

Abstract
Aims
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is characterized by right ventricular myocardial replacement and life-threatening ventricular arrhythmias. Desmosomal gene mutations are sometimes identified, but clinical and genetic diagnosis remains challenging. Desmosomal skin disorders can be caused by desmosomal gene mutations or autoantibodies. We sought to determine if anti-desmosome antibodies are present in subjects with ARVC.
Methods and results
We evaluated ARVC subjects and controls for antibodies to cardiac desmosomal cadherin proteins. Desmoglein-2 (DSG2), desmocollin-2, and N-cadherin proteins on western blots were exposed to sera, in primary and validation cohorts of subjects and controls, as well as the naturally occurring Boxer dog model of ARVC. We identified anti-DSG2 antibodies in 12/12 and 25/25 definite ARVC cohorts and 7/8 borderline subjects. Antibody was absent in 11/12, faint in 1/12, and absent in 20/20 of two control cohorts. Anti-DSG2 antibodies were present in 10/10 Boxer dogs with ARVC, and absent in 18/18 without. In humans, the level of anti-DSG2 antibodies correlated with the burden of premature ventricular contractions (r = 0.70), and antibodies caused gap junction dysfunction, a common feature of ARVC, in vitro. Anti-DSG2 antibodies were present in ARVC subjects regardless of whether an underlying mutation was identified, or which mutation was present. A disease-specific DSG2 epitope was identified.
Conclusion
Anti-DSG2 antibodies are a sensitive and specific biomarker for ARVC. The development of autoimmunity as a result of target-related mutations is unique. Anti-DSG2 antibodies likely explain the cardiac inflammation that is frequently identified in ARVC and may represent a new therapeutic target.

Keywords: Arrhythmogenic right ventricular cardiomyopathy, Biomarker, Autoantibody, Human desmoglein-2, Boxer dog
Introduction
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a heritable heart muscle disease characterized by replacement of myocardium by fat and/or fibrosis, and arrhythmias arising usually from the right ventricular free wall.1 It is a major contributor to sudden cardiac deaths (SCDs) in young adults,2 athletes,3 and children and adolescents age 10–18 years,4 accounting for 11%, 22%, and 25% of SCD in these groups, respectively. Arrhythmogenic right ventricular cardiomyopathy is estimated to occur in one in 2000 individuals,5 and sudden deaths during sport are often attributed to this disorder.
Arrhythmogenic right ventricular cardiomyopathy is difficult to diagnose, but a set of Task Force criteria developed in 19946 by the European Society of Cardiology, and revised in 2010,7 provides the current standard for identification. These criteria evaluate detailed imaging, pathology, electrocardiography, family history and genetics assessments, and the total number of major and minor criteria characterize patients as having definite, borderline, possible, or no ARVC. Task Force criteria assessments are complex and costly, must be repeated regularly and are still considered to miss 30% of affected individuals. Once diagnosed, risk stratification and therapy also pose clinical challenges that are addressed by additional guidelines.8
Mutations in 12 genes are associated with the development of ARVC or ARVC mimics9–19; however, the genetic cause remains unidentified in approximately 50% of cases. Arrhythmogenic right ventricular cardiomyopathy is often characterized by incomplete penetrance and variable expression, with the disease appearing to skip generations or appear in isolated individuals. Activity and exercise clearly modify disease expression.20 Where identified, genetic mutations most typically affect proteins of the desmosome, a structure that links heart muscle cells together at their intercalated discs. These abnormal desmosomes fail to mechanically link cardiomyocytes, resulting in myocardial replacement with fat and scar tissue either directly, or as a result of nuclear signalling altering cell fate.21 A common feature identified in ARVC, regardless of the gene cause, is secondary failure of gap junction electrical connections at the cardiomyocyte intercalated discs, resulting in cardiac conduction delay and ventricular arrhythmias.22 Frequently, there is also evidence for inflammation in the pathogenesis of ARVC. Lymphocytic infiltrates are frequently seen in myocardial specimens,23 and the disease often demonstrates flares or hot phases of disease.24
Outside of the heart, desmosomes also provide mechanical links between epithelial cells. Since skin desmosomal disease is caused by either mutations of desmosomal proteins or autoantibodies against these proteins,25 we hypothesized that anti-desmosomal autoantibodies might contribute to ARVC as well.
