

Abstract
Galectin-1 (Gal-1), an evolutionarily conserved β-galactoside-binding lectin, controls immune cell homeostasis and tempers acute and chronic inflammation by blunting proinflammatory cytokine synthesis, engaging T-cell apoptotic programs, promoting expansion of T regulatory (Treg) cells, and deactivating antigen-presenting cells. In addition, this lectin promotes angiogenesis by co-opting the vascular endothelial growth factor receptor (VEGFR) 2 signaling pathway. Since a coordinated network of immunomodulatory and proangiogenic mediators controls cardiac homeostasis, this lectin has been proposed to play a key hierarchical role in cardiac pathophysiology via glycan-dependent regulation of inflammatory responses. Here, we discuss the emerging roles of Gal-1 in cardiovascular diseases including acute myocardial infarction, heart failure, Chagas cardiomyopathy, pulmonary hypertension, and ischemic stroke, highlighting underlying anti-inflammatory mechanisms and therapeutic opportunities. Whereas Gal-1 administration emerges as a potential novel treatment option in acute myocardial infarction and ischemic stroke, Gal-1 blockade may contribute to attenuate pulmonary arterial hypertension.
1. Introduction
Galectins are a family of β-galactoside-binding lectins widely expressed in a variety of cells and tissues [1]. These animal lectins play critical roles in the regulation of both innate and adaptive immunity, inflammation, wound healing, and angiogenesis [2–6]. To date, 15 different members have been identified, which are classified according to their biochemical structure into three groups: (a) prototype galectins, including galectin- (Gal-) 1, -2, -5, -7, 10, -11, -13, -14, and -15, display one carbohydrate-recognition domain (CRD) and can dimerize; (b) tandem-repeat galectins, including Gal-4, -6, -8, -9, and -12, exhibit two CRDs associated with a linker peptide; and (c) the unique chimera-type Gal-3 contains one CRD and an additional nonlectin domain and can oligomerize [1]. Emerging evidence demonstrates that galectins play important roles in cardiovascular pathophysiology by controlling acute and chronic inflammatory responses [7]. Despite structural similarities, individual members of the galectin family play distinct roles during inflammatory processes by controlling innate and adaptive immune compartments [8]. This includes modulation of proinflammatory cytokine production, promotion of T regulatory (Treg) cell expansion, and control of immune cell differentiation and survival [9].
1.1. Galectin-1 (Gal-1)
Gal-1, the first galectin identified, functions mostly as an anti-inflammatory mediator that represses innate and adaptive immune programs [1, 9]. Expression of Gal-1 is prominent in inflammatory macrophages [10, 11], activated T lymphocytes [12], Treg cells [13], and tolerogenic dendritic cells [14]. From a structural viewpoint, Gal-1 consists of two subunits of 14.5 kDa (135 amino acids) present in a dynamic dimerization equilibrium [15]. Because of an unusual number of six cysteine residues, this lectin is highly susceptible to oxidative inactivation, which hampers its immunoregulatory activity [16]. Gal-1 binds to multiple galactose-β1-4-N-acetyl-glucosamine (N-acetyl-lactosamine; LacNAc) units present on the branches of N- or O-linked glycans on a variety of cell surface receptors and modulates their segregation, endocytosis, and signaling [4, 15]. These glycan structures are generated by the coordinated action of a set of glycosyltransferases including N-acetylglucosaminyltransferase 5 (MGAT5), an enzyme that creates β1,6-N-acetylglucosamine-branched complex N-glycans, and core-2 β1-6-N-acetylglucosaminyltransferase 1 (C2GNT1), an enzyme that catalyzes branching of core-2 O-glycan structures. Conversely, Gal-1 binding is prevented when LacNAc is modified by α2,6-linked sialic acid incorporated by α2,6-sialyltransferase 1 (ST6GAL1) [3]. Gal-1 controls biological processes through extracellular mechanisms by cross-linking cell surface glycoconjugates or intracellularly by influencing a variety of signaling events [1, 2]. Through glycosylation-dependent mechanisms, this lectin modulates cellular activation, proliferation, migration, and survival, playing a critical role in the resolution of acute and chronic inflammation [4–6, 15]. In addition, Gal-1 promotes neovascularization by signaling through vascular endothelial growth factor receptor- (VEGFR-) 2 [17, 18]. Because of its prominent anti-inflammatory, proresolving, and proangiogenic activities, Gal-1 has been proposed to be a key mediator of cardiovascular homeostasis [19].
Here, we discuss the relevance of Gal-1 in cardiovascular disorders including acute myocardial infarction (AMI), heart failure (HF), Chagas cardiomyopathy, pulmonary hypertension (PAH), and ischemic stroke and highlight cellular and molecular mechanisms underlying these effects.
1.2. Acute Myocardial Infarction
Acute myocardial infarction (AMI) is caused by a sudden interruption in blood oxygen supply mostly due to atherothrombosis; this effect is accompanied by a storm of proinflammatory cytokines and a dysregulation of effector and regulatory immune cell mechanisms [20]. AMI is the leading cause of death worldwide, and those who survive are at higher risk for adverse ventricular remodeling and heart failure [21].
Gal-1 is expressed in the normal heart of many species [22–24]. Cardiomyocytes constitutively express Gal-1 at the cytosolic compartment in an organized striated pattern close to sarcomeric actin but not myosin [23] (Figure 1(a)). However, Gal-1 expression is further upregulated during AMI, with a peak as early as 20 min after experimental coronary artery ligation [24], followed by a second peak at 7 days [19]. This bimodal expression pattern may reflect different cells that express Gal-1 in AMI either ischemic cardiomyocytes (20 min) or leukocytes that promote resolution of inflammation (7 days).
Figure 1.

Role of Gal-1 in acute myocardial infarction. (a) Gal-1 is constitutively expressed in cardiomyocytes close to sarcomeric actin and is increased and secreted after acute myocardial infarction (AMI), inflammation, and hypoxia. (b) Mice lacking Gal-1 (Lgals1−/−) show adverse ventricular remodeling after AMI associated with increased inflammation and reduced proportion of regulatory T (Treg) cells.
Cultured cardiomyocytes upregulate and secrete Gal-1 early after hypoxia and in response to proinflammatory cytokines [19], as seen in the early phase of AMI in vivo (Figure 1(a)). Whereas early upregulation of Gal-1 may serve as a homeostatic safeguard mechanism to prevent a dysregulated inflammatory response that could otherwise promote adverse remodeling [20], augmented Gal-1 expression later in the course of AMI could influence the resolution of cardiac inflammation and restore homeostasis [25].
Interestingly, Gal-1 was found to be upregulated 7 days after nonreperfused AMI, associated with a peak in infiltrating dendritic cells, lymphocytes, and macrophages [26]. Since Gal-1 promotes differentiation of tolerogenic dendritic cells and M2-type macrophages [14, 27] which play protective roles in AMI [28, 29], Gal-1-glycan interactions may contribute to reparative functions mediated by these cells.
Within the T-cell compartment, Gal-1 promotes apoptosis of CD8 and Th1 and Th17 lymphocytes, promoting a shift toward a Th2-dominant cytokine profile [3, 30, 31]. This effect is highly relevant in cardiovascular diseases as upregulation of Th1 and Th17 cytokines is a typical hallmark observed in patients with acute coronary syndromes [32–34] and is associated with adverse remodeling after AMI [32]. On the other hand, healthy patients exhibiting Th2-dominant responses may be protected from cardiovascular disease [35]. Thus, Gal-1 emerges as an attractive therapeutic candidate to limit innate and adaptive responses early or late during cardiovascular inflammation. Remarkably, mice lacking Gal-1 (Lgals1−/−) showed adverse ventricular remodeling after AMI with increased cardiac dilation associated with dysregulated uncontrolled inflammation (Table 1) [19]. Absence of Gal-1 led to an increase in cardiac infiltration by T lymphocytes, macrophages, and natural killer (NK) cells, while anti-inflammatory Treg cells were significantly reduced in this setting (Figure 1(b)) [19]. As Treg cells are protective in AMI [36], and Gal-1 is also a critical mediator of Treg function [13, 37], Gal-1-driven inhibitory circuits may also operate in Treg-mediated protection during AMI.
