

Abstract
Prexasertib is a novel inhibitor of checkpoint kinase 1. The primary objective of this study was to evaluate prexasertib tolerability in Japanese patients with advanced solid tumors. This nonrandomized single‐arm open‐label phase 1 study of prexasertib consisted of 2 dose levels, 80 mg/m2 and the global‐recommended dose based on a US study of 105 mg/m2, administered intravenously once every 14 days (n = 6 for each dose). Transition to the higher dose proceeded if the frequency of dose‐limiting toxicity observed in cycle 1 was <33% at the lower dose. Safety measures, pharmacokinetics and antitumor activity were assessed. A total of 12 patients were treated. Two patients, one in each dose group, experienced dose‐limiting toxicities of febrile neutropenia, one grade 4 and the other grade 3; both patients recovered and continued the study treatment. The grade 4 treatment‐emergent adverse events related to study treatment were neutropenia (6 patients [50.0%]), leukopenia (4 patients [33.3%]), and 1 instance each (8.3%) of anemia, febrile neutropenia and thrombocytopenia. Neutropenia was generally transient and reversible; 11 patients (91.7%) required granulocyte colony‐stimulating factor treatment during the study. There were no discontinuations due to adverse events or deaths. The prexasertib pharmacokinetics displayed dose‐independent and time‐independent behavior across both dose levels, similar to the profile observed in the US‐based phase 1 study. Eight patients had a best overall response of stable disease. These data are consistent with the known safety profile for prexasertib and confirm its tolerability in Japanese patients with advanced solid tumors.
Keywords: Checkpoint kinase 1, Japan, phase I clinical trial, prexasertib, solid tumors
1. INTRODUCTION
Checkpoint kinase 1 (CHK1) is a multifunctional serine/threonine‐specific protein kinase that has a critical role in regulating response to DNA damage.1, 2 The proteins ataxia telangiectasia mutated and RAD3‐related (ATR) and/or ataxia telangiectasia mutated (ATM) are activated in response to DNA damage, which in turn phosphorylate CHK1 and checkpoint kinase 2 (CHK2), respectively.3 p53 is another critical mediator of the response to DNA damage. It is activated through phosphorylation by CHK2 and ATM, and participates in a parallel pathway to ATR and CHK1.3 CHK1 can also regulate DNA replication checkpoints by maintaining or enhancing the inhibitory phosphorylation of cyclin‐dependent kinases, which are key drivers of cell cycle progression.2 In addition, CHK1 can stabilize replication forks and promote DNA repair, control initiation of DNA replication, and coordinate mitosis.2 Inhibitors of CHK1 have been developed as combination therapy to augment the efficacy of DNA‐damaging chemotherapeutics.4, 5, 6, 7 Given the central role that CHK1 has in DNA replication and regulation of the cell cycle, inhibitors of CHK1 are also being developed as single‐agent therapies. 3, 8
Prexasertib (LY2606368) is an inhibitor of CHK1 and, to a lesser extent, CHK2 that can disrupt DNA replication, induce DNA damage, and prevent DNA repair, eventually leading to cell death via interruption of DNA replication.1 Preclinical studies in solid tumor models have shown that prexasertib as a single agent, or in combination with other agents, has both in vitro and in vivo antitumor activity.1, 9, 10 Prexasertib, in combination with cetuximab and irradiation, has also been shown to have antitumor effects in head and neck squamous cell carcinoma (SCC) in vitro and in vivo.11
A phase 1 nonrandomized, open‐label, dose‐escalation study of prexasertib monotherapy, conducted in the US in 45 patients with advanced solid tumors, resulted in a recommended prexasertib dose of 105 mg/m2 administered once every 14 days for further evaluation.8 The most common drug‐related grade 3 or 4 treatment‐emergent adverse events (TEAE) in the dose‐escalation portion of this phase 1 study were reversible neutropenia, leukopenia, anemia, thrombocytopenia and fatigue.8 Two patients (4%) had a partial response, 1 with SCC of the anus and 1 with SCC of the head and neck, and 15 patients (33%) with various cancer types had a best overall response (BOR) of stable disease.8 In a phase 2 single‐center study of prexasertib in 28 women with BRCA wild‐type recurrent high‐grade serous ovarian cancer, partial responses were reported for 8 patients (29%) in the intention‐to‐treat population, with the most common grade 3 or 4 TEAE being neutropenia, leukopenia, thrombocytopenia and anemia.12
This phase 1 study was conducted to investigate the safety, tolerability, pharmacokinetics (PK) and antitumor activity of prexasertib in Japanese patients with advanced solid tumors. This study is registered with ClinicalTrials.gov, identifier NCT02514603.
2. PATIENTS AND METHODS
2.1. Eligibility
Japanese patients at least 20 years of age with advanced and/or metastatic solid tumors who experienced treatment failure with standard therapies and who had an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1 and adequate organ and coagulation function, including absolute neutrophil count ≥1.5 × 109/L, platelets ≥120 × 109/L and hemoglobin ≥9 g/dL, were enrolled. Patients had to have discontinued previous treatments for cancer and to have recovered from the acute effects of therapies before enrollment. Exclusion criteria included serious preexisting medical condition(s), active infection(s), a second primary malignancy, a corrected QT interval of >470 ms on screening electrocardiogram in repeated (at least 2) measurements, or a family history of long‐QT syndrome.