In this study, we report the presence of anti-desmoglein-2 (DSG2) autoantibodies as a highly sensitive and specific biomarker identifying Task Force positive ARVC human subjects, as well as ARVC affected Boxer dogs. Our functional analyses suggest that anti-DSG2 autoantibodies are intrinsically involved in the pathogenesis of ARVC.
Materials and methods
Subjects and animals
The study protocol followed the ethical guidelines of the Declaration of Helsinki. The Research Ethics Board of the Hospital for Sick Children and the Cantonal Ethical Committee Zurich, Switzerland, and all human participants gave written informed consent. Patients referred to these clinics for assessment of ARVC were evaluated using the 2010 ARVC Task Force criteria [including electrocardiograms (ECGs), signal-averaged ECGs (SAECGs), 24-h ambulatory monitoring, echocardiography, and magnetic resonance imaging] and only those meeting Definite or Borderline criteria (Hospital for Sick Children) or Definite criteria (Zurich validation cohort) were included. Sera were collected and frozen at −80°C until analysed. Sera from Zurich were transported on ice for evaluation. Normal Control sera were purchased from two commercial sources: Innovative Research Inc. (Novi, MI, USA) and from BBI Solutions (Cardiff, UK).
Optimization of western blot
We used a recombinant fusion protein of human DSG2 Fc Chimera (a portion of Fc region of human IgG attached to full DSG2 protein). This structure is commonly used in antibody studies to improve solubility and presentation of the protein. Though the attached Fc region is not the native Fc region of IgG, it may give some weak binding to the secondary antibody [Goat anti-human IgG-horseradish peroxidase (IgG-HRP) conjugate] and thus can appear as a false positive for negative samples. Therefore, we optimized the conditions for the western blot to avoid this weak positive reaction by testing different amounts of antigen, dilutions of Goat anti-human IgG-HRP, and exposure times for the enhanced chemiluminescence reaction (see Supplementary material online).
Western blot analysis
Western blot procedure was adopted from Chatterjee-Chakraborty and Chatterjee.26 The analysis of serum from controls and patients were carried out using recombinant human DSG2, DSC2, and N-Cadherin proteins. The conditions used for our study to prevent false positive reactions for the attached Fc fragment in the fusion proteins were: (i) keeping the recombinant DSG2 fusion protein per lane to 100 ng, (ii) using the dilution of Goat anti-human IgG-HRP to 1:10 000, and (iii) maintaining the exposure time to X-ray film at 5 s. We have checked the specificity of the blotting method used by different prior control experiments as described (see Supplementary material online).
Desmoglein-2 antibody ELISA protocol
A direct ELISA was performed by first coating a micro-titre plate with recombinant human DSG2 protein and then exposing each well to diluted human sera (100 μL of 1:100 dilution) followed by washing. The resultant bound anti-DSG2 antibody was then assayed by anti-human IgG-HRP using tetramethylbenzidine chromophore to measure optical density (O.D.) at 450 nm wavelength. Details of the solutions, dilutions, incubations, and washing are provided in the Supplementary material online.
Western blot analysis of synthetic desmoglein-2 oligopeptide library
Western blots with serum from control samples and patient samples were carried out using synthetic oligopeptides, made on the basis of the known sequence of human DSG2 protein. We used 112 sequential peptides of 15 amino acid (a.a.) residues (overlapping by five residues) to completely cover the DSG2 sequence from the N-terminal to C-terminal, and characterized the resultant binding as strong, moderate, weak, or faint (see also Supplementary material online).
Statistical analysis
To correlate the phenotype to the band intensities of the patients, the ambulatory burden of premature ventricular contractions (PVCs) was plotted with the pixels calculated from the band intensities. Statistical calculations and graphs were completed with Prism 5.0 for Mac OS X Version 5.0 of 31 July 2012 (Graphpad Software, Inc.).