Table 1.
Preclinical models of cardiovascular disease in Lgals1−/− mice.
| Model | Effects | Mechanisms | Reference |
|---|---|---|---|
| AMI | Worse remodeling | Increased inflammation | [19] |
| HF | Spontaneous CMP | Increased inflammation | [19] |
| Chagas disease | Reduced cardiac inflammation and parasite infection | Not fully understood. Controversial results on survival | [52, 54] |
| PAH | Less PAH and reduced RV hypertrophy | Decreased vasoreactivity | [65] |
AMI = acute myocardial infarction; HF = heart failure; PAH = pulmonary arterial hypertension; CMP = cardiomyopathy; RV = right ventricle.
As mentioned above, Gal-1 is expressed at the cytosolic compartment of cardiomyocytes in association with actin and is secreted in response to injury. Since Lgals1−/− mice lack both the intracellular and extracellular Gal-1 activities, it is difficult to infer which regulatory function of this lectin controls adverse remodeling after AMI. However, treatment with recombinant Gal-1 has been used to address the extracellular versus intracellular roles of this lectin. Mice treated with a single dose of recombinant Gal-1 during AMI showed a significant improvement in ventricular function and remodeling (Table 2) [19]. These effects support the concept that extracellular activities of Gal-1 prevail in cardioprotection and highlight the therapeutic potential of this lectin in patients with AMI. Interestingly, exogenous Gal-1 also prevented renal ischemia-reperfusion injury through anti-inflammatory mechanisms [38]. Nevertheless, as Gal-1 can be taken up by cells devoid of this lectin [39], this alternative mechanism could also operate in cardiomyocytes. Thus, in addition to the anti-inflammatory effects of the exogenous protein, Gal-1-driven nonimmunological events may also take place.
Table 2.
Effects of Gal-1 treatment on cardiovascular disease.
| Model | Effects | Mechanisms | Reference |
|---|---|---|---|
| AMI (reperfused) | Improved remodeling | Inflammation? (not evaluated) | [19] |
| Stroke | Improved sensorimotor dysfunction | Increased neurogenesis. No effect on infarct volume | [72] |
AMI = acute myocardial infarction; HF = heart failure; PAH = pulmonary arterial hypertension.
Atherosclerosis is the underlying pathology for most AMIs. Atherosclerotic plaque rupture leads to coronary thrombosis and myocardial necrosis. Inflammation plays a key role in both development and fate of atherosclerosis [40]. A recent large clinical study showed that inhibition of atherosclerosis improved clinical outcomes in patients with previous AMI [41]. Experimental atherosclerosis in ApoE−/− mice fed with cholesterol showed increased Gal-1 expression in atherosclerotic plaques both in the media and in the intima layer [42]. However, in broad contrast to Gal-3, Gal-1 expression was not increased over time. Moreover, statin treatment led to inhibition of Gal-3 but had no effect on Gal-1 expression [42]. Although the role of Gal-1 in atherosclerosis has not yet been examined in detail, Gal-3 blockade led to reduced atherosclerosis in ApoE−/− mice [43, 44].
1.3. Heart Failure
Patients who experience adverse ventricular remodeling after AMI are at increased risk of developing heart failure (HF). Although mortality after AMI decreased over the last decades, HF experienced a significant increase [21]. HF may also result from dilated nonischemic cardiomyopathy in the absence of AMI. The worldwide burden of HF is increasing, representing a global health problem. Despite advances in both pharmacological approaches and left ventricle assisting devices, the only available treatment for patients with end-stage HF is cardiac transplantation. Given the role of Gal-1 as an important regulator of immune responses, Gal-1 expression was investigated in patients with advanced HF. Heart samples explanted from HF patients undergoing transplantation showed increased Gal-1 expression compared to control hearts [19]. As expected, Gal-1 was localized within the inflammatory infiltrate and the interstitium, but was also found in cardiomyocytes. Accordingly, Gal-1 was upregulated in hearts from Chagas cardiomyopathy patients [45]. Whether Gal-1 expression in the heart of HF patients is part of the pathogenic mechanisms of the disease or may represent a compensatory effect in response to enhanced inflammation is still not clear. Further studies in patients with ischemic as well as nonischemic HF are warranted to better understand the role of Gal-1 in both etiology and prognosis of this disease. Moreover, as different environmental factors may influence Gal-1 expression including hypoxia, inflammation, aging, and metabolic status [3, 17], further work is needed to dissect the role of these factors in regulating the activity of this lectin.
Interestingly, mice lacking Gal-1 showed mild ventricular dilation, reduced contractility, and an enhanced inflammatory infiltrate composed of lymphocytes, macrophages, and NK cells, as well as reduced number of Treg cells, indicative of autoimmune myocarditis (Table 1) [19]. Moreover, Lgals1−/− mice showed increased levels of circulating Th1 and Th17 cytokines [3] which might contribute to ventricular dysfunction and dilation, similar to the dysfunction observed in septic patients [46], as well as experimental inflammation induced by interleukin- (IL-) 1β and IL-18 [47, 48]. The upregulated expression of Gal-1 in patients with HF may therefore represent a homeostatic mechanism that controls autoimmune myocarditis in response to injury. Whether intracellular Gal-1 participates in cardiomyocyte contraction and may contribute to cardiomyopathy and HF is still uncertain.
1.4. Chagas Disease
Chagas disease is caused by infection with the protozoan parasite Trypanosoma cruzi (T. cruzi). Acute infection is usually mild and undiagnosed; however, 20–30% of patients develop dilated cardiomyopathy several years later [49]. Although the precise mechanisms underlying Chagas cardiomyopathy are not completely understood, they involve (a) injury to cardiomyocytes induced by T. cruzi, (b) cardiac inflammation, (c) microvascular dysfunction, and (d) autonomic disorders [50].
The T. cruzi parasite itself does not express Gal-1, but anti-Gal-1 antibodies have been observed in patients with Chagas disease: whereas IgM and IgE antibodies raise with acute infection, anti-Gal-1 IgG antibodies are present in the chronic phase of the disease [45]. The source of this humoral response was mainly associated to antigen release from host cells, since no cross-reactivity was found between Gal-1 and T cruzi proteins. Likewise, autoantibodies against other host cell proteins have been observed in Chagas disease patients [51]. In fact, serum Gal-1 increases in patients with Chagas disease irrespective of cardiac involvement [52].
Infection with T. cruzi induces the release of interferon-γ (IFN-γ), which promotes death of intracellular parasites, thus limiting the extent of infection [50]. However, IFN-γ and uncontrolled inflammation may also lead to host damage and myocarditis [53]. Thus, regulation of Gal-1 expression by IFN-γ [19] may contribute as a homeostatic mechanism to counterbalance cardiac inflammation. Whereas a study found that Gal-1 binds weekly to T. cruzi with no direct effect [54], another study showed no significant binding to the parasite [52]. On the contrary, Gal-1 appears to be upregulated after infection and contributes to parasite-immune escape. Activated B lymphocytes from infected mice secreted high levels of Gal-1 which promoted apoptosis of activated T lymphocytes [55]. Moreover, infected dendritic cells also secreted Gal-1 which contributed to expansion of the Treg cell compartment, leading to inhibition of CD8+ T-cells. These results suggest that Gal-1 may limit the clearance of T. cruzi parasite by triggering tolerogenic circuits [54]. However, the direct effect of T. cruzi on cardiomyocytes involves different mechanisms. Whereas T cruzi infects cardiomyocytes and induces apoptosis [56], infected cardiomyocytes may secrete Gal-1 as a cardioprotective mechanism, as Gal-1 affinity to cardiomyocytes prevents T. cruzi infection via glycosylation-dependent mechanisms [52]. Whether this pathway involves inhibition of T. cruzi binding to cardiomyocytes through hindrance of glycan-binding sites remains uncertain. Interestingly, infected cardiomyocytes show an altered glycophenotype which prevents Gal-1 binding, promoting parasite escape [52]. Thus, Gal-1 may control the fate and function of cardiomyocytes through direct or indirect mechanisms involving immune-dependent or independent pathways.
