of Gcn5-catalyzed posttranslational modifications (PTM) that are relevant to fungal pathogenicity, are not fully known. Autophagy was also demonstrated as essential for M. oryzae infection, likely by serving a function in programmed cell death during appressorium maturation (22) or in maintaining lipid body integrity (23).
Here, we present the identification of potential target proteins of M. oryzae Gcn5 via quantitative acetylome analysis. We noticed that carbon metabolism, oxidative phosphorylation, and cell death are mediated directly or indirectly by Gcn5-catalyzed acetylation on nonhistone, cytosolic proteins. Furthermore, our experimental results revealed that Gcn5 regulated autophagy induction by a combination of posttranslational modifications and epigenetic regulation of gene expression and regulated autophagic degradation of pyruvate kinase (Pk) likely via acetylation of Pk protein. These findings provide an integrative regulation of morphogenesis and/or infection-related autophagy and its potential substrate(s) for degradation for a fungal pathogen.
RESULTS AND DISCUSSION
Identification of phototropic induced lysine acetylation sites.
To identify more Gcn5 substrates and investigate their function in M. oryzae pathogenicity, we developed a robust workflow for quantitative acetylome as depicted in Fig. S1 in the supplemental material. Mycelia from GCN5OX (overexpression [OX]) and wild-type (WT) strains were exposed to light for 12 h, to maximize phototropic induction of protein acetylation, before total protein lysis. To control for process variability, three independent sets of mycelial samples per strain were prepared in parallel for MS analysis. Each sample was digested with trypsin, and peptides containing AcK were immunoprecipitated using PTMScan Acetyl-Lysine Motif (Ac-K) kit (Cell Signaling Technology, catalog no. 13416S). The enriched AcK peptides were analyzed in triplicate by LC-MS/MS on a Q Exactive mass spectrometer (Thermo Fisher Scientific, US), and data were searched against the M. oryzae proteome.
Experimental flow of comparative acetylome analysis with WT and OX strains. Mycelia were harvested from WT or OX colonies grown on 10 PA solid medium plates for total protein lysis and trypsin digestion. Three independent biological replications were sampled for each strain. The acK peptides were immunoprecipitated using PTMScan Acetyl-Lysine Motif (Ac-K) kit (Cell Signaling Technology, catalog no. 13416S). Enriched peptides were separated and analyzed in duplicate by LC-MS/MS, followed by bioinformatic analyses to identify differentially acetylated proteins between WT and OX strain. Download FIG S1, TIF file, 0.8 MB (858KB, tif) .
Copyright © 2018 Liang et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
By using a false discovery rate (FDR) cutoff of less than or equal to 1%, we identified 1,551 unique AcK sites across 704 proteins (see Data Set S1 in the supplemental material), with an overlap of 94% and 93%, respectively, between the WT and OX (Fig. 1A). Almost half of these 704 proteins were multiacetylated, with 47% having two or more AcK sites (Fig. 1B). The most acetylated proteins were the Hsp70-like protein (MGG_06958), heat shock protein 90 (Hsp90; MGG_06759), glyceraldehyde-3-phosphate dehydrogenase (G3PD; MGG_01084), and an uncharacterized protein (MGG_11259), with 13 acetylated lysines, respectively. We also found abundant acetylation on histone proteins (Data Set S1), including H2A.Z (5 of 18 lysines acetylated), H3 (8 of 14 lysines acetylated), H2A (5 of 12 lysines acetylated), H2B (12 of 23 lysines acetylated), H4 (MGG_06093; 1 of 12 lysines acetylated), H3-like centromeric protein cse-4 (1 of 12 lysines acetylated), and H4 (MGG_01160; 6 of 11 lysines acetylated). This confirmed the role of Gcn5 as a chromatin modifier that epigenetically regulates global gene transcription through modification on histone proteins in M. oryzae. However, most of these proteins or lysine residues did not show significant differences in acetylation levels between the OX and WT strains, and therefore are unlikely to be the specific substrates of Gcn5.
FIG 1.

Identification of acetylated peptides and proteins by LC-MS/MS. (A) Overlap of identified acetylated proteins and peptides from WT and OX mycelia. (B) Distribution of acK sites identified per protein. The numbers of proteins with different numbers of acetylation sites (1 to 13) are shown above the bars. (C) Gene Ontology (GO) term (level 2) distribution for the acetylome of WT and OX mycelia. The proteins were classified by GO annotation based on three categories: biological process, cellular component, and molecular function. (D) KEGG pathway-based enrichment analysis of the acetylated proteins. The KEGG pathway database (http://www.genome.jp/kegg/pathway.html) was used to annotate the pathways of the differentially acetylated proteins.
Acetyl proteins/residuals identified by acetylome analysis. Download Data Set S1, XLS file, 1.9 MB (1.9MB, xls) .
Copyright © 2018 Liang et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Identification and enrichment of lysine acetylation sites potentially dependent on Gcn5.
To identify true substrates of Gcn5 among all acetylated proteins and sites, we compared the relative levels of AcK peptide abundances between WT and OX mycelia using label-free quantitation. Of the 1,551 acK sites identified, we found that acetylation at 92 sites present in 82 proteins changed more than twofold (P ≤ 0.05) in OX mycelia at an FDR of 0.04 (Data Set S2).
Differential acetyl proteins/residuals identified by label-free quantification. Download Data Set S2, XLSX file, 0.4 MB (370KB, xlsx) .
Copyright © 2018 Liang et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Gene Ontology (GO) annotation and enrichment with the acetylated proteins significantly different between the WT and OX samples were as listed in Data Set S3 and schematically represented in three ontologies as molecular function, cellular component, and biological process, as in Fig. 1C. KEGG pathway annotation and enrichment were as listed in Data Set S4 and schematically represented as in Fig. 1D. These pathway analyses revealed the enrichment of differentially acetylated proteins involved in carbon metabolism, including glycolysis, pentose phosphate pathway, starch and sucrose metabolism, TCA cycle, and carbon fixation pathway, in the OX samples compared to the WT samples. The second enriched pathway in the OX samples was the fatty acid metabolism and fatty acid degradation pathway. Another group of enriched proteins in the OX samples were shown to be involved in the signaling pathway of mitochondrial oxidative phosphorylation, which is likely leading to programmed cell death (24). Thus, GCN5OX mycelia show a profound altered protein acetylation across metabolic and signaling pathways.
GO annotation and enrichment with the acetyl proteins significantly different between the WT and the OX samples. Download Data Set S3, XLS file, 2.0 MB (2MB, xls) .
Copyright © 2018 Liang et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
KEGG pathway annotation and enrichment with the acetyl proteins significantly different between the WT and OX samples. Download Data Set S4, XLSX file, 0.04 MB (41.9KB, xlsx) .
Copyright © 2018 Liang et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Gcn5 regulates M. oryzae autophagy induction via histone and nonhistone protein acetylation.
Because there have been limited reports of substrates and sites of Gcn5 regulation, we compared our data with the data previously reported for histone H3. There were 8 acetylated lysine residues detected in H3, namely, H3 K9, K14, K18, K23, K27, and K36 as reported (25, 26), and H3 K4, K56, and K79 that have not been reported in other organisms. No change was detected between the WT and OX strains in the AcK levels of most of the H3 acetylated lysines, except for H3 K79, which was absent in the OX strain (Fig. S2A and Data Set S2). We inferred that phototropic induction of H3 acetylation may reach a saturated level; therefore, overexpression of GCN5 could not further increase H3 acetylation, at most of the AcKs. H3K79 acetylation may be catalyzed by another histone acetyltransferase, whose function may be suppressed by Gcn5; thus, H3K79ac was lost in the OX strain. To further confirm this hypothesis, a single set of acetylome analysis using OX and KO (gcn5Δ) mutants (Data Set S5) also identified multiple acetylation sites, and H3 K79 showed higher acetylation in the KO mutant compared to the OX mutant.
Acetylation profiles of histone H3 and Atg7 and PK expression assessed by qRT-PCR. (A) Acetylation profiles of histone H3. (B) Acetylation profiles of Atg7. **, significant difference (P < 0.05). (C) qRT-PCR to assess expression of WT or mutated forms of PK. Relative fold changes were calculated using the −ΔΔCt method. The ACTIN gene (MGG_03982) was used as an internal control. Asterisks denote significant difference (P < 0.05) compared to the WT values. There were no significant differences between the values for the three overexpression strains. Download FIG S2, TIF file, 0.4 MB (422.6KB, tif) .
Copyright © 2018 Liang et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Differential acetyl proteins identified in KO versus OX samples and Gcn5-associated proteins identified by LC-MS/MS. Download Data Set S5, XLSX file, 0.5 MB (482.2KB, xlsx) .
