

Abstract
Genetic conditions, even those associated with identical gene mutations, can present with variable clinical manifestations. One widely accepted explanation for this phenomenon is the existence of genetic factors capable of modifying the consequences of disease-causing mutations (modifier genes). Here, we address the concepts and principles by which genetic factors may be involved in modifying risk for cardiac arrhythmia, then discuss the current knowledge and interpretation of their contribution to clinical heterogeneity. We illustrate these concepts in the context of two important clinical conditions associated with risk for sudden cardiac death including a monogenic disorder (congenital long QT syndrome) in which the impact of modifier genes has been established, and a complex trait (life-threatening arrhythmias in acute myocardial infarction) for which the search for genetic modifiers of arrhythmic risk is more challenging. Advances in understanding the contribution of modifier genes to a higher or lower propensity towards sudden death should improve patient-specific risk stratification and be a major step towards precision medicine.
Keywords: Genetics , Long QT syndrome , Acute myocardial infarction , Genetic variants , Genetic modifiers
Introduction
‘Why is it that within one family, among members sharing the same disease-causing mutation, some suffer cardiac arrest and die suddenly while others go through life without any symptoms?’. This is one of the most intriguing questions faced by any experienced cardiologist managing patients affected by genetic disorders associated with life-threatening cardiac arrhythmias. Given an identical, disease-causing mutation one would expect a similar clinical course, but reality is often different. While it is probably true that different behaviours or environments may impact on the development of a phenotype, most current efforts to address this question are focused on the role of modifier genes. This term is used to describe genetic factors capable of modifying in either direction the consequences of disease-causing mutations.
The search for modifier genes is challenging because not all diseases are equally suitable for a successful investigation. It is much more difficult to identify a modifier in a complex genetic trait than in a monogenic disorder as illustrated by the different progress made in the congenital long QT syndrome (LQTS)1 vs. in ischaemic heart disease. Indeed, the incomplete penetrance and variable expressivity of LQTS2 associated with congenital arrhythmia susceptibility has contributed to the emerging concept that even Mendelian disorders can have genetic complexity. Unravelling the underlying causes for this variability in disease expression may offer new ways to stratify and mitigate sudden cardiac death (SCD) risk in genetically susceptible hosts.
The genetic basis of SCD in general terms has been reviewed previously.3,4 Our objective here is to review the current knowledge about modifier genes related or potentially related to SCD including their mechanism(s) of action where known, and their impact on cardiovascular diseases associated with life-threatening arrhythmias with emphasis on their value for risk stratification and clinical management.5 We will focus particularly on the areas where progress has been made or is being meaningfully attempted, namely LQTS and ischaemic heart disease, exemplified by acute myocardial infarction (AMI).
Concepts and principles of modifier genes
Clinical heterogeneity is a common feature of many monogenic disorders including those associated with congenital arrhythmia susceptibility. In some families segregating a monogenic disorder, members that share an identical primary disease-causing mutation may have no evidence of the disease or have clinical presentations of varying severity. These phenomena are referred to as incomplete penetrance and variable disease expressivity.2 Penetrance refers to the proportion of people with a specific mutation who exhibit signs and symptoms of a genetic disease. Incomplete penetrance is said to exist when some people with the mutation do not develop the disorder. Variable expressivity refers to the diversity of signs and symptoms, including severity, that occur in different people with the same genetic disease. Incomplete penetrance and variable expressivity may result from combinations of known and unknown genetic, environmental and lifestyle factors. These phenomena confound interpretation of a discovered genotype and make prediction of an individual’s genetic risk more challenging.
The concept of modifier genes has been put forth to partially explain incomplete penetrance and variable expressivity of monogenic disorders. Modifier genes may promote higher risk for symptomatic disease and/or more severe disease expression, or might protect an individual from developing the disease. Furthermore, different genetic variants within the same gene might confer opposite effects. Genetic factors that can impact arrhythmia risk may include genes encoding determinants of the primary cellular substrate for abnormal cardiomyocyte excitability such as proteins contributing to the balance of inward and outward currents operating during the cardiac action potential, those governing intracellular calcium cycling or involved with the trafficking of cellular and membrane proteins. Some candidate modifier genes are themselves monogenic arrhythmia susceptibility genes,5 such as cardiac ion channel subunits or regulatory proteins.
Inter-individual differences in the propensity for arrhythmia triggers may also have a genetic basis and certain genes involved are candidate modifiers. The balance between sympathetic and parasympathetic tone as well as autonomic reflexes that are crucial to determining the onset of life-threatening arrhythmias, and the magnitude of catecholamine responses to stress and exercise varies among individuals. Some of this variability may have a genetic basis, and genes that participate in these autonomic responses are logical candidate modifiers of arrhythmia susceptibility.
Genetic variation with the potential for modifying arrhythmia susceptibility may occur within the coding or noncoding regions of a gene (intragenic) or fall within the vast genomic landscape between genes (intergenic). Further, genetic variability occurs in many forms from single nucleotide and copy number variants to larger chromosomal disruptions. Genetic modifiers may exist in populations at widely different frequency from ultra-rare (e.g. less than 1 in 20 000 persons) to common (e.g. greater than 1% of a population). Genetic variants that modify arrhythmia predisposition could have any population allele frequency. As a general rule, rare variants including disease-causing mutations have greater impact (e.g. effect size) on disease expression than common variants. However, the probability and feasibility of identifying common variants as disease modifiers is much greater than that for rare or ultra-rare variants.
Genetic modifiers of long QT syndrome
The congenital LQTS offers special opportunities to investigate the contributions of modifier genes to genotype–phenotype correlation for several reasons. With a prevalence of approximately one in 2000, LQTS is uncommon but not rare.6 The diagnosis of LQTS is relatively straightforward.1 The disorder is clinically characterized by two easily determined features, the QT interval and occurrence of cardiac events (syncope, cardiac arrest, or sudden death), which represent hard endpoints useful for assessing the impact of modifiers.1 In 4–8% of LQTS cases, more than one primary disease-causing mutation help explain exaggerated disease severity compared with other family members carrying single alleles,7,8 but other explanations are needed to understand why some mutation-positive subjects have less arrhythmia risk. One approach used to identify genetic modifiers of LQTS has focused on candidate genes that fall into two categories: (i) genes and genomic variants that modify the underlying arrhythmogenic substrate including genes associated with variability of QT interval in healthy populations and (ii) genetic factors that affect the probability of arrhythmia-triggering events. By design, candidate gene approaches are biased by currently available knowledge and specific hypotheses. In contrast, genome-wide association studies (GWAS) are best for discovery of previously unsuspected genetic markers of disease risk without the bias of prior knowledge. Unbiased approaches using combinations of genomic and physiological investigations have also emerged recently as successful strategies.9Table 1 summarizes the known genetic modifiers of LQTS according to their deleterious or protective effect.
Table 1.
List of SNPs regarded as genetic modifiers for the long QT syndrome
| SNP | Gene | Localization | Nucleotide Substitution | Modifier Allele | Effect of the Modifier Allele | References |
|---|---|---|---|---|---|---|
| Detrimental SNPs | ||||||
| rs4657139 | NOS1AP | Intronic | A > T | A | ↑ QT ↑ ACA, SD | 37,45 |
| rs16847548 | NOS1AP | Intronic | T > C | C | ↑ QT ↑ CA, SCD↑ risk of cardiac events | 23,37,45 |
| rs10494366 | NOS1AP | Intronic | T>G | G | ↑cardiac events | 45 |
| rs12143842 | NOS1AP | Intronic | C>T | T | ↑ QT | 23 |
| rs2880058 | NOS1AP | Intronic | A>G | G | ↑ QT | 23 |
| rs2519184 | KCNQ1 | 3′ UTR | G>A | A | Inconsistently associated with ↑ QT and↑ risk of cardiac events | 25–28 |
| rs8234 | KCNQ1 | 3′ UTR | A>G | G | Inconsistently associated with ↑ QT and↑ risk of cardiac events | 25–28 |
| rs10798 | KCNQ1 | 3′ UTR | A>G | G | Inconsistently associated with ↑ QT and↑ risk of cardiac events | 25–28 |
| rs1805123 | KCNH2 | Exonic (c.269A>C: p.Lys897Thr) | A > C | C | ↑ symptoms in LQT2↑ IKr current reduction↑ QT in LQT1-G589D founder population | 10–12 |
| rs1805128 | KCNE1 | Exonic (c.253G>A: p.Asp85Asn) | G > A | A | ↑ QT↑ risk of cardiac events | 18,23,20–22 |
| rs11772585 | AKAP9 | Intronic | C > T | T | ↑ risk of cardiac events | 39 |
| rs7808587 | AKAP9 | Intronic | A > G | G | ↑ risk of cardiac events | 39 |
| rs2961024 | AKAP9 | Intronic | T > G | G | ↑ QT | 39 |
| rs8014119 | REM2 | Exonic (Gly96Ala) | G > C | C | ↑ QT ↑ risk of cardiac events | 9 |
| Protective SNPs | ||||||
| rs2282972 | AKAP9 | Intronic | C > T | T | ↓ risk of cardiac events | 39 |
| rs2074238 | KCNQ1 | Intronic | C > T | T | ↓ QT ↓ symptoms | 23,41 |
| rs1805124 | SCN5A | Exonic (c.1673A>G: p.His558Arg) | A > G | G | Rescue normal phenotype in LQT3 | 13,14 |
| rs10947804 | KCNK17 | Exonic (Ser21Gly) | T > C | C | ↓ QT ↓ symptoms | 9 |
The table shows for each SNP the corresponding gene, the alternative alleles (the minor allele in the general population, according to NCBI-dbSNP, is reported as second), the modifier allele and the corresponding associated effect. The references in the last column refer to the published studies describing the modifier effect of the SNPs.