Despite the immunosuppressive effects of Gal-1, studies in T. cruzi-infected Lgals1−/− mice showed reduced cardiac inflammation [52] and lower myocardial parasite infiltration (Table 1) [54]. Results on survival and parasitemia in those mice were however controversial: as one study showed longer survival despite increased parasitemia [52], another study indicated reduced survival with similar parasitemia [54]. Differences in these parameters may depend on the T. cruzi strain: in vitro cardiomyocyte infection with Brazil, but not Tulahuen strain of T. cruzi, showed an early mild reduction of Gal-1 expression [52]. In this regard, in vivo infection with T. cruzi Y strain showed higher cardiac inflammation and mortality than the VFRA and Sc43 strains [57]. Thus, different parasite strains may differentially affect Gal-1 expression, selective binding to glycosylated receptors on host cells, and clinical outcome of in vivo infection. Of note, all studies were performed in T. cruzi-infected mice and may not fully recapitulate the pathophysiology of Chagas cardiomyopathy, which takes place several years after infection and may not be associated with parasitemia or myocardial parasite infiltration. Finally, survival of these septic mice should not be considered as a surrogate for Chagas cardiomyopathy. Unfortunately, no animal models have completely recapitulated the pathophysiology and clinical course of human Chagas cardiomyopathy.
1.5. Pulmonary Arterial Hypertension
Pulmonary arterial hypertension (PAH) is defined as a sustained increase in mean pulmonary artery pressure in the absence of an elevation in pulmonary artery wedge pressure [58]. Symptoms consist of dyspnea and right ventricle (RV) overload leading to heart failure and death. The hallmark of PAH is proliferation of vascular smooth muscle cells (VSMCs) that cause arterial narrowing leading to an increase in vascular resistance [59]. However, endothelial cells (ECs) control VSMC function, contraction, and proliferation through soluble mediators, direct contact, and extracellular vesicles [60].
Gal-1 is expressed and secreted by both ECs and VSMCs. This lectin modulates VSMC attachment, spreading, and migration through affinity to extracellular matrix glycoproteins including laminin, fibronectin, and α1β1 integrin [61]. Both vascular deposits of laminin and fibronectin are typically observed in PAH [59, 62]. Interestingly, the precise effects of Gal-1 on VSMC are controversial: whereas some studies showed inhibition of spreading [60] and migration [63], one study showed increased DNA replication following exposure to this lectin [64].
Chronic hypobaric hypoxia, an established model of PAH, leads to upregulation of lung Gal-1 expression [65]. Mice lacking Gal-1 showed reduced PAH in this model with ameliorated RV hypertrophy (Table 1) [65]. Despite the in vitro effects of Gal-1 on VSMCs, arterial muscularization and microvessel density were not affected in Gal-1-deficient mice. However, absence of Gal-1 in the lungs was associated with decreased vasoreactivity after acute hypoxia, with no effects in vasopressor response after 5-hydroxytryptamine and potassium chloride [65]. Interestingly, effects on vascular reactivity could be associated to calcium influx in VSMCs, since intracellular Gal-1 has been shown to inhibit ICa,L modifying the Cav1.2 calcium channel in a splice variant-dependent manner [66]. In this regard, Gal-1 promotes Cav1.2 degradation in smooth muscle cells controlling vascular reactivity and blood pressure [67]. Moreover, Gal-1 has also been implicated in the pathogenesis of preeclampsia, which is characterized by hypertension and proteinuria during pregnancy [68], suggesting new roles for this lectin in pulmonary, systemic, and pregnancy-induced arterial hypertension.
Remarkably, PAH increases RV afterload leading to contractile dysfunction and Gal-1 expression in the normal heart has been shown to be more pronounced in RV [24]. The contribution of cardiomyopathy to the pathogenesis of PAH in Gal-1-deficient mice warrants further consideration. Moreover, the phenotype of Lgals1−/− PAH mice appears to be the combination of the absence of this lectin in the lung vasculature, the RV, and the contribution of the immune system. Further experiments are warranted to investigate the translational and therapeutic implications of these findings.
1.6. Stroke
Similar to AMI, stroke is characterized by a sudden drop in oxygen blood supply to the brain. In the normal brain, Gal-1 is expressed by both neurons and glia [69, 70] and modulate astrocyte function [71]. Gal-1 upregulation is observed in vitro after anoxia [71] and in vivo in the infarcted and penumbra area after experimental stroke [72]. Gal-1 has been shown to induce astrocyte differentiation and brain-derived neurotrophic factor (BDNF) secretion [71]. However, Gal-1 treatment inhibited normal astrocyte proliferation in vitro [73] and promoted microglial deactivation [27]. In vivo, Gal-1 treatment promoted neurogenesis after stroke in a carbohydrate-dependent manner, and antibody-mediated Gal-1 blockade prevented this effect (Table 2) [72]. Similar to Gal-1 treatment, transfer of stem cells overexpressing Gal-1 significantly reduced infarct volume [74]. Stroke confers an oxidative environment which inactivates Gal-1 impairing some carbohydrate-binding activities. However, oxidized Gal-1 stimulates axonal regeneration [75]. Besides its role in neuronal proliferation after injury, Gal-1 showed a neuroprotective effect through inhibition of central nervous system (CNS) inflammation. Interestingly, Gal-1 treatment induced an M2 microglia phenotype, which prevented inflammation-induced neurodegeneration [27]. On the contrary, an M1 “inflammatory” phenotype was associated with CNS toxicity [76]. Gal-1 binds with higher affinity to M1 microglia skewing the balance toward an M2 phenotype [27]. In addition, Gal-1 modifies the glutamate receptor NMDA preventing binding of this neurotransmiter [77]. Furthermore, this lectin participates in neuronal development, since Lgals1−/− mice display altered olfactory neurons [78] and reduced thermal sensitivity [79].
Serum Gal-1 levels have been evaluated in patients with stroke. Whereas one study showed normal values of this protein [80], a larger cohort showed increased serum Gal-1 after ischemic stroke [81]. Both studies, however, found no association between Gal-1 levels and functional recovery. Thus, serum Gal-1 does not appear to be a reliable biomarker of local activation or neuroprotection.
1.7. Other Galectins
In addition to Gal-1, other members of the galectin family may control cardiovascular pathophysiology. Although their functions are beyond the scope of the present review, we will briefly summarize their most important contributions to cardiovascular diseases. Remarkably, Gal-3 has been widely studied in the context of cardiovascular disease and its effects and potential diagnostic and prognostic values have been extensively reviewed elsewhere [7, 8]. This “chimera-type” lectin is highly expressed by macrophages, promoting inflammation, and fibrosis in various organs [82]. In AMI, Gal-3 is upregulated early and released to the extracellular compartment. Mice lacking Gal-3 (Lgals3−/−) showed improved cardiac function and reduced fibrosis after pressure overload and during hypertension [83]. On the contrary, adverse remodeling with increased infarct size and reduced scar fibrosis was observed in Lgals3−/− mice after AMI, probably due to ventricular rupture [84]. Gal-3 promotes cardiac as well as renal, hepatic, vascular, and pulmonary fibrosis [8, 85]. Several clinical studies reported increased serum levels of Gal-3 in cardiovascular diseases, and this lectin is currently available as a cardiovascular risk biomarker for heart failure [86]. Moreover, Gal-3 has been proposed as a biomarker that links oxidative stress and inflammation in patients with atherothrombosis [87]. In addition, Gal-3 levels are also increased in patients with AMI although the clinical value of this lectin is still a matter of debate [8].