Results
Study population
We analysed sera from both definite and borderline affected adult and childhood cases of ARVC based on 2010 Task Force criteria followed in the inherited arrhythmia clinic of the Hospital for Sick Children. These include cases referred for ventricular arrhythmias, resuscitated sudden cardiac arrest or family history of ARVC. Subjects had undergone an ECG, SAECG, ambulatory ECG, echocardiogram, cardiac magnetic resonance imaging, and testing for genetic causes of ARVC. When these investigations did not yield a diagnosis of ARVC, an endomyocardial biopsy was also performed. Eleven of these subjects had mutations in known desmosomal genes (PKP2 in eight, DSC2 in one, DSG2 in one, and DSP in one). Eight had no pathogenic mutation detected and one patient did not have genetic testing performed. A validation cohort of adults with definite ARVC from the Zurich ARVC programme was also assessed. Nineteen of these subjects had mutations in known ARVC or ARVC mimic genes: PKP2 in six, DSG2 in five, DSP in two, SCN5A in two, TTN in two, DSC2 in one, LMNA in one, RYR2 in one. One subject had a digenic mutation, one subject had a homozygous DSG2 mutation and six had no pathogenic mutations detected. All subjects are summarized according to ARVC Task Force criteria in Table 1. Six subjects had neither a Family History criterion nor pathogenic ARVC mutation, although two had suspicious deaths in second degree relatives (not meeting criteria) and one had a rare conserved DSC2 variant (DSC2 c.236T>G). Thus three subjects in total could be considered to have sporadic ARVC.
Table 1.
Phenotypic and genetic data for patients included in the original and validation cohorts
| Patients | Agea | Sex | Ib | IIc | IIId | IVe | Vf | VIg | Total (major, minor) | Diagnosis | Biomarker | Therapyh | PVCi | Geneticsj |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HCG001 | 15 | M | 1,0 | 0,0 | 0,0 | 0,1 | 0,1 | 0,0 | 1,2 | ARVC | Yes | Nadolol | 2600k | No Mutl |
| HRC0168 | 12 | M | 0,1 | 0,0 | 0,0 | 0,0 | 0,1 | 1,0 | 1,2 | ARVC | Yes | Levothyroxin | 921 | DSC2 |
| HRC0160 | 34 | F | 1,0 | 0,0 | 0,0 | 0,0 | 0,0 | 1,0 | 2,0 | ARVC | Yes | Bisoprolol | 0k | No Mut |
| HRC0118 | 34 | F | 1,0 | 0,0 | 1,0 | — | — | 1,0 | 3,0 | ARVC | Yes | Metoprolol, Sotalol | 854 | PKP2 |
| HRC0101 | 37 | F | 1,0 | 0,0 | 1,0 | — | 0,0 | 0,1 | 2,1 | ARVC | Yes | Bisoprolol | 114k | No Mut |
| HRC0099 | 17 | F | 1,0 | 0,0 | 1,0 | 0,0 | —,1 | 1,0 | 3,1 | ARVC | Yes | Nadolol | 10 266 | PKP2 |
| HRC0098 | 54 | F | 1,0 | 0,0 | 0,0 | 0,0 | 0,0 | 1,0 | 2,0 | ARVC | Yes | — | — | PKP2 |
| HRC0085 | 13 | M | 0,1 | 0,0 | 0,0 | 0,1 | 0,0 | 1,0 | 1,2 | ARVC | Yes | — | PKP2 | |
| HRC0084 | 16 | F | 1,0 | 0,0 | 1,0 | 0,0 | 0,1 | 1,0 | 3,1 | ARVC | Yes | Atenolol | 3091 | PKP2 |
| HRC0083 | 37 | M | — | 0,0 | 0,1 | 0,1 | — | 1,0 | 1,2 | ARVC | Yes | Sotalol, L-thyroxin | — | DSP |