Common non-synonymous coding variants in the same gene as the primary mutation can act as intragenic modifiers. For example, we reported a family segregating a moderate loss-of-function mutation in KCNH2 (p.A1116V) associated with a latent form of LQTS.10 Whereas most family members with this mutation were asymptomatic, a 44-year-old woman had cardiac arrest. In addition to the primary KCNH2 mutation, she also carried a common nonsynonymous coding variant resulting in the amino acid substitution p.Lys897Thr (K897T) on the opposite KCNH2 allele. The heterologous co-expression of the primary mutation with K897T in an in vitro expression assay demonstrated a markedly lower level of rapid delayed rectifier potassium current (IKr) consistent with LQTS pathogenesis. A similar observation was made for another KCNH2 mutation (c.2775insG; p.P926AfsX14) discovered in a woman with asymptomatic LQTS who was evaluated for early postnatal death of a child and a separate occurrence of intrauterine foetal demise.11 The deceased offspring both inherited the primary KCNH2 mutation in combination with the K897T variant transmitted from the father. KCNH2-K897T also aggravates LQTS associated with KCNQ1 mutation. In a group of Finnish LQTS cases caused by mutation KCNQ1-G589D, the presence of KCNH2-K897T was associated with a longer QT interval during maximal exercise.12 A common variant in the cardiac sodium channel gene (SCN5A-H558R) may also impact the severity of LQTS when present on the same allele as a primary mutation.13,14 The latter observations led to the discovery of intersubunit interactions in voltage-gated sodium channels.15
The aforementioned anecdotal observations suggest that KCNH2-K897T may impair repolarization reserve in the setting of LQT1 or LQT2 mutations. The impact of this common variant in the general population may be more complex as evidence by three reports demonstrating an association of this variant with shorter QT intervals in European subjects16–18 and one report showing association with longer QT intervals in the Framingham study population.19
Common variants in genes encoding ion channel modulators or auxiliary subunits have also been implicated as genetic modifiers of arrhythmia syndromes. In particular, a common nonsynonymous coding variant in KCNE1 (p.Asp85Asn, also known as D85N) encoding a regulatory subunit of the KCNQ1-encoded potassium channel has been demonstrated to predispose to both congenital and drug-induced LQTS.20,21 This variant has also been associated with variation in QT interval duration among healthy individuals.18 In Finnish LQTS families segregating the founder mutation KCNQ1-p.Gly589Asp, KCNE1-p.Asp85Asn was associated with greater QT interval prolongation in males but not females suggesting that it acts as a sex-specific genetic modifier.22 Similarly, KCNE1-p.Asp85Asn was nominally associated with longer QT intervals and higher probability of cardiac events in cases of type 2 LQTS caused by KCNH2 mutations.23 These observations can be explained by impaired repolarizing currents (IKs, IKr) when this KCNE1 variant interacts with KCNQ1 or HERG channels.20 The clinical counterpart is that, when in asymptomatic individuals presenting exclusively with borderline QT prolongation only this variant is found, we do not make diagnosis of LQTS but believe that avoidance of IKr blocking drugs should be strongly considered.
Interindividual variability in the level of LQTS gene expression may also contribute to variable impact of the mutant allele. Specifically, variants affecting transcriptional control or mRNA stability of the normal allele may partially offset the contribution of a mutation by changing the ratio of wild-type and mutant proteins. Many autosomal dominant LQTS-associated mutations, especially those in multimeric potassium channel genes, are known to exert dominant-negative effects on the wild-type subunit.24 Hence, the balance between transcription or translation of wild-type and mutant alleles may modify the probability of forming dysfunctional ion channel complexes.
This intriguing concept has been explored for variants within the 3′-untranslated region (3′-UTR) of the KCNQ1 gene. It is widely known that the 3′-UTR of genes may contain noncoding elements important for mRNA stability and translation. An international collaborative study examined the impact of three common KCNQ1 variants affecting nucleotides within the 3′-UTR on QTc duration and symptoms in several LQT1 families.25 The investigation sought to determine if 3′-UTR variants were associated with opposite effects on arrhythmic risk and QT interval duration depending on whether they were in cis or trans to the mutant allele. In vitro analyses demonstrated that variant 3′-UTR haplotypes promoted lower expression of a coupled reporter gene (luciferase). Genetic evaluation documented significant associations with longer QT intervals when 3′-UTR variants were in trans with the wild type KCNQ1 allele, which suggested a lower ratio of wild type to mutant KCNQ1 subunits. Extrapolation to the general population did not reveal that 3′-UTR variants in KCNQ1 impact the QT interval suggesting that this effect may only be relevant in the context of heterozygous KCNQ1 mutation. Attempts to replicate these findings in three LQT1 founder populations failed26 possibly because of differences in genetic architecture of the 3′-UTR, different statistical approaches or the nature of the primary mutations.27,28 Additional investigations are needed to determine the robustness of these findings.
Discovery of genetic modifiers in long QT syndrome founder populations
Even though serendipitous observations within a single family may provide important clues regarding the identity of genetic modifiers, as it was the case for the KCNH2-K897T variant discussed above,10 a structured approach exploiting founder populations29–33 has offered additional opportunities for modifier gene discovery. Given that genetic modifiers have quantitative effects smaller than that of a disease-causing mutation, their discovery is facilitated by avoiding the confounding effect of different mutations (each of which would have its own and variable impact) and by examining instead a large numbers of individuals carrying the same mutation. An approach is the study of members of founder populations, which are characterized by a large number of individuals and families related to a single ancestor affected by the disease and who thereby carry the same disease-causing mutation.30 Founder populations are not found everywhere in the world; there are countries or selected areas where they are more easily encountered, and this almost always relates to specific historic or geographic aspects.33 For example, founder populations are more prevalent in South Africa,29,30 in Finland,31 Sweden,32 Quebec,34 and the Netherlands.35
We previously investigated a large LQT1 cohort comprised of approximately 500 members of whom approximately 200 are heterozygous for the KCNQ1-A341V mutation. An ancestor of this founder population moved from the Netherlands to South Africa in A.D. 1690 and genetic mapping studies of several LQT1 families in the region eventually connected an extended pedigree.29KCNQ1-A341V confers a highly penetrant, clinically severe mutation in the South African cohort and in other mutation-positive families throughout the world.36 However, phenotypic heterogeneity was evident in the South African population with respect to the presence or absence of cardiac events and highly variable QT interval duration (Figure 1), thus indicating that something besides the primary KCNQ1 mutation influenced disease expression and penetrance.
Figure 1.
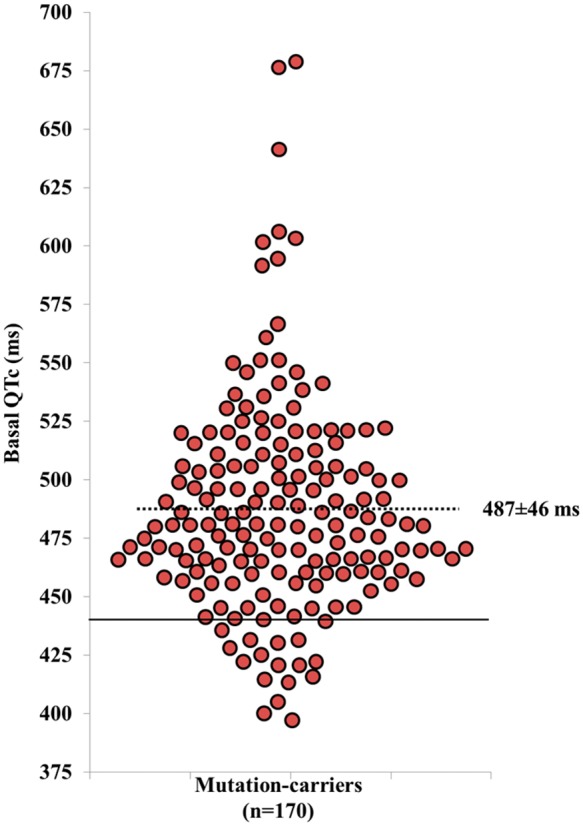
Basal QTc in KCNQ1-A341V mutation carriers. The uninterrupted line represents the upper limit of normal values for men (440 ms). The dotted line represents the mean QTc value of the 170 mutation carriers. If the QTc prolongation were determined only by the A341V mutation, the QTc values should be uniformly prolonged with modest variability. The large spectrum of QTc values points to the presence of additional variants affecting ventricular repolarization. Modified from Brink et al.29 with permission.