Copyright © 2018 Liang et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Consistent with our previous finding (21), an autophagy protein Atg7 was found to be acetylated at two sites, K107 and K338. Atg7 K338ac was significantly higher in the OX than in the WT, while K107ac was changed slightly (Fig. S2B and Data Set S2). This result confirmed that Atg7 K338 acetylation is Gcn5 dependent. However, our point mutation of Atg7 (K338R) could not restore phototropic induction of autophagy in the OX strain (unpublished data). It has been reported that histone H3 hyperacetylation in Drosophila melanogaster is associated with transcriptional downregulation of several autophagy-essential ATG genes (ATG5, ATG7, and ATG14), which may restrict autophagic activities (27–29). A recent report displayed that deacetylation of H3 may increase autophagy genes ATG6, ATG15, and ATG16 and ATG22 expression and thus also modulate M. oryzae autophagy (16). As mentioned above, H3 acetylation in OX was at a comparable level with WT, for most of the detected H3 acK sites, except for H3 K79. Therefore, we designed and expressed H3 K79Q (acetylation mimic) mutation in the GCN5OX strain, alone or together with the Atg7 K338R mutation. Interestingly, we observed that a combination of H3 K79Q and Atg7 K338R mutation could successfully restore autophagy induction in the GCN5OX mutant in response to nitrogen starvation, while either single point mutations could not (Fig. 2A to C and Fig. 3). We therefore conclude that Gcn5 regulates M. oryzae autophagy induction at both the epigenetic (through H3 modification) and posttranslational (Atg7 modification) levels.
FIG 2.

Epigenetic and posttranslational regulation of nitrogen-induced autophagy in M. oryzae. (A) Autophagic activity visualized by RFP-Atg8 signal under nitrogen starvation. Representative images for WT or GCN5OX ATG7M H3M mycelia are presented. Vacuolar RFP signal (white arrows), autophagosome (white arrowheads) are indicated. Bar = 5 μm. (B) Quantification of autophagy activity by calculating the percentage (mean ± standard error [SE]) of mycelial area labeled by RFP-Atg8 as autophagosomes or autophagic vacuoles by using software ImageJ (https://imagej.nih.gov/ij/) and the “Analyze>Analyze Particles” function. More than 10 pieces of mycelia were used for such quantification in each instance. (C) Total protein lysates from WT, GCN5OX, GCN5OX H3M, and GCN5OX ATG7M H3M strain were analyzed by immunoblotting with anti-RFP antibodies (rabbit; 1:1,000; Clontech, catalog no. R10367). The presence of free RFP band indicates induction of autophagy. The extent of autophagy was estimated by calculating the amount of free RFP compared with the total amount of intact RFP-Atg8 and free RFP and presented in the bar chart for WT and GCN5OX ATG7M H3M strains. Densitometric analysis was performed by ImageJ (https://imagej.nih.gov/ij/). Coomassie blue stain of the total protein lysates serves as a loading control.
FIG 3.

Epigenetic and posttranslational regulation of nitrogen-induced autophagy in M. oryzae. Autophagic activity visualized by RFP-Atg8 signal in CM- or MM-N-cultured mycelia of the WT, OX, OX (H3M). and OX (Atg7M H3M) mutants. GFP-Gcn5 signal appeared in the strains carrying the overexpressed GCN5 gene. DIC, differential interference contrast. Bar = 5 μm.
Gcn5 regulates pyruvate kinase protein stability via acetylation.
Our comparative acetylome analysis also identified that lysine 188 (K188) on pyruvate kinase (PK), the key enzyme of glycolysis, was slightly more acetylated in the OX strain than in the WT strain (Data Set S2), while it was significantly reduced in the KO strain (Data Set S5), and was among Gcn5-associated proteins isolated by immunoprecipitation and identified also by LC-MS/MS (Data Set S5). The GFP-Gcn5 strain we used in immunoprecipitation is the same OX strain used in our comparative acetylome analysis.
First, we verified that the elevated acetylation level of Pk protein in the OX strain compared to the level in the WT strain did not result from the potential increased Pk protein. Using anti-Pk antibody (Abcam, ab34554), we detected the endogenous Pk level in the WT, OX, and KO strains, and our results showed no obvious difference in the Pk protein level in these three strains (Fig. 4A), confirming that Pk acetylation was indeed increased with GCN5 overexpression. Next, we verified the direct interaction between Gcn5 and Pk by pulling down the GFP-Gcn5-associated proteins using GFP-Trap kit (Chromo Tek, gta-20) and detecting the presence of Pk protein using anti-Pk antibody. Our results showed that Pk was detected in the GFP-trapped samples from the GFP-Gcn5 (OX) strain while absent in that from Tfb5-GFP strain (serving as a negative control; Fig. 4B). Overall, we inferred that Pk may be a potential substrate of Gcn5 and may be possibly related to M. oryzae carbon metabolism. As the PK gene encodes a critical enzyme in glycolysis and may be essential for cell viability, we were unable to obtain deletion mutants of it, although repeated attempts have been made. To investigate its function, we overexpressed this gene in the WT form and two mutation forms, K188R (M1, unacetylable) and K188Q (M2, acetylated mimic), respectively, in the wild-type M. oryzae. The transcription level of PK in the wild-type M. oryzae and in three forms of PK overexpression strains was verified by qRT-PCR (Fig. S2C). The fused GFP-Pk protein (WT or mutated varieties) was detected by anti-PK antibody (Fig. 4C). To our surprise, at the protein level, the WT and M1 forms of Pk seemed more stable than the M2 form of Pk, as an increase in the Pk peptide alone was detected in the M2 mycelia, indicative of enhanced degradation of GFP-Pk fusion protein (Fig. 4C). Similarly, when we detected the fusion protein with anti-GFP antibody, we also observed a higher level of GFP peptide accumulation in the M2 mycelia (Fig. 5A). When expressed in the gcn5Δ background, the wild-type GFP-Pk fusion protein seemed more abundant than in the WT background, indicating that loss or reduced Pk acetylation mediated by Gcn5 may stabilize Pk. In other word, acetylated Pk (likely on K188) is subject to degradation.
FIG 4.

Gcn5 interacts with Pk and may be responsible for Pk stability. (A) Total protein lysates were extracted from wild-type (WT), GCN5OX (OX), and gcn5Δ (KO) strains, and then analyzed by immunoblotting with anti-PK antibodies (rabbit; 1:500; Abcam, ab34554). Detection of a single band higher than the 50-kDa molecular marker band corresponds to Pk protein. Coomassie blue staining of SDS-PAGE of total proteins served as a loading control. (B) Coimmunoprecipitation assays among Gcn5 and Pk proteins. The M. oryzae strains carrying GFP-Gcn5 or GFP-Tfb5 were grown in liquid CM at 28°C for 48 h. Total proteins extracted from the strains were subjected to immunoprecipitation with GFP-Trap (Chromo Tek, gta-20), and the IP proteins were separated by 12% SDS-PAGE and detected by anti-Pk antibody (α-Pk). Coomassie blue staining of SDS-PAGE of total proteins served as a loading control for input proteins. (C) Total protein lysates were extracted from the M. oryzae strains carrying PKOX, PK(M1)OX, and PK(M2)OX and then analyzed by immunoblotting with anti-PK antibodies. The fused GFP-Pk protein and the Pk peptide alone were detected, judged by the molecular weight (Mw) as shown. Coomassie blue staining of SDS-PAGE of total proteins served as a loading control. (D) Total protein lysates were extracted from the M. oryzae strains carrying PKOX, PK(M1)OX, and PK(M2)OX, in both WT or gcn5Δ background, in a time course of 0, 4, and 8 h after treatment with the protein synthesis inhibitor cycloheximide (Sigma-Aldrich, catalog no. C104450). Detection of the fused GFP-Pk protein and GFP peptide alone was labeled. The extent of Pk degradation was estimated by calculating the amount of free GFP compared with the total amount of intact GFP-Pk and free GFP (the numbers appear below the blot). Densitometric analysis was performed using ImageJ (https://imagej.nih.gov/ij/). Coomassie blue staining of SDS-PAGE of total proteins served as a loading control.
FIG 5.