2.2. Study design and treatment
All eligible patients in this nonrandomized single‐arm open‐label phase 1 study received prexasertib at 80 or 105 mg/m2, administered intravenously on day 1 of every 14‐day cycle, with each dose level consisting of a maximum of 6 patients. The primary objective was to evaluate tolerability, which was confirmed if the dose‐limiting toxicity (DLT) frequency observed in cycle 1 was <33%, of prexasertib in Japanese patients with advanced solid tumors. The secondary objectives were the characterization of the prexasertib safety and toxicity profile, PK, and documentation of antitumor activity.
This study was conducted in accordance with the International Conference on Harmonization Guidelines for Good Clinical Practice and the Declaration of Helsinki, and with approval from each institution's ethical review board. Patients provided written informed consent. This study is registered with ClinicalTrials.gov, identifier NCT02514603.
2.3. Safety evaluations
A DLT was defined as an adverse event (AE) during cycle 1 (the first 14 days of treatment) that was possibly related to prexasertib and fulfilled any of the following criteria using the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) version 4.03: grade ≥3 nonhematologic toxicity, except (i) nausea, vomiting, constipation, diarrhea, fatigue or anorexia that was manageable with appropriate care, lasting ≤2 days; (ii) transient (≤7 days) grade 3 elevations of alanine aminotransferase and/or aspartate aminotransferase without evidence of other hepatic injury in the setting of preexisting hepatic metastasis; or (iii) asymptomatic electrolyte disturbance treatable with oral substitution therapy; grade 3 thrombocytopenia with bleeding, grade 4 thrombocytopenia, or thrombocytopenia that required platelet transfusion; grade 4 anemia or neutropenia >5 days duration; anemia that required packed red blood cell transfusion; febrile neutropenia; toxicity that prevented the start of cycle 2 for >2 weeks from the end of cycle 1; and any other significant toxicity deemed to be dose limiting. Primary prophylaxis with granulocyte colony‐stimulating factor (G‐CSF), including pegylated G‐CSF, was not permitted during the DLT evaluation period, although G‐CSF could be used for treatment‐emergent neutropenia as well as secondary prophylaxis.
2.4. Pharmacokinetic analyses
Blood samples for PK analyses were collected on days 1, 2 and 8 of cycles 1 and 2, and on day 1 of cycle 3. Prexasertib plasma concentrations were quantified using a validated liquid chromatography/tandem mass spectrometry method. The PK parameter estimates for prexasertib were calculated by standard noncompartmental methods of analysis using Phoenix WinNonlin 6.4 (Certara; Princeton, NJ, USA). The maximum plasma concentration (C max), the area under the drug plasma concentration versus time curve (AUC), the AUC from time zero to 24 hours (AUC[0‐24]) and the AUC from time zero to 72 hours (AUC[0‐72]) within cycles 1 and 2 were calculated to assess intercycle variability in exposure and the intracycle and intercycle accumulation of prexasertib. In addition, the average plasma concentration over a 72‐hour interval after prexasertib infusion (C av,72), terminal elimination half‐life (t 1/2), volume of distribution at steady state (V ss) and systemic clearance (CL) were reported from the noncompartmental PK analyses.
2.5. Efficacy evaluations
Patients were assessed for tumor measurement by computed tomography scan or mRI using RECIST version 1.1. The BOR was defined as the best response recorded from the start of the treatment until disease progression. The best response of stable disease was defined as disease that did not meet the criteria for complete response, partial response or progressive disease and had been evaluated approximately 4 weeks (defined as ≥28 days) after the date of the first dose.
3. RESULTS
3.1. Patients
Between 16 October 2015 and 25 January 2017, 12 eligible patients were enrolled and treated, 6 with prexasertib 80 mg/m2 and 6 with prexasertib 105 mg/m2. Two‐thirds of patients (66.7%) were male, the median age was 62 years, and most patients (58.3%) had an ECOG performance status of 1 (Table 1). All except 1 patient had received at least 1 (median, 4; range, 0‐10) prior systemic treatment (91.7%), two‐thirds had received prior surgery (66.7%), and half had received prior radiotherapy (50.0%). Twenty‐five percent of patients had esophageal SCC. The cancer types of malignant neoplasm of thymus, rhabdomyosarcoma, small intestine carcinoma and transitional cell carcinoma had not been studied in the previous phase 1 study of prexasertib treatment in the US.8
Table 1.
