

Abstract
Background
Gray leaf spot is a devastating disease caused by Stemphylium lycopersici that threatens tomato-growing areas worldwide. Typically, many pathogenesis-related and unrelated secreted proteins can be predicted in genomes using bioinformatics and computer-based prediction algorithms, which help to elucidate the molecular mechanisms of pathogen-plant interactions.
Results
S. lycopersici-secreted proteins were predicted from 8997 proteins using a set of internet-based programs, including SignalP v4.1 TMHMM v2.0, big-PI Fungal Predictor, ProtComp V9.0 and TargetP v1.1. Analysis showed that 511 proteins are predicted to be secreted. These proteins vary from 51 to 600 residues in length, with signal peptides ranging from 14 to 30 residues in length. Functional analysis of differentially expressed proteins was performed using Blast2GO. Gene ontology analysis of 305 proteins classified them into 8 groups in biological process (BP), 6 groups in molecular function (MF), and 10 groups in cellular component (CC). Pathogen-host interaction (PHI) partners were predicted by performing BLASTp analysis of the predicted secreted proteins against the PHI database. In total, 159 secreted proteins in S. lycopersici might be involved in pathogenicity and virulence pathways. Scanning S. lycopersici-secreted proteins for the presence of carbohydrate-active enzyme (CAZyme)-coding gene homologs resulted in the prediction of 259 proteins. In addition, 12 of the 511 proteins predicted to be secreted are small cysteine-rich proteins (SCRPs).
Conclusions
S. lycopersici secretory proteins have not yet been studied. The study of S. lycopersici genes predicted to encode secreted proteins is highly significant for research aimed at understanding the hypothesized roles of these proteins in host penetration, tissue necrosis, immune subversion and the identification of new targets for fungicides.
Electronic supplementary material
The online version of this article (10.1186/s12866-018-1329-y) contains supplementary material, which is available to authorized users.
Keywords: Tomato leaf gray spot disease, Signal peptide, Effector, Pathogen-host interaction, CAZymes, SCRSPs
Background
The fungus Stemphylium lycopersici is distributed worldwide and causes gray leaf spots on tomatoes and other crops, resulting in a great decrease in fruit quality and production. On tomato leaves, the disease first appears as circular to elongated dark specks. As the spots enlarge, they become gray or dark brown. Severely infected leaves turn yellow and then die and drop (Fig 1) [1–5].
Fig. 1.

Symptoms of S. lycopersici in field on tomato leave and fruit
Various plant pathogens, including fungi, oomycetes, bacteria, and nematodes, contains an ancient and conserved mechanism which secreted proteins and other molecules into cells of the hosts to colonize in plants or against the plant’s immune system [6–9]. Studies of the functions of these secreted proteins and molecules are critical for understanding the mechanisms of potential host colonization and pathogenicity [9]. Interactions between plants and pathogens involve the sensing and secretion of many signal molecules, elicitins and pathogenic factors that interact with plant receptor proteins [6, 7]. A number of avirulence (AVR) proteins, pathogenic factors, and hydrolases are secreted proteins [10–12]. Some elicitins, which function as AVR factors, are a family of structurally related extracellular proteins that induce hypersensitive cell death and other biochemical changes associated with defense responses in plants. For example, INF1, INF2A, INF2B, and Avr3a in potato late blight Phytophthora infestans [13, 14]; and Avr-Pita, AvrPiz-t and PWL2 in rice blast fungus caused by Magnaporthe oryzae [8, 15–17] exhibit pathogenic functions during pathogen infection. PsXEG1, a glycoside hydrolase 12 (GH12) from P. sojae, is a novel pathogen-associated molecular pattern (PAMP) [18].
The availability of public genomic sequencing data has significantly benefited the research community in the study of fungal genetics, fungal biology, gene function and plant pathology to investigate and control pathogen-host interactions (PHIs). Many scholars have used bioinformatics tools to predict secreted proteins from Aspergillus nidulans, Saccharomyces cerevisiae, Agrobacterium tumefaciens, Fusarium graminearum, M. oryzae, Puccinia helianthi, P. infestans, and Neurospora crassa [19–23]. However, S. lycopersici-secreted proteins have not yet been analyzed. In this study, internet-based tools, such as SignalP v4.1, Transmembrane Helices Hidden Markov Model (TMHMM) v2.0, big-PI Fungal Predictor, ProtComp V9.0 and TargetP v1.1, were used to predict typical S. lycopersici-secreted proteins, resulting in the identification of 511 proteins predicted to be secreted among 8997 S. lycopersici proteins. This research provides important information on the systematic analysis of the S. lycopersici elicitins and pathogenic factors to reveal the molecular mechanisms and interactions between S. lycopersici and its hosts.
Methods
Sequence data and preparation
The S. lycopersici proteome was obtained from the National Center for Biotechnology Information (NCBI) database: ftp://ftp.ncbi.nih.gov/genomes/genbank/fungi/Stemphylium_lycopersici/latest_assembly_versions/GCA_001191545.1_ASM119154v1/GCA_001191545.1_ASM119154v1_protein.faa.gz [24]. Secreted proteins were predicted from the N-terminal amino acid (aa) sequences of 8997 proteins.
Prediction of secreted proteins
The computational secretome should have the following characteristics: (a) an N-terminal signal peptide; (b) no transmembrane domains; (c) no glycosyl phosphatidyl inositol (GPI)-anchor site; and (d) no localization signal predicted to target the protein to the mitochondria or other intracellular organelles. Open reading frames (ORFs) fulfilling these four criteria were included in the set of secreted proteins that we defined as the computational secretome.
In this study, we used five internet-based programs that were selected for their applicability to high throughput analysis and their ability to predict secreted proteins in S. lycopersici (Table 1). We queried the S. lycopersici ORF set with SignalP v4.1 to identify N-terminal signal peptides, setting the default D-cutoff for SignalP-noTM networks at 0.45 and the D-cutoff for SignalP-TM networks at 0.5 [25]. Next, TMHMM v2.0 was used to predict transmembrane domains with the default parameters [26], and big-PI Fungal Predictor was used to identify potential GPI-anchor sites [20, 27, 28]. ProtComp V9.0 was used to predict the subcellular localization for fungal proteins [29], and TargetP v1.1 was used to identify secretory pathway signal peptide sequences. In this study, custom Python scripts were used to preprocess text, extract sequences from the S. lycopersici proteome for each step of prediction and compare sequences with the PHI database.