| HRC0179 | 46 | F | 1,0 | — | 1,0 | 0,1 | 0,1 | 1,0 | 3,2 | ARVC | Yes | — | 667 | PKP2 |
| HRC0066 | 42 | F | 1,0 | — | 1,0 | 0,0 | 0,1 | — | 2,1 | ARVC | Yes | None | 9126 | — |
| HRC0159 | 9 | M | 0,0 | 0,0 | 0,0 | 0,1 | 0,0 | 1,0 | 1,1 | Bord.m ARVC | Yes | None | 0 | No Mut |
| HRC0086 | 37 | F | — | 0,0 | 0,0 | 1,0 | 0,0 | 1,0 | 1,1 | Bord. ARVC | No | — | PKP2 | |
| HRC0090 | 11 | F | 0,0 | 0,0 | 0,0 | 0,1 | — | 1,0 | 1,1 | Bord. ARVC | Yes | None | 0 | DSG2 |
| HRC0174 | 44 | M | 0,0 | 0,0 | 0,0 | 0,1 | 0,0 | 1,0 | 1,1 | Bord. ARVC | Yes | None | 26 | PKP2 |
| HRC0189 | 12 | M | 0,1 | — | 0.0 | 0,0 | 0,0 | 1,0 | 1,1 | Bord. ARVC | Yes | None | 1 | No Mut |
| HRC0109 | 15 | M | 0,0 | 0,0 | 0,0 | 0,1 | 0,0 | 1,0 | 1,1 | Bord. ARVC | Yes | None | 0 | No Mut |
| HRC0108 | 40 | M | 0,0 | 0,0 | 0,1 | 0,0 | 0,0 | 1,0 | 1,1 | Bord. ARVC | Yes | None | — | No Mut |
| HRC0087 | 36 | F | — | — | — | 0,1 | 0,0 | 1,0 | 1,1 | Bord. ARVC | Yes | — | 0 | No Mut |
| ZH-001 | 60 | M | 1,0 | 0,0 | 1,0 | 0,1 | 0,1 | 1,0 | 3,2 | ARVC | Yes | Amiodarone | 124k | DSG2 |
| ZH-007 | 64 | M | 1,0 | 0,0 | 1,0 | 0,1 | 1,0 | 0,0 | 3,1 | ARVC | Yes | Betablocker, Amiodarone | 238k | No Mut |
| ZH-013 | 50 | M | 1,0 | 0,0 | 1,0 | 1,0 | 0,1 | 1,0 | 4,1 | ARVC | Yes | Betablocker, Amiodarone | 1673k | PKP2 |
| ZH-015 | 26 | M | 1,0 | 0,0 | 1,0 | 0,0 | 0,1 | 1,0 | 3,1 | ARVC | Yes | Sotalol | 2511k | PKP2 |
| ZH-016 | 46 | M | 1,0 | 1,0 | 0,1 | 1,0 | 0,1 | 0,0 | 3,2 | ARVC | Yes | Sotalol, ACE-I/ARB, Aldosterone antagonist | 90 | TTN |
| ZH-017 | 52 | M | 1,0 | 0,0 | 0,1 | 0,1 | 1,0 | 0,0 | 2,2 | ARVC | Yes | Betablocker | 13k | No Mut |
| ZH-019 | 28 | M | 1,0 | 0,0 | 1,0 | 0,0 | 0,1 | 0,0 | 2,1 | ARVC | Yes | Betablocker | 7899k | DSG2 |
| ZH-025 | 60 | M | 1,0 | 1,0 | 0,1 | 0,0 | 0,0 | 1,0 | 3,1 | ARVC | Yes | Betablocker; Amiodarone | <500 | DSP |
| ZH-027 | 53 | F | 1,0 | 0,0 | 0,0 | 0,1 | 0,1 | 1,0 | 2,2 | ARVC | Yes | N/A | 8769k | PKP2 |
| ZH-028 | 59 | M | 1,0 | 0,0 | 0,0 | 0,1 | 0,1 | 1,0 | 2,2 | ARVC | Yes | Betablocker; ACE-I/ARB, Aldosterone antagonist | 92k | DSP |
| ZH-038 | 54 | M | 1,0 | 0,0 | 0,1 | 1,0 | 0,1 | 0,0 | 2,2 | ARVC | Yes | None | 138 | No Mut |
| ZH-051 | 43 | M | 1,0 | 0,0 | 1,0 | 1,0 | 1,0 | 0,0 | 4,0 | ARVC | Yes | None | 2203 | TTN, SCN5A |
| ZH-053 | 44 | M | 1,0 | 0,0 | 0,1 | 0,0 | 1,0 | 0,0 | 2,1 | ARVC | Yes | None | 6275k | LMNA |
| ZH-081 | 66 | F | 1,0 | 0,0 | 1,0 | 0,0 | 0,0 | 1,0 | 3,0 | ARVC | Yes | Betablocker, ACE-I/ARB | <500k | DSC2 |
| ZH-087 | 62 | F | 1,0 | 0,0 | 1,0 | 0,0 | 0,1 | 1,0 | 3,1 | ARVC | Yes | Betablocker | 1063k | No Mut |
| ZH-092 | 52 | F | 1,0 | 0,0 | 1,0 | 0,0 | 1,0 | 1,0 | 4,0 | ARVC | Yes | Sotalol | 901k | PKP2 |
| ZH-101 | 26 | M | 1,0 | 0,0 | 0,0 | 0,0 | 0,1 | 1,0 | 2,1 | ARVC | Yes | None | 1373k | PKP2 |