Baseline demographic and clinical characteristics
| Prexasertib 80 mg/m2 N = 6 | Prexasertib 105 mg/m2 N = 6 | Total N = 12 | |
|---|---|---|---|
| Age, years | |||
| Median (range) | 63 (35‐74) | 62 (47‐78) | 62 (35‐78) |
| Gender | |||
| Female | 2 (33.3) | 2 (33.3) | 4 (33.3) |
| Male | 4 (66.7) | 4 (66.7) | 8 (66.7) |
| Race | |||
| Asian | 6 (100) | 6 (100) | 12 (100) |
| Weight, kg | |||
| Median (range) | 52 (43‐70) | 62 (46‐85) | 57 (43‐85) |
| Body surface area, m2 | |||
| Median (range) | 1.6 (1.3‐1.9) | 1.7 (1.4‐1.9) | 1.6 (1.3‐1.9) |
| Cancer type | |||
| Endometrial adenocarcinoma | 0 | 1 (16.7) | 1 (8.3) |
| Liposarcoma | 0 | 1 (16.7) | 1 (8.3) |
| Malignant neoplasm of thymus | 0 | 1 (16.7) | 1 (8.3) |
| Esophageal squamous cell carcinoma | 2 (33.3) | 1 (16.7) | 3 (25.0) |
| Rhabdomyosarcoma | 1 (16.7) | 0 | 1 (8.3) |
| Skin cancer | 0 | 1 (16.7) | 1 (8.3) |
| Small intestine carcinoma | 1 (16.7) | 0 | 1 (8.3) |
| Squamous cell carcinoma of unknown origin | 0 | 1 (16.7) | 1 (8.3) |
| Squamous cell carcinoma of lung | 1 (16.7) | 0 | 1 (8.3) |
| Transitional cell carcinoma | 1 (16.7) | 0 | 1 (8.3) |
| ECOG PS | |||
| 0 | 3 (50.0) | 2 (33.3) | 5 (41.7) |
| 1 | 3 (50.0) | 4 (66.7) | 7 (58.3) |
| Prior interventions | |||
| ≥1 surgery | 4 (66.7) | 4 (66.7) | 8 (66.7) |
| ≥1 radiotherapy | 3 (50.0) | 3 (50.0) | 6 (50.0) |
| ≥1 prior systemic regimens | 6 (100) | 5 (83.3) | 11 (91.7) |
ECOG PS, Eastern Cooperative Oncology Group performance status.
Data are n (%) unless otherwise indicated.
3.2. Safety
A total of 6 patients in each dose group were included in the DLT evaluation. One patient in each dose group reported a DLT of treatment‐related febrile neutropenia during cycle 1 (1 grade 4 in the 80 mg/m2 group and the other grade 3 in the 105 mg/m2 group). Both patients recovered and continued the study treatment at reduced doses of 60 and 80 mg/m2.
Among all 12 patients in the study, frequently‐reported TEAE were leukopenia (100%), neutropenia (91.7%) and thrombocytopenia (66.7%). Treatment‐related grade 4 TEAE reported were neutropenia in 6 patients (50.0%), leukopenia in 4 patients (33.3%), and 1 instance each (8.3% of all patients) of anemia, febrile neutropenia and thrombocytopenia. There were no reported nonhematologic grade 3 or 4 treatment‐related TEAE. Two patients receiving prexasertib 80 mg/m2 had treatment‐related grade 2 prolongations in QT interval using Fridericia's correction. One of these patients also experienced grade 1 atrial fibrillation on cycle 1, day 2. No clinically significant cardiac toxicities or AE related to QT prolongation were observed. One patient experienced grade 1 supraventricular extrasystoles. A summary of treatment‐related TEAE is presented in Table 2.
Table 2.
Treatment‐emergent adverse events related to study treatment
| Prexasertib 80 mg/m2 N = 6 | Prexasertib 105 mg/m2 N = 6 | Total N = 12 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Any grade | Grade 3 | Grade 4 | Grade 3 + 4 | Any grade | Grade 3 | Grade 4 | Grade 3 + 4 | Any grade | Grade 3 | Grade 4 | Grade 3 + 4 | |
| Any | 6 (100) | 2 (33.3) | 4 (66.7) | 6 (100) | 6 (100) | 3 (50.0) | 3 (50.0) | 6 (100) | 12 (100) | 5 (41.7) | 7 (58.3) | 12 (100) |
| Neutropenia | 5 (83.3) | 1 (16.7) | 3 (50.0) | 4 (66.7) | 6 (100) | 3 (50.0) | 3 (50.0) | 6 (100) | 11 (91.7) | 4 (33.3) | 6 (50.0) | 10 (83.3) |
| Leukopenia | 6 (100) | 1 (16.7) | 3 (50.0) | 4 (66.7) | 6 (100) | 4 (66.7) | 1 (16.7) | 5 (83.3) | 12 (100) | 5 (41.7) | 4 (33.3) | 9 (75.0) |
| Thrombocytopenia | 4 (66.7) | 2 (33.3) | 1 (16.7) | 3 (50.0) | 4 (66.7) | 1 (16.7) | 0 | 1 (16.7) | 8 (66.7) | 3 (25.0) | 1 (8.3) | 4 (33.3) |
| Anemia | 3 (50.0) | 1 (16.7) | 1 (16.7) | 2 (33.3) | 0 | 0 | 0 | 0 | 3 (25.0) | 1 (8.3) | 1 (8.3) | 2 (16.7) |
| Febrile neutropenia | 1 (16.7) | 0 | 1 (16.7) | 1 (16.7) | 1 (16.7) | 1 (16.7) | 0 | 1 (16.7) | 2 (16.7) | 1 (8.3) | 1 (8.3) | 2 (16.7) |
| Lymphocytopenia | 2 (33.3) | 1 (16.7) | 0 | 1 (16.7) | 0 | 0 | 0 | 0 | 2 (16.7) | 1 (8.3) | 0 | 1 (8.3) |
| Atrial fibrillation | 1 (16.7) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (8.3) | 0 | 0 | 0 |