Although less studied, Gal-2 has also been shown to play an important role in cardiovascular disease. Similar to Gal-1, Gal-2 is a prototype lectin that promotes apoptosis of activated CD8+ T cells [88, 89]. However, in contrast to Gal-1, Gal-2 also showed proinflammatory activity within the monocyte/macrophage compartment promoting a shift towards an M1 phenotype [90] and inhibiting arteriogenesis [91]. Interestingly, Gal-2 is elevated in atherosclerotic plaques and colocalized with lymphotoxin-α [91, 92], suggesting a galectin-cytokine interaction that favors proinflammmatory responses. Whereas a study in the Japanese population showed a functional single-nucleotide polymorphism (SNP) rs7291467 (C3279C➔T) in the Gal-2 gene (LGALS2) that was associated with increased risk of AMI [92], these results could not be confirmed in Caucasian individuals, and a meta-analysis of seven studies showed no association between LGALS2-C3279T and coronary artery disease [93].
Gal-9, a tandem-repeat member of the galectin family, functions mostly as an anti-inflammatory lectin [94] that promotes Th1 and Th17 apoptosis through interaction with T-cell immunoglobulin domain and mucin domain protein 3 (TIM-3) [2, 4]. Gal-9 has shown to prevent renal ischemia/reperfusion injury, to attenuate experimental viral myocarditis [95], and to improve survival after cardiac transplantation [96]. Further studies are needed to unravel the precise roles of Gal-9 in AMI and other cardiovascular diseases.
Finally, Gal-12, a tandem-repeat galectin preferentially expressed in adipose tissue, contributes to differentiation and turnover of preadipocytes [4, 97]. Targeted disruption of Gal-12 gene (Lgals12) in mice increased insulin sensitivity and glucose tolerance and contributed to diabetes and metabolic syndrome [4, 97], suggesting the potential role of this lectin in cardiovascular diseases.
Although a preferential set of receptors have been described for individual members of the galectin family [98], some glycosylated receptors may be shared by some galectins. This includes CD45 which may be engaged by both Gal-1 and Gal-3 on T lymphocytes [99] and VEGFR2 which mediates the function of those galectins on ECs [18, 100]. Since different galectins may induce antagonic effects (e.g., proinflammatory versus anti-inflammatory effects), potential cross-talk between these glycan-binding proteins and competition for similar receptors or glycan structures should also be taken into consideration.
2. Conclusions
Galectins, a family of β-galactoside-binding lectins, have emerged as key modulators of inflammatory processes [1–4]. Gal-1, a highly conserved member of this family, is widely expressed in different tissues and contributes to immunosuppression in cancer, infection, and autoimmune diseases [1–4]. Recently, an emerging role for Gal-1 in cardiovascular processes has been proposed (Figure 2). Gal-1 is constitutively expressed in cardiomyocytes complexed with cytosolic actin. Interestingly, Gal-1 expression is increased in the heart of patients with AMI, HF, and Chagas cardiomyopathy. Mice lacking Gal-1 show increased susceptibility to chronic inflammatory diseases and develop autoimmune myocarditis and cardiac dysfunction, suggesting its central nonredundant roles in cardiac homeostasis. Through binding to cell surface ligands, Gal-1 not only controls innate and adaptive immune programs that lead to resolution of inflammatory responses, but also may control cardiomyocyte survival leading to potential applications of this lectin in the treatment of AMI and other cardiovascular disorders. Accordingly, Gal-1 has been associated to the pathogenesis of PAH and ischemic stroke (Figure 2). Further studies should be aimed at defining specific roles of Gal-1 in the heart, identifying its candidate receptors within cardiac tissue and unveiling potential interplay with other members of the galectin family, with the ultimate goal of designing and implementing galectin-based therapies. Several strategies have been proposed to modulate Gal-1 expression and function, including anti-Gal-1 antibodies in cancer and infection settings [17, 101] as well as recombinant Gal-1 in autoimmune and chronic inflammatory disorders [3, 31]. From a translational standpoint, recombinant Gal-1 administration emerges as the most attractive approach for treatment of cardiovascular diseases, particularly AMI [19]. On the other hand, as Gal-1 cross-links and signals via glycosylated receptors, an alternative strategy could potentially involve modulation of glycans generated by the concerted action of glycosyltransferases, including MGAT5, C2GNT1, and ST6GAL1 [1]. Although mice lacking these glycosyltransferases display clear immunological phenotypes, there is still no information on their role in cardiovascular inflammation. Moreover, no studies have been performed to evaluate the impact of glycosyltransferase inhibitors in these pathologies. Although distinct Gal-1 receptors have been identified on the surface of different cell types, including CD45 and CD43 on T cells [99], CD45 on microglia [27], and VEGFR2 on ECs [18], its potential counter-receptors and glycosylated ligands on the surface of cardiomyocytes have not yet been explored. Finally, further studies should be conducted to validate the above mentioned mechanisms in clinical settings. Although animal models represent an invaluable tool for understanding cardiovascular pathology, clinical studies are essential to definitely validate galectin-driven circuits as patients may exhibit other pathologic conditions which could also influence Gal-1 expression and function.
Figure 2.
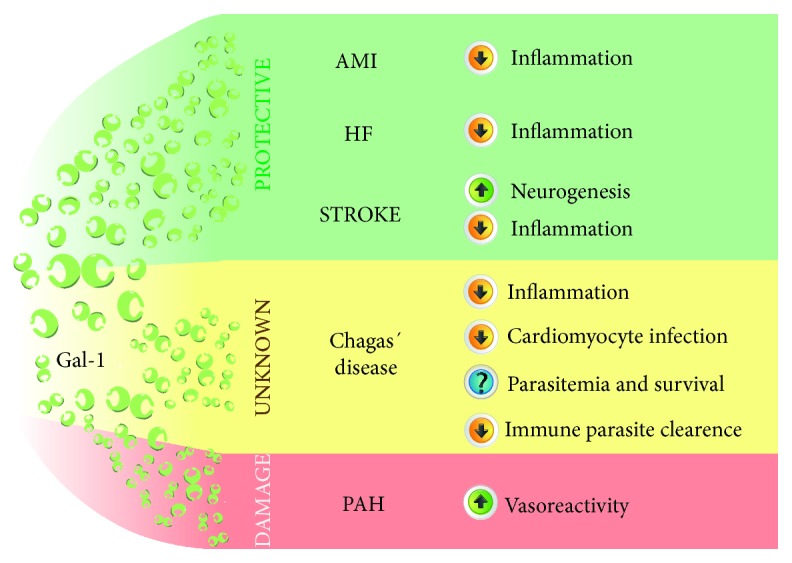
Proposed roles of Gal-1 in cardiovascular disease. Galectin-1 (Gal-1) has been implicated in a number of cardiovascular disorders. The roles of this lectin vary according to different clinical contexts. Gal-1 = galectin-1; AMI = acute myocardial infarction; HF = heart failure; PAH = pulmonary arterial hypertension.
Acknowledgments
This work was supported by grants from the Argentinean Agency for Promotion of Science and Technology (PICT V 2014-367 and 2014-2320 to G.A.R.'s laboratory and G.E.G., respectively), Sales and Bunge & Born Foundations (to G.A.R.'s laboratory), research grant from University of Buenos Aires Science and Technology (UBACyT 20020170100619BA to G.E.G.), research grant from Allende Foundation and the National Academy of Medicine of Argentina (to I.M.S.), Hospital Italiano de Buenos Aires (to D.O.B.), and grants (HL121402 and HL136816) from the National Heart, Lung, and Blood Institute, Bethesda, Maryland, USA (to A.A.).