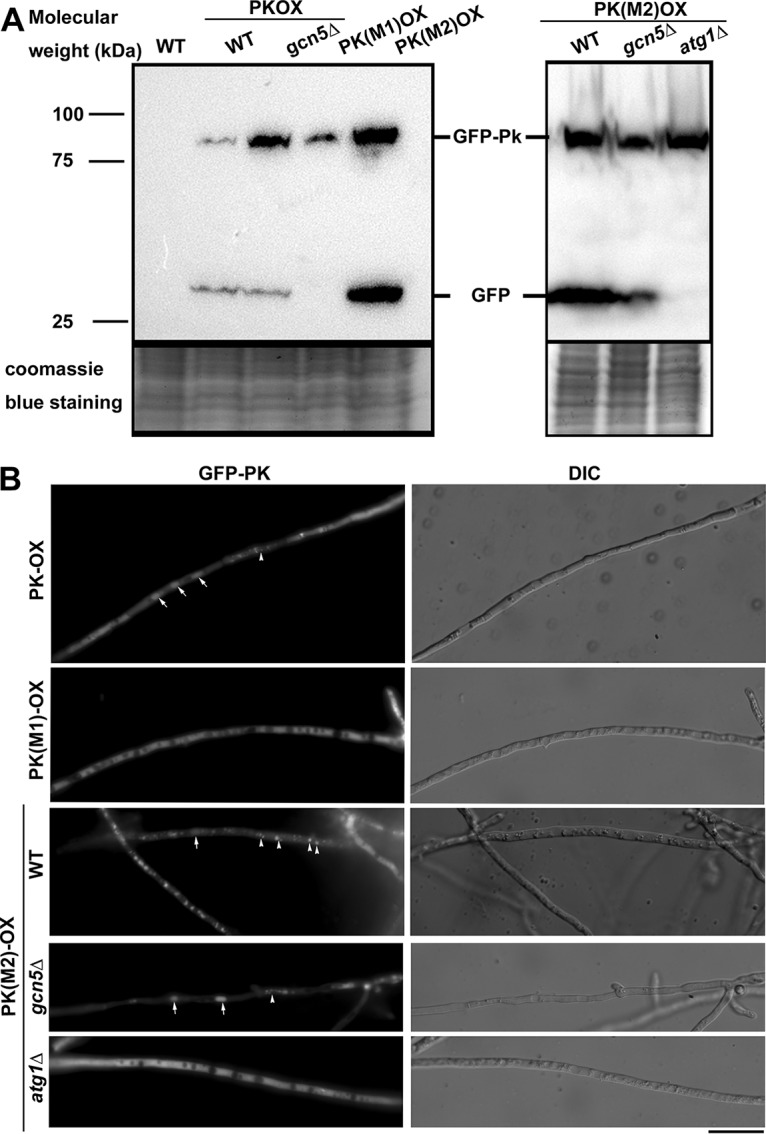
Gcn5-mediated acetylation on Pk lysine 188 may contribute to autophagy degradation of Pk. (A) Total protein lysates were extracted from the wild-type (WT) strain, three versions of PK-overexpressed strains in the WT background, PKOX in the gcn5Δ background, and WT, gcn5Δ, and atg1Δ (autophagy-deficient) strains expressing M2 (K188Q) version of PK. Total protein lysates were analyzed by immunoblotting with anti-GFP antibodies (rabbit; 1:5,000; Invitrogen Molecular Probes, catalog no. A6455). The presence of the free GFP band indicates cleavage between GFP and Pk protein, and likely degradation of fusion proteins. Coomassie blue staining of SDS-PAGE of total proteins served as a loading control. (B) Epifluorescence microscopy with PKOX, PK(M1)OX (K188R, unacetylated mimic) strain, and WT, gcn5Δ, and atg1Δ (autophagy-deficient) strains expressing M2 (K188Q) version of PK. Arrows denote spherical vacuoles and arrowheads indicate vesicular puncta or filamentous vacuoles. Bar = 5 μm.
To verify that Gcn5-mediated Pk acetylation contributes to Pk degradation, we performed a time course detection of cleavage of GFP peptide from the GFP-Pk fusion protein in these three overexpressed Pk varieties in both the WT or gcn5Δ background. Our results showed that cleavage of GFP peptide from the GFP-Pk fusion protein, indicative of GFP-Pk degradation, was obviously suppressed in the PK(M1)OX (K188R) mutant, but not in the PK(M2)OX (K188Q) mutant, and such degradation could be effectively suppressed with the loss of GCN5 in the PKOX strain (Fig. 4D). We observed that cleavage of GFP-Pk(M2) (K188Q, acetylation mimic) was comparable in the WT or gcn5Δ background, further confirming a role of acetylation in Pk degradation.
Interestingly, we found that such degradation of the GFP-Pk(M2) protein (K188Q mutation) was completely lost in the autophagy-deficient mutant atg1Δ background, but not in the gcn5Δ background (Fig. 5A). Correspondingly, we observed that the M2 mutated form of GFP-Pk fusion protein accumulated in vacuoles of M. oryzae mycelia, while the M1 (unacetylated mimic) form of GFP-Pk was cytosolic (Fig. 5B). Vacuolar localization of M2 GFP-Pk was lost in the atg1Δ mutant, but not in the gcn5Δ mutant (Fig. 5B).
Overall, we infer that Gcn5 may catalyze acetylation on Pk lysine 188, which facilitates its degradation in an autophagy-dependent manner. This observation is consistent with a recent report for cancer cells that acetylation of the embryonic M2 isoform of pyruvate kinase (named PKM2) promotes its degradation via chaperone-mediated autophagy, which links metabolic shift and tumor growth (30). However, unlike in animal cells, M. oryzae has only one Pk protein but not with any other isoform. By acetylation- and autophagy-dependent degradation of the only Pk protein, M. oryzae could modulate its carbon metabolism (glycolysis) and likely also determine its cell fate.
Pyruvate kinase plays an important role in M. oryzae pathogenicity.
We further investigate the biological significance of Pk acetylation (and degradation) in M. oryzae by characterizing conidiation (production of asexual spores) and infection of the strains carrying these overexpressed PK variants. For conidiation, we observed no obvious difference between the wild-type (WT) strain and the PKOX variants (P > 0.05; Fig. 6A), except for the PK(M2)OX mutant (K188Q mutation; P < 0.05). This suggests that Pk protein acetylation may promote M. oryzae conidiation. We have reported that the gcn5Δ mutant produced more conidia than the wild-type strain did, likely due to promotion of autophagy induction (21), and here we further assessed conidiation of the gcn5Δ mutant for each PKOX variant. We found that as in the wild-type background, deletion of the GCN5 gene in the PKOX strain or in the PK(M1)OX strain (K188R mutation) resulted in a hyperconidiation phenotype (P < 0.05; Fig. 6A), while such hyperconidiation was not observed in the PK(M2)OX mutant (K188Q mutation; P > 0.05; Fig. 6A). This result could be viewed as an indication that the overexpressed wild-type form or the R or Q mutation of Pk variants is functional. We further infer that both autophagy induction and Pk acetylation play a positive role in M. oryzae conidiation and are subject to Gcn5 regulation.
FIG 6.
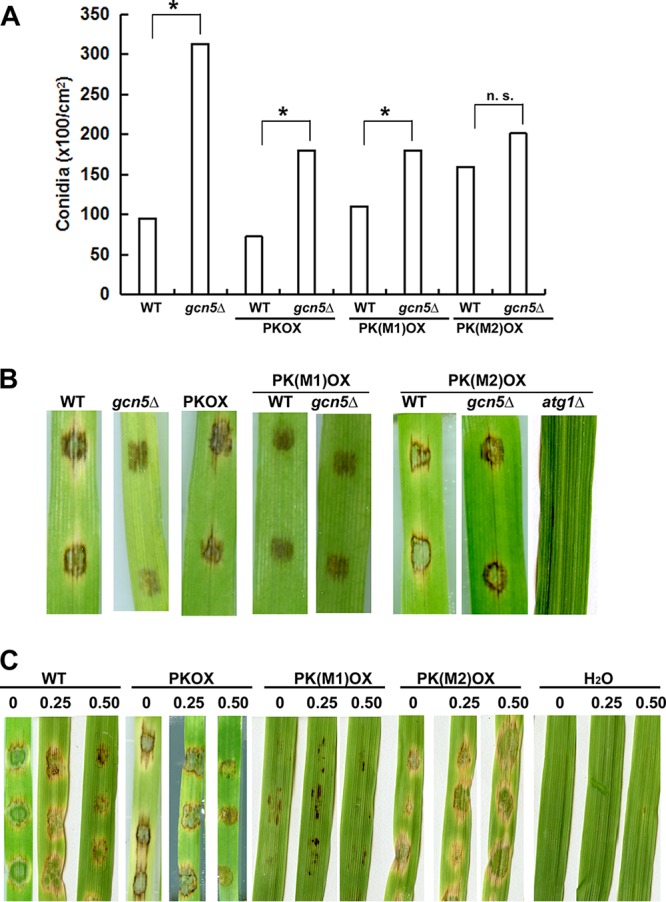
Characterization of mutant strains carrying overexpressed PK variants. (A) Bar chart depicting quantitatively assessed conidiation in the PKOX variants compared with the WT strain, grown on prune agar (PA) medium. Mean values (±SE) were derived from three independent experiments (n = 4 colonies for each sample). Assessments were performed 3 days after photoinduction. n. s., not significant. (B) Disease symptoms on cut leaves of barley inoculated with WT (B157), gcn5Δ mutant, PKOX, and PK(M1)OX, PK(M2)OX conidia in WT and different mutant backgrounds, respectively, were photographed at 7 day postinoculation. (C) Disease symptoms on cut leaves of barley inoculated with WT (B157), PKOX, PK(M1)OX, and PK(M2)OX conidia with or without pyruvate at different concentrations (labeled on top of the images of inoculated leaves), were photographed at 7 days postinoculation. Inoculation with water (H2O) with 0.25 and 0.50 mM pyruvate on barley leave explants served as a negative control.