Table 1.
The bioinformatics tools adopted for the prediction of secreted proteins from S. lycopersici
| Prediction algorithms | Objects predicted | References |
|---|---|---|
| SignalP v4.1 | Signal peptides | http://www.cbs.dtu.dk/services/SignalP/ |
| TMHMM v2.0 | Transmembrane domains | http://www.cbs.dtu.dk/services/TMHMM/ |
| big-PI Fungal Predictor | GPI-anchor site | http://mendel.imp.ac.at/gpi/fungi_server.html |
| ProtComp v9.0 | sub-cellular localization | http://www.softberry.com/ |
| TargetP v1.1 | localization | http://www.cbs.dtu.dk/services/TargetP/ |
Functional annotation of secreted proteins
Gene ontology (GO) classification of the identified proteins was performed using the web-accessible Blast2GO v4.1 annotation system (https://www.blast2go.com/) [30]. The first step in Blast2Go is to search siminar sequences against the NCBI non-redundant (nr) database by Basic Local Alignment Search Tool protein/nucleotide (BLASTp/BLASTn) with an expectation value of 10− 3. Next, mapping and annotation were performed on Blast2GO using default parameters. By applying this methodology, the identified proteins will be divided into three main categories which are biological processes (BP), molecular functions (MF) and cellular components (CC) [30]. Briefly, the FASTA sequences of the secreted proteins were uploaded to Blast2GO, and analysis was performed in three steps as follows: 1) BLAST analysis to identify homologous sequences using a BLAST e-value cutoff of 10− 3; 2) mapping to retrieve GO terms; and 3) annotation of the sequence to select reliable functions using a GO weight of 50.
Prediction of pathogenicity-associated secreted proteins
PHI partners were identified by subjecting predicted secreted proteins to BLASTp against the PHI database (E-value: 10− 10) [31].
Fungal extracellular carbohydrate-active enzymes (CAZymes) help break down the components of the plant cell wall, such as complex carbohydrates, allowing fungi to access the host and facilitate infection. To identify S. lycopersici CAZymes involved in these processes, we applied the dbCAN2 (http://cys.bios.niu.edu/dbCAN2/) with an e-value cutoff of 10− 10 for secreted proteins [32].
Small cysteine-rich secreted proteins (SCRSPs) were predicted based on their expected sequence characteristics and typically consist of 20 to 200 aa residues with an N-terminal signal peptide and at least four cysteine residues. Secreted S. lycopersici proteins with these characteristics were identified as putative SCRSPs. Searches for conserved domains of SCRSPs were performed using an online tool Conserved Domain Database (CDD; e-value cutoff of 10− 3) (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) [33].
Results
Secreted protein prediction of the S. lycopersici proteome
A total of 1053 (11.70%) out of 8997 ORFs were predicted to be classical secreted proteins using SignalP v4.1. The number of transmembrane helices was predicted using TMHMM. Out of the 1053 total predicted secreted proteins, 860 have no predicted transmembrane domain (TMD), and 193 have at least one predicted transmembrane helix. Protcomp v9.0 was used to predict the subcellular localization of the 860 proteins, resulting in 554 extracellular proteins. The big-PI Predictor identified 528 proteins with no GPI modification sites and 26 proteins with one or more GPI-anchored sites. To further confirm that these predicted proteins were secreted from the cell, we performed subcellular localization predictions using TargetP-v1.1. Finally, TargetP v1.1 identified 511 proteins (5.68% of the proteome) that were selected as candidate secreted pathways with signal peptides (Fig 2, Additional file 1).
Fig. 2.

Methods used to predict secreted proteins in S. lycopersici
Characteristics of secreted proteins and their signal peptides
The 511 predicted secreted proteins ranged between 64 and 2260 aa in length, with most (415 proteins) being between 100 and 600 aa in length (81.21% of predicted secreted proteins, mean = 402 aa) (Fig 3).
Fig. 3.
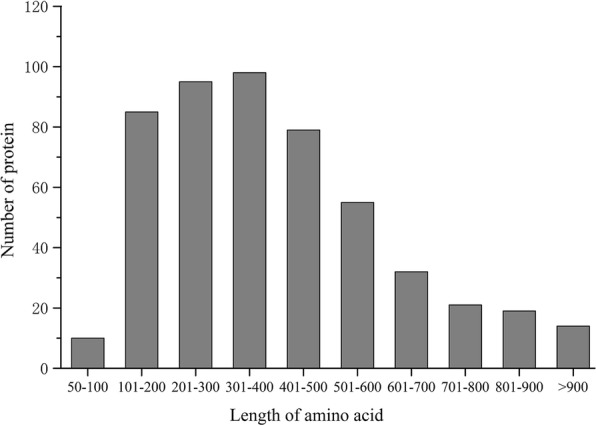
Analysis length of the predicted secreted proteins amino acid residues in S. lycopersici
The analysis of the signal peptides of the 511 predicted secreted proteins identified signal peptides ranging from 14 to 30 residues in length, with an average of 19 residues. The most common signal peptide length was 19 aa, accounting for 30% of the total (Fig 4).
Fig. 4.