Studying the South African LQT1 founder population led us to demonstrate that NOS1AP is a genetic modifier of LQTS.37 This discovery and related work in the field are discussed separately below. This unique LQT1 population was also valuable for demonstrating that variants in AKAP9, encoding the protein kinase A anchoring protein yotiao important for regulating IKs,38 are associated with greater risk for cardiac events in KCNQ1-A341V carriers and longer rate-corrected QT interval duration.39 The specific variants tested in this study have uncertain effects on gene expression or protein function but may exist in linkage disequilibrium with functional variation in the gene.
The biological basis for the modifier effects of AKAP9 variants may involve dysregulation of IKs by cAMP-dependent pathways such as adrenergic receptor-mediated signalling. Genetic variation in modulating the probability of triggered arrhythmic events may also be important in this LQT1 cohort. Two variants in genes encoding adrenergic receptors (ADRA2C-Del322-325 and ADRB1-R389) that have been implicated with exaggerated adrenergic responses were associated with enhanced autonomic reflexes, faster heart rates, and a trend for more cardiac events in the South African LQT1 founder population.40 These findings correlated with clinical evidence that carriers of the KCNQ1-A341V mutation who exhibited faster resting heart rates and brisk autonomic responses had a greater probability of being symptomatic.40 These preliminary observations suggest that quantitatively greater release of epinephrine, norepinephrine, or even acetylcholine, may predispose to a higher probability of initiating either re-entrant or triggered arrhythmias in LQT1.
A conceptually important departure from the previous observations, in which variants were associated with increased arrhythmic risk, came from an international case–control study examining 112 LQTS duos.41 The duos were discordant pairs of one symptomatic and one asymptomatic first degree relatives, both carriers of the same KCNQ1 or KCNH2 mutation. Twenty five variants previously associated with QTc duration in healthy subjects or variable adrenergic responsiveness were investigated as potential modifiers of QTc duration and cardiac events. The minor allele of one intronic KCNQ1 variant (rs2074238, T allele) was associated with a lower arrhythmic risk and a shorter QTc. These findings were validated in 336 LQT1 subjects from two founder populations (South African, Finnish) and provided evidence for a protective genetic modifier.41 An independent study that considered KCNQ1 rs2074238 in a LQT2 cohort demonstrated a nominal association with shorter QTc duration but no association with cardiac events.23
NOS1AP is an important genetic modifier of long QT syndrome
Genome wide association studies previously identified single nucleotide variants in NOS1AP encoding a nitric oxide synthase adaptor protein associated with QT interval duration in the general population and these findings contributed to the initial search for modifier genes in LQTS.42–44 In the South African LQT1 founder population, we demonstrated that two common noncoding NOS1AP variants were associated with elevated risk of life-threatening arrhythmias.37 Our finding was subsequently validated in heterogeneous populations of LQTS patients45 and in a cohort of LQT2 families studied in the Netherlands.23 The consistency of these findings also indicates that NOS1AP can be regarded as a valid genetic modifier of LQTS severity. Interestingly, NOS1AP variants are also associated with the risk for developing drug-induced long-QT syndrome.46 These studies collectively demonstrate that modifier genes discovered in a founder population may be relevant to heterogeneous groups with the same or related disorders.
Despite the strong genetic evidence of association with QT duration and LQTS severity, there are conflicting data regarding the physiological effects of NOS1AP variants on cardiomyocyte repolarization. The minor alleles of common NOS1AP variants associated with longer QT duration in populations have been correlated with higher levels of gene expression in human ventricular myocardium obtained opportunistically from extracted pacemaker or defibrillator leads.47 Similar findings were reported for putative functional NOS1AP variants within a defined cis-regulatory (‘enhancer’) element within in the gene.48 Modulating expression levels of NOS1AP in experimental systems have provided conflicting results. Two groups have reported shortening of action potential duration (APD) with over expression of NOS1AP in either isolated adult guinea pig ventricular myocytes49 or cultured neonatal rat ventricular cardiomyocytes.48 In contrast, shortened APD was evoked by suppressing NOS1AP expression in embryonic zebrafish heart.50 These contradictory biological effects of NOS1AP expression on cardiomyocyte repolarization may be due to species differences, but which model system best represents human cells is unclear. A conundrum exists with these observations in that NOS1AP variant alleles that are associated with longer QT intervals in populations are also correlated with elevated NOS1AP expression in human ventricular tissue. Therefore, shortened APD evoked by overexpression in guinea pig or rat myocytes is not consistent with human data. Reconciling these observations may require human cardiomyocyte model systems.
Discovery of long QT syndrome modifiers using a physiological genomics approach
While candidate gene approaches have had some success in identifying plausible modifier genes in LQTS, additional unbiased approaches are needed to discover previously unrecognized genetic factors. An example of a new approach is illustrated by recent work of Chai et al.9 that combined whole exome sequencing and electrophysiological investigations of induced pluripotent stem cell derived cardiomyocytes (iPSC-CM) from members of a large family with type 2 LQTS. In this family, LQTS was associated with a pathogenic mutation in KCNH2 but there were notable differences in disease expression among mutation carriers. Electrophysiological measurements made from iPSC-CM from severely affected and mildly affected family members recapitulated this genotype–phenotype discordance. Specifically, only iPSC-CM from severely affected individuals exhibited prolonged APD despite the fact that all cells had lower levels of rapid delayed rectifier current (IKr) consistent with a loss of function mutation in KCNH2. This finding implied that there was either a protective factor blunting the effect of KCNH2 mutation on APD or that an aggravating factor was necessary to express impaired repolarization. Unexpectedly, iPSC-CM from the severely affected subjects exhibited larger L-type calcium current (ICa,L) amplitude, which was pharmacologically confirmed when nislodipine shortened the APD in these cells. Hence, genetic factors responsible for enhanced ICa,L were considered as potential LQTS modifiers.
In parallel investigations, exome sequencing revealed variants in the 2-pore domain potassium channel gene KCNK17 and in a GTP-binding protein encoded by REM2, a suspected physiological modulator of ICa,L. The KCNK17 variant was demonstrated to be gain-of-function in heterologous cells, and was absent in all severely affected family members. Silencing this gene in iPSC-CM from a mildly affected subject caused APD prolongation. This finding suggested that this KCNK17 gain-of-function variant was a protective allele. In contrast, the REM2 variant was carried by all severely affected subjects and overexpression of the variant in control iPSC-CM potentiated ICa,L. Hence, this was considered a plausible aggravating allele. Correction of the REM2 variant in affected cells using genome editing technology (CRISPR/Cas9) reversed the cellular phenotype. This elegant study demonstrates the synergy of combining modern genetic and cellular technologies to discover novel modifier genes in LQTS.
The practical importance of ‘protective modifiers’ goes beyond their relevance for risk stratification as it suggests the potential of identifying, through the understanding of their mechanism of action, totally novel approaches to therapy.
Applying genetic modifiers in clinical practice
The search for modifier genes in monogenetic congenital arrhythmia susceptibility represents an important research endeavour that may pay benefits in the future for management of affected individuals. Conceptually, subjects with a primary LQT1 mutation who are also carriers of well-validated NOS1AP risk alleles may require more vigilant monitoring for arrhythmic events or more aggressive therapy than those without these variants. The next step in evaluating the significance of modifier genes and their impact on clinical practice will be a prospective analysis to determine long-term outcomes and their association with risk alleles. Efforts to derive and test multilocus genetic risk scores42–44,51 may be valuable in this regard. However, there are challenges in applying genetic markers of risk deduced from population studies to individuals unless validated by a prospective analysis. The study by Strauss et al.51 provides an example of prospective testing to determine the value of a population-based genetic risk score applied to individuals. In this randomized, double-blind and placebo-controlled crossover trial of 3 QT-prolonging drugs, a genetic QT risk score comprising 61 common genetic variants previously identified by GWAS correlated with drug-induced QTc prolongation. Applying this genetic QT risk score to LQTS mutation carriers in a similar prospective study might help determine the clinical value of this predictive index in congenital arrhythmia susceptibility.
Acute myocardial infarction
Sudden cardiac death in association with an AMI represents a major public health problem and a condition for which there are likely genetic risk modifiers. Even though approximately 5% of subjects with a first AMI do not arrive alive to the hospital due to ventricular fibrillation (VF),52,53 the clinical features that might distinguish those with from those without VF have not been identified. Clinical features including infarct size, location, pump function, age, or sex fail to differentiate the two groups. The hypothesis that genetic variation might contribute to modifying the VF risk in the setting of an AMI has been supported by clinical and experimental studies. In the Paris Prospective Study involving almost 7000 French men followed for 26 years, SCD of one parent increased the relative risk of dying suddenly by 2.6 times, whereas SCD of both parents increased this risk by 9.4 times.54 A Dutch case–control study showed that familial SCD had occurred significantly more frequently among patients with a first myocardial infarction (MI) complicated by VF than in patients with a first MI without VF (43.1% and 25.1%, respectively; odds ratio 2.72, 95% confidence interval 1.84–4.03).55 A Finnish study with an almost identical design provided similar results.56 These three studies together provided evidence that SCD during a first MI has an important familial and likely genetic component. Further support for this concept comes from a canine model for SCD in which depressed baroreflex sensitivity (BRS) allows the early identification of post-MI dogs at high risk for SCD,57 a finding confirmed in >1000 post-MI patients.58 As the depression in BRS was evident even prior to the experimental MI, this suggests a genetic component.