Conflicts of Interest
Dr. Abbate has served as a consultant and has received research support from Novartis (Basel, Switzerland), Swedish Orphan Biovitrum (Stockholm, Sweden), and Olatec Therapeutics (New York, USA).
References
- 1.Cerliani J. P., Blidner A. G., Toscano M. A., Croci D. O., Rabinovich G. A. Translating the ‘sugar code’ into immune and vascular signaling programs. Trends in Biochemical Sciences. 2017;42(4):255–273. doi: 10.1016/j.tibs.2016.11.003. [DOI] [PubMed] [Google Scholar]
- 2.Méndez-Huergo S. P., Blidner A. G., Rabinovich G. A. Galectins: emerging regulatory checkpoints linking tumor immunity and angiogenesis. Current Opinion in Immunology. 2017;45:8–15. doi: 10.1016/j.coi.2016.12.003. [DOI] [PubMed] [Google Scholar]
- 3.Toscano M. A., Martínez Allo V. C., Cutine A. M., Rabinovich G. A., Mariño K. V. Untangling galectin-driven regulatory circuits in autoimmune inflammation. Trends in Molecular Medicine. 2018;24(4):348–363. doi: 10.1016/j.molmed.2018.02.008. [DOI] [PubMed] [Google Scholar]
- 4.Brinchmann M. F., Patel D. M., Iversen M. H. The role of galectins as modulators of metabolism and inflammation. Mediators of Inflammation. 2018;2018:11. doi: 10.1155/2018/9186940.9186940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cedeno-Laurent F., Dimitroff C. J. Galectin-1 research in T cell immunity: past, present and future. Clinical Immunology. 2012;142(2):107–116. doi: 10.1016/j.clim.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smetana K., Jr, André S., Kaltner H., Kopitz J., Gabius H. J. Context-dependent multifunctionality of galectin-1: a challenge for defining the lectin as therapeutic target. Expert Opinion on Therapeutic Targets. 2013;17(4):379–392. doi: 10.1517/14728222.2013.750651. [DOI] [PubMed] [Google Scholar]
- 7.van der Hoeven N. W., Hollander M. R., Yıldırım C., et al. The emerging role of galectins in cardiovascular disease. Vascular Pharmacology. 2016;81:31–41. doi: 10.1016/j.vph.2016.02.006. [DOI] [PubMed] [Google Scholar]
- 8.Meijers W. C., van der Velde A. R., Pascual-Figal D. A., de Boer R. A. Galectin-3 and post-myocardial infarction cardiac remodeling. European Journal of Pharmacology. 2015;763(Part A):115–121. doi: 10.1016/j.ejphar.2015.06.025. [DOI] [PubMed] [Google Scholar]
- 9.Ilarregui J. M., Bianco G. A., Toscano M. A., Rabinovich G. A. The coming of age of galectins as immunomodulatory agents: impact of these carbohydrate binding proteins in T cell physiology and chronic inflammatory disorders. Annals of the Rheumatic Diseases. 2005;64(Supplement_4):iv96–iv103. doi: 10.1136/ard.2005.044347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rabinovich G., Castagna L., Landa C., Riera C. M., Sotomayor C. Regulated expression of a 16-kd galectin-like protein in activated rat macrophages. Journal of Leukocyte Biology. 1996;59(3):363–370. doi: 10.1002/jlb.59.3.363. [DOI] [PubMed] [Google Scholar]
- 11.Cerliani J. P., Stowell S. R., Mascanfroni I. D., Arthur C. M., Cummings R. D., Rabinovich G. A. Expanding the universe of cytokines and pattern recognition receptors: galectins and glycans in innate immunity. Journal of Clinical Immunology. 2011;31(1):10–21. doi: 10.1007/s10875-010-9494-2. [DOI] [PubMed] [Google Scholar]
- 12.Fuertes M. B., Molinero L. L., Toscano M. A., et al. Regulated expression of galectin-1 during T-cell activation involves Lck and Fyn kinases and signaling through MEK1/ERK, p 38 MAP kinase and p70S6 kinase. Molecular and Cellular Biochemistry. 2004;267(1/2):177–185. doi: 10.1023/B:MCBI.0000049376.50242.7f. [DOI] [PubMed] [Google Scholar]
- 13.Garín M. I., Chu C. C., Golshayan D., Cernuda-Morollón E., Wait R., Lechler R. I. Galectin-1: a key effector of regulation mediated by CD4+CD25+ T cells. Blood. 2007;109(5):2058–2065. doi: 10.1182/blood-2006-04-016451. [DOI] [PubMed] [Google Scholar]
- 14.Ilarregui J. M., Croci D. O., Bianco G. A., et al. Tolerogenic signals delivered by dendritic cells to T cells through a galectin-1-driven immunoregulatory circuit involving interleukin 27 and interleukin 10. Nature Immunology. 2009;10(9):981–991. doi: 10.1038/ni.1772. [DOI] [PubMed] [Google Scholar]
- 15.Sundblad V., Morosi L. G., Geffner J. R., Rabinovich G. A. Galectin-1: a Jack-of-all-trades in the resolution of acute and chronic inflammation. Journal of Immunology. 2017;199(11):3721–3730. doi: 10.4049/jimmunol.1701172. [DOI] [PubMed] [Google Scholar]
- 16.Guardia C. M., Caramelo J. J., Trujillo M., et al. Structural basis of redox-dependent modulation of galectin-1 dynamics and function. Glycobiology. 2014;24(5):428–441. doi: 10.1093/glycob/cwu008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Croci D. O., Salatino M., Rubinstein N., et al. Disrupting galectin-1 interactions with N-glycans suppresses hypoxia-driven angiogenesis and tumorigenesis in Kaposi’s sarcoma. The Journal of Experimental Medicine. 2012;209(11):1985–2000. doi: 10.1084/jem.20111665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Croci D. O., Cerliani J. P., Dalotto-Moreno T., et al. Glycosylation-dependent lectin-receptor interactions preserve angiogenesis in anti-VEGF refractory tumors. Cell. 2014;156(4):744–758. doi: 10.1016/j.cell.2014.01.043. [DOI] [PubMed] [Google Scholar]
- 19.Seropian I. M., Cerliani J. P., Toldo S., et al. Galectin-1 controls cardiac inflammation and ventricular remodeling during acute myocardial infarction. The American Journal of Pathology. 2013;182(1):29–40. doi: 10.1016/j.ajpath.2012.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Westman P. C., Lipinski M. J., Luger D., et al. Inflammation as a driver of adverse left ventricular remodeling after acute myocardial infarction. Journal of the American College of Cardiology. 2016;67(17):2050–2060. doi: 10.1016/j.jacc.2016.01.073. [DOI] [PubMed] [Google Scholar]
- 21.Velagaleti R. S., Pencina M. J., Murabito J. M., et al. Long-term trends in the incidence of heart failure after myocardial infarction. Circulation. 2008;118(20):2057–2062. doi: 10.1161/CIRCULATIONAHA.108.784215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ashraf G. M., Rizvi S., Naqvi S., et al. Purification, characterization, structural analysis and protein chemistry of a buffalo heart galectin-1. Amino Acids. 2010;39(5):1321–1332. doi: 10.1007/s00726-010-0574-7. [DOI] [PubMed] [Google Scholar]
- 23.Dias-Baruffi M., Stowell S. R., Song S. C., et al. Differential expression of immunomodulatory galectin-1 in peripheral leukocytes and adult tissues and its cytosolic organization in striated muscle. Glycobiology. 2010;20(5):507–520. doi: 10.1093/glycob/cwp203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Al-Salam S., Hashmi S. Galectin-1 in early acute myocardial infarction. PLoS One. 2014;9(1, article e86994) doi: 10.1371/journal.pone.0086994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen B., Frangogiannis N. G. Immune cells in repair of the infarcted myocardium. Microcirculation. 2017;24(1) doi: 10.1111/micc.12305. [DOI] [PubMed] [Google Scholar]