| Constipation | 2 (33.3) | 0 | 0 | 0 | 1 (16.7) | 0 | 0 | 0 | 3 (25.0) | 0 | 0 | 0 |
| ECG QT prolonged | 2 (33.3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 (16.7) | 0 | 0 | 0 |
| Fatigue | 1 (16.7) | 0 | 0 | 0 | 1 (16.7) | 0 | 0 | 0 | 2 (16.7) | 0 | 0 | 0 |
| Headache | 0 | 0 | 0 | 0 | 1 (16.7) | 0 | 0 | 0 | 1 (8.3) | 0 | 0 | 0 |
| Hypocalcemia | 0 | 0 | 0 | 0 | 1 (16.7) | 0 | 0 | 0 | 1 (8.3) | 0 | 0 | 0 |
| Lung infection | 1 (16.7) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (8.3) | 0 | 0 | 0 |
| Nausea | 2 (33.3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 (16.7) | 0 | 0 | 0 |
| Pyrexia | 1 (16.7) | 0 | 0 | 0 | 1 (16.7) | 0 | 0 | 0 | 2 (16.7) | 0 | 0 | 0 |
| Rash maculopapular | 1 (16.7) | 0 | 0 | 0 | 1 (16.7) | 0 | 0 | 0 | 2 (16.7) | 0 | 0 | 0 |
| Rash | 1 (16.7) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (8.3) | 0 | 0 | 0 |
| Stomatitis | 1 (16.7) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (8.3) | 0 | 0 | 0 |
| SVE | 1 (16.7) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (8.3) | 0 | 0 | 0 |
Treatment‐emergent adverse events related to study treatment by maximum National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) grade are listed by preferred term in decreasing frequency of grade 3 + 4 events.
Data are n (%).
ECG, electrocardiogram; SVE, supraventricular extrasystoles.
All 11 patients (5 patients in the 80 mg/m2 group and 6 patients in the 105 mg/m2 group) who had the TEAE of neutropenia received G‐CSF at some point during the study. Three patients received secondary prophylactic G‐CSF subsequent to the TEAE of febrile neutropenia, neutropenia and leukopenia, respectively. The most commonly used G‐CSF was filgrastim (9 patients [75.0%]). In general, neutrophil counts in patients with grade 4 drug‐related neutropenia reached a nadir on approximately day 8, and grade 4 decreases in most cases were transient and lasted <5 days. G‐CSF was commonly used to treat neutropenia around the nadir period (median start date of G‐CSF treatment, day 8; range, days 4‐10) during cycle 1. The incidence of infection was low, with 1 grade 2 drug‐related TEAE of lung infection reported in 1 patient in the 80 mg/m2 group.
One patient receiving prexasertib 80 mg/m2 reported 3 serious AE, 2 of which (anemia and thrombocytopenia) were considered related to the study treatment, whereas the other (constipation) was not. The patient required hospitalization, a red blood cell transfusion and a platelet transfusion. The thrombocytopenia was grade 4 in severity and occurred between study days 51 and 53. No bleeding events were reported for this patient during the study. No patient deaths were reported during the study. Furthermore, no patients discontinued the study due to an AE and no other patterns or trends were found in the laboratory assessments or vital signs.
Among all patients in the study, 8 patients (66.7%; 5 patients in the 80 mg/m2 group and 3 patients in the 105 mg/m2 group) experienced at least 1 AE that led to a dose adjustment. Six patients (50.0%; 3 patients in each group) experienced at least 1 AE that led to a dose delay. The most common reason for dose delay was neutropenia (33.3%; 2 patients in each group). Five patients (41.7%; 4 patients in the 80 mg/m2 group and 1 patient in the 105 mg/m2 group) experienced at least 1 AE that led to dose reduction. The dose was reduced from 80 to 60 mg/m2 for 4 patients, and 105 to 80 mg/m2 for 1 patient. The most common reasons for dose reductions were febrile neutropenia (16.7%; 1 patient in the 80 mg/m2 group and 1 patient in the 105 mg/m2 group) and neutropenia (16.7%; 2 patients in the 80 mg/m2 group and no patients in the 105 mg/m2 group). No patients experienced AE that led to treatment interruption.
3.3. Pharmacokinetics
The PK profile of prexasertib displayed a multiexponential decline in plasma concentrations with dose‐dependent increases in systemic exposure and consistent PK profiles after single‐dose and multiple‐dose administration across the dose range investigated.