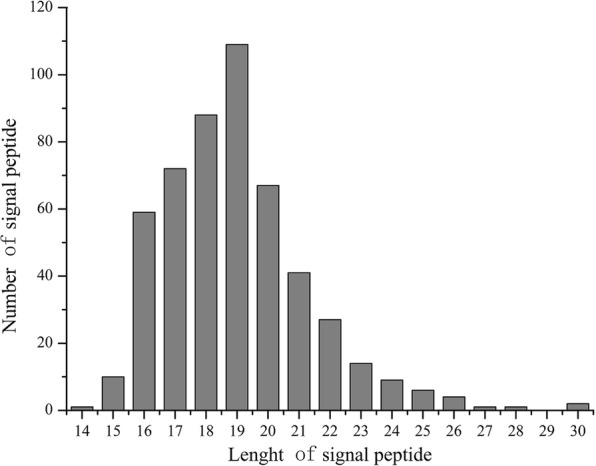
Analysis of the candidate secreted proteins with different length of signal peptide in S. lycopersici
The abundances of 20 aa in the signal peptides were analyzed, and the frequencies of these aa (in descending order) were A-L-S-T-V-M-F-I-G-P-R-K-Q-C-Y-H-N-W-E-D. Alanine (A) was the most abundant aa (22.71%) in the assayed signal peptides, followed by leucine (L) (18.29%), while aspartate (D) was the least common (0.22%). Statistical analysis of the aa in the signal peptides showed that nonpolar hydrophobic aa residues (A, L, V, I, G, and P) represented 57.08% of aa, polar noncharged aa residues (S, T, M, Q, C, and N) represented 27.85% of aa, positively charged aa residues (R, K, and H) represented 5.18% of aa, aromatic aa residues (W, F, and Y) represented 7.34% of aa, and negatively charged aa residues (D and E) represented only 0.56% aa (Fig 5).
Fig. 5.
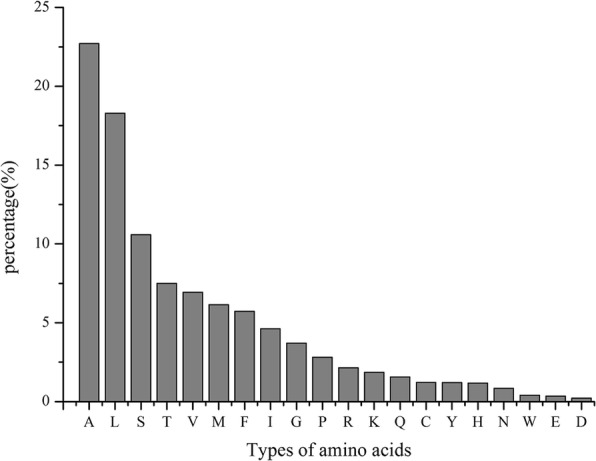
Percentage of 20 amino acid residues in S. lycopersici candidate secreted protein signal peptides
All aa in the signal peptide cleavage sites were analyzed using a custom Python2.7 script. We defined the three aa N-terminus to the cleavage sites as − 3, − 2, − 1 and the three aa C-terminus to the cleavage sites as + 1, + 2, + 3. The aa with the highest probability of being located at sites between − 3 and 3 were A, S, A, A, P, and T; the frequencies of these aa were 49.32, 20.35, 83.37, 24.07, 35.62, and 12.92%, respectively. An A-S-A motif was most likely to occupy the − 3 to − 1 site and accounted for 51 motifs observed at this site. An A-X-A motif, a typical signal peptidase I (SPase I) cleavage site [34, 35], accounted for 216 motifs (Table 2). SPase I is a serine protease with a catalytic dimer of serine lysine or serine histidine at the active site. The recognition site of SPase I for signal peptide cleavage is determined mainly by an A-X-A motif at the C-terminal of the signal peptide from the cleavage site [35].
Table 2.
Amino acids frequency and distribution in cleavage site of signal peptide of the predicted secreted proteins in S. lycopersici
| Kinds of | −3 | −2 | −1 | 1 | 2 | 3 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Amino acid | No. | pct.(%) | No. | pct.(%) | No. | pct.(%) | No. | pct.(%) | No. | pct.(%) | No. | pct.(%) |
| A | 252 | 49.32% | 72 | 14.09% | 426 | 83.37% | 123 | 24.07% | 29 | 5.68% | 44 | 8.61% |
| V | 133 | 26.03% | 21 | 4.11% | 0 | 0.00% | 20 | 3.91% | 30 | 5.87% | 47 | 9.20% |
| T | 48 | 9.39% | 35 | 6.85% | 6 | 1.17% | 28 | 5.48% | 42 | 8.22% | 66 | 12.92% |
| S | 33 | 6.46% | 104 | 20.35% | 28 | 5.48% | 47 | 9.20% | 34 | 6.65% | 49 | 9.59% |
| I | 20 | 3.91% | 15 | 2.94% | 0 | 0.00% | 18 | 3.52% | 8 | 1.57% | 48 | 9.39% |
| G | 11 | 2.15% | 6 | 1.17% | 38 | 7.44% | 23 | 4.50% | 21 | 4.11% | 17 | 3.33% |
| C | 7 | 1.37% | 10 | 1.96% | 5 | 0.98% | 4 | 0.78% | 5 | 0.98% | 11 | 2.15% |
| L | 5 | 0.98% | 92 | 18.00% | 0 | 0.00% | 36 | 7.05% | 22 | 4.31% | 51 | 9.98% |
| H | 1 | 0.20% | 16 | 3.13% | 0 | 0.00% | 42 | 8.22% | 7 | 1.37% | 14 | 2.74% |
| R | 1 | 0.20% | 6 | 1.17% | 0 | 0.00% | 17 | 3.33% | 5 | 0.98% | 13 | 2.54% |
| D | 0 | 0.00% | 5 | 0.98% | 0 | 0.00% | 7 | 1.37% | 29 | 5.68% | 17 | 3.33% |
| E | 0 | 0.00% | 15 | 2.94% | 0 | 0.00% | 9 | 1.76% | 13 | 2.54% | 6 | 1.17% |
| F | 0 | 0.00% | 33 | 6.46% | 0 | 0.00% | 15 | 2.94% | 9 | 1.76% | 25 | 4.89% |
| K | 0 | 0.00% | 1 | 0.20% | 0 | 0.00% | 10 | 1.96% | 5 | 0.98% | 6 | 1.17% |
| M | 0 | 0.00% | 12 | 2.35% | 0 | 0.00% | 7 | 1.37% | 8 | 1.57% | 6 | 1.17% |
| N | 0 | 0.00% | 21 | 4.11% | 0 | 0.00% | 14 | 2.74% | 26 | 5.09% | 18 | 3.52% |
| Q | 0 | 0.00% | 30 | 5.87% | 3 | 0.59% | 76 | 14.87% | 18 | 3.52% | 20 | 3.91% |
| W | 0 | 0.00% | 1 | 0.20% | 0 | 0.00% | 7 | 1.37% | 5 | 0.98% | 6 | 1.17% |
| Y | 0 | 0.00% | 16 | 3.13% | 0 | 0.00% | 8 | 1.57% | 13 | 2.54% | 10 | 1.96% |
| P | 0 | 0.00% | 0.00% | 5 | 0.98% | 0 | 0.00% | 182 | 35.62% | 37 | 7.24% | |
pct percentage, A alanine, C cysteine, D aspartic acid, E glutamic acid, F phenylalanine, G glycine, H histidine, I isoleucine, K lysine, L leucine, M methionine, N asparagine, P proline, Q glutamine, R arginine, S serine, T threonine, V valine, W tryptophan, Y tyrosine
The aa residues at the − 3 to + 3 sites were compared between S. lycopersici and other plant pathogens. Alanine exhibited the highest frequency of aa residues at the − 3, − 1 and + 1 sites, which are relatively conserved in most species. The S. lycopersici aa residues at − 3 to + 3 of the secreted proteins were exactly the same as those in two Pleosporaceae pathogens, Curvularia lunata and Cochliobolus heterostrophus (Table 3) [36]. These conserved residues are important for the recognition and cleavage of the signal peptide.