- 26.Vandervelde S., van Amerongen M. J., Tio R. A., Petersen A. H., van Luyn M. J. A., Harmsen M. C. Increased inflammatory response and neovascularization in reperfused vs. non-reperfused murine myocardial infarction. Cardiovascular Pathology. 2006;15(2):83–90. doi: 10.1016/j.carpath.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 27.Starossom S. C., Mascanfroni I. D., Imitola J., et al. Galectin-1 deactivates classically activated microglia and protects from inflammation-induced neurodegeneration. Immunity. 2012;37(2):249–263. doi: 10.1016/j.immuni.2012.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choo E. H., Lee J. H., Park E. H., et al. Infarcted myocardium-primed dendritic cells improve remodeling and cardiac function after myocardial infarction by modulating the regulatory T cell and macrophage polarization. Circulation. 2017;135(15):1444–1457. doi: 10.1161/CIRCULATIONAHA.116.023106. [DOI] [PubMed] [Google Scholar]
- 29.Hulsmans M., Sam F., Nahrendorf M. Monocyte and macrophage contributions to cardiac remodeling. Journal of Molecular and Cellular Cardiology. 2016;93:149–155. doi: 10.1016/j.yjmcc.2015.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rubinstein N., Alvarez M., Zwirner N. W., et al. Targeted inhibition of galectin-1 gene expression in tumor cells results in heightened T cell-mediated rejection; a potential mechanism of tumor-immune privilege. Cancer Cell. 2004;5(3):241–251. doi: 10.1016/S1535-6108(04)00024-8. [DOI] [PubMed] [Google Scholar]
- 31.Toscano M. A., Bianco G. A., Ilarregui J. M., et al. Differential glycosylation of TH1, TH2 and TH-17 effector cells selectively regulates susceptibility to cell death. Nature Immunology. 2007;8(8):825–834. doi: 10.1038/ni1482. [DOI] [PubMed] [Google Scholar]
- 32.Cheng X., Liao Y. H., Ge H., et al. TH1/TH2 functional imbalance after acute myocardial infarction: coronary arterial inflammation or myocardial inflammation. Journal of Clinical Immunology. 2005;25(3):246–253. doi: 10.1007/s10875-005-4088-0. [DOI] [PubMed] [Google Scholar]
- 33.Lluberas N., Trías N., Brugnini A., et al. Lymphocyte subpopulations in myocardial infarction: a comparison between peripheral and intracoronary blood. Springerplus. 2015;4(1):p. 744. doi: 10.1186/s40064-015-1532-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Methe H., Brunner S., Wiegand D., Nabauer M., Koglin J., Edelman E. R. Enhanced T-helper-1 lymphocyte activation patterns in acute coronary syndromes. Journal of the American College of Cardiology. 2005;45(12):1939–1945. doi: 10.1016/j.jacc.2005.03.040. [DOI] [PubMed] [Google Scholar]
- 35.Engelbertsen D., Andersson L., Ljungcrantz I., et al. T-helper 2 immunity is associated with reduced risk of myocardial infarction and stroke. Arteriosclerosis, Thrombosis, and Vascular Biology. 2013;33(3):637–644. doi: 10.1161/ATVBAHA.112.300871. [DOI] [PubMed] [Google Scholar]
- 36.Wang Y. P., Xie Y., Ma H., et al. Regulatory T lymphocytes in myocardial infarction: a promising new therapeutic target. International Journal of Cardiology. 2016;203:923–928. doi: 10.1016/j.ijcard.2015.11.078. [DOI] [PubMed] [Google Scholar]
- 37.Dalotto-Moreno T., Croci D. O., Cerliani J. P., et al. Targeting galectin-1 overcomes breast cancer-associated immunosuppression and prevents metastatic disease. Cancer Research. 2013;73(3):1107–1117. doi: 10.1158/0008-5472.CAN-12-2418. [DOI] [PubMed] [Google Scholar]
- 38.Carlos C. P., Silva A. A., Gil C. D., Oliani S. M. Pharmacological treatment with galectin-1 protects against renal ischaemia-reperfusion injury. Scientific Reports. 2018;8(1):p. 9568. doi: 10.1038/s41598-018-27907-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thijssen V. L., Barkan B., Shoji H., et al. Tumor cells secrete galectin-1 to enhance endothelial cell activity. Cancer Research. 2010;70(15):6216–6224. doi: 10.1158/0008-5472.CAN-09-4150. [DOI] [PubMed] [Google Scholar]
- 40.Libby P., Ridker P. M., Hansson G. K., Leducq Transatlantic Network on Atherothrombosis Inflammation in atherosclerosis: from pathophysiology to practice. Journal of the American College of Cardiology. 2009;54(23):2129–2138. doi: 10.1016/j.jacc.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ridker P. M., Everett B. M., Thuren T., et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. The New England Journal of Medicine. 2017;377(12):1119–1131. doi: 10.1056/NEJMoa1707914. [DOI] [PubMed] [Google Scholar]
- 42.Lee Y. J., Koh Y. S., Park H. E., et al. Spatial and temporal expression, and statin responsiveness of galectin-1 and galectin-3 in murine atherosclerosis. The Korean Circulation Journal. 2013;43(4):223–230. doi: 10.4070/kcj.2013.43.4.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.MacKinnon A. C., Liu X., Hadoke P. W., Miller M. R., Newby D. E., Sethi T. Inhibition of galectin-3 reduces atherosclerosis in apolipoprotein E-deficient mice. Glycobiology. 2013;23(6):654–663. doi: 10.1093/glycob/cwt006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lu Y., Zhang M., Zhao P., et al. Modified citrus pectin inhibits galectin-3 function to reduce atherosclerotic lesions in apoE-deficient mice. Molecular Medicine Reports. 2017;16(1):647–653. doi: 10.3892/mmr.2017.6646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Giordanengo L., Gea S., Barbieri G., Rabinovich G. A. Anti-galectin-1 autoantibodies in human Trypanosoma cruzi infection: differential expression of this β-galactoside-binding protein in cardiac Chagas’ disease. Clinical and Experimental Immunology. 2001;124(2):266–273. doi: 10.1046/j.1365-2249.2001.01512.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Parrillo J. E., Burch C., Shelhamer J. H., Parker M. M., Natanson C., Schuette W. A circulating myocardial depressant substance in humans with septic shock. Septic shock patients with a reduced ejection fraction have a circulating factor that depresses in vitro myocardial cell performance. The Journal of Clinical Investigation. 1985;76(4):1539–1553. doi: 10.1172/jci112135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Van Tassell B. W., Seropian I. M., Toldo S., Mezzaroma E., Abbate A. Interleukin-1β induces a reversible cardiomyopathy in the mouse. Inflammation Research. 2013;62(7):637–640. doi: 10.1007/s00011-013-0625-0. [DOI] [PubMed] [Google Scholar]
- 48.Toldo S., Mezzaroma E., O'Brien L., et al. Interleukin-18 mediates interleukin-1-induced cardiac dysfunction. American Journal of Physiology. Heart and Circulatory Physiology. 2014;306(7):H1025–H1031. doi: 10.1152/ajpheart.00795.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bern C. Chagas' Disease. The New England Journal of Medicine. 2015;373(5):456–466. doi: 10.1056/NEJMra1410150. [DOI] [PubMed] [Google Scholar]
- 50.Bocchi E. A., Bestetti R. B., Scanavacca M. I., Cunha Neto E., Issa V. S. Chronic Chagas heart disease management: From etiology to cardiomyopathy treatment. Journal of the American College of Cardiology. 2017;70(12):1510–1524. doi: 10.1016/j.jacc.2017.08.004. [DOI] [PubMed] [Google Scholar]