Figure 1 shows the arithmetic mean plasma concentration vs time profiles of prexasertib after a single dose in cycle 1 (Figure 1A) and multiple doses in cycle 2 (Figure 1B). There was no intercycle accumulation of prexasertib between cycle 1 and cycle 2 in both treatment groups (Table 3). Mean t 1/2 values were consistent across different dose levels (Table 3). A small‐to‐moderate degree of interpatient variability (mean percent coefficient of variation range: 7%‐25%) in prexasertib CL was observed in both treatment groups. The CL and V ss were consistent after repeat administration at both dose levels (Table 3), indicating dose‐independent and time‐independent PK behavior.
Figure 1.
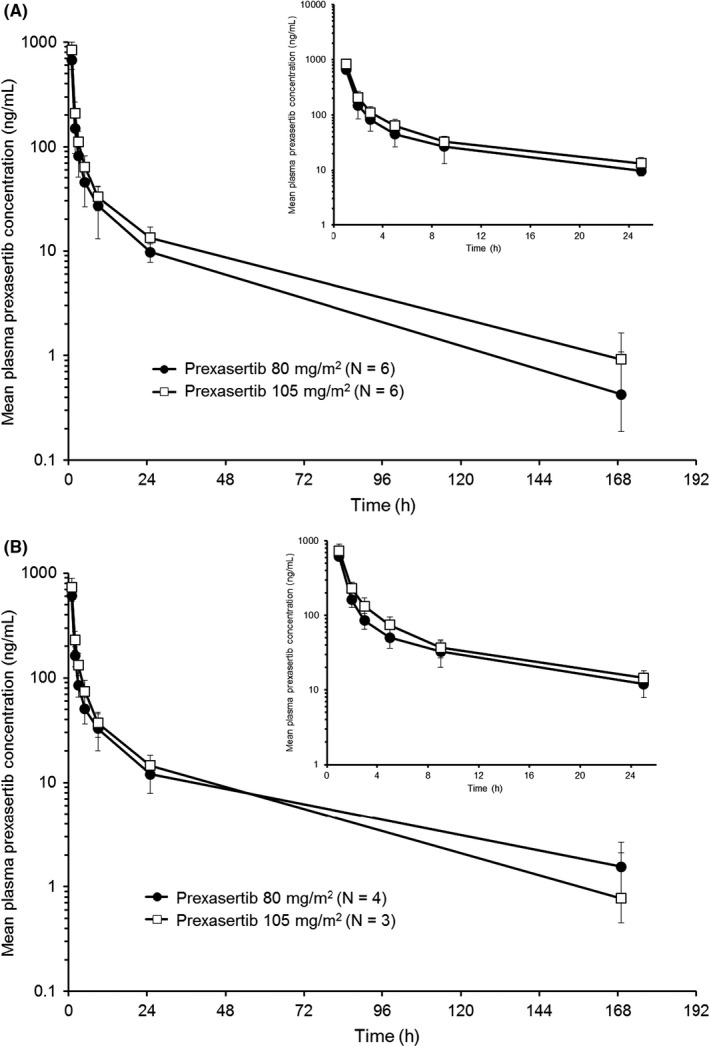
Arithmetic mean (±SD) plasma concentration vs time profiles of prexasertib in Japanese patients on (A) day 1 of cycle 1 and (B) day 1 of cycle 2, following a 1‐h intravenous infusion of prexasertib 80 or 105 mg/m2. Insets depict the first 24 h
Table 3.
Noncompartmental pharmacokinetic parameters of prexasertib
| Prexasertib 80 mg/m2 | Prexasertib 105 mg/m2 | |||
|---|---|---|---|---|
| Cycle 1, day 1 | Cycle 2, day 1 | Cycle 1, day 1 | Cycle 2, day 1 | |
| N | 6 | 4 | 6 | 3 |
| C max (ng/mL) | 661 (18) | 608 (12) | 820 (22) | 721 (23) |
| C av,72 (ng/mL) | 23.1 (22) | 24.8 (16) | 30.0 (13) | 31.5 (22) |
| AUC(0‐24) (ng h/mL) | 1300 (23) | 1360 (13) | 1690 (11) | 1720 (23) |
| AUC(0‐72) (ng h/mL) | 1670 (22) | 1780 (16) | 2160 (13) | 2270 (22) |
| CL (L/h) | 78.6 (25) | 63.0 (23) | 77.1 (9) | 79.5 (7) |
| V ss (L) | 1000 (89) | 1590 (79) | 1380 (93) | 938 (132) |
| t 1/2 a (h) | 16.0 (8.15‐47.3) | 27.1 (7.15‐44.3) | 21.7 (6.87‐45.4) | 13.3 (7.10‐39.9) |
| R A b | NC | 0.959 (19) | NC | 0.960 (8) |
Data are geometric mean (CV%) unless otherwise indicated.
Geographic mean (range).
R A = intercycle accumulation (Cycle 2, Day 1 AUC[0‐24]/Cycle 1, Day 1 AUC[0‐24]).