More interestingly, infection on the barley leaf explants with PK(M1)OX conidia was significantly reduced compared to that of WT conidia, and similar to that of the gcn5Δ conidia (Fig. 6B). Our previous study reported that Gcn5 is required for full pathogenicity, but the mechanism is not fully understood (21). Here we added a piece of evidence that the pathogenicity defect of the gcn5Δ mutant may due to loss/reduced acetylation on Pk protein, which leads to stabilization of this protein. To further support our hypothesis, the PK(M2)OX conidia in the gcn5Δ background could partially restore its pathogenicity, but not in the atg1Δ (autophagy-deficient) background (Fig. 6B). This result demonstrates that autophagy-dependent degradation of acetylated Pk is crucial for M. oryzae pathogenicity. Therefore, our study identified a Gcn5 substrate, Pk, with biological significance in M. oryzae pathogenicity.
As Pk acetylation was reported to be responsible for a metabolic shift in cancer cells (30), we suspected that the pathogenicity defect in the PK(M1)OX or gcn5Δ mutant may result from excessive production of pyruvate, the direct product of Pk-catalyzed glycolysis, as a consequence of the loss of Pk acetylation (on lysine 188 residue) and thus, block/delay Pk degradation via the autophagy pathway. To test this hypothesis, we assessed the effect of pyruvate on M. oryzae pathogenicity by inoculating conidia from the WT strain and the strain carrying overexpressed PK variants, with or without pyruvate, on barley leaf explants. We observed that with increased concentrations of pyruvate, the pathogenicity of WT or PKOX conidia gradually decreased (Fig. 6C). PK(M1)OX conidia were very weak in pathogenicity with or without pyruvate treatment, while PK(M2)OX conidia remained pathogenic even in the presence of 0.50 mM pyruvate (Fig. 6C). Furthermore, when inoculated on an artificial surface, 0.5 mM pyruvate could effectively suppress appressorium formation in the wild-type strain and in the three PK overexpression mutants, while 0.25 mM pyruvate could not (Table 1). Pyruvate was shown to be essential for M. oryzae appressorium formation and pathogenicity, and it could be produced as an intermediate of the glyoxylate cycle by a peroxisomal alanine:glyoxylate aminotransferase, Agt1 (31). Here our infection assay revealed that excessive pyruvate could also suppress M. oryzae appressorium formation and pathogenicity, and therefore, the endogenous pyruvate level needs to be fine-tuned. Besides Agt1, Pk is another critical enzyme controlling pyruvate production and therefore, it plays an important role in M. oryzae pathogenicity.
TABLE 1.
Pyruvate suppressed M. oryzae appressorium formation
| Strain | AP%a
|
||
|---|---|---|---|
| Untreated | 0.25 mM pyruvate | 0.50 mM pyruvate | |
| Wild type | 87.2 ± 3.5 | 90.65 ± 9.2 | 7.5 ± 0.7** |
| PKOX | 91.5 ± 9.2 | 74.3 ± 17.1 | 13.1 ± 8.5** |
| PK(M1)OX | 94.7 ± 3.4 | 91.6 ± 3.5 | 18.0 ± 24.1** |
| PK(M2)OX | 92.0 ± 5.6 | 87.5 ± 13.4 | 53.0 ± 16.8** |
AP%, percent appressorium formation. The values are means ± standard errors (SE) derived from three independent repeats (n ≥ 50 for each instance). Values for pyruvate-treated conidia that are significantly different from the values for the untreated condition for the same strain are indicated by a pair of asterisks.
Gcn5 regulates gene transcription.
As Gcn5 also modulates (directly or indirectly) histone protein H3’s acetylation, we infer that Gcn5 could also epigenetically regulate global gene transcription in M. oryzae. Therefore, we performed transcriptome analysis with OX samples versus WT samples under the same conditions as we performed comparative acetylome analysis to identify potential target genes subject to epigenetic regulation by Gcn5 function. In total, we identified 147 differentially expressed genes (DEGs) (absolute log2 fold change of >1 and FDR of ≤0.05), 60 upregulated genes and 87 downregulated genes (Data Set S6). Among the DEGs listed in Data Set S6, we noticed that genes encoding components of stress response, particularly oxidative stress response (MGG_03329, MGG_05719, MGG_09226, MGG_10760, MGG_10239, MGG_01941, MGG_09162, and MGG_09212), carbohydrate/fatty acid/amino acid metabolism (MGG_09190, MGG_17103, MGG_03995, and MGG_08537), membrane transporters (MGG_09680, MGG_15435, MGG_15331, MGG_15411, MGG_09119, MGG_01864, MGG_04248, MGG_04864, MGG_14824, and MGG_05880), transcriptional factors (MGG_10575, MGG_08536, and MGG_09273), cell wall biosynthesis (MGG_17153, MGG_08527, and MGG_07677), mycelial/conidial pigmentation (MGG_07219 and MGG_05059), and secondary metabolism (MGG_00806, MGG_10671, and MGG_08527), were upregulated in the OX samples. GCN5 overexpression was also verified by the transcriptome analysis, as it (MGG_03677) was found among the upregulated genes (Data Set S6; 2.6-fold, P ≈ 0.0005). In addition, we were interested in the finding that a bacteriorhodopsin-like protein-encoding gene (MGG_09015) was upregulated in the OX strain, as bacteriorhodopsin serves as light-driven proton pump in halobacteria, eubacteria, or fungi (32–34). Given that the functioning photosensor or photoreceptor for phototropic conidiation has not yet identified, this bacteriorhodopsin-like protein may be a potential candidate. In the list of downregulated genes, we also found abundant genes encoding transcriptional factors/regulators (MGG_13350, MGG_04213, MGG_13360, MGG_04171, MGG_06312, and MGG_10422), membrane transport proteins (MGG_03341, MGG_10893, MGG_04234, MGG_07639, and MGG_10869), enzymes involved in cellular metabolism (MGG_10023, MGG_09150, MGG_12228, MGG_03392, MGG_01924, MGG_10274, MGG_04345, MGG_01925, MGG_07626, MGG_13239, MGG_09994, MGG_09647, MGG_08072, MGG_00311, MGG_02261, and MGG_08989), and enzymes involved in cell wall biosynthesis (MGG_14966, MGG_09167, MGG_07214, MGG_06585, and MGG_00110). A serine/threonine protein kinase encoded by MGG_11636 was found to be downregulated in the OX, which is the only kinase identified to be differentially regulated by GCN5 in our transcriptome analysis. A SET protein-encoding gene (MGG_00019) was repressed by GCN5, which is reasonable, as SET proteins inhibit acetylation of nucleosomes by histone acetylases (HAT), especially on histone H3 and H4 (35, 36). Vegetative (heterokaryon) incompatibility responses, which usually result in cell death, may also be suppressed by GCN5, probably via downregulation of MGG_09107, encoding a heterokaryon incompatibility protein. Correlating with this aspect, genes whose products may be involved in cell toxicity and/or death, including a prion inhibition and propagation protein-encoding gene (MGG_00203), an NACHT domain-containing protein (MGG_09355), a BRCT domain-containing protein (MGG_01032), were all repressed by GCN5. However, three autophagy genes, ATG5 (MGG_09262), ATG7 (MGG_07297), and ATG14 (MGG_03698), were not among the DEGs between WT and OX strains under photoinduction. Overall, our transcriptome analysis reveals GCN5-dependent regulation, either directly or indirectly, of global transcription of genes involved in cellular metabolism, stress response, cell toxicity, and death.
Differentially expressed genes (DEGs) between the WT and OX samples identified by transcriptome analysis. Download Data Set S6, XLS file, 0.08 MB (84.5KB, xls) .