These observations fostered the design of studies aiming at identifying either rare or common genetic variants that favour or hinder the development of VF in AMI. The first such studies were based on a candidate-gene approach. Small studies tested the role of rare variants in arrhythmia-susceptibility genes and suggested a role for SCN5A rare variants.59,60 Other studies evaluated the role of candidate SNPs. Specifically, NOS1AP variants were associated with higher risk for SCD in white adults61; an association confirmed in the Rotterdam Study, which however did not exclusively include AMI.62 The KCNH2-K897T polymorphism, previously identified as a genetic modifier of arrhythmic risk in LQTS,10,11 was subsequently associated with life-threatening arrhythmias following AMI.63 More recently, 257 ST-elevation myocardial infarction cases with VF and 537 controls (ST-elevation myocardial infarction without VF) of the Danish GEVAMI cohort were analysed for 27 SNPs previously associated with SCD and VF in different cohorts of patients not limited to AMI. One SNP located in intron 1 of SCN5A (rs11720524) showed an association with arrhythmic risk.64
However, when the field moved from candidate genes to GWAS, none of the SNPs or the genes previously associated with life-threatening arrhythmias in different clinical settings and during AMI showed a significant signal. Two GWAS reported an association of common genetic variants with VF/SCD at a genome-wide statistical significance (P < 5 × 10−8). One study, which compared patients with or without VF during a first AMI (AGNES population), identified a significant signal at a SNP near the CXADR gene.65 This gene, encoding the coxsackie and adenovirus receptor, is highly expressed in heart tissue. Mice haploinsufficient for this gene have delayed cardiac conduction and greater arrhythmia vulnerability during ischaemia.66 The other study detected an association signal in BAZ2B, which encodes the bromodomain adjacent zinc finger domain 2B.67BAZ2B and other genes in proximity of the risk variant are expressed in the heart, however, it is not yet known how they might influence arrhythmic risk. Although these two GWAS uncovered associations at genome-wide statistical significance, replication efforts have been limited and findings have been inconsistent.68
The significant complexities inherent with such studies include the multiplicity of factors that may contribute to the development of VF during AMI, e.g. autonomic activation, extent of ischaemia, electrolyte imbalance. Among them, a most important one is the activation of sympathetic reflexes that occurs during acute myocardial ischaemia,69 and which through the localized release of norepinephrine at the neural endings in the ventricles, accentuates the heterogeneity of ventricular repolarization to promote re-entrant arrhythmias.70 Indeed, the Paris prospective study identified excessive sympathetic activation and abnormal heart rate responses as major risk factors for SCD in victims of post-MI SCD.71 Similarly, it has been repeatedly demonstrated that prolongation of the QT interval is a major risk factor for SCD in post-MI patients72,73 and in the general population.74 This, in turn, has led to the suggestion that – if medically appropriate – drugs devoid of IKr activity should be preferred always and not just in LQTS patients.75
The recognition of these complexities has impacted the design of three similar ongoing studies in Europe seeking to identify genetic risk factors of SCD in the setting of AMI. These studies focus on the cleanest and most well defined phenotypes (first MI with or without VF) and have avoided reliance on surrogate markers (e.g. ICD discharges) that may be confounded by concomitant heart disease independently promoting arrhythmia risk through non-genetic mechanisms. These ongoing prospective studies (AGNES,65,66 GEVAMI,64 and PREDESTINATION76) have already enrolled 1400 cases and 2200 controls for a GWAS and, having now joined their databases in an EU Horizon 2020 programme project (ESCAPE-NET),77 they represent excellent opportunities to identify genetic modifiers of SCD risk in ischaemic heart disease.
Conclusions
Remarkable progress has been made during the last 20 years in identifying modifier genes in clinical situations with elevated risk for SCD. Clinical observations that were previously difficult to reconcile with the presence of a given disease-causing mutation are now conceptually more easily explained. This has resulted in a concept relevant to both monogenic and polygenic diseases, recently expressed in a consensus statement on SCD.78 Namely, that if an individual inherits both protective and arrhythmogenic SNPs, but their sum acts predominantly in one direction, there may be an important effect on clinical outcome (Figure 2). Conversely, if by the play of chance another individual inherits by the parents not only a disease-causing mutation but also multiple variants each with even modest detrimental effect (i.e. losses in repolarization reserve), this could create the setting for life-threatening arrhythmias (Figure 3).
Figure 2.
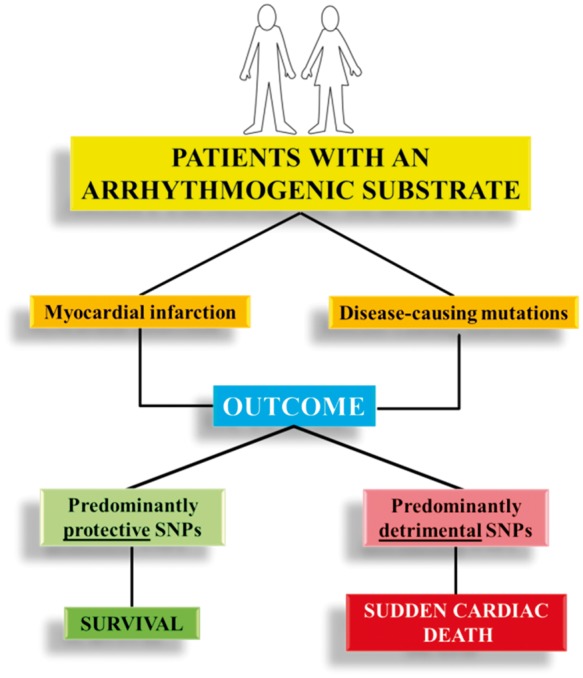
Potential impact on outcome (survival vs. sudden death) of the interaction between two arrhythmogenic substrates (mutations causing arrhythmogenic diseases or acute myocardial infarction) and predominantly protective or detrimental clusters of common genetic variants.
Figure 3.
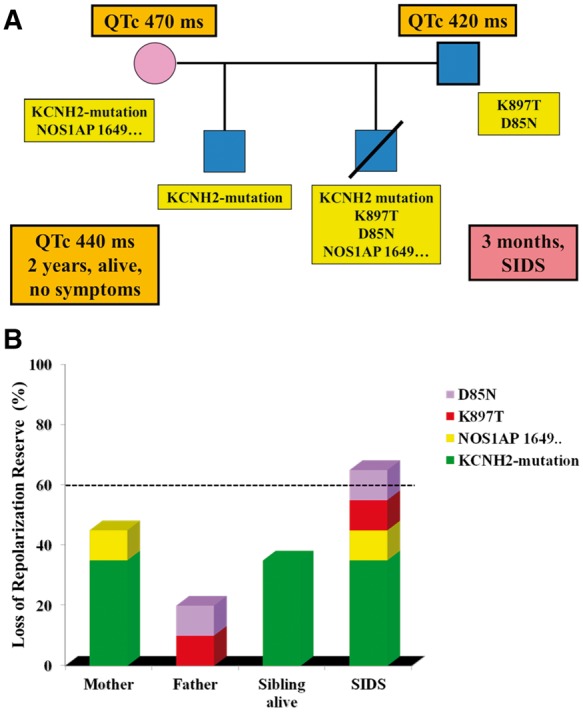
The importance of the additive weight of multiple variants impacting on repolarization reserve. In (A) the pedigree represents an imaginary situation in which an infant dies (diagnosed as Sudden Infant Death Syndrome) and the genetic analysis of the family identifies the presence of a long QT syndrome-causing mutation (reducing repolarization reserve by 35%) and of common variants each of which reduces repolarization reserve by 10%. In (B) the horizontal dotted line at 60% loss of repolarization reserve is an arbitrary threshold for the occurrence of life-threatening arrhythmias in the same family. The cartoon shows that while the mutation alone can prolong the QT interval, it is not sufficient to trigger arrhythmias. It is the combination of the mutation with multiple variants, each adding their small damaging effect, that brings vulnerability over the threshold.
The growing recognition that certain specific, and relatively common, genetic variants are able to modulate arrhythmic risk is promoting a more patient-specific risk stratification with the potential for clinical management to become progressively better tailored to the entire genetic background of an individual patient. The progress is more rapid for monogenic diseases and will take more time for complex polygenic disorders, but it should now be clear that, thanks to the smart use of genetic information, clinical management of arrhythmic disorders has taken a turn and precision medicine is in sight.79
Acknowledgements
The authors are grateful to Pinuccia De Tomasi for expert editorial support.