AUC, area under the plasma concentration vs time curve; AUC(0‐24), AUC from time zero to 24 h; AUC(0‐72), AUC from time zero to 72 h; C av,72, average plasma concentration over 72 h after prexasertib infusion; CL, systemic clearance; C max, maximum plasma concentration; CV, coefficient of variation; N, number of pharmacokinetic observations; NC, not calculated; R A, accumulation ratio; t 1/2, terminal elimination half‐life; V ss, volume of distribution at steady state.
3.4. Efficacy
A total of 8 of 11 patients with baseline and post‐baseline tumor measurements had stable disease as a BOR. Of the 6 patients in the 80 mg/m2 group, the BOR was stable disease for 4 patients (66.7%) and progressive disease for 2 patients (33.3%). The 4 patients in the 80 mg/m2 group with a BOR of stable disease remained on the study for between 68 and 93 days. Of the 5 patients in the 105 mg/m2 group with baseline and postbaseline tumor measurements, the BOR was stable disease for 4 patients (80.0%) and progressive disease for 1 patient (20.0%). The 4 patients in the 105 mg/m2 group with a BOR of stable disease remained in the study for between 53 and 76 days. No patients in the study had a complete response or partial response. Of 6 evaluable patients receiving prexasertib 80 mg/m2, 4 showed maximum tumor shrinkage, ranging from approximately 10% to 24% (Figure 2), and 3 of 5 evaluable patients receiving prexasertib 105 mg/m2 showed maximum tumor shrinkage, ranging from approximately 7% to ‐13% (Figure 2). Two patients receiving prexasertib 80 mg/m2, both of whom had esophageal SCC and a BOR of stable disease, had maximum tumor shrinkage from baseline of 20% and 13%.
Figure 2.
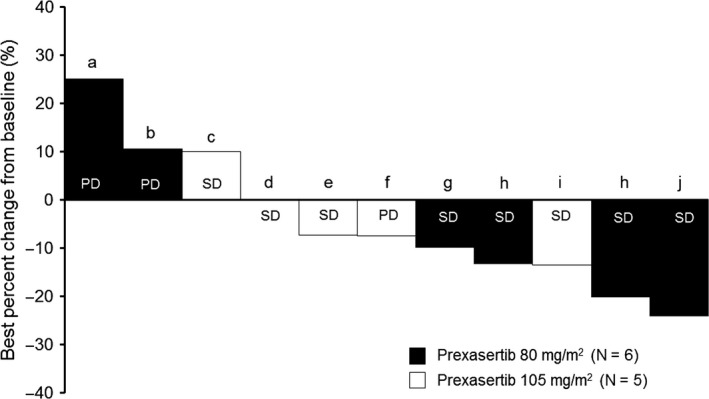
Best percentage change in tumor size from baseline with prexasertib 80 mg/m2 and prexasertib 105 mg/m2. Subjects with both baseline and post‐baseline values are included. Tumor types: a, small intestine carcinoma; b, rhabdomyosarcoma; c, malignant neoplasm of thymus; d, liposarcoma; e, squamous cell carcinoma of unknown origin; f, skin cancer; g, squamous cell carcinoma of lung; h, esophageal squamous cell carcinoma; i, endometrial adenocarcinoma; j, transitional cell carcinoma. PD, progressive disease; SD, stable disease
4. DISCUSSION
The results of this study confirm the tolerability of prexasertib administered at 80 and 105 mg/m2 in Japanese patients with advanced solid tumors. The observed safety and tolerability profile, including DLT, in Japanese patients was consistent with the known safety profile for prexasertib in the US phase 1 study.8 Prexasertib PK displayed dose‐independent and time‐independent behavior across both dose levels investigated, and was consistent with the PK in non‐Japanese patients.8 Of 11 patients, 8 had a BOR of stable disease and no responders were reported. The limitations of this study include the relatively small patient cohorts and the heterogeneity of the tumor types investigated.
The most common toxicities associated with prexasertib were hematologic, with a similar incidence and severity to that observed in non‐Japanese patients.8 Although there were 2 patients with DLT of febrile neutropenia, both recovered and continued the study treatment at a reduced dose. Six patients reported grade 4 neutropenia related to the study drug. Neutrophil counts in these patients reached a nadir with almost the exact same timing, indicating that the onset of neutropenia was predictable. A similar incidence of neutropenia with prexasertib was reported in the previous phase 1 trial conducted in non‐Japanese patients.8 The incidence of neutropenia observed with prexasertib is higher than that reported for AZD7762 (CHK1/CHK2 inhibitor) and MK‐8776 (CHK1 inhibitor).4, 5, 13 However, non‐hematologic TEAE occurred at lower rates and at a lower severity with prexasertib compared to other CHK1 inhibitors. In addition, consistent with the previous phase 1 trial conducted in non‐Japanese patients,8 there were no clinically significant cardiac toxicities or AE related to QT prolongation with prexasertib, whereas other CHK1 inhibitors have been associated with cardiotoxicity, including myocardial infarction and significant QTc changes.4, 13 Regardless, direct comparisons are difficult because the majority of experience with other CHK1 inhibitors is in combination with cytotoxic agents and not as single‐agent treatments.