Copyright © 2018 Liang et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
A subset of candidate genes from the DEGs were selected for further verification. The list of selected genes and the respective primer sequences are shown in Table S1. We noticed that two FAD-binding protein-encoding genes (MGG_07219 and MGG_05059) were upregulated in the OX (GCN5OX) strain compared to the WT (Fig. 7), indicating that the OX strain may be more resistant/tolerant to the oxidative stress, which is consistent with the sensitivity test results reported previously (21). Also, a stress-responsive transcriptional factor, MGG_09273, was upregulated in the OX strain while reduced in the KO (gcn5Δ) mutant (Fig. 7). Pigmentation occurs during M. oryzae conidiation and infection (37), and two pigment-related genes (MGG_07219 and MGG_05059) were identified as upregulated in the OX strain (Data Set S6) and were verified by RT-PCR (Fig. 7). A gene encoding a bacteriorhodopsin-like protein (MGG_09015) was found highly upregulated in the OX strain compared to the WT strain (Data Set S6). Surprisingly, we detected even higher expression levels of this gene in the KO strain than in either the WT or OX strain (Fig. 7). It is worth following up to confirm the expression pattern of this gene, and more importantly, the functional analysis of this gene as a potential light sensor involved in phototropism of M. oryzae. For the observation that some candidate genes, including two pigmentation genes and a gene encoding bacteriorhodopsin-like protein were upregulated in the OX strain compared to the WT strain, while they were upregulated to an even greater extent in the KO strain, we do not know the exact reason at present. The HAT Gcn5 may regulate gene expression in both a direct manner (via acetylating histone proteins) or indirect manner (via acetylating other HAT or HDAC enzymes and modulating their activity and thus yielding an effect on gene expression through other histone modifiers). Therefore, overexpression of Gcn5 may result in upregulation of gene expression, while deletion of Gcn5 may change the activity of another histone modifier(s) as a nonhistone substrate(s) of Gcn5, and may also lead to induction of the same set of genes.
FIG 7.
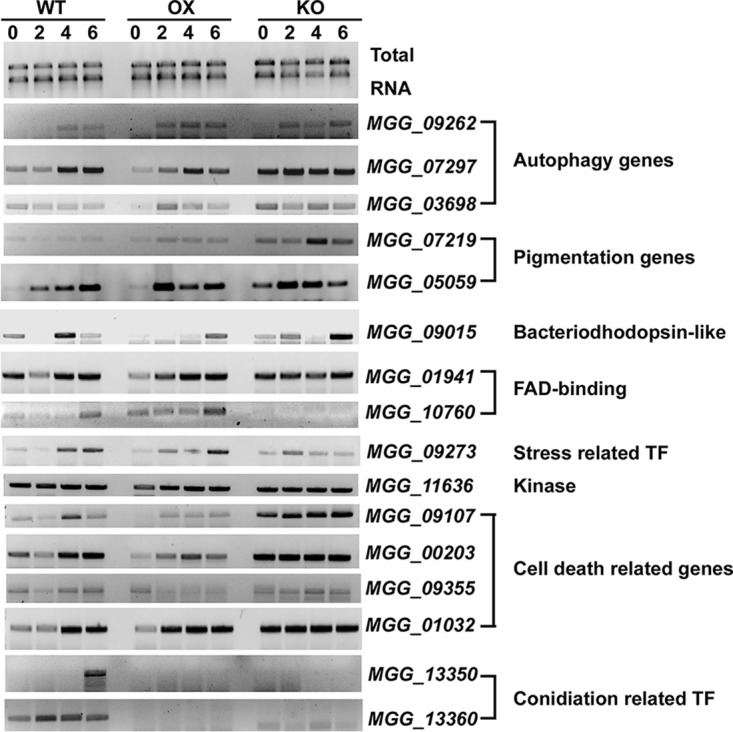
Transcriptional profiling of selected genes by RT-PCR. Total RNA was extracted from the wild-type (WT), GCN5OX (OX), or gcn5Δ (KO) mycelia exposed to light for 0, 2, 4, and 6 h and used for RT-PCR analysis with selected candidate genes. A 1% denaturing agarose gel of total RNA showing 18S and 28S rRNA served as a loading control. The experiments were repeated three times, and representative results are displayed here. TF, transcription factor.
Selected differentially expressed genes for verification by RT-PCR. Download Table S1, DOCX file, 0.02 MB (16.3KB, docx) .
Copyright © 2018 Liang et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
It was also interesting to observe that four genes (MGG_09107, MGG_00203, MGG_09355, and MGG_01032) involved in cell death were all significantly downregulated in the OX strain (Data Set S6). Our RT-PCR verified that MGG_09107, MGG_00203, and MGG_09355 were indeed expressed at lower levels in the OX strain, while they were expressed at higher levels in the KO strain than in the WT strain (Fig. 7). However, the differential expression level of MGG_01032 was not prominent in these three strains (Fig. 7). Transcription of autophagy genes ATG5, ATG7, and ATG14 were comparable between the OX and WT strains as verified by RT-PCR (Fig. 7). We could not rule out the possibility that epigenetic regulation of transcription of another autophagy gene(s) contributes to regulation of autophagy in M. oryzae. Overall, we verified that Gcn5 is indeed involved in epigenetic regulation of several candidate genes involved in cellular development and/or cell and death, as well as stress response, likely via histone protein acetylation. However, the exact function of such candidate genes and the regulatory mode by Gcn5 await further investigation.
Sequence motif analysis of Gcn5-regulated acetylation sites in M. oryzae.
To determine whether there is a common sequence motif required for acetylation by Gcn5, we compared the amino acid sequences of all significantly increased acetylated sites in the OX strain by using Motif-x (38) (Table S2). A preference was observed for leucine, phenylalanine, aspartic acid, or glutamic acid at the −1 position and for leucine, phenylalanine, arginine or lysine at the +1 position. Interestingly, Gcn5 substrates showed a stronger preference for K at the +1 and +4 positions (Table S2).
Statistical analysis of the acK motif significantly increased in the OX strain. Download Table S2, DOCX file, 0.2 MB (228.9KB, docx) .
Copyright © 2018 Liang et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The histone acetyltransferase (HAT) Gcn5 was shown to be essential for regulating M. oryzae conidiation and pathogenicity (21), but the underlying mechanism, which may be pleiotropic, is not fully understood. By utilizing comparative acetylome and transcriptome analyses, we identified a set of histone and nonhistone proteins as potential substrates of Gcn5-mediated acetylation in M. oryzae. By genetic and biochemistry analyses, we revealed the cell biology and biological function of some of these substrates. Our results reveal the roles of Gcn5 in epigenetically regulating gene transcription as a histone modifier and in executing broad regulation of multiple proteins and sites across several key metabolism and stress response pathways involved in nutrient signaling and energy homeostasis. Proteins involved in regulating carbon metabolism were particularly targeted by Gcn5, which indicates the potential for regulation during changes in nutrient availability. Our large-scale inventory of acetylated proteins and sites in the M. oryzae mycelia significantly expands our understanding of how Gcn5 may regulate fungal pathogenic differentiation via epigenetic regulation of global gene expression as well as posttranslational modifications (PTM) of nonhistone proteins. We summarized the metabolic pathways potentially subject to Gcn5 regulation via acetylation in Fig. 8. Multiple substrates of Gcn5 were identified distributing across numerous cellular compartments and carrying a broad range of cellular functions, such as epigenetic regulation of gene expression via histone acetylation in nuclei, regulating carbohydrate metabolism (glycolysis and PPP), fatty acid metabolism, autophagy, and cell wall β-glucan biosynthesis in the cytosol, regulating oxidative phosphorylation and TCA cycle in mitochondria, and regulating protein synthesis in ribosomes. This may at least partially explain the pleiotropic effects observed in the gcn5Δ (KO) mutant. In this study, we verified that Pk is subjected to Gcn5 acetylation, and likely on the lysine 188 residue critical for its degradation in an autophagy-dependent manner. Regulation of Pk turnover via acetylation and autophagic degradation contributes to proper conidiation and host infection, likely through fine-tuning glycerol production through glycolysis to suit different physiological/environmental conditions. Further verification of more Gcn5 substrates and the functional role of acetylation on such a substrate(s) during phototropic induction of asexual differentiation and/or host infection will broaden our understanding of fungal pathogenicity and the corresponding regulatory mechanisms. Future studies will also be needed to address the stoichiometry of acetylation at different sites and possible competition between different acyl modifications.
FIG 8.

Schematic depicting the Gcn5-regulated metabolic pathways. Circles represent constituent proteins or protein complexes within each pathway. Abbreviations: G1P, glucose 1-phosphate; G6P, glucose 6-phosphate; F1,6-2P, fructose 1,6-bisphosphate; 1,3-DPGA, 1,3-bisphosphoglyceric acid; 2-PGA, 2-phosphoglyceric acid; PEP, phosphoenolpyruvic acid; T6P, trehalose phosphate; SuccCoA, succinyl-CoA; OX Phos, oxidative phosphorylation; FA, fatty acid; TCA, tricarboxylic acid.
MATERIALS AND METHODS
Growth conditions and strains used in this study.
Magnaporthe oryzae wild-type strain B157 (field isolate, mat1-2) was obtained from the Fungal Patho-Biology Group (Temasek Life Sciences Laboratory, Singapore). The GCN5OX strain was generated and verified as described previously (21). M. oryzae strains were cultivated on solid prune agar (PA) medium (39) at 28°C for 6 days, followed by 12-h light exposure.
LC-MS/MS analysis.