Funding
This work was supported by US NIH [HL122010 and HL131914 to A.L.G.]; Italian Ministry of Health [GR-2009-1472102 ‘Sudden cardiac death during myocardial ischaemia: unmasking genetic factors contributing to ventricular fibrillation’ to L.C.]; Italian Ministry of Health Grant ‘Predisposizione genetica alla fibrillazione ventricolare (FV) in pazienti con infarto miocardico acuto’ (to P.J.S.); Italian Ministry for Foreign Affairs 2008–2010 Grant ‘Identificazione di “geni modificatori” modulanti il rischio di morte improvvisa in malattie cardiache aritmogene ereditarie’ (to P.J.S.); ESCAPE-NET project (European Union’s Framework Horizon 2020 programme under Grant Agreement No. 733381) (to P.J.S., L.C.).
Conflict of interest: none declared.
References
- 1. Schwartz PJ, Ackerman MJ.. The long QT syndrome: a transatlantic clinical approach to diagnosis and therapy. Eur Heart J 2013;34:3109–3116. [DOI] [PubMed] [Google Scholar]
- 2. Priori SG, Napolitano C, Schwartz PJ.. Low penetrance in the long QT syndrome: clinical impact. Circulation 1999;99:529–533. [DOI] [PubMed] [Google Scholar]
- 3. Bezzina CR, Lahrouchi N, Priori SG.. Genetics of sudden cardiac death. Circ Res 2015;116:1919–1926. [DOI] [PubMed] [Google Scholar]
- 4. George AL., Jr. Molecular and genetic basis of sudden cardiac death. J Clin Invest 2013;123:75–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schwartz PJ, Ackerman MJ, George AL Jr, Wilde AM.. Impact of genetics on the clinical management of channelopathies. J Am Coll Cardiol 2013;62:169–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana A, Bosi G, Gabbarini F, Goulene K, Insolia R, Mannarino S, Mosca F, Nespoli L, Rimini A, Rosati E, Salice P, Spazzolini C.. Prevalence of the congenital long-QT syndrome. Circulation 2009;120:1761–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schwartz PJ, Priori SG, Napolitano C.. How really rare are rare diseases? The intriguing case of independent compound mutations in the long QT syndrome. J Cardiovasc Electrophysiol 2003;14:1120–1121. [DOI] [PubMed] [Google Scholar]
- 8. Westenskow P, Splawski I, Timothy KW, Keating MT, Sanguinetti MC.. Compound mutations: a common cause of severe long-QT syndrome. Circulation 2004;109:1834–1841. [DOI] [PubMed] [Google Scholar]
- 9. Chai S, Wan X, Ramirez-Navarro A, Tesar PJ, Kaufman ES, Ficker E, George AL Jr, Deschênes I.. Physiological genomics identifies genetic modifiers of long QT syndrome type 2 severity. J Clin Invest 2018;128:1043–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Crotti L, Insolia R, Barajas-Martinez H, Pollevick GD, Oliva A, Guerchicoff A, De FG, Dagradi F, Schwartz PJ, Viskin S, Antzelevitch C.. KCNH2-K897T is a genetic modifier of latent congenital long QT syndrome. Circulation 2005;112:1251–1258. [DOI] [PubMed] [Google Scholar]
- 11. Nof E, Cordeiro JM, Pérez GJ, Scornik FS, Calloe K, Love B, Burashnikov E, Caceres G, Gunsburg M, Antzelevitch C.. A common single nucleotide polymorphism can exacerbate long-QT type 2 syndrome leading to sudden infant death. Circ Cardiovasc Genet 2010;3:199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Paavonen KJ, Chapman H, Laitinen PJ, Fodstad H, Piippo K, Swan H, Toivonen L, Viitasalo M, Kontula K, Pasternack M.. Functional characterization of the common amino acid 897 polymorphism of the cardiac potassium channel KCNH2 (HERG). Cardiovasc Res 2003;59:603–611. [DOI] [PubMed] [Google Scholar]
- 13. Ye B, Valdivia CR, Ackerman MJ, Makielski JC.. A common human SCN5A polymorphism modifies expression of an arrhythmia causing mutation. Physiol Genomics 2003;12:187–193. [DOI] [PubMed] [Google Scholar]
- 14. Shinlapawittayatorn K, Du XX, Liu H, Ficker E, Kaufman ES, Deschênes I.. A common SCN5A polymorphism modulates the biophysical defects of SCN5A mutations. Heart Rhythm 2011;8:455–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Clatot J, Hoshi M, Wan X, Liu H, Jain A, Shinlapawittayatorn K, Marionneau C, Ficker E, Ha T, Deschênes I.. Voltage-gated sodium channels assemble and gate as dimers. Nat Commun 2017;8:2077.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bezzina CR, Verkerk AO, Busjahn A, Jeron A, Erdmann J, Koopmann TT, Bhuiyan ZA, Wilders R, Mannens MM, Tan HL, Luft FC, Schunkert H, Wilde AA.. A common polymorphism in KCNH2 (HERG) hastens cardiac repolarization. Cardiovasc Res 2003;59:27–36. [DOI] [PubMed] [Google Scholar]
- 17. Pfeufer A, Jalilzadeh S, Perz S, Mueller JC, Hinterseer M, Illig T, Akyol M, Huth C, Schöpfer-Wendels A, Kuch B, Steinbeck G, Holle R, Näbauer M, Wichmann HE, Meitinger T, Kääb S.. Common variants in myocardial ion channel genes modify the QT interval in the general population: results from the KORA study. Circ Res 2005;96:693–701. [DOI] [PubMed] [Google Scholar]
- 18. Gouas L, Nicaud V, Berthet M, Forhan A, Tiret L, Balkau B, Guicheney P, Study Group DESIR.. Association of KCNQ1, KCNE1, KCNH2 and SCN5A polymorphisms with QTc interval length in a healthy population. Eur J Hum Genet 2005;13:1213–1222. [DOI] [PubMed] [Google Scholar]
- 19. Newton-Cheh C, Guo CY, Larson MG, Musone SL, Surti A, Camargo AL, Drake JA, Benjamin EJ, Levy D, D'Agostino RB Sr, Hirschhorn JN, O'donnell CJ.. Common genetic variation in KCNH2 is associated with QT interval duration: the Framingham Heart Study. Circulation 2007;116:1128–1136. [DOI] [PubMed] [Google Scholar]
- 20. Nishio Y, Makiyama T, Itoh H, Sakaguchi T, Ohno S, Gong YZ, Yamamoto S, Ozawa T, Ding WG, Toyoda F, Kawamura M, Akao M, Matsuura H, Kimura T, Kita T, Horie M.. D85N, a KCNE1 polymorphism, is a disease-causing gene variant in long QT syndrome. J Am Coll Cardiol 2009;54:812–819. [DOI] [PubMed] [Google Scholar]
- 21. Kääb S, Crawford DC, Sinner MF, Behr ER, Kannankeril PJ, Wilde AA, Bezzina CR, Schulze-Bahr E, Guicheney P, Bishopric NH, Myerburg RJ, Schott JJ, Pfeufer A, Beckmann BM, Martens E, Zhang T, Stallmeyer B, Zumhagen S, Denjoy I, Bardai A, Van Gelder IC, Jamshidi Y, Dalageorgou C, Marshall V, Jeffery S, Shakir S, Camm AJ, Steinbeck G, Perz S, Lichtner P, Meitinger T, Peters A, Wichmann HE, Ingram C, Bradford Y, Carter S, Norris K, Ritchie MD, George AL Jr, Roden DM.. A large candidate gene survey identifies the KCNE1 D85N polymorphism as a possible modulator of drug-induced torsades de pointes. Circ Cardiovasc Genet 2012;5:91–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lahtinen AM, Marjamaa A, Swan H, Kontula K.. KCNE1 D85N polymorphism—a sex-specific modifier in type 1 long QT syndrome? BMC Med Genet 2011;12:11.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kolder ICRM, Tanck MWT, Postema PG, Barc J, Sinner MF, Zumhagen S, Husemann A, Stallmeyer B, Koopmann TT, Hofman N, Pfeufer A, Lichtner P, Meitinger T, Beckmann BM, Myerburg RJ, Bishopric NH, Roden DM, Kääb S, Wilde AAM, Schott JJ, Schulze-Bahr E, Bezzina CR.. Analysis for genetic modifiers of disease severity in patients with long-QT syndrome type 2. Circ Cardiovasc Genet 2015;8:447–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sanguinetti MC. Dysfunction of delayed rectifier potassium channels in an inherited cardiac arrhythmia. Ann N Y Acad Sci 1999;868:406–413. [DOI] [PubMed] [Google Scholar]