In contrast to the phase 1 trial conducted in non‐Japanese patients, which reported 2 partial responses (4% of patients) with prexasertib,8 no patients had an observed complete response or partial response in this study. Furthermore, the phase 2 study of prexasertib in BRCA wild‐type recurrent high‐grade serous ovarian cancer showed partial responses in 8 of 28 patients (29%).12 Other CHK1/CHK2 inhibitors have shown limited responses, even when combined with other agents such as pemetrexed and cisplatin, gemcitabine, and irinotecan,4, 5, 7, 13, 14, 15 although it is not currently known whether these potential differences in efficacy are due to dissimilarities in the demographics and clinical characteristics of the study populations, such as tumor types, or differences in the drugs. Pretreatment biopsies were not mandated in this phase 1 study, so it is not known whether any patients had tumor characteristics that may have decreased the likelihood of responding to prexasertib. It is also possible that no responses were observed in Japanese patients due to the smaller number of patients evaluated for response compared to the phase 1 trial conducted in the US (11 patients vs 43 patients), with only 5 patients evaluable at the global‐recommended phase 2 dose of 105 mg/m2.
Although the tumor types varied, 2 patients with esophageal SCC receiving prexasertib 80 mg/m2 had a BOR of stable disease with maximum tumor shrinkage from baseline of 20% and 13%. The phase 1 trial conducted in non‐Japanese patients observed tumor shrinkage in 10 of 34 patients, with many of these cases occurring in SCC.8 Squamous cell carcinoma may harbor increased levels of replication stress and/or DNA damage response, which may contribute to an increased response to CHK1/2 inhibitors.3 On the basis of these findings, cohorts in SCC are being evaluated in the dose‐expansion phase of the phase 1 study in non‐Japanese patients.8
Prexasertib PK in Japanese patients displayed dose‐independent and time‐independent PK behavior at both dose levels investigated. After administration of prexasertib 105 mg/m2, the geometric mean AUC(0‐72) achieved the median AUC(0‐72) of 1896 ng h/mL predicted from the nonclinical Calu‐6 xenograft PK/pharmacodynamic model to achieve the maximal tumor response following prexasertib monotherapy.8 In addition, after administration of prexasertib105 mg/m2 in cycle 1 and cycle 2, 8 of 9 (89%) of the noncompartmental AUC(0‐72) values were greater than the median AUC(0‐72) predicted to correlate with the maximum tumor response after prexasertib monotherapy.8 The mean average prexasertib plasma concentrations over the first 72 hours (C av,72) after administration of 105 mg/m2 in cycle 1 and cycle 2 were also greater than the IC50 (14.1 ng/mL) derived from the single‐agent Calu‐6 xenograft model.8 Prexasertib PK are, therefore, suitable for achieving the median human exposure predicted from the nonclinical Calu‐6 xenograft PK/pharmacodynamic model.8 Prexasertib PK for Japanese patients are also consistent with the PK reported in the US‐based phase 1 study in non‐Japanese patients.8 Taken together, these data indicate that prexasertib PK in Japanese patients are suitable for minimizing intercycle accumulation and for achieving the median predicted maximum efficacious systemic exposure following intravenous administration of 105 mg/m2 once every 14 days.
In conclusion, the overall safety findings in this study were consistent with the known safety profile for prexasertib, and tolerability at the recommended dose of 105 mg/m2 was confirmed in Japanese patients with advanced solid tumors, including those with cancer types not previously investigated with prexasertib treatment.
CONFLICT OF INTEREST
Hiroya Asou, Zhihong Cai, Koichi Inoue, Kanako Saito, Yuko Shibasaki, Masaomi Tajimi, and Hiroki Takai are full‐time employees of Eli Lilly Japan K.K. Hiroya Asou owns stock in Eli Lilly and Company. Aimee Bence Lin is a full‐time employee of, and owns stock in, Eli Lilly and Company. Burch Lin (husband of Aimee Bence Lin) is a full‐time employee of, and owns stock in, Eli Lilly and Company. Noboru Yamamoto reports advisory roles with Eisai, Takeda, OncoTherapy Science, Otsuka, and Boehringer Ingelheim. Noboru Yamamoto received honoraria from Bristol‐Myers Squibb, Pfizer, AstraZeneca, Eli Lilly and Company, Ono, and Chugai. Noboru Yamamoto also received research funding from Quintiles, Astellas, Chugai, Eisai, Taiho, Bristol‐Myers Squibb, Pfizer, Novartis, Daiichi Sankyo, Bayer, Boehringer Ingelheim, Kyowa Hakko Kirin, Takeda, and Ono. Toshihiko Doi received research funding from Eli Lilly and Company. All remaining authors have declared no conflicts of interest.