Table 3.
The highest frequency of amino acid residue around the cleavage site of signal peptide in S. lycopersici and other organisms [36]
| Organisms | −3 | −2 | −1 | + 1 | + 2 | + 3 |
|---|---|---|---|---|---|---|
| S. lycopersici | A | S | A | A | P | T |
| Curvularia lunata | A | S | A | A | P | T |
| Cochliobolus heterostrophus | A | S | A | A | P | T |
| Agrobacterium tumefaciens | A | L | A | A | D | L |
| Phytophthora infestans | A | A | A | A | A | A |
| Neurospora crassa | A | L | A | A | P | – |
| Verticillium dahliae | A | S | A | A | P | L |
| Ctenopharyngodon idellus | A | N | A | A | S | S/W |
| Caenorthaditis elegans | V | S | A | Q | P | I |
| Puccinia helianthi | V | S | A | Q | P | L |
-: No statistics
Annotation and classification of S. lycopersici-secreted proteins
Blast2GO is an all-in-one platform for high-quality protein functional prediction and and the genome-wide analysis of annotation data. Using Blast2GO, the 305 identified proteins were potentially classified by their 8 BP groups, 6 MF groups, and 10 CC groups. The functional annotation for the secreted proteins illustrated the following: 1) most representative biological processes—the following categories were highly represented: metabolic processes (GO: 0008152, 179), cellular processes (GO: 0009987, 56) and single-organism processes (GO: 0044699, 48); 2) most representative molecular functions—the following categories dominated: catalytic activity (GO: 0003824, 240) and binding activity (GO: 0005488, 72); and 3) most representative cellular components—the following categories were represented: extracellular region (GO:0005576, 72), cell (GO:0005623, 41) and cell part (GO:00444464, 40) (Fig 6, Additional file 2).
Fig. 6.

Gene ontology (GO) annotation of the predicted secreted proteins of S. lycopersici. The best 305 identified proteins hits were aligned to the GO database and assigned to GO term
Pathogenicity-associated secreted proteins
The identification of pathogenic-related genes is important to understand the mechanisms of PHIs. According to PHI-base catalogs, 4775 genes and 8610 interactions were predicted to be involved in pathogenicity. In our analysis, a search against PHI-base predicted 159 secreted proteins in S. lycopersici that may be involved in pathogenicity and virulence pathways. Of these proteins, 74 secreted proteins could be correlated with “pathogenicity”, 58 secreted proteins were predicted to “reduce virulence”, 13 secreted proteins were predicted to be an “effector”, 7 secreted proteins were predicted to result in “loss of pathogenicity”, and 7 secreted proteins were predicted to result in “increased virulence” (Fig 7, Additional file 3).
Fig. 7.

Predicted PHI proteins of the candidate secreted proteins of S. lycopersici
Scanning S. lycopersici-secreted proteins for the presence of CAZyme-coding gene homologs resulted in the prediction of a set of 259 sequences. The glycoside hydrolase (GH) superfamily was the most highly represented, containing 98 homologs distributed among 38 families. Glycosyl transferases (GT), polysaccharide lyases (PL), carbohydrate esterases (CE), carbohydrate-binding modules (CBM) and auxiliary activities (AA) superfamilies had 1, 22, 40, 42 and 56 homologs each, representing 1, 5, 8, 17 and 6 families, respectively (Table 4, Additional file 4). Comparing our data with those from two other tomato pathogens, Phytophthora parasitica and P. infestans, demonstrated the variation in CAZyme annotation. In addition to the GT, in terms of the numbers of GH, PL, CE, CBM, and AA CAZyme families, the ratios were quite similar.
Table 4.
A summary of the carbohydrate-active proteins predicted in S. lycopesici and comparison of these data with those from P. parasitica and P. infestans
| S. lycopesici | P. parasitica | P.infestans | ||||
|---|---|---|---|---|---|---|
| CAZyme families | CAZyme modules | CAZyme families | CAZyme modules | CAZyme families | CAZyme modules | |
| GH | 38 | 98 | 37 | 293 | 36 | 275 |
| GT | 1 | 1 | 29 | 169 | 30 | 158 |
| PL | 5 | 22 | 3 | 47 | 5 | 67 |
| CE | 8 | 40 | 12 | 113 | 12 | 90 |
| CBM | 17 | 42 | 14 | 85 | 16 | 74 |
| AA | 7 | 56 | 4 | 43 | 8 | 42 |
Twelve potential SCRSPs were predicted among the secreted proteins, ranging from 90 to 170 aa residues in size (Table 5). Of these SCRSPs, 8 were annotated in GenBank and had important functions in S. lycopersici. KNG49607, KNG48427 and KNG47745 were annotated as “hypothetical proteins”, and we observed that some of these SCRSPs had common in fungal extracellular membrane proteins (CFEM) domains or lysine motif (LysM) domains [33].