- 25. Amin AS, Giudicessi JR, Tijsen AJ, Spanjaart AM, Reckman YJ, Klemens CA, Tanck MW, Kapplinger JD, Hofman N, Sinner MF, Müller M, Wijnen WJ, Tan HL, Bezzina CR, Creemers EE, Wilde AA, Ackerman MJ, Pinto YM.. Variants in the 3' untranslated region of the KCNQ1-encoded Kv7.1 potassium channel modify disease severity in patients with type 1 long QT syndrome in an allele-specific manner. Eur Heart J 2012;33:714–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Crotti L, Lahtinen AM, Spazzolini C, Mastantuono E, Monti MC, Morassutto C, Parati G, Heradien M, Goosen A, Lichtner P, Meitinger T, Brink PA, Kontula K, Swan H, Schwartz PJ.. Genetic modifiers for the Long QT Syndrome. How important is the role of variants in the 3' untranslated region of KCNQ1? Circ Cardiovasc Genet 2016;9:581–589. [DOI] [PubMed] [Google Scholar]
- 27. Amin AS, Pinto YM, Ackerman MJ, Wilde AA.. Letter by Amin Regarding Article, “Genetic modifiers for the long-QT syndrome: how important is the role of variants in the 3' untranslated region of KCNQ1?”. Circ Cardiovasc Genet 2016;9:580. [DOI] [PubMed] [Google Scholar]
- 28. Crotti L, Lahtinen AM, Spazzolini C, Mastantuono E, Monti MC, Morassutto C, Parati G, Heradien M, Goosen A, Lichtner P, Meitinger T, Brink PA, Kontula K, Swan H, Schwartz PJ.. Response by Crotti et al to Letter Regarding Article, “Genetic modifiers for the long-QT syndrome: how important is the role of variants in the 3' untranslated region of KCNQ1?”. Circ Cardiovasc Genet 2016;9:581–582. [DOI] [PubMed] [Google Scholar]
- 29. Brink PA, Crotti L, Corfield V, Goosen A, Durrheim G, Hedley P, Heradien M, Geldenhuys G, Vanoli E, Bacchini S, Spazzolini C, Lundquist AL, Roden DM, George AL Jr, Schwartz PJ.. Phenotypic variability and unusual clinical severity of congenital long QT Syndrome in a founder population. Circulation 2005;112:2602–2610. [DOI] [PubMed] [Google Scholar]
- 30. Brink PA, Schwartz PJ.. Of founder populations, long QT syndrome, and destiny. Heart Rhythm 2009;6:S25–S33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Norio R. The Finnish Disease Heritage III: the individual diseases. Hum Genet 2003;112:470–526. [DOI] [PubMed] [Google Scholar]
- 32. Winbo A, Diamant UB, Rydberg A, Persson J, Jensen SM, Stattin EL.. Origin of the Swedish long QT syndrome Y111C/KCNQ1 founder mutation. Heart Rhythm 2011;8:541–547. [DOI] [PubMed] [Google Scholar]
- 33. Schwartz PJ. Sudden cardiac death, founder populations and mushrooms. What is the link with gold mines and modifier genes? Heart Rhythm 2011;8:548–550. [DOI] [PubMed] [Google Scholar]
- 34. Laberge AM, Michaud J, Richter A, Lemyre E, Lambert M, Brais B, Mitchell GA.. Population history and its impact on medical genetics in Quebec. Clin Genet 2005;68:287–301. [DOI] [PubMed] [Google Scholar]
- 35. Ter Bekke RMA, Isaacs A, Barysenka A, Hoos MB, Jongbloed JDH, Hoorntje JCA, Patelski ASM, Helderman-van den Enden ATJM, van den Wijngaard A, Stoll M, Volders PGA.. Heritability in a SCN5A-mutation founder population with increased female susceptibility to non-nocturnal ventricular tachyarrhythmia and sudden cardiac death. Heart Rhythm 2017;14:1873–1881. [DOI] [PubMed] [Google Scholar]
- 36. Crotti L, Spazzolini C, Schwartz PJ, Shimizu W, Denjoy I, Schulze-Bahr E, Zaklyazminskaya EV, Swan H, Ackerman MJ, Moss AJ, Wilde AA, Horie M, Brink PA, Insolia R, De Ferrari GM, Crimi G.. The common Long QT Syndrome mutation KCNQ1/A341V causes unusually severe clinical manifestations in patients with different ethnic backgrounds: toward a mutation-specific risk stratification. Circulation 2007;116:2366–2375. [DOI] [PubMed] [Google Scholar]
- 37. Crotti L, Monti MC, Insolia R, Peljto A, Goosen A, Brink PA, Greenberg DA, Schwartz PJ, George AL Jr.. NOS1AP is a genetic modifier of the long-QT syndrome. Circulation 2009;120:1657–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kurokawa J, Motoike HK, Rao J, Kass RS.. Regulatory actions of the A-kinase anchoring protein Yotiao on a heart potassium channel downstream of PKA phosphorylation. Proc Natl Acad Sci USA 2004;101:16374–16378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. de Villiers CP, van der Merwe L, Crotti L, Goosen A, George AL Jr, Schwartz PJ, Brink PA, Moolman-Smook JC, Corfield VA.. AKAP9 is a genetic modifier of congenital long-QT syndrome type 1. Circ Cardiovasc Genet 2014;7:599–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schwartz PJ, Vanoli E, Crotti L, Spazzolini C, Ferrandi C, Goosen A, Hedley P, Heradien M, Bacchini S, Turco A, La Rovere MT, Bartoli A, George AL Jr, Brink PA.. Neural control of heart rate is an arrhythmia risk modifier in long QT syndrome. J Am Coll Cardiol 2008;51:920–929. [DOI] [PubMed] [Google Scholar]
- 41. Duchatelet S, Crotti L, Peat R, Denjoy I, Itoh H, Berthet M, Ohno S, Fressart V, Monti MC, Crocamo C, Pedrazzini M, Dagradi F, Vicentini A, Klug D, Brink PA, Goosen A, Swan H, Toivonen L, Lahtinen A, Kontula K, Shimizu W, Horie M, George AL, Trégouët DA, Guicheney P, Schwartz PJ.. Identification of a KCNQ1 polymorphism acting as a protective modifier against arrhythmic risk in the long QT syndrome. Circ Cardiovasc Genet 2013;6:354–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Newton-Cheh C, Eijgelsheim M, Rice KM, de Bakker PI, Yin X, Estrada K, Bis JC, Marciante K, Rivadeneira F, Noseworthy PA, Sotoodehnia N, Smith NL, Rotter JI, Kors JA, Witteman JC, Hofman A, Heckbert SR, O'Donnell CJ, Uitterlinden AG, Psaty BM, Lumley T, Larson MG, Stricker BH.. Common variants at ten loci influence QT interval duration in the QTGEN Study. Nat Genet 2009;41:399–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pfeufer A, Sanna S, Arking DE, Müller M, Gateva V, Fuchsberger C, Ehret GB, Orrú M, Pattaro C, Köttgen A, Perz S, Usala G, Barbalic M, Li M, Pütz B, Scuteri A, Prineas RJ, Sinner MF, Gieger C, Najjar SS, Kao WH, Mühleisen TW, Dei M, Happle C, Möhlenkamp S, Crisponi L, Erbel R, Jöckel KH, Naitza S, Steinbeck G, Marroni F, Hicks AA, Lakatta E, Müller-Myhsok B, Pramstaller PP, Wichmann HE, Schlessinger D, Boerwinkle E, Meitinger T, Uda M, Coresh J, Kääb S, Abecasis GR, Chakravarti A.. Common variants at ten loci modulate the QT interval duration in the QTSCD Study. Nat Genet 2009;41:407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Arking DE, Pulit SL, Crotti L, van der Harst P, Munroe PB, Koopmann TT, Sotoodehnia N, Rossin EJ, Morley M, Wang X, Johnson AD, Lundby A, Gudbjartsson DF, Noseworthy PA, Eijgelsheim M, Bradford Y, Tarasov KV, Dörr M, Müller-Nurasyid M, Lahtinen AM, Nolte IM, Smith AV, Bis JC, Isaacs A, Newhouse SJ, Evans DS, Post WS, Waggott D, Lyytikäinen L-P, Hicks AA, Eisele L, Ellinghaus D, Hayward C, Navarro P, Ulivi S, Tanaka T, Tester DJ, Chatel S, Gustafsson S, Kumari M, Morris RW, Naluai ÅT, Padmanabhan S, Kluttig A, Strohmer B, Panayiotou AG, Torres M, Knoflach M, Hubacek JA, Slowikowski K, Raychaudhuri S, Kumar RD, Harris TB, Launer LJ, Shuldiner AR, Alonso A, Bader JS, Ehret G, Huang H, Kao WHL, Strait JB, Macfarlane PW, Brown M, Caulfield MJ, Samani NJ, Kronenberg F, Willeit J, Smith JG, Greiser KH, Meyer Zu Schwabedissen H, Werdan K, Carella M, Zelante L, Heckbert SR, Psaty BM, Rotter JI, Kolcic I, Polašek O, Wright AF, Griffin M, Daly MJ, Arnar DO, Hólm H, Thorsteinsdottir U, Denny JC, Roden DM, Zuvich RL, Emilsson V, Plump AS, Larson MG, O'Donnell CJ, Yin X, Bobbo