ACKNOWLEDGMENTS
We thank the patients, their families, the study sites, and the study personnel who participated in this clinical trial. We also thank Hisaki Fujii for contributions to the interpretation of the data in the manuscript. Medical writing support was provided by Andrew Sakko, PhD, CMPP, and editorial support was provided by Noelle Gasco, of Syneos Health and funded by Eli Lilly and Company in accordance with Good Publication Practice (GPP3) guidelines (http://www.ismpp.org/gpp3).
Iwasa S, Yamamoto N, Shitara K, et al. Dose‐finding study of the checkpoint kinase 1 inhibitor, prexasertib, in Japanese patients with advanced solid tumors. Cancer Sci. 2018;109:3216–3223. 10.1111/cas.13750
This study is registered with ClinicalTrials.gov, identifier NCT02514603.
REFERENCES
- 1. King C, Diaz HB, McNeely S, et al. LY2606368 causes replication catastrophe and antitumor effects through CHK1‐dependent mechanisms. Mol Cancer Ther. 2015;14:2004‐2013. [DOI] [PubMed] [Google Scholar]
- 2. McNeely S, Beckmann R, Bence Lin AK. CHEK again: revisiting the development of CHK1 inhibitors for cancer therapy. Pharmacol Ther. 2014;142:1‐10. [DOI] [PubMed] [Google Scholar]
- 3. Lin AB, McNeely SC, Beckmann RP. Achieving precision death with cell‐cycle inhibitors that target DNA replication and repair. Clin Cancer Res. 2017;23:3232‐3240. [DOI] [PubMed] [Google Scholar]
- 4. Sausville E, Lorusso P, Carducci M, et al. Phase I dose‐escalation study of AZD7762, a checkpoint kinase inhibitor, in combination with gemcitabine in US patients with advanced solid tumors. Cancer Chemother Pharmacol. 2014;73:539‐549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Daud AI, Ashworth MT, Strosberg J, et al. Phase I dose‐escalation trial of checkpoint kinase 1 inhibitor MK‐8776 as monotherapy and in combination with gemcitabine in patients with advanced solid tumors. J Clin Oncol. 2015;33:1060‐1066. [DOI] [PubMed] [Google Scholar]
- 6. Scagliotti G, Kang JH, Smith D, et al. Phase II evaluation of LY2603618, a first‐generation CHK1 inhibitor, in combination with pemetrexed in patients with advanced or metastatic non‐small cell lung cancer. Invest New Drugs. 2016;34:625‐635. [DOI] [PubMed] [Google Scholar]
- 7. Infante JR, Hollebecque A, Postel‐Vinay S, et al. Phase I study of GDC‐0425, a checkpoint kinase 1 inhibitor, in combination with gemcitabine in patients with refractory solid tumors. Clin Cancer Res. 2017;23:2423‐2432. [DOI] [PubMed] [Google Scholar]
- 8. Hong D, Infante J, Janku F, et al. Phase I study of LY2606368, a checkpoint kinase 1 inhibitor, in patients with advanced cancer. J Clin Oncol. 2016;34:1764‐1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lowery CD, VanWye AB, Dowless M, et al. The checkpoint kinase 1 inhibitor prexasertib induces regression of preclinical models of human neuroblastoma. Clin Cancer Res. 2017;23:4354‐4363. [DOI] [PubMed] [Google Scholar]
- 10. Brill E, Yokoyama T, Nair J, Yu M, Ahn YR, Lee JM. Prexasertib, a cell cycle checkpoint kinases 1 and 2 inhibitor, increases in vitro toxicity of PARP inhibition by preventing Rad51 foci formation in BRCA wild type high‐grade serous ovarian cancer. Oncotarget. 2017;8:111026‐111040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zeng L, Beggs RR, Cooper TS, Weaver AN, Yang ES. Combining Chk1/2 inhibition with cetuximab and radiation enhances in vitro and in vivo cytotoxicity in head and neck squamous cell carcinoma. Mol Cancer Ther. 2017;16:591‐600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lee JM, Nair J, Zimmer A, et al. Prexasertib, a cell cycle checkpoint kinase 1 and 2 inhibitor, in BRCA wild‐type recurrent high‐grade serous ovarian cancer: a first‐in‐class proof‐of‐concept phase 2 study. Lancet Oncol. 2018;19:207‐215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Seto T, Esaki T, Hirai F, et al. Phase I, dose‐escalation study of AZD7762 alone and in combination with gemcitabine in Japanese patients with advanced solid tumours. Cancer Chemother Pharmacol. 2013;72:619‐627. [DOI] [PubMed] [Google Scholar]
- 14. Ho AL, Bendell JC, Cleary JM, et al. Phase I, open‐label, dose‐escalation study of AZD7762 in combination with irinotecan (irino) in patients (pts) with advanced solid tumors. J Clin Oncol. 2011;29(suppl): Abstract 3033. [Google Scholar]
- 15. Calvo E, Chen VJ, Marshall M, et al. Preclinical analyses and phase I evaluation of LY2603618 administered in combination with pemetrexed and cisplatin in patients with advanced cancer. Invest New Drugs. 2014;32:955‐968. [DOI] [PubMed] [Google Scholar]