Table 5.
conserved domains search of the predicted small cysteine-rich secreted proteins (SCRSPs) against CDD database
| Gene code | Hit type | Length | Math site | E-Value | Accession | Description of SCRSP |
|---|---|---|---|---|---|---|
| KNG51615.1 | specific | 156 | 28–151 | 8.60E-49 | cd12798 | alt_a1 major allergen |
| KNG51272.1 | superfamily | 138 | 38–137 | 2.27E-54 | cl03937 | polysaccharide lyase family 3 protein |
| KNG49607.1 | specific | 90 | 21–79 | 0.000154 | pfam05730 | hypothetical protein TW65_03780 |
| KNG48427.1 | specific | 153 | 46–116 | 2.38E-05 | pfam05730 | hypothetical protein TW65_04690 |
| KNG46663.1 | specific | 143 | 42–139 | 4.84E-43 | cd00606 | guanyl-specific ribonuclease f1 |
| KNG46057.1 | superfamily | 170 | 73–155 | 4.97E-07 | cl09109 | snoal-like polyketide cyclase family protein |
| KNG45713.1 | specific | 147 | 6–128 | 5.00E-53 | COG1956 | gaf domain nucleotide-binding protein |
| KNG50949.1 | specific | 154 | 44–147 | 1.45E-36 | cd00448 | translation initiation inhibitor |
| KNG48201.1 | superfamily | 141 | 22–140 | 5.36E-65 | cl06331 | eliciting plant response protein |
| KNG47345.1 | superfamily | 160 | 24–153 | 5.19E-31 | cl17157 | major allergen alt |
| KNG47745.1 | specific | 36 | 51–86 | 1.75E-07 | cd00118 | hypothetical protein TW65_05484 |
| KNG46314.1 | specific | 101 | 29–89 | 1.04E-20 | pfam06766 | hypothetical protein TW65_86951 |
Discussion
In nature, plant pathogens have evolved quite distinct and specialised strategies for attacking plants. Many pathogens secrete a number of proteins to facilitate infection by interfering with host cellular functions and by inducing host responses [37–40]. It is of great importance to study the quantity, type and characteristics of secreted proteins in pathogens. Advances in genomic information have provided great opportunities to identify putative secreted proteins in different fungal species.
Based on the 8997 ORFs in the S. lycopersici protein database, 511 (5.68%) proteins were predicted to be secreted using a set of bioinformatics tools. These putative secretory proteins were small proteins, and most were proteins of 100 to 600 aa with signal peptides of 16 to 21 aa. The highly conserved signal peptide length distribution suggested that their function is mediated by small differences in the type and sequences of the aa residues.
The abundances of 20 aa in S. lycopersici signal peptides were highly similar to those reported in several other pathogenic fungi, including C. lunata, Verticillium dahliae, Saccharomyces cerevisiae, P. infestans, and A. tumefaciens [19, 21, 36, 41]. Three aa, A, L, and S, were highly represented in the signal peptides. Numerous hydrophobic aa were present in the signal peptides of the putative secreted proteins. This kind of motif may be related to the characteristics of secreted proteins that facilitate signal peptide transport across the membrane [35]. Four major classes of amino-terminal signal peptides can be distinguished on the basis of the SPase recognition sequence. This sequence can help transport proteins to different parts of the cell. Thus, the aa sequence of the cleavage site is essential for SPase recognition. In this study, these sequences included 216 proteins that have a potential signal peptide with a SPase I cleavage site with an A-X-A motif. SPase I, also known as the leader peptidase (Lep), is essential for cell viability, and SPase deficiency results in the accumulation of precursors of secreted proteins [42, 43]. Although the cleavage sites were conserved, the signal peptides were highly evolved. The analysis showed that all 511 signal peptides were not identical in the aa sequence (data not shown), suggesting that each signal peptide may have specific functions.
BLAST2GO is a bioinformatics platform for high-quality functional annotation and analysis of genomic datasets. This program allows for analysis and visualization of newly sequenced genomes by combining state-of-the-art methodologies, standard resources and algorithms [30]. The large number of observed “metabolic process” proteins indicated that these secreted proteins might participate in metabolic processes that include both biosynthetic and catabolic processes. “Catalytic activity” and “binding activity” were the most represented stress-responsive categories, thus indicating that metabolic adjustments may be involved in the PHI process.
PHI-base catalogs experimentally verified pathogenic, virulence and effector genes into a web-accessible database [7, 8, 31]. This database can be used to find novel pathogenic genes in important pathogens, which may be potential targets for fungicides [31]. We predicted 159 PHI-related proteins using BLASTp. Thirteen genes were annotated as an “effector” using BLASTp with PHI-base, and the “effector” was reportedly required for direct or indirect recognition of a pathogen only in the resistant host genotype, which possesses the corresponding disease resistance gene [44]. Some fungal effectors were identified that directly and specifically contributed to eliciting immune responses, perturbing host cellular processes and causing programmed cell death [13, 37, 39, 45].
Plant pathogens may initially use cell wall-degrading enzymes to digest the surface layers of cell walls to facilitate penetration [46, 47]. CAZymes, which are grouped into six functional classes (GH, GT, PL, CE, CBM and AA), are involved in the biosynthesis and degradation of glycoconjugates and oligo- and polysaccharides. In addition, CAZymes play a central role in the synthesis and breakdown of the plant cell wall [46, 48]. The results of the analysis of S. lycopersici classical secretory proteins showed that 259 secretory proteins are predicted as CAZymes, accounting for 50.68% of the total secreted proteins. The GH, PL, and CE superfamilies, which accounted for 31.31% of the total secreted proteins, are also known as cell-wall-degrading enzymes (CWDE) due to their role in the disintegration of the plant cell wall by bacterial and fungal pathogens. Given the complexity of carbohydrate biochemistry and the broad range of hydrolytic activities involved in this process, it is unsurprising that the examined genome exhibits a considerable number of GHs, which have extremely detailed enzyme entries in the database [48, 49]. S. lycopersici-secreted proteins were especially rich in family GH5 protein models (33 homologs), which act on β-linked oligo- and polysaccharides and glycoconjugates [32, 49]. Most of these CAZymes were unequivocally involved in the biochemical pathways aimed at maintaining fungal metabolism.