| ZH-102 | 20 | F | 1,0 | 0,0 | 1,0 | 0,0 | 1,0 | 1,0 | 4,0 | ARVC | Yes | None | 2005k | DSG2 |
| ZH-107 | 74 | M | 1,0 | 0,0 | 1,0 | 0,0 | 0,1 | 0,0 | 2,1 | ARVC | Yes | Amiodarone, ACE-I/ARB, Aldosterone antagonist | 209k | No Mut |
| ZH-121 | 45 | F | 1,0 | 0,0 | 1,0 | 1,0 | 1,0 | 0,0 | 4,0 | ARVC | Yes | Betablocker. Amiodarone | 26k | No Mut |
| ZH-123 | 51 | M | 1,0 | 0,0 | 0,1 | 0,0 | 1,0 | 0,0 | 1,2 | ARVC | Yes | Amiodarone | >500k | RYR2 |
| ZH-148 | 19 | F | 1,0 | 0,0 | 1,0 | 0,0 | 0,1 | 0,0 | 2,1 | ARVC | Yes | Amiodarone | 4499k | SCN5A |
| ZH-206 | 45 | M | 1,0 | 0,0 | 1,0 | 0,1 | 0,1 | 0,0 | 2,2 | ARVC | Yes | Betablocker, Aldosterone antagonist | 11 875k | DSG2 |
| ZH-213 | 38 | M | 1,0 | 0,0 | 1,0 | 0,0 | 1,0 | 1,0 | 4,0 | ARVC | Yes | Sotalol | <500k | DSG2 |
| ZH-222 | 37 | F | 1,0 | 0,0 | 1,0 | 0,0 | 1,0 | 1,0 | 4,0 | ARVC | Yes | Amiodarone | >500k | PKP2 |
Age at time of blood draw.
Global or regional dysfunction and structural alterations.
Tissue characterization of wall.
Repolarization abnormalities.
Depolarization/conduction abnormalities.
Arrhythmias.
Family history.
Medications/therapies patient was on during time of blood draw.
Premature ventricular contractions recorded from 24 h-Holter ECG closest to time of blood draw.
Gene in which mutation has been identified for patient (according to www.arvcdatabase.info, the American College of Medical Genetics and Genomics, and previous literature).
Patient was on medical therapy during Holter from which PVC count was recorded.
No mutations were identified in patient.
Borderline ARVC as defined by meeting one major and one minor criterion, or three minor criteria as defined by the 2010 Task Force Criteria for the diagnosis of ARVC.
Normal control sera were obtained from two commercial sources: 12 from Innovative Research Inc. Novi, MI, USA and 20 from BBI Solutions, Cardiff, UK. In addition, a cohort of subjects from Mayo clinic with hypertrophic cardiomyopathy (see Supplementary material online, Table S1) and a cohort of subjects from Zurich with dilated cardiomyopathy (see Supplementary material online, Table S2) served as non-ARVC cardiomyopathic controls. Finally, as additional validation, we assessed sera from 10 Boxer dogs (a naturally occurring animal model of ARVC)27 with manifest disease (see Supplementary material online, Table S3) compared with 18 unaffected dogs (2 mongrels and 16 Boxers) with no pedigree history of ARVC.
Autoantibody assessments
In 12 of 12 cases of definite ARVC in our human samples, and seven of eight cases of Borderline ARVC in our human samples, we found that autoantibodies to the desmosomal protein DSG2 were present on western blots, whereas these autoantibodies were absent (in 11) or very faint (in one) from sera of 12 control subjects from our first commercial source (Figure 1). No antibodies to desmocollin-2 or N-cadherin were identified in any sample.