Total protein lysates (10 mg) from WT and OX strain (with three independent biological replications) were digested with trypsin for 20 h at 37°C, followed by enrichment of acetylated peptides with the PTMScan Acetyl-Lysine Motif (Ac-K) kit (Cell Signaling Technology, catalog no. 13416S). The peptides were separated on Easy-nLC1000 liquid chromatograph (Thermo Fisher Scientific, US) with a trap column (Thermo Fisher Scientific Acclaim PepMap100; 100 μm by 2 cm; nanoViper C18) and analysis column (Thermo Fisher Scientific EASY column; 10 cm; ID 75 μm; 3 μm; C18-A2). The peptides were analyzed in a Q Exactive mass spectrometer (Thermo Fisher Scientific, US), eluted from the column with a linear solvent gradient (buffer A is 0.1% formic acid [FA]; buffer B is 84% ACN/0.1% FA) for 120 min at a flow rate of 300 nl/min (from 0 to 110 min, f gradient from 0 to 55% buffer B; from 110 to 115 min, gradient from 55 to100% buffer B; from 115 to 120 min, 100% buffer B). This LC gradient was used for all mobile phase compositions. The mass spectrometer was operated in the positive ion mode at 2.1 kV, and the capillary temperature was set at 320°C. The full scan was performed in the enhanced mode, 300 to 1,800 m/z.
Database search and bioinformatic analyses.
Raw LC-MS/MS data files were processed with Maxquant software (version 1.3.0.5) for database searching, using uniprot_Magnaporthe_oryzae_39372_20160315.fasta and with the following parameters: Main search ppm, 6 ppm; Missed cleavage, 2; MS/MS tolerance ppm, 20 ppm; De-Isotopic, True; enzyme, trypsin; Fixed modification, Carbamidomethyl(C); Variable modification, Oxidation(M), Acetyl (Protein N-term), Acetyl(K); Decoy database pattern, Reverse; iBAQ, True; Match between runs, 2 min. The criteria for inclusion of protein or peptide were set as follows: a peptide FDR of ≤0.01 or a protein FDR of ≤0.01.
The comparative proteomics strategy was conducted by employing data-dependent nano-LC–MS/MS analysis (40), using Perseus (version 1.3.0.4) (41). Quantitation of peptides was performed on the processed data using Perseus, which uses MS intensities from raw LC-MS data to find statistical proteomic differences between two samples. Prior to ratio quantitation in Perseus, peak intensities from all six LC-MS/MS runs were normalized by the total ion chromatogram intensity. Student’s t test was performed for statistical analysis, and statistical filters were set with a P value of ≤0.05 to detect differential protein ratios between two samples. Statistical analysis of the AcK motif was performed with motif-x (38) and a well-established methodology (42).
Functional annotation and protein interaction network of acetylated proteins.
Annotation of identified proteins from the Perseus processing was performed with Blast2go suite (43) under a trial license, and Interpro scan (44) (http://www.ebi.ac.uk/InterProScan/). Analysis of Gene Ontology (GO) terms and KEGG pathway enrichment of differentially acetylated proteins was performed by Applied Protein Technology Co. Ltd. (Shanghai, China).
Epifluorescence microscopy.
M. oryzae cells expressing fluorescent protein-fused chimera were grown under requisite conditions. Epifluorescence microscopy was performed using Observer Z1 and a sCMOS camera (PCO Edge, Germany).
Immunoblotting and immunoprecipitation.
Total protein extraction follows the established protocol (21). Total protein concentration was measured using the Bio-Rad Protein Assay (catalog no. 500-0006). Samples were resolved by 12% SDS-PAGE, followed by Western blotting with anti-RFP (rabbit; 1:1,000; Clontech, catalog no. R10367), or anti-GFP (rabbit; 1:5,000; Invitrogen Molecular Probes, catalog no. A6455). The secondary antibody was anti-rabbit (1:20,000; HiSecTMHRP-conjugated, Ab202) followed by detection using the SuperSignal West Pico chemiluminescent substrate (Pierce, catalog no. 34080). Total lysates from the GFP-Gcn strain were extracted with the lysis buffer (10 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.5 mM EDTA, 0.5% Nonidet P-40 substitute [Sigma-Aldrich, IGEPAL CA-630, I3021], with 2 mM PMSF and proteinase inhibitor cocktail [Sigma-Aldrich, cOmplete, catalog no. 11836170001]). Such nondenatured lysates were subject to immunoprecipitation with GFP-Trap (Chromo Tek, gta-20). The IP proteins (about 30 μg) were digested in solution using trypsin (Sigma-Aldrich, catalog no. T7409) for 20 h at 37°C, and the digested peptides were subjected to Nano-HPLC/ESI-ion trap-MS/MS analysis with a Q Exactive mass spectrometer (Thermo Fisher Scientific, US). Raw MS data files were processed and analyzed using Mascot2.2 (Matrix Science, UK) for database search with the following parameters: Database, UniProt; Taxonomy, Magnaporthe oryzae (39372); Enzyme, trypsin; Dynamical modifications, Oxidation (M); Fixed modifications, Carbamidomethyl (C); Max Missed Cleavages, 2; ProteomicsTools, 3.1.6; Filter by score, ≥20.
RNA-seq and transcriptome analyses.
RNeasy mini kit (Qiagen, catalog no. 74104) was used for total RNA extraction from WT and OX strains. Next-generation sequencing library preparations were constructed according to the manufacturer’s protocol (NEBNext Ultra RNA Library Prep kit for Illumina). The poly(A) mRNA isolation was performed using the NEBNext Poly(A) mRNA Magnetic Isolation Module (New England BioLabs [NEB]). mRNA fragmentation and priming were performed using NEBNext First Strand Synthesis Reaction Buffer and NEBNext Random Primers. First-strand cDNA was synthesized using ProtoScript II Reverse Transcriptase and the second-strand cDNA was synthesized using Second Strand Synthesis Enzyme Mix. The purified double-stranded cDNA(by AxyPrep Mag PCR Clean-up [Axygen]) was then treated with End Prep Enzyme Mix to repair both ends and add a dA tail in one reaction, followed by a T-A ligation to add adaptors to both ends. Size selection of adaptor-ligated DNA was then performed using AxyPrep Mag PCR Clean-up (Axygen), and fragments of ∼360 bp (with the approximate insert size of 300 bp) were recovered. Each sample was then amplified by PCR for 11 cycles using Illumina P5 and P7 adaptor primers, both carrying sequences which can anneal with the flow cell to perform bridge PCR and P7 primer carrying a six-base index allowing for multiplexing. The PCR products were cleaned up using AxyPrep Mag PCR Clean-up (Axygen), validated using an Agilent 2100 bioanalyzer (Agilent Technologies, Palo Alto, CA, USA), and quantified with a Qubit 2.0 fluorometer (Invitrogen, Carlsbad, CA, USA). Then libraries with different indices were multiplexed and loaded on an Illumina HiSeq instrument according to the manufacturer’s instructions (Illumina, San Diego, CA, USA). Sequencing was carried out using a 2 × 150-bp paired-end (PE) configuration; image analysis and base calling were conducted by the HiSeq Control Software (HCS) plus OLB plus GAPipeline-1.6 (Illumina) on the HiSeq instrument.
In order to remove technical sequences, including adaptors, PCR primers, or fragments thereof, and quality of bases lower than 20, pass filter data of fastq format were processed by Trimmomatic (v0.30) to be high-quality clean data. Then, the clean data were aligned to the reference genome via software Hisat2 (v2.0.1).
Genes with a fold change of ≥2 and a false discovery rate (FDR) of <0.05 in a comparison of significant differentially expressed genes (DEGs) were subjected to enrichment analysis of GO functions and KEGG pathways. GO-TermFinder was used identifying Gene Ontology (GO) terms that annotate a list of enriched genes with a significant P value of less than 0.05. The formula used to calculate the P value was:
where N is the number of all genes with GO annotation, n is the number of DEGs in N, M is the number of all genes annotated to specific pathways, and m is the number of DEGs in M. The calculated P value went through FDR Correction with a FDR of ≤0.05 as the threshold.
KEGG (Kyoto Encyclopedia of Genes and Genomes) is a collection of databases dealing with genomes, biological pathways, diseases, drugs, and chemical substances (http://en.wikipedia.org/wiki/KEGG) (45). Enrich significant differential expression gene in KEGG pathways was performed with in-house script of the Genewiz company. The calculating formula is the same as that in GO analysis.
Reverse transcriptase PCR.
Reverse transcriptase PCR (RT-PCR) was performed using a one-step RT-PCR kit (Qiagen, catalog no. 210212). The primers for RT-PCR are shown in Table S1 in the supplemental material.
ACKNOWLEDGMENTS
We thank the Shanghai Applied Protein Technology Co. Ltd. for technology support in quantitative acetylome analysis and the Genewiz company for RNA-seq and transcriptome analyses.
This work was supported by the National Basic Research Program of China (973 Program, grant 2015CB150600; URL http://program.most.gov.cn/) to L.-H. Zhang and Y.Z. Deng.