Fungal effector proteins are typically small in size. Hydrophobins, small and cysteine-rich hydrophobic proteins, assemble on the surface of hyphae and are required as effectors by pathogens that attach to hydrophobic surfaces [50]. SCRSPs are secreted directly into host plant cells and perform multiple biological functions, such as host recognition or colonization, hypersensitive response (HR) induction and pathogenicity. In this study, we predicted 12 SCRSPs from the S. lycopersici secretome. These SCRSPs contain CFEM domains, which typically contain eight cysteine residues, and are fungal-specific extracellular membrane proteins, such as Pth11p of M. grisea. Pth11p plays important roles in appressorium formation and fungal pathogenesis [24]. Therefore, hydrophobins or SCRSPs predicted in S. lycopersici also likely have key functions in pathogenesis and serve as important candidate proteins for the study of PHI mechanisms.
Conclusion
In conclusion, bioinformatics tools have been widely applied in molecular biology experiments, promoting the investigation and selection of genes or proteins of interest. Many bioinformatics tools are very efficient at predicting the secretion of proteins in fungi. With the development of next-generation sequencing technology, substantial amounts of plant pathogenic fungal, bacterial and other genomic data have been released. However, S. lycopersici secretory proteins have not yet been studied. The release of the S. lycopersici whole genome sequence provided some important data for studying the pathogenic factors of S. lycopersici. The study of S. lycopersici genes predicted to encode secreted proteins is highly significant for research aimed at understanding the potential roles of these proteins in host penetration, tissue necrosis, immune subversion and the identification of new targets for fungicides.
Additional files
Sequences of 511 predicted secreted proteins. (FASTA 249 kb)
Blast2go details. (XLSX 14 kb)
PHI details. (XLSX 14 kb)
CAZyme details. (XLSX 74 kb)
Acknowledgements
We would like to express our gratitude to Franco ME et al. for their jobs in “draft genome sequence and gene annotation of Stemphylium lycopersici”. We appreciate the help provided by Dr. Liao in data analysis.
Funding
This work, including the design of the study and collection, analysis, and interpretation of data and in writing the manuscript, was financially supported by The National Key Research and Development Program of China (Grant No. 2016YFD0201008), The Youth Talent Development Plan of Shanghai Municipal Agricultural System (Grant No. 20160119), SAAS Program for Excellent Research Team (Grant No. 2018(B-01)), and Natural Science Foundation of Shanghai (16ZR1424100).
Availability of data and materials
Operating System: Linux.
Programming Language: Python 2.7
Abbreviations
- AVR
Avirulence
- BP
Biological Process
- CAZymes
Carbohydrate-active Enzymes
- CC
Cellular Component
- CE
Carbohydrate Esterases
- CFEM
Common in Fungal Extracellular Membrane Proteins
- GH
Glycoside Hydrolase
- GO
Gene Ontology
- GPI
Glycosyl Phosphatidyl Inositol
- GT
Glycosyltransferases
- LysM
Lysine Motif
- MF
Molecular Function
- ORF
Open Reading Frame
- PAMPs
Pathogen-associated Molecular Patterns
- PHI
Pathogen-host Interaction
- PL
Polysaccharide Lyases
- SCRPs
Secreted Small Cysteine-rich Proteins
- SPase
Signal Peptidase
- TMD
Transmembrane Domain
Authors’ contributions
ZR, DFM, XLH and LX designed the study and wrote and revised the manuscript. ZR and GSG performed the tests and analyzed the results. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Rong Zeng, Email: zengrong@saas.sh.cn.
Shigang Gao, Email: shggao@163.com.
Lihui Xu, Email: xulihui@saas.sh.cn.
Xin Liu, Email: liuxinyesno@163.com.
Fuming Dai, Email: fumingdai@163.com.
References
- 1.Enjoji S. Two diseases of tomato (2) J Plant Prot. 1931;18:48–53. [Google Scholar]
- 2.Kurose D, Hoang LH, Furuya N, Takeshita M, Sato T, Tsushima S, Tsuchiya K. Pathogenicity of Stemphylium lycopersici isolated from rotted tobacco seeds on seedlings and leaves. J Gen Plant Pathol. 2014;80(2):147–152. doi: 10.1007/s10327-013-0501-3. [DOI] [Google Scholar]
- 3.Li XQ, Tian ZL, Wang H, Li HB, Zhou YF, Zheng JR. A pathogen causing grey leaf spot of tomato in Zhejiang Province. Mycosystema. 2015;35(9):1151–1156. [Google Scholar]
- 4.Al-Amri K, Al-Sadi AM, Al-Shihi A, Nasehi A, Al-Mahmooli I, Deadman ML. Population structure of Stemphylium lycopersici associated with leaf spot of tomato in a single field. Springerplus. 2016;5(1):1642. doi: 10.1186/s40064-016-3324-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nasehi A, Kadir J, Nasresfahani M, Abedashtiani F, Golkhandan E, Ashkani S. Identification of the new pathogen (Stemphylium lycopersici) causing leaf spot on Pepino (Solanum muricatum) J Phytopathol. 2016;164(6):421–426. doi: 10.1111/jph.12431. [DOI] [Google Scholar]
- 6.Abramovitch RB, Anderson JC, Martin GB. Bacterial elicitation and evasion of plant innate immunity. Nat Rev Mol Cell Biol. 2006;7(8):601–611. doi: 10.1038/nrm1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Birch PRJ, Rehmany AP, Pritchard L, Kamoun S, Beynon JL. Trafficking arms: Oomycete effectors enter host plant cells. Trends Microbiol. 2006;14(1):8–11. doi: 10.1016/j.tim.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 8.Kamoun S. A catalogue of the effector secretome of plant pathogenic oomycetes. Annu Rev Phytopathol. 2006;44(1):41–60. doi: 10.1146/annurev.phyto.44.070505.143436. [DOI] [PubMed] [Google Scholar]