- 51.Medei E. H., Nascimento J. H. M., Pedrosa R. C., Carvalho A. C. C. d. Envolvimento de auto-anticorpos na fisiopatologia da doença de Chagas. Arquivos Brasileiros de Cardiologia. 2008;91(4):281–286. doi: 10.1590/S0066-782X2008001600012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Benatar A. F., García G. A., Bua J., et al. Galectin-1 prevents infection and damage induced by Trypanosoma cruzi on cardiac cells. PLoS Neglected Tropical Diseases. 2015;9(10, article e0004148) doi: 10.1371/journal.pntd.0004148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reifenberg K., Lehr H. A., Torzewski M., et al. Interferon-γ induces chronic active myocarditis and cardiomyopathy in transgenic mice. The American Journal of Pathology. 2007;171(2):463–472. doi: 10.2353/ajpath.2007.060906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Poncini C. V., Ilarregui J. M., Batalla E. I., et al. Trypanosoma cruzi infection imparts a regulatory program in dendritic cells and T cells via galectin-1-dependent mechanisms. Journal of Immunology. 2015;195(7):3311–3324. doi: 10.4049/jimmunol.1403019. [DOI] [PubMed] [Google Scholar]
- 55.Zuñiga E., Rabinovich G. A., Iglesias M. M., Gruppi A. Regulated expression of galectin-1 during B-cell activation and implications for T-cell apoptosis. Journal of Leukocyte Biology. 2001;70(1):73–79. [PubMed] [Google Scholar]
- 56.Stahl P., Ruppert V., Meyer T., et al. Trypomastigotes and amastigotes of Trypanosoma cruzi induce apoptosis and STAT3 activation in cardiomyocytes in vitro. Apoptosis. 2013;18(6):653–663. doi: 10.1007/s10495-013-0822-x. [DOI] [PubMed] [Google Scholar]
- 57.Rodriguez H. O., Guerrero N. A., Fortes A., Santi-Rocca J., Gironès N., Fresno M. Trypanosoma cruzi strains cause different myocarditis patterns in infected mice. Acta Tropica. 2014;139:57–66. doi: 10.1016/j.actatropica.2014.07.005. [DOI] [PubMed] [Google Scholar]
- 58.Galiè N., Humbert M., Vachiery J. L., et al. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: the Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT) The European Respiratory Journal. 2015;46(4):903–975. doi: 10.1183/13993003.01032-2015. [DOI] [PubMed] [Google Scholar]
- 59.Humbert M., Morrell N. W., Archer S. L., et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. Journal of the American College of Cardiology. 2004;43(12):S13–S24. doi: 10.1016/j.jacc.2004.02.029. [DOI] [PubMed] [Google Scholar]
- 60.Gao Y., Chen T., Raj J. U. Endothelial and smooth muscle cell interactions in the pathobiology of pulmonary hypertension. American Journal of Respiratory Cell and Molecular Biology. 2016;54(4):451–460. doi: 10.1165/rcmb.2015-0323TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Moiseeva E. P., Spring E. L., Baron J. H., de Bono D. P. Galectin-1 modulates attachment, spreading and migration of cultured vascular smooth muscle cells via interactions with cellular receptors and components of extracellular matrix. Journal of Vascular Research. 1999;36(1):47–58. doi: 10.1159/000025625. [DOI] [PubMed] [Google Scholar]
- 62.Miserocchi G., Passi A., Negrini D., Del Fabbro M., De Luca G. Pulmonary interstitial pressure and tissue matrix structure in acute hypoxia. American Journal of Physiology. Lung Cellular and Molecular Physiology. 2001;280(5):L881–L887. doi: 10.1152/ajplung.2001.280.5.L881. [DOI] [PubMed] [Google Scholar]
- 63.Tsai M. S., Chiang M. T., Tsai D. L., et al. Galectin-1 restricts vascular smooth muscle cell motility via modulating adhesion force and focal adhesion dynamics. Scientific Reports. 2018;8(1, article 11497) doi: 10.1038/s41598-018-29843-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Moiseeva E. P., Javed Q., Spring E. L., de Bono D. P. Galectin 1 is involved in vascular smooth muscle cell proliferation. Cardiovascular Research. 2000;45(2):493–502. doi: 10.1016/S0008-6363(99)00276-X. [DOI] [PubMed] [Google Scholar]
- 65.Case D., Irwin D., Ivester C., et al. Mice deficient in galectin-1 exhibit attenuated physiological responses to chronic hypoxia-induced pulmonary hypertension. American Journal of Physiology. Lung Cellular and Molecular Physiology. 2007;292(1):L154–L164. doi: 10.1152/ajplung.00192.2006. [DOI] [PubMed] [Google Scholar]
- 66.Wang J., Thio S. S. C., Yang S. S. H., et al. Splice variant specific modulation of CaV1.2 calcium channel by galectin-1 regulates arterial constriction. Circulation Research. 2011;109(11):1250–1258. doi: 10.1161/CIRCRESAHA.111.248849. [DOI] [PubMed] [Google Scholar]
- 67.Hu Z., Li G., Wang J. W., et al. Regulation of blood pressure by targeting CaV1.2-galectin-1 protein interaction. Circulation. 2018;138(14):1431–1445. doi: 10.1161/circulationaha.117.031231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Freitag N., Tirado-Gonzalez I., Barrientos G., et al. Interfering with Gal-1-mediated angiogenesis contributes to the pathogenesis of preeclampsia. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(28):11451–11456. doi: 10.1073/pnas.1303707110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Akazawa C., Nakamura Y., Sango K., Horie H., Kohsaka S. Distribution of the galectin-1 mRNA in the rat nervous system: its transient upregulation in rat facial motor neurons after facial nerve axotomy. Neuroscience. 2004;125(1):171–178. doi: 10.1016/j.neuroscience.2004.01.034. [DOI] [PubMed] [Google Scholar]
- 70.Sakaguchi M., Okano H. Neural stem cells, adult neurogenesis, and galectin-1: from bench to bedside. Developmental Neurobiology. 2012;72(7):1059–1067. doi: 10.1002/dneu.22023. [DOI] [PubMed] [Google Scholar]
- 71.Qu W. S., Wang Y. H., Wang J. P., et al. Galectin-1 enhances astrocytic BDNF production and improves functional outcome in rats following ischemia. Neurochemical Research. 2010;35(11):1716–1724. doi: 10.1007/s11064-010-0234-z. [DOI] [PubMed] [Google Scholar]
- 72.Ishibashi S., Kuroiwa T., Sakaguchi M., et al. Galectin-1 regulates neurogenesis in the subventricular zone and promotes functional recovery after stroke. Experimental Neurology. 2007;207(2):302–313. doi: 10.1016/j.expneurol.2007.06.024. [DOI] [PubMed] [Google Scholar]
- 73.Sasaki T., Hirabayashi J., Manya H., Kasai K., Endo T. Galectin-1 induces astrocyte differentiation, which leads to production of brain-derived neurotrophic factor. Glycobiology. 2004;14(4):357–363. doi: 10.1093/glycob/cwh043. [DOI] [PubMed] [Google Scholar]
- 74.Wang J., Xia J., Zhang F., et al. Galectin-1-secreting neural stem cells elicit long-term neuroprotection against ischemic brain injury. Scientific Reports. 2015;5(1):p. 9621. doi: 10.1038/srep09621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kadoya T., Horie H. Structural and functional studies of galectin-1: a novel axonal regeneration-promoting activity for oxidized galectin-1. Current Drug Targets. 2005;6(4):375–383. doi: 10.2174/1389450054022007. [DOI] [PubMed] [Google Scholar]