M, D'Adamo AP, Iorio A, Sinagra G, Carracedo A, Cummings SR, Nalls MA, Jula A, Kontula KK, Marjamaa A, Oikarinen L, Perola M, Porthan K, Erbel R, Hoffmann P, Jöckel K-H, Kälsch H, Nöthen MM, den Hoed M, Loos RJF, Thelle DS, Gieger C, Meitinger T, Perz S, Peters A, Prucha H, Sinner MF, Waldenberger M, de Boer RA, Franke L, van der Vleuten PA, Beckmann BM, Martens E, Bardai A, Hofman N, Wilde AAM, Behr ER, Dalageorgou C, Giudicessi JR, Medeiros-Domingo A, Barc J, Kyndt F, Probst V, Ghidoni A, Insolia R, Hamilton RM, Scherer SW, Brandimarto J, Margulies K, Moravec CE, del Greco M F, Fuchsberger C, O'Connell JR, Lee WK, Watt GCM, Campbell H, Wild SH, El Mokhtari NE, Frey N, Asselbergs FW, Mateo Leach I, Navis G, van den Berg MP, van Veldhuisen DJ, Kellis M, Krijthe BP, Franco OH, Hofman A, Kors JA, Uitterlinden AG, Witteman JCM, Kedenko L, Lamina C, Oostra BA, Abecasis GR, Lakatta EG, Mulas A, Orrú M, Schlessinger D, Uda M, Markus MRP, Völker U, Snieder H, Spector TD, Ärnlöv J, Lind L, Sundström J, Syvänen A-C, Kivimaki M, Kähönen M, Mononen N, Raitakari OT, Viikari JS, Adamkova V, Kiechl S, Brion M, Nicolaides AN, Paulweber B, Haerting J, Dominiczak AF, Nyberg F, Whincup PH, Hingorani AD, Schott J-J, Bezzina CR, Ingelsson E, Ferrucci L, Gasparini P, Wilson JF, Rudan I, Franke A, Mühleisen TW, Pramstaller PP, Lehtimäki TJ, Paterson AD, Parsa A, Liu Y, van Duijn CM, Siscovick DS, Gudnason V, Jamshidi Y, Salomaa V, Felix SB, Sanna S, Ritchie MD, Stricker BH, Stefansson K, Boyer LA, Cappola TP, Olsen JV, Lage K, Schwartz PJ, Kääb S, Chakravarti A, Ackerman MJ, Pfeufer A, de Bakker PIW, Newton-Cheh C.. Genetic association study of QT interval highlights role for calcium signaling pathways in myocardial repolarization. Nat Genet 2014;46:826–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tomás M, Napolitano C, De Giuli L, Bloise R, Subirana I, Malovini A, Bellazzi R, Arking DE, Marban E, Chakravarti A, Spooner PM, Priori SG.. Polymorphisms in the NOS1AP gene modulate QT interval duration and risk of arrhythmias in the long QT syndrome. J Am Coll Cardiol 2010;55:2745–2752. [DOI] [PubMed] [Google Scholar]
- 46. Jamshidi Y, Nolte IM, Dalageorgou C, Zheng D, Johnson T, Bastiaenen R, Ruddy S, Talbott D, Norris KJ, Snieder H, George AL, Marshall V, Shakir S, Kannankeril PJ, Munroe PB, Camm AJ, Jeffery S, Roden DM, Behr ER.. Common variation in the NOS1AP gene is associated with drug-induced QT prolongation and ventricular arrhythmia. J Am Coll Cardiol 2012;60:841–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Saba S, Mehdi H, Shah H, Islam Z, Aoun E, Termanini S, Mahjoub R, Aleong R, McTiernan CF, London B.. Cardiac levels of NOS1AP RNA from right ventricular tissue recovered during lead extraction. Heart Rhythm 2012;9:399–404. [DOI] [PubMed] [Google Scholar]
- 48. Kapoor A, Sekar RB, Hansen NF, Fox-Talbot K, Morley M, Pihur V, Chatterjee S, Brandimarto J, Moravec CS, Pulit SL QT Interval-International GWAS Consortium Pfeufer A, Mullikin J, Ross M, Green ED, Bentley D, Newton-Cheh C, Boerwinkle E, Tomaselli GF, Cappola TP, Arking DE, Halushka MK, Chakravarti A. An enhancer polymorphism at the cardiomyocyte intercalated disc protein NOS1AP locus is a major regulator of the QT interval. Am J Hum Genet 2014;94:854–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chang KC, Barth AS, Sasano T, Kizana E, Kashiwakura Y, Zhang Y, Foster DB, Marbán E.. CAPON modulates cardiac repolarization via neuronal nitric oxide synthase signaling in the heart. Proc Natl Acad Sci USA 2008;105:4477–4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Milan DJ, Kim AM, Winterfield JR, Jones IL, Pfeufer A, Sanna S, Arking DE, Amsterdam AH, Sabeh KM, Mably JD, Rosenbaum DS, Peterson RT, Chakravarti A, Kääb S, Roden DM, MacRae CA.. Drug-sensitized zebrafish screen identifies multiple genes, including GINS3, as regulators of myocardial repolarization. Circulation 2009;120:553–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Strauss DG, Vicente J, Johannesen L, Blinova K, Mason JW, Weeke P, Behr ER, Roden DM, Woosley R, Kosova G, Rosenberg MA, Newton-Cheh C.. Common genetic variant risk score is associated with drug-induced QT prolongation and torsade de pointes risk: a pilot study. Circulation 2017;135:1300–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jabbari R, Engstrøm T, Glinge C, Risgaard B, Jabbari J, Winkel BG, Terkelsen CJ, Tilsted HH, Jensen LO, Hougaard M, Chiuve SE, Pedersen F, Svendsen JH, Haunsø S, Albert CM, Tfelt-Hansen J.. Incidence and risk factors of ventricular fibrillation before primary angioplasty in patients with first ST-elevation myocardial infarction: a nationwide study in Denmark. J Am Heart Assoc 2015;4:e001399.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Karam N, Bataille S, Marijon E, Giovannetti O, Tafflet M, Savary D, Benamer H, Caussin C, Garot P, Juliard JM, Pires V, Boche T, Dupas F, Le Bail G, Lamhaut L, Laborne F, Lefort H, Mapouata M, Lapostolle F, Spaulding C, Empana JP, Jouven X, Lambert Y; e-MUST Study Investigators. Identifying patients at risk for prehospital sudden cardiac arrest at the early phase of myocardial infarction: the e-MUST Study (Evaluation en Médecine d’Urgence des Stratégies Thérapeutiques des infarctus du myocarde). Circulation 2016;134:2074–2083. [DOI] [PubMed] [Google Scholar]
- 54. Jouven X, Desnos M, Guerot C, Ducimetière P.. Predicting sudden death in the population: the Paris Prospective Study I. Circulation 1999;99:1978–1983. [DOI] [PubMed] [Google Scholar]
- 55. Dekker LR, Bezzina CR, Henriques JP, Tanck MW, Koch KT, Alings MW, Arnold AE, de BMJ, Gorgels AP, Michels HR, Verkerk A, Verheugt FW, Zijlstra F, Wilde AA.. Familial sudden death is an important risk factor for primary ventricular fibrillation: a case-control study in acute myocardial infarction patients. Circulation 2006;114:1140–1145. [DOI] [PubMed] [Google Scholar]
- 56. Kaikkonen KS, Kortelainen ML, Linna E, Huikuri HV.. Family history and the risk of sudden cardiac death as a manifestation of an acute coronary event. Circulation 2006;114:1462–1467. [DOI] [PubMed] [Google Scholar]
- 57. Schwartz PJ, Vanoli E, Stramba-Badiale M, De Ferrari GM, Billman GE, Foreman RD.. Autonomic mechanisms and sudden death. New insights from analysis of baroreceptor reflexes in conscious dogs with and without a myocardial infarction. Circulation 1988;78:969–979. [DOI] [PubMed] [Google Scholar]
- 58. La Rovere MT, Bigger JT Jr, Marcus FI, Mortara A, Schwartz PJ.. for the ATRAMI (Autonomic Tone and Reflexes After Myocardial Infarction) Investigators Baroreflex sensitivity and heart-rate variability in prediction of total cardiac mortality after myocardial infarction. Lancet 1998;351:478–484. [DOI] [PubMed] [Google Scholar]
- 59. Hu D, Viskin S, Oliva A, Carrier T, Cordeiro JM, Barajas-Martinez H, Wu Y, Burashnikov E, Sicouri S, Brugada R, Rosso R, Guerchicoff A, Pollevick GD, Antzelevitch C.. Novel mutation in the SCN5A gene associated with arrhythmic storm development during acute myocardial infarction. Heart Rhythm 2007;4:1072–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Boehringer T, Bugert P, Borggrefe M, Elmas E.. SCN5A mutations and polymorphisms in patients with ventricular fibrillation during acute myocardial infarction. Mol Med Rep 2014;10:2039–2044. [DOI] [PubMed] [Google Scholar]