- 9.Hogenhout SA, van der Hoorn RAL, Terauchi R, Kamoun S. Emerging concepts in effector biology of plant-associated organisms. Mol Plant-Microbe Interact. 2009;22(2):115. doi: 10.1094/MPMI-22-2-0115. [DOI] [PubMed] [Google Scholar]
- 10.Kelley BS, Lee SJ, Damasceno CMB, Chakravarthy S, Kim BD, Martin GB, Rose JKC. A secreted effector protein (SNE1) from Phytophthora infestans is a broadly acting suppressor of programmed cell death. Plant J. 2010;62(3):357–366. doi: 10.1111/j.1365-313X.2010.04160.x. [DOI] [PubMed] [Google Scholar]
- 11.Lee SA, Wormsley S, Kamoun S, Lee AFS, Joiner K, Wong B. An analysis of the Candida albicans genome database for soluble secreted proteins using computer-based prediction algorithms. Yeast. 2003;20(7):595–610. doi: 10.1002/yea.988. [DOI] [PubMed] [Google Scholar]
- 12.Khang CH, Park SY, Lee YH, Valent B, Kang S. Genome organization and evolution of the AVR-Pita avirulence gene family in the Magnaporthe grisea species complex. Mol Plant-Microbe Interact. 2008;21(5):658–670. doi: 10.1094/MPMI-21-5-0658. [DOI] [PubMed] [Google Scholar]
- 13.Kamoun S, Lindqvist H, Govers F. A novel class of elicitin-like genes from Phytophthora infestans. Mol Plant-Microbe Interact. 1997;10(8):1028–1030. doi: 10.1094/MPMI.1997.10.8.1028. [DOI] [PubMed] [Google Scholar]
- 14.Kamoun S, van West P, de Jong AJ, de Groot KE, Vleeshouwers VG, Govers F. A gene encoding a protein elicitor of Phytophthora infestans is down-regulated during infection of potato. Mol Plant-Microbe Interact. 1997;10(1):13–20. doi: 10.1094/MPMI.1997.10.1.13. [DOI] [PubMed] [Google Scholar]
- 15.Orbach MJ, Farrall L, Sweigard JA, Chumley FG, Valent B. A telomeric avirulence gene determines efficacy for the rice blast resistance gene pi-ta. Plant Cell. 2000;12(11):2019–2032. doi: 10.1105/tpc.12.11.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li W, Wang B, Wu J, Lu G, Hu Y, Zhang X, Zhang Z, Zhao Q, Feng Q, Zhang H, et al. The Magnaporthe oryzae avirulence gene AvrPiz-t encodes a predicted secreted protein that triggers the immunity in rice mediated by the blast resistance gene Piz-t. Mol Plant-Microbe Interact. 2009;22(4):411–420. doi: 10.1094/MPMI-22-4-0411. [DOI] [PubMed] [Google Scholar]
- 17.Sweigard JA, Carroll AM, Kang S, Valent B. Identification, cloning, and characterization of PWL2, a gene for host species specificity in the rice blast fungus. Plant Cell. 1995;7(8):1221–1223. doi: 10.1105/tpc.7.8.1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma Z, Zhu L, Song T, Wang Y, Zhang Q, Xia Y, Qiu M, Lin Y, Li H, Kong L, et al. A paralogous decoy protects Phytophthora sojae apoplastic effector PsXEG1 from a host inhibitor. Science. 2017;355(6326):710–714. doi: 10.1126/science.aai7919. [DOI] [PubMed] [Google Scholar]
- 19.Fan CM, Li CY, Zhao MF, He YQ. Analysis of signal peptides of the secreted proteins in Agrobacterium tumefaciens C58. Acta Microbiol Sin. 2005;45(4):561–566. [PubMed] [Google Scholar]
- 20.Eisenhaber B, Schneider G, Wildpaner M, Eisenhaber F. A sensitive predictor for potential GPI lipid modification sites in fungal protein sequences and its application to genome-wide studies for Aspergillus nidulans, Candida albicans, Neurospora crassa, Saccharomyces cerevisiae and Schizosaccharomyces pombe. J Mol Bio. 2004;337(2):243–253. doi: 10.1016/j.jmb.2004.01.025. [DOI] [PubMed] [Google Scholar]
- 21.Sun HC, Yang F, Xu JM, Yuan WA. Analysis of the secreted proteins in genome of signal peptides in grass carp Ctenopharyngodon idellus. Fish Sci. 2011;30(3):164–167. [Google Scholar]
- 22.Wu HZ, Li CY, Zhu YY, Bi YF. Computational analysis of signal peptide-dependent secreted protein in Caenorthaditis elegans ws123. Hereditas. 2006;28(4):470–478. [PubMed] [Google Scholar]
- 23.Zhou XG, Hou SM, Chen DW, Tao N, Sun YMDML, Zhang SS. Genome-wide analysis of the secreted proteins of Phytophthora infestans. Hereditas. 2011;33(7):785–793. doi: 10.3724/SP.J.1005.2011.00785. [DOI] [PubMed] [Google Scholar]
- 24.DeZwaan TM, Carroll AM, Valent B, Sweigard JA. Magnaporthe grisea pth11p is a novel plasma membrane protein that mediates appressorium differentiation in response to inductive substrate cues. Plant Cell. 1999;11(10):2013–2030. doi: 10.1105/tpc.11.10.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Petersen TN, Brunak S, Von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8(10):785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 26.Krogh A, Larsson B, Heijne G, Sonnhammer ELL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305(3):567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 27.Eisenhaber B, Bork P, Eisenhaber F. Post-translational GPI lipid anchor modification of proteins in kingdoms of life: analysis of protein sequence data from complete genomes. Protein Eng. 2001;14(1):17–25. doi: 10.1093/protein/14.1.17. [DOI] [PubMed] [Google Scholar]
- 28.Eisenhaber B, Bork P, Eisenhaber F. Prediction of potential GPI-modification sites in proprotein sequences. J Mol Biol. 1999;292(3):741–758. doi: 10.1006/jmbi.1999.3069. [DOI] [PubMed] [Google Scholar]
- 29.Emanuelssona O, Nielsenb H, Brunakb S, Heijne G. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J Mol Biol. 2000;300(4):1005. doi: 10.1006/jmbi.2000.3903. [DOI] [PubMed] [Google Scholar]