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Imhof A, Yang XJ, Ogryzko VV, Nakatani Y, Wolffe AP, Ge H. 1997. Acetylation of general transcription factors by histone acetyltransferases. Curr Biol 7:689–692. doi: 10.1016/S0960-9822(06)00296-X. [DOI] [PubMed] [Google Scholar]
- 2.Moraga F, Aquea F. 2015. Composition of the SAGA complex in plants and its role in controlling gene expression in response to abiotic stresses. Front Plant Sci 6:865. doi: 10.3389/fpls.2015.00865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gaupel AC, Begley TJ, Tenniswood M. 2015. Gcn5 modulates the cellular response to oxidative stress and histone deacetylase inhibition. J Cell Biochem 116:1982–1992. doi: 10.1002/jcb.25153. [DOI] [PubMed] [Google Scholar]
- 4.Rardin MJ, Newman JC, Held JM, Cusack MP, Sorensen DJ, Li B, Schilling B, Mooney SD, Kahn CR, Verdin E, Gibson BW. 2013. Label-free quantitative proteomics of the lysine acetylome in mitochondria identifies substrates of SIRT3 in metabolic pathways. Proc Natl Acad Sci U S A 110:6601–6606. doi: 10.1073/pnas.1302961110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sebti S, Prebois C, Perez-Gracia E, Bauvy C, Desmots F, Pirot N, Gongora C, Bach AS, Hubberstey AV, Palissot V, Berchem G, Codogno P, Linares LK, Liaudet-Coopman E, Pattingre S. 2014. BAT3 modulates p300-dependent acetylation of p53 and autophagy-related protein 7 (ATG7) during autophagy. Proc Natl Acad Sci U S A 111:4115–4120. doi: 10.1073/pnas.1313618111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin SY, Li TY, Liu Q, Zhang C, Li X, Chen Y, Zhang SM, Lian G, Liu Q, Ruan K, Wang Z, Zhang CS, Chien KY, Wu J, Li Q, Han J, Lin SC. 2012. GSK3-TIP60-ULK1 signaling pathway links growth factor deprivation to autophagy. Science 336:477–481. doi: 10.1126/science.1217032. [DOI] [PubMed] [Google Scholar]
- 7.Tscherner M, Zwolanek F, Jenull S, Sedlazeck FJ, Petryshyn A, Frohner IE, Mavrianos J, Chauhan N, von Haeseler A, Kuchler K. 2015. The Candida albicans histone acetyltransferase Hat1 regulates stress resistance and virulence via distinct chromatin assembly pathways. PLoS Pathog 11:e1005218. doi: 10.1371/journal.ppat.1005218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang P, Fan X, Chen J. 2015. Function and subcellular localization of Gcn5, a histone acetyltransferase in Candida albicans. Fungal Genet Biol 81:132–141. doi: 10.1016/j.fgb.2015.01.011. [DOI] [PubMed] [Google Scholar]
- 9.González-Prieto JM, Rosas-Quijano R, Domínguez A, Ruiz-Herrera J. 2014. The UmGcn5 gene encoding histone acetyltransferase from Ustilago maydis is involved in dimorphism and virulence. Fungal Genet Biol 71:86–95. doi: 10.1016/j.fgb.2014.09.002. [DOI] [PubMed] [Google Scholar]
- 10.Xie J, Jenull S, Tscherner M, Kuchler K. 2016. The paralogous histone deacetylases Rpd3 and Rpd31 play opposing roles in regulating the white-opaque switch in the fungal pathogen Candida albicans. mBio 7:e01807-16. doi: 10.1128/mBio.01807-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elías-Villalobos A, Fernández-Álvarez A, Moreno-Sánchez I, Helmlinger D, Ibeas JI. 2015. The Hos2 histone deacetylase controls Ustilago maydis virulence through direct regulation of mating-type genes. PLoS Pathog 11:e1005134. doi: 10.1371/journal.ppat.1005134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ding SL, Liu W, Iliuk A, Ribot C, Vallet J, Tao A, Wang Y, Lebrun MH, Xu JR. 2010. The tig1 histone deacetylase complex regulates infectious growth in the rice blast fungus Magnaporthe oryzae. Plant Cell 22:2495–2508. doi: 10.1105/tpc.110.074302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Izawa M, Takekawa O, Arie T, Teraoka T, Yoshida M, Kimura M, Kamakura T. 2009. Inhibition of histone deacetylase causes reduction of appressorium formation in the rice blast fungus Magnaporthe oryzae. J Gen Appl Microbiol 55:489–498. doi: 10.2323/jgam.55.489. [DOI] [PubMed] [Google Scholar]
- 14.Canovas D, Marcos AT, Gacek A, Ramos MS, Gutierrez G, Reyes-Dominguez Y, Strauss J. 2014. The histone acetyltransferase GcnE (GCN5) plays a central role in the regulation of Aspergillus asexual development. Genetics 197:1175–1189. doi: 10.1534/genetics.114.165688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fernandez J, Marroquin-Guzman M, Nandakumar R, Shijo S, Cornwell KM, Li G, Wilson RA. 2014. Plant defence suppression is mediated by a fungal sirtuin during rice infection by Magnaporthe oryzae. Mol Microbiol 94:70–88. doi: 10.1111/mmi.12743. [DOI] [PubMed] [Google Scholar]
- 16.He M, Xu Y, Chen J, Luo Y, Lv Y, Su J, Kershaw MJ, Li W, Wang J, Yin J, Zhu X, Liu X, Chern M, Ma B, Wang J, Qin P, Chen W, Wang Y, Wang W, Ren Z, Wu X, Li P, Li S, Peng Y, Lin F, Talbot NJ, Chen X. 2018. MoSnt2-dependent deacetylation of histone H3 mediates MoTor-dependent autophagy and plant infection by the rice blast fungus Magnaporthe oryzae. Autophagy 14:1543–1561. doi: 10.1080/15548627.2018.1458171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Talbot NJ. 2003. On the trail of a cereal killer: exploring the biology of Magnaporthe grisea. Annu Rev Microbiol 57:177–202. doi: 10.1146/annurev.micro.57.030502.090957. [DOI] [PubMed] [Google Scholar]
- 18.Lee K, Singh P, Chung WC, Ash J, Kim TS, Hang L, Park S. 2006. Light regulation of asexual development in the rice blast fungus, Magnaporthe oryzae. Fungal Genet Biol 43:694–706. doi: 10.1016/j.fgb.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 19.Deng YZ, Naqvi NI. 2010. A vacuolar glucoamylase, Sga1, participates in glycogen autophagy for proper asexual differentiation in Magnaporthe oryzae. Autophagy 6:455–461. doi: 10.4161/auto.6.4.11736. [DOI] [PubMed] [Google Scholar]
- 20.Deng YZ, Ramos-Pamplona M, Naqvi NI. 2009. Autophagy-assisted glycogen catabolism regulates asexual differentiation in Magnaporthe oryzae. Autophagy 5:33–43. doi: 10.4161/auto.5.1.7175. [DOI] [PubMed] [Google Scholar]
- 21.Zhang S, Liang M, Naqvi NI, Lin C, Qian W, Zhang LH, Deng YZ. 2017. Phototrophy and starvation-based induction of autophagy upon removal of Gcn5-catalyzed acetylation of Atg7 in Magnaporthe oryzae. Autophagy 13:1318–1330. doi: 10.1080/15548627.2017.1327103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Veneault-Fourrey C, Barooah M, Egan M, Wakley G, Talbot NJ. 2006. Autophagic fungal cell death is necessary for infection by the rice blast fungus. Science 312:580–583. doi: 10.1126/science.1124550. [DOI] [PubMed] [Google Scholar]
- 23.Liu XH, Lu JP, Zhang L, Dong B, Min H, Lin FC. 2007. Involvement of a Magnaporthe grisea serine/threonine kinase gene, MgATG1, in appressorium turgor and pathogenesis. Eukaryot Cell 6:997–1005. doi: 10.1128/EC.00011-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vakifahmetoglu-Norberg H, Ouchida AT, Norberg E. 2017. The role of mitochondria in metabolism and cell death. Biochem Biophys Res Commun 482:426–431. doi: 10.1016/j.bbrc.2016.11.088. [DOI] [PubMed] [Google Scholar]
- 25.Cieniewicz AM, Moreland L, Ringel AE, Mackintosh SG, Raman A, Gilbert TM, Wolberger C, Tackett AJ, Taverna SD. 2014. The bromodomain of Gcn5 regulates site specificity of lysine acetylation on histone H3. Mol Cell Proteomics 13:2896–2910. doi: 10.1074/mcp.M114.038174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuo YM, Andrews AJ. 2013. Quantitating the specificity and selectivity of Gcn5-mediated acetylation of histone H3. PLoS One 8:e54896. doi: 10.1371/journal.pone.0054896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eisenberg T, Schroeder S, Andryushkova A, Pendl T, Kuttner V, Bhukel A, Marino G, Pietrocola F, Harger A, Zimmermann A, Moustafa T, Sprenger A, Jany E, Buttner S, Carmona-Gutierrez D, Ruckenstuhl C, Ring J, Reichelt W, Schimmel K, Leeb T, Moser C, Schatz S, Kamolz LP, Magnes C, Sinner F, Sedej S, Frohlich KU, Juhasz G, Pieber TR, Dengjel J, Sigrist SJ, Kroemer G, Madeo F. 2014. Nucleocytosolic depletion of the energy metabolite acetyl-coenzyme A stimulates autophagy and prolongs lifespan. Cell Metab 19:431–444. doi: 10.1016/j.cmet.2014.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eisenberg T, Schroeder S, Buttner S, Carmona-Gutierrez D, Pendl T, Andryushkova A, Marino G, Pietrocola F, Harger A, Zimmermann A, Magnes C, Sinner F, Sedej S, Pieber TR, Dengjel J, Sigrist S, Kroemer G, Madeo F. 2014. A histone point mutation that switches on autophagy. Autophagy 10:1143–1145. doi: 10.4161/auto.28767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eisenberg T, Knauer H, Schauer A, Buttner S, Ruckenstuhl C, Carmona-Gutierrez D, Ring J, Schroeder S, Magnes C, Antonacci L, Fussi H, Deszcz L, Hartl R, Schraml E, Criollo A, Megalou E, Weiskopf D, Laun P, Heeren G, Breitenbach M, Grubeck-Loebenstein B, Herker E, Fahrenkrog B, Frohlich KU, Sinner F, Tavernarakis N, Minois N, Kroemer G, Madeo F. 2009. Induction of autophagy by spermidine promotes longevity. Nat Cell Biol 11:1305–1314. doi: 10.1038/ncb1975. [DOI] [PubMed] [Google Scholar]