Figure 1.

(A) Western blot images of serum interaction with cadherin proteins from 12 subjects with definite arrhythmogenic right ventricular cardiomyopathy, demonstrating anti-desmoglein-2 antibodies in all. (B) Western blot images of serum interaction with cadherin proteins from 8 subjects with borderline arrhythmogenic right ventricular cardiomyopathy, demonstrating anti-desmoglein-2 antibodies in seven of eight. (C) Western blot images of serum interaction of 12 control subjects with cadherin proteins, demonstrating only faint detection in one. For all panels, as we are assessing for the presence of serum antibodies, each set of proteins is exposed to one patient serum as an individual blot. ARVC, arrhythmogenic right ventricular cardiomyopathy.
In a subset of five subject and three control sera, we isolated the immunoglobulin G fraction, which also demonstrated similar antibody staining to DSG2 only (see Supplementary material online, Figure S1). A subset of sera was also assessed for antibodies to non-cadherin desmosomal proteins, as well as to skin cadherin proteins, with none found (see Supplementary material online, Figure S2). Signal levels for anti-DSG2 antibody varied between ARVC samples and were measured from western blots using quantitative analysis of the band densities as previously described.26,28
Anti-DSG2 antibodies were absent on western blot of serum from one subject with asymptomatic borderline ARVC. This individual was the mother of an identified ARVC subject with one borderline abnormal measurement of high frequency, low amplitude signal duration on SAECG. She has a very rare novel missense variant of PKP2 that has been classified as pathogenic (www.arvcdatabase.info) based on moderately reduced co-localization of connexin-43 gap junction protein to the cardiac intercalated disk.19 Anti-DSG2 antibodies were weakly present in one control sample from a 53-year-old Hispanic female, but did not reach the level seen in the 12 definite ARVC subjects with antibodies based on quantitation of western blot density, nor the seven of eight borderline ARVC cases that demonstrated antibody.
Within our validation cohort of 25 additional adult subjects with definite ARVC from the Zurich ARVC Program, we identified anti-DSG2 antibodies in all 25 (Figure 2A). We assessed a second set of 20 clinically well characterized control sera from another commercial source (BBI Solutions, Cardiff, UK) and did not identify anti-DSG2 antibodies in any (Figure 2B). In addition, no anti-DSG2 antibodies were identified among 10 subjects with hypertrophic cardiomyopathy or among six subjects with dilated cardiomyopathy (see Supplementary material online, Figure S3). Assessing the naturally occurring Boxer dog model of ARVC, we identified anti-DSG2 antibodies in 10 of 10 Boxers presenting with manifest disease, but in none of 18 healthy dogs with no pedigree history of ARVC (2 mongrel and 16 Boxer dogs; Figure 3).
Figure 2.

(A) Western blot images of serum interaction with cadherin proteins from 25 additional subjects with definite arrhythmogenic right ventricular cardiomyopathy from our validation cohort. (B) Western blot images of serum interaction with cadherin proteins from 20 additional control sera.
Figure 3.

(Top) Western blot images of serum interaction with cadherin proteins from 10 Boxer dogs affected by arrhythmogenic right ventricular cardiomyopathy. (Bottom) Western blot images of serum interaction with cadherin proteins from 18 healthy dogs with no pedigree history of arrhythmogenic right ventricular cardiomyopathy.
We also created an ELISA assay and characterized antibody concentration by O.D. in our control, borderline, and definite (primary and validation) subjects (Figure 4). Our ELISA assay provided comparable results, with a cut-point of O.D. = 0.1060 providing 97.78% sensitivity (95% confidence interval = 88.23–99.94%) and 96.88% specificity (95% confidence interval = 83.78–99.92%). A Receiver Operator Characteristic (ROC) curve (see Supplementary material online, Figure S4) demonstrates an area under the curve of 0.99.
Figure 4.

Dot plot of ELISA optical density for controls, borderline arrhythmogenic right ventricular cardiomyopathy subjects, and definite arrhythmogenic right ventricular cardiomyopathy subjects from our primary and validation cohorts. Error bars represent the mean value of each cohort (mid horizontal line) and standard deviation within each cohort. The two-tailed P-values between the indicated groups are exact values calculated using a Mann–Whitney U-test.