- 61. Kao WH, Arking DE, Post W, Rea TD, Sotoodehnia N, Prineas RJ, Bishe B, Doan BQ, Boerwinkle E, Psaty BM, Tomaselli GF, Coresh J, Siscovick DS, Marbán E, Spooner PM, Burke GL, Chakravarti A.. Genetic variations in nitric oxide synthase 1 adaptor protein are associated with sudden cardiac death in US white community-based populations. Circulation 2009;119:940–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Eijgelsheim M, Newton-Cheh C, Aarnoudse AL, van Noord C, Witteman JC, Hofman A, Uitterlinden AG, Stricker BH.. Genetic variation in NOS1AP is associated with sudden cardiac death: evidence from the Rotterdam Study. Hum Mol Genet 2009;18:4213–4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Crotti L, Hu D, Barajas-Martinez H, De Ferrari GM, Oliva A, Insolia R, Pollevick GD, Dagradi F, Guerchicoff A, Greco F, Schwartz PJ, Viskin S, Antzelevitch C.. Torsades de pointes following acute myocardial infarction: evidence for a deadly link with a common genetic variant. Heart Rhythm 2012;9:1104–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jabbari R, Glinge C, Jabbari J, Risgaard B, Winkel BG, Terkelsen CJ, Tilsted HH, Jensen LO, Hougaard M, Haunsø S, Engstrøm T, Albert CM, Tfelt-Hansen J.. A common variant in SCN5A and the risk of ventricular fibrillation caused by first ST-segment elevation myocardial infarction. PLoS One 2017;12:e0170193.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bezzina CR, Pazoki R, Bardai A, Marsman RF, de Jong JSSG, Blom MT, Scicluna BP, Jukema JW, Bindraban NR, Lichtner P, Pfeufer A, Bishopric NH, Roden DM, Meitinger T, Chugh SS, Myerburg RJ, Jouven X, Kääb S, Dekker LRC, Tan HL, Tanck MWT, Wilde AAM.. Genome-wide association study identifies a susceptibility locus at 21q21 for ventricular fibrillation in acute myocardial infarction. Nat Genet 2010;42:688–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Marsman RF, Bezzina CR, Freiberg F, Verkerk AO, Adriaens ME, Podliesna S, Chen C, Purfürst B, Spallek B, Koopmann TT, Baczko I, Dos Remedios CG, George AL Jr, Bishopric NH, Lodder EM, de Bakker JM, Fischer R, Coronel R, Wilde AA, Gotthardt M, Remme CA.. Coxsackie and adenovirus receptor is a modifier of cardiac conduction and arrhythmia vulnerability in the setting of myocardial ischemia. J Am Coll Cardiol 2014;63:549–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Arking DE, Junttila MJ, Goyette P, Huertas-Vazquez A, Eijgelsheim M, Blom MT, Newton-Cheh C, Reinier K, Teodorescu C, Uy-Evanado A, Carter-Monroe N, Kaikkonen KS, Kortelainen ML, Boucher G, Lagacé C, Moes A, Zhao X, Kolodgie F, Rivadeneira F, Hofman A, Witteman JC, Uitterlinden AG, Marsman RF, Pazoki R, Bardai A, Koster RW, Dehghan A, Hwang SJ, Bhatnagar P, Post W, Hilton G, Prineas RJ, Li M, Köttgen A, Ehret G, Boerwinkle E, Coresh J, Kao WH, Psaty BM, Tomaselli GF, Sotoodehnia N, Siscovick DS, Burke GL, Marbán E, Spooner PM, Cupples LA, Jui J, Gunson K, Kesäniemi YA, Wilde AA, Tardif JC, O'Donnell CJ, Bezzina CR, Virmani R, Stricker BH, Tan HL, Albert CM, Chakravarti A, Rioux JD, Huikuri HV, Chugh SS.. Identification of a sudden cardiac death susceptibility locus at 2q24.2 through genome-wide association in European ancestry individuals. PLoS Genet 2011;7:e1002158.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bugert P, Elmas E, Stach K, Weiss C, Kälsch T, Dobrev D, Borggrefe M.. No evidence for an association between the rs2824292 variant at chromosome 21q21 and ventricular fibrillation during acute myocardial infarction in a German population. Clin Chem Lab Med 2011;49:1237–1239. [DOI] [PubMed] [Google Scholar]
- 69. Malliani A, Schwartz PJ, Zanchetti A.. A sympathetic reflex elicited by experimental coronary occlusion. Am J Physiol 1969;217:703–709. [DOI] [PubMed] [Google Scholar]
- 70. Han J, Moe GK.. Nonuniform recovery of excitability in ventricular muscle. Circ Res 1964;14:44–60. [DOI] [PubMed] [Google Scholar]
- 71. Jouven X, Empana JP, Schwartz PJ, Desnos M, Courbon D, Ducimetière P.. Heart rate profile during exercise as a predictor of sudden death. N Engl J Med 2005;352:1951–1958. [DOI] [PubMed] [Google Scholar]
- 72. Schwartz PJ, Wolf S.. QT interval prolongation as predictor of sudden death in patients with myocardial infarction. Circulation 1978;57:1074–1077. [DOI] [PubMed] [Google Scholar]
- 73. Aro AL, Reinier K, Rusinaru C, Uy-Evanado A, Darouian N, Phan D, Mack WJ, Jui J, Soliman EZ, Tereshchenko LG, Chugh SS.. Electrical risk score beyond the left ventricular ejection fraction: prediction of sudden cardiac death in the Oregon Sudden Unexpected Death Study and the Atherosclerosis Risk in Communities Study. Eur Heart J 2017;38:3017–3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ashar FN, Mitchell RN, Albert CM, Newton-Cheh C, Brody JA, Muller-Nurasyid M, Moes A, Meitinger T, Mak A, Huikuri H, Junttila MJ, Goyette P, Pulit SL, Pazoki R, Tanck MW, Blom MT, Zhao XQ, Havulinna AS, Jabbari R, Glinge C, Tragante V, Escher SA, Chakravarti A, Ehret G, Coresh J, Li M, Prineas RJ, Franco OH, Kwok P-Y, Lumley T, Dumas F, MD, McKnight B, Rotter JI, Lemaitre RN, Heckbert SR, O’Donnell CJ, Hwang S-J, Tardif J-C, VanDenburgh M, Uitterlinden AG, Hofman A, Stricker BHC, de Bakker PIW, Franks PW, Jansson J-H, Asselbergs FW, Halushka MK, Maleszewski JJ, Tfelt-Hansen J, Engstrom T, Salomaa V, Virmani R, Kolodgie F, Wilde AAM, Tan HL, Bezzina CR, Eijgelsheim M, Rioux JD, Jouven X, Kääb S, Psaty BM, Siscovick DS, Arking DE, Sotoodehnia N for the SCD working group of the CHARGE Consortium . A comprehensive evaluation of the genetic architecture of sudden cardiac arrest. Eur Heart J 2018; in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Schwartz PJ, Gentilini G. Can genetics predict risk for sudden cardiac death? The relentless search for the Holy Grail. Eur Heart J doi:10.1093/eurheartj/ehy508 (in press). [DOI] [PubMed] [Google Scholar]
- 76. De Ferrari GM, De Regibus V, Gionti V, Civardi D, Insolia R, Pedrazzini M, Gentilini D, Di Blasio A, Crotti L, Schwartz PJ.. PREDESTINATION: PRimary vEntricular fibrillation and suDden dEath during a firST myocardIal iNfArcTION: genetic basis In Grieco N, Marzegalli M, Paganoni AM, eds. New Diagnostic, Therapeutic and Organizational Strategies for Acute Coronary Syndromes Patients. Italia: Springer; 2013, pp. 85–96. [Google Scholar]
- 77. Tan HL, Dagres N, Böttiger BW, Schwartz PJ; on behalf of the ESCAPE-NET Investigators. European Sudden Cardiac Arrest network: towards Prevention, Education and New Effective Treatments (ESCAPE-NET). Eur Heart J 2018;39:86–88. [Google Scholar]
- 78. Wellens HJJ, Schwartz PJ, Lindemans FW, Buxton AE, Goldberger JJ, Hohnloser SH, Huikuri HV, Kääb S, La Rovere MT, Malik M, Myerburg RJ, Simoons ML, Swedberg K, Tijssen J, Voors AA, Wilde AA.. Risk stratification for sudden cardiac death: current status and challenges for the future. Eur Heart J 2014;35:1642–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Mehta A, Ramachandra CJA, Singh P, Chitre A, Lua C-H, Mura M, Crotti L, Wong P, Schwartz PJ, Gnecchi M, Shim W. Identification of a targeted and testable antiarrhythmic therapy for LQT2 using a patient-specific cellular model. Eur Heart J 2018;39:1446–1455. [DOI] [PubMed] [Google Scholar]