- 30.Lv L, Li D, Zhao D, Lin R, Chu Y, Zhang H, Zha Z, Liu Y, Li Z, Xu Y, Wang G, Huang Y, Xiong Y, Guan KL, Lei QY. 2011. Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol Cell 42:719–730. doi: 10.1016/j.molcel.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bhadauria V, Banniza S, Vandenberg A, Selvaraj G, Wei Y. 2012. Peroxisomal alanine: glyoxylate aminotransferase AGT1 is indispensable for appressorium function of the rice blast pathogen, Magnaporthe oryzae. PLoS One 7:e36266. doi: 10.1371/journal.pone.0036266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klare JP, Chizhov I, Engelhard M. 2008. Microbial rhodopsins: scaffolds for ion pumps, channels, and sensors. Results Probl Cell Differ 45:73–122. doi: 10.1007/400_2007_041. [DOI] [PubMed] [Google Scholar]
- 33.Brown LS. 2014. Eubacterial rhodopsins - unique photosensors and diverse ion pumps. Biochim Biophys Acta 1837:553–561. doi: 10.1016/j.bbabio.2013.05.006. [DOI] [PubMed] [Google Scholar]
- 34.Brown LS. 2004. Fungal rhodopsins and opsin-related proteins: eukaryotic homologues of bacteriorhodopsin with unknown functions. Photochem Photobiol Sci 3:555–565. doi: 10.1039/b315527g. [DOI] [PubMed] [Google Scholar]
- 35.Saavedra F, Rivera C, Rivas E, Merino P, Garrido D, Hernandez S, Forne I, Vassias I, Gurard-Levin ZA, Alfaro IE, Imhof A, Almouzni G, Loyola A. 2017. PP32 and SET/TAF-Ibeta proteins regulate the acetylation of newly synthesized histone H4. Nucleic Acids Res 45:11700–11710. doi: 10.1093/nar/gkx775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ginsburg DS, Anlembom TE, Wang J, Patel SR, Li B, Hinnebusch AG. 2014. NuA4 links methylation of histone H3 lysines 4 and 36 to acetylation of histones H4 and H3. J Biol Chem 289:32656–32670. doi: 10.1074/jbc.M114.585588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Howard RJ, Valent B. 1996. Breaking and entering: host penetration by the fungal rice blast pathogen Magnaporthe grisea. Annu Rev Microbiol 50:491–512. doi: 10.1146/annurev.micro.50.1.491. [DOI] [PubMed] [Google Scholar]
- 38.Chou MF, Schwartz D. 2011. Biological sequence motif discovery using motif-x. Curr Protoc Bioinformatics Chapter 13:Unit 13.15-24. doi: 10.1002/0471250953.bi1315s35. [DOI] [PubMed] [Google Scholar]
- 39.Ramos-Pamplona M, Naqvi NI. 2006. Host invasion during rice-blast disease requires carnitine-dependent transport of peroxisomal acetyl-CoA. Mol Microbiol 61:61–75. doi: 10.1111/j.1365-2958.2006.05194.x. [DOI] [PubMed] [Google Scholar]
- 40.Cholewa BD, Pellitteri-Hahn MC, Scarlett CO, Ahmad N. 2014. Large-scale label-free comparative proteomics analysis of polo-like kinase 1 inhibition via the small-molecule inhibitor BI 6727 (Volasertib) in BRAF(V600E) mutant melanoma cells. J Proteome Res 13:5041–5050. doi: 10.1021/pr5002516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J. 2016. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 13:731–740. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- 42.Schwartz D, Gygi SP. 2005. An iterative statistical approach to the identification of protein phosphorylation motifs from large-scale data sets. Nat Biotechnol 23:1391–1398. doi: 10.1038/nbt1146. [DOI] [PubMed] [Google Scholar]
- 43.Gotz S, Garcia-Gomez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ, Robles M, Talon M, Dopazo J, Conesa A. 2008. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res 36:3420–3435. doi: 10.1093/nar/gkn176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Quevillon E, Silventoinen V, Pillai S, Harte N, Mulder N, Apweiler R, Lopez R. 2005. InterProScan: protein domains identifier. Nucleic Acids Res 33:W116–W120. doi: 10.1093/nar/gki442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T, Yamanishi Y. 2008. KEGG for linking genomes to life and the environment. Nucleic Acids Res 36:D480–D484. doi: 10.1093/nar/gkm882. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental flow of comparative acetylome analysis with WT and OX strains. Mycelia were harvested from WT or OX colonies grown on 10 PA solid medium plates for total protein lysis and trypsin digestion. Three independent biological replications were sampled for each strain. The acK peptides were immunoprecipitated using PTMScan Acetyl-Lysine Motif (Ac-K) kit (Cell Signaling Technology, catalog no. 13416S). Enriched peptides were separated and analyzed in duplicate by LC-MS/MS, followed by bioinformatic analyses to identify differentially acetylated proteins between WT and OX strain. Download FIG S1, TIF file, 0.8 MB (858KB, tif) .
Copyright © 2018 Liang et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Acetyl proteins/residuals identified by acetylome analysis. Download Data Set S1, XLS file, 1.9 MB (1.9MB, xls) .
Copyright © 2018 Liang et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Differential acetyl proteins/residuals identified by label-free quantification. Download Data Set S2, XLSX file, 0.4 MB (370KB, xlsx) .
Copyright © 2018 Liang et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
GO annotation and enrichment with the acetyl proteins significantly different between the WT and the OX samples. Download Data Set S3, XLS file, 2.0 MB (2MB, xls) .
Copyright © 2018 Liang et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
KEGG pathway annotation and enrichment with the acetyl proteins significantly different between the WT and OX samples. Download Data Set S4, XLSX file, 0.04 MB (41.9KB, xlsx) .
Copyright © 2018 Liang et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Acetylation profiles of histone H3 and Atg7 and PK expression assessed by qRT-PCR. (A) Acetylation profiles of histone H3. (B) Acetylation profiles of Atg7. **, significant difference (P < 0.05). (C) qRT-PCR to assess expression of WT or mutated forms of PK. Relative fold changes were calculated using the −ΔΔCt method. The ACTIN gene (MGG_03982) was used as an internal control. Asterisks denote significant difference (P < 0.05) compared to the WT values. There were no significant differences between the values for the three overexpression strains. Download FIG S2, TIF file, 0.4 MB (422.6KB, tif) .
Copyright © 2018 Liang et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Differential acetyl proteins identified in KO versus OX samples and Gcn5-associated proteins identified by LC-MS/MS. Download Data Set S5, XLSX file, 0.5 MB (482.2KB, xlsx) .
Copyright © 2018 Liang et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Differentially expressed genes (DEGs) between the WT and OX samples identified by transcriptome analysis. Download Data Set S6, XLS file, 0.08 MB (84.5KB, xls) .
Copyright © 2018 Liang et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Selected differentially expressed genes for verification by RT-PCR. Download Table S1, DOCX file, 0.02 MB (16.3KB, docx) .
Copyright © 2018 Liang et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Statistical analysis of the acK motif significantly increased in the OX strain. Download Table S2, DOCX file, 0.2 MB (228.9KB, docx) .
Copyright © 2018 Liang et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.