Correlation with disease severity
We assessed whether antibody density as measured by pixel count of the western blot or ELISA O.D. correlated with disease severity as measured by 24-h burden of PVCs within the groups of definite ARVC. The Pearson correlation coefficient (r) was 0.70 (P = 0.0001) for PVCs vs. pixel count of the western band, indicating that 49% of the variation in PVC count could be accounted for by its linear relationship with antibody density (Figure 5).
Figure 5.

Premature ventricular contraction burden per 24 h vs. antibody density, as measured by pixel count of the density of the western blot.
Antibody effects on gap junction function
To assess whether anti-DSG2 antibody might cause gap junction dysfunction, a common feature of ARVC,19 we tested purified IgG from two randomly selected ARVC patients as well as commercial anti-DSG2 antibodies for effects on cell-to-cell dye transfer. Using automated microinjection of fluorescent dye molecules into single cells (Figure 6)29 of confluent normal human iPSC-derived cardiomyocytes (I-Cell®, Cellular Dynamics International), we identified reduced dye transfer distances from each injected cell to its adjacent cells in those cultures exposed to patient-derived purified antibodies or commercial anti-DSG2 antibody, compared with those exposed to purified antibody from control sera (Figure 7). We injected between 37 and 42 cells for each assessment.
Figure 6.
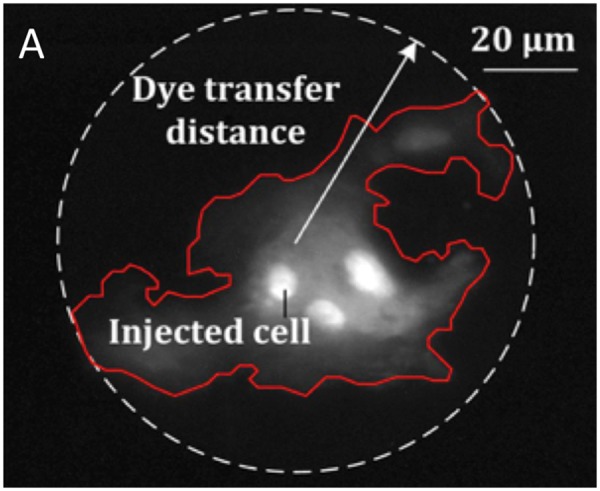
Gap junction function measurement. The gap junction function was assessed via microinjection of single cells with fluorescent molecules, measuring dye transfer to adjacent cells.
Figure 7.

Purified IgG antibodies from the serum of two patients with arrhythmogenic right ventricular cardiomyopathy, as well as commercial anti-DSG-2 IgG demonstrate reduced cell-to-cell dye transfer distance compared with cells exposed to IgG from normal human serum (P ≤ 0.0001 and P = 0.0025, respectively). The two-tailed P-values between the indicated groups are exact values calculated using a Mann–Whitney U-test. Error bars indicate standard deviation.
Identification of the antigen epitope
To identify the DSG2 epitope bound by ARVC serum autoantibodies, we assessed two random patient sera against a peptide library (Sigma-Aldrich Canada) of sequential 15 a.a. peptides (with 5 a.a. overlap) derived from the human DSG2 protein sequence. Strong binding of serum autoantibodies was identified against a.a. 485–531 and a.a. 586–610, within the fourth extracellular domain and extracellular anchor regions, respectively (Figure 8).
Figure 8.

Serum binding intensity to desmoglein-2 oligopeptides is presented as colour grades onto a Protter (http://wlab.ethz.ch/protter) plot of the desmoglein-2 protein (red, strong binding; orange, moderate biding; yellow, weak binding; green, faint binding). Strongest binding is in regions of the fourth extracellular cadherin and extracellular anchor domains.