- 76.Kigerl K. A., Gensel J. C., Ankeny D. P., Alexander J. K., Donnelly D. J., Popovich P. G. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. The Journal of Neuroscience. 2009;29(43):13435–13444. doi: 10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lekishvili T., Hesketh S., Brazier M. W., Brown D. R. Mouse galectin-1 inhibits the toxicity of glutamate by modifying NR1 NMDA receptor expression. The European Journal of Neuroscience. 2006;24(11):3017–3025. doi: 10.1111/j.1460-9568.2006.05207.x. [DOI] [PubMed] [Google Scholar]
- 78.Tenne-Brown J., Puche A. C., Key B. Expression of galectin-1 in the mouse olfactory system. The International Journal of Developmental Biology. 1998;42(6):791–799. [PubMed] [Google Scholar]
- 79.McGraw J., Gaudet A. D., Oschipok L. W., et al. Altered primary afferent anatomy and reduced thermal sensitivity in mice lacking galectin-1. Pain. 2005;114(1):7–18. doi: 10.1016/j.pain.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 80.He X. W., Li W. L., Li C., et al. Serum levels of galectin-1, galectin-3, and galectin-9 are associated with large artery atherosclerotic stroke. Scientific Reports. 2017;7(1, article 40994) doi: 10.1038/srep40994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dong H., Wang Z. H., Zhang N., Liu S. D., Zhao J. J., Liu S. Y. Serum galectin-3 level, not galectin-1, is associated with the clinical feature and outcome in patients with acute ischemic stroke. Oncotarget. 2017;8(65):109752–109761. doi: 10.18632/oncotarget.18211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Liu F. T., Rabinovich G. A. Galectins: regulators of acute and chronic inflammation. Annals of the New York Academy of Sciences. 2010;1183(1):158–182. doi: 10.1111/j.1749-6632.2009.05131.x. [DOI] [PubMed] [Google Scholar]
- 83.Yu L., Ruifrok W. P. T., Meissner M., et al. Genetic and pharmacological inhibition of galectin-3 prevents cardiac remodeling by interfering with myocardial fibrogenesis. Circulation. Heart Failure. 2013;6(1):107–117. doi: 10.1161/CIRCHEARTFAILURE.112.971168. [DOI] [PubMed] [Google Scholar]
- 84.González G. E., Rhaleb N. E., D'Ambrosio M. A., et al. Cardiac-deleterious role of galectin-3 in chronic angiotensin II-induced hypertension. American Journal of Physiology-Heart and Circulatory Physiology. 2016;311(5):H1287–H1296. doi: 10.1152/ajpheart.00096.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Henderson N. C., Sethi T. The regulation of inflammation by galectin-3. Immunological Reviews. 2009;230(1):160–171. doi: 10.1111/j.1600-065X.2009.00794.x. [DOI] [PubMed] [Google Scholar]
- 86.Chow S. L., Maisel A. S., Anand I., et al. Role of biomarkers for the prevention, assessment, and management of heart failure: a scientific statement from the American Heart Association. Circulation. 2017;135(22):e1054–e1091. doi: 10.1161/cir.0000000000000490. [DOI] [PubMed] [Google Scholar]
- 87.Madrigal-Matute J., Lindholt J. S., Fernandez-Garcia C. E., et al. Galectin-3, a biomarker linking oxidative stress and inflammation with the clinical outcomes of patients with atherothrombosis. Journal of the American Heart Association. 2014;3(4) doi: 10.1161/jaha.114.000785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sturm A., Lensch M., André S., et al. Human galectin-2: novel inducer of T cell apoptosis with distinct profile of caspase activation. Journal of Immunology. 2004;173(6):3825–3837. doi: 10.4049/jimmunol.173.6.3825. [DOI] [PubMed] [Google Scholar]
- 89.Loser K., Sturm A., Voskort M., et al. Galectin-2 suppresses contact allergy by inducing apoptosis in activated CD8+ T cells. Journal of Immunology. 2009;182(9):5419–5429. doi: 10.4049/jimmunol.0802308. [DOI] [PubMed] [Google Scholar]
- 90.Yıldırım C., Vogel D. Y. S., Hollander M. R., et al. Galectin-2 induces a proinflammatory, anti-arteriogenic phenotype in monocytes and macrophages. PLoS One. 2015;10(4, article e0124347) doi: 10.1371/journal.pone.0124347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.van der Laan A. M., Schirmer S. H., de Vries M. R., et al. Galectin-2 expression is dependent on the rs 7291467 polymorphism and acts as an inhibitor of arteriogenesis. European Heart Journal. 2012;33(9):1076–1084. doi: 10.1093/eurheartj/ehr220. [DOI] [PubMed] [Google Scholar]
- 92.Ozaki K., Inoue K., Sato H., et al. Functional variation in LGALS2 confers risk of myocardial infarction and regulates lymphotoxin-alpha secretion in vitro. Nature. 2004;429(6987):72–75. doi: 10.1038/nature02502. [DOI] [PubMed] [Google Scholar]
- 93.Li W., Xu J., Wang X., et al. Lack of association between lymphotoxin-α, galectin-2 polymorphisms and coronary artery disease: a meta-analysis. Atherosclerosis. 2010;208(2):433–436. doi: 10.1016/j.atherosclerosis.2009.08.014. [DOI] [PubMed] [Google Scholar]
- 94.John S., Mishra R. Galectin-9: from cell biology to complex disease dynamics. Journal of Biosciences. 2016;41(3):507–534. doi: 10.1007/s12038-016-9616-y. [DOI] [PubMed] [Google Scholar]
- 95.Zhang Y., Zhang M., Li X., Tang Z., He L., Lv K. Expansion of CD11b+Ly-6C+ myeloid-derived suppressor cells (MDSCs) driven by galectin-9 attenuates CVB3-induced myocarditis. Molecular Immunology. 2017;83:62–71. doi: 10.1016/j.molimm.2017.01.013. [DOI] [PubMed] [Google Scholar]
- 96.Tao Y. F., Lin F., Yan X. Y., et al. Galectin-9 in combination with EX-527 prolongs the survival of cardiac allografts in mice after cardiac transplantation. Transplantation Proceedings. 2015;47(6):2003–2009. doi: 10.1016/j.transproceed.2015.04.091. [DOI] [PubMed] [Google Scholar]
- 97.Wan L., Yang R. Y., Liu F. T. Galectin-12 in cellular differentiation, apoptosis and polarization. International Journal of Molecular Sciences. 2018;19(1) doi: 10.3390/ijms19010176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Elola M. T., Chiesa M. E., Alberti A. F., Mordoh J., Fink N. E. Galectin-1 receptors in different cell types. Journal of Biomedical Science. 2005;12(1):13–29. doi: 10.1007/s11373-004-8169-5. [DOI] [PubMed] [Google Scholar]
- 99.Stillman B. N., Hsu D. K., Pang M., et al. Galectin-3 and galectin-1 bind distinct cell surface glycoprotein receptors to induce T cell death. Journal of Immunology. 2006;176(2):778–789. doi: 10.4049/jimmunol.176.2.778. [DOI] [PubMed] [Google Scholar]
- 100.Markowska A. I., Jefferies K. C., Panjwani N. Galectin-3 protein modulates cell surface expression and activation of vascular endothelial growth factor. Receptor 2 in human endothelial cells. The Journal of Biological Chemistry. 2011;286(34):29913–29921. doi: 10.1074/jbc.M111.226423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Davicino R. C., Méndez-Huergo S. P., Eliçabe R. J., et al. Galectin-1–driven tolerogenic programs aggravate Yersinia enterocolitica infection by repressing antibacterial immunity. Journal of Immunology. 2017;199(4):1382–1392. doi: 10.4049/jimmunol.1700579. [DOI] [PubMed] [Google Scholar]