- 30.Götz S, García-Gómez J, Terol J, Williams T, Nagaraj S, Nueda M, Robles M, Talón M, Dopazo J, Conesa A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008;36(10):3420–3435. doi: 10.1093/nar/gkn176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Winnenburg R, Baldwin TK, Urban M, Rawlings C, Köhler J, Hammond-Kosack KE. PHI-base: a new database for pathogen host interactions. Nucleic Acids Res. 2006;34(Database issue):459–464. doi: 10.1093/nar/gkj047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Han, Yohe Tanner, Huang Le, Entwistle Sarah, Wu Peizhi, Yang Zhenglu, Busk Peter K, Xu Ying, Yin Yanbin. dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Research. 2018;46(W1):W95–W101. doi: 10.1093/nar/gky418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, de Weese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, et al. CDD: a conserved domain database for the functional annotation of proteins. Nucleic Acids Res. 2011;39(Database issue):225–229. doi: 10.1093/nar/gkq1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tjalsma H, Bolhuis A, Jongbloed JDH, Bron S, van Dijl JM. Signal peptide-dependent protein transport in Bacillus subtilis: a genome-based survey of the secretome. Microbiol Mol Biol Rev. 2000;64(3):515–517. doi: 10.1128/MMBR.64.3.515-547.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zalucki YM, Jennings MP. Signal peptidase I processed secretory signal sequences: selection for and against specific amino acids at the second position of mature protein. Biochem Biophys Res Commun. 2017;483(3):972–977. doi: 10.1016/j.bbrc.2017.01.044. [DOI] [PubMed] [Google Scholar]
- 36.Gao J, Gao S, Li Y, Chen J. Genome-wide prediction and analysis of the classical secreted proteins of Curvularia lunata. J Plant Protection. 2015;42(6):869–876. [Google Scholar]
- 37.Chen SB, Songkumarn P, Venu RC, Gowda M, Bellizzi M, Hu J, Liu W, Ebbole D, Meyers B, Mitchell T, et al. Identification and characterization of in planta–expressed secreted effector proteins from Magnaporthe oryzae that induce cell death in Rice. Mol Plant-Microbe Interact. 2013;26(2):191–202. doi: 10.1094/MPMI-05-12-0117-R. [DOI] [PubMed] [Google Scholar]
- 38.Sharpee W, Oh Y, Yi M, Franck W, Eyre A, Okagaki LH, Valent B, Dean RA. Identification and characterization of suppressors of plant cell death (SPD) effectors from Magnaporthe oryzae. Mol Plant Pathol. 2016;18(6):850–863. doi: 10.1111/mpp.12449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Redkar A, Hoser R, Schilling L, Zechmann B, Krzymowsk M, Walbot V, Doehlemann G. A secreted effector protein of Ustilago maydis guides maize leaf cells to form tumors. Plant Cell. 2015;27(4):1332–1351. doi: 10.1105/tpc.114.131086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu H, Zhang S, Schell MA, Denny TP. Pyramiding unmarked deletions in Ralstonia solanacearum shows that secreted proteins in addition to plant cell-wall-degrading enzymes contribute to virulence. Mol Plant-Microbe Interact. 2005;18(12):1296–1305. doi: 10.1094/MPMI-18-1296. [DOI] [PubMed] [Google Scholar]
- 41.Tian L, Chen JY, Chen XY, Wang JN, Dai XF. Prediction and analysis of Verticillium dahliae VdLs.17 Secretome. Scientia Agricultura Sinica. 2011;44(15):3142–3153. [Google Scholar]
- 42.Date T. Demonstration by a novel genetic technique that leader peptidase is an essential enzyme of Escherichia coli. J Bacteriol. 1983;154(1):76–83. doi: 10.1128/jb.154.1.76-83.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Luo J, Walsh E, Naik A, Zhuang W, Zhang K, Cai L, Zhang N. Temperate pine barrens and tropical rain forests are both rich in undescribed fungi. PLoS One. 2014;9(7):e103753. doi: 10.1371/journal.pone.0103753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stergiopoulos I, de Wit PJGM. Fungal effector proteins. Annu Rev Phytopathol. 2009;47(1):233–263. doi: 10.1146/annurev.phyto.112408.132637. [DOI] [PubMed] [Google Scholar]
- 45.Xiang J, Li X, Yin L, Liu Y, Zhang Y, Qu J, Lu J. A candidate RxLR effector from Plasmopara viticola can elicit immune responses in Nicotiana benthamiana. BMC Plant Bio. 2017;17(1):75. doi: 10.1186/s12870-017-1016-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ospina-Giraldo MD, Griffith JG, Laird EW, Mingora C. The CAZyome of Phytophthora spp.: a comprehensive analysis of the gene complement coding for carbohydrate-active enzymes in species of the genus Phytophthora. BMC Genomics. 2010;11:525. doi: 10.1186/1471-2164-11-525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brouwer H, Coutinho PM, Henrissat B, RPd V. Carbohydrate-related enzymes of important Phytophthora plant pathogens. Fungal Genet Biol. 2014;72:192–200. doi: 10.1016/j.fgb.2014.08.011. [DOI] [PubMed] [Google Scholar]
- 48.Battaglia E, Benoit I, van den Brink J, Wiebenga A, Coutinho PM, Henrissat B, de Vries RP. Carbohydrate-active enzymes from the zygomycete fungus Rhizopus oryzae: a highly specialized approach to carbohydrate degradation depicted at genome level. BMC Genomics. 2011;12:38. doi: 10.1186/1471-2164-12-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aspeborg H, Coutinho PM, Wang Y, Brumer H, Henrissat B. Evolution, substrate specificity and subfamily classification of glycoside hydrolase family 5 (GH5) BMC Evol Biol. 2012;12:186. doi: 10.1186/1471-2148-12-186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Elliot MA, Talbot NJ. Building filaments in the air: aerial morphogenesis in bacteria and fungi. Curr Opin Microbiol. 2004;7(6):594–601. doi: 10.1016/j.mib.2004.10.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sequences of 511 predicted secreted proteins. (FASTA 249 kb)
Blast2go details. (XLSX 14 kb)
PHI details. (XLSX 14 kb)
CAZyme details. (XLSX 74 kb)
Data Availability Statement
Operating System: Linux.
Programming Language: Python 2.7