Discussion
This study identified autoantibodies to the cardiac DSG2 protein as a common feature in the sera of patients with ARVC. These autoantibodies were specific to ARVC, as they were essentially absent in two independent sets of control sera, as well as sera from subjects with other forms of heritable cardiomyopathy. Assessment of anti-DSG2 antibodies may provide a reliable assay for detection of ARVC patients undergoing evaluation. Anti-DSG2 antibodies were confirmed within a validation cohort and were present independent of whether an underlying genetic defect had been identified, and independent of the specific ARVC gene when known. Anti-DSG2 antibodies were also demonstrated to be specific and sensitive for disease in the naturally occurring Boxer dog model of ARVC, providing the potential for a veterinary test for this disease, as well as a potential large animal model for assessment of immune therapies for ARVC. In contrast to desmosomal disorders of the skin, which are caused by either mutations or autoantibodies, our ARVC subjects had anti-DSG2 antibodies usually in the presence of familial and/or genetic disease.
Prognostically, the level of anti-DSG2 antibodies correlated with disease burden as measure by PVC count, and purified antibodies resulted in reduced gap junction function, an identified common pathway in ARVC pathophysiology. This latter finding is consistent with a previous study that demonstrated that anti-N-cadherin antibody Fab fragments result in failure to form adherens junctions and inhibit gap junction dye transfer in an experimental cell model.30 As such, our test provides a quantitative result that may contribute to the evaluation of risk within diagnosed ARVC patients, or predict hot phases of disease.
Desmoglein-2 is one of several cardiac cadherin proteins. Cadherins are calcium-dependent adhering molecules providing mechanical attachment between cells in multiple tissues.31 Typically, three calcium ions (12 in total) are pocketed into binding motifs present between each pair of five successive extracellular cadherin (EC) domains, providing the crescent shape required to bring cadherin domains from opposing cells into a 90° conformation for trans binding. This 90° conformation is critical for binding as it allows for tryptophan residues on each EC1 domain to be inserted simultaneously into the hydrophobic pocket of the alternate cadherin. Interfering with this orientation, such as through missense mutations, or antibody binding to proximal extracellular DSG2 domains, might be expected to reduce binding and adhesion.
Recently, it has been recognized that cell-to-cell binding at desmosomal structures of the cardiac intercalated disk is heterophilic, with desmogleins typically forming adhesive dimers with desmocollins.32 The ontogeny of the expression of intercalated disk structures from 15 weeks gestation through 11 years postnatal has been assessed, with structural (adherens junction and desmosomal) proteins localizing to the intercalated disk at 1 year after birth, followed by the cardiac sodium channel at 2 years and gap junction connexin 43 protein at 7 years.33 Within proteins of the intercalated disk, DSG2 does not appear to localize to the intercalated disk until 6 weeks of postnatal life, whereas DSC2 is present in the intercalated disk prenatally.34
We do not yet fully understand the pathobiology responsible for an autoimmune response to DSG2 in ARVC patients. We also do not know to what degree autoimmunity participates in the pathophysiology of ARVC. It is plausible that just as catenins are released into the sarcoplasm in the face of mutations that disrupt the desmosome, DSG2 protein may include ‘cryptic’ epitopes,35 which are also exposed or released into the intercellular space and/or circulation and as a result of desmosomal mutations. Such released DSG2 proteins may link with an antigen-presenting cell to stimulate a T-cell response, generating the observed autoantibodies. Unmasking of cryptic epitopes by gene mutations could contribute to other forms of autoimmunity. Alternately, the failure of tolerance for the DSG2 epitope may be related to its relatively late expression at or around 6 weeks of infancy.34 Although three subjects had sporadic ARVC, it would be premature to suggest that autoantibodies cause ARVC in isolation, as ARVC is recognized to be incompletely penetrant, variably expressed and often gene-elusive.
Our results are promising for the development of a serological test for the diagnosis of ARVC. The high sensitivity and specificity of this assay is anticipated to improve the diagnosis of ARVC, possibly even in the pre-symptomatic period. Cardiac tissue sections have been used by others to assess for the presence of anti-heart or anti-intercalated disk antibodies in myocarditis and dilated cardiomyopathy,36 as well as ARVC.37 The frequencies of anti-heart or anti-intercalated disk antibodies were higher (31%; 15%) in ARVC, than in non-ischaemic cardiomyopathy, ischaemic heart failure, or normal subjects.38 Antibodies were associated with PKP2 and DSP mutation-positive individuals, and with those with reduced left ventricular function. However, the intercalated disk is known to contain more than 700 different proteins based on Human Protein Atlas.39 Our technique assays autoantibodies to a single-specific isolated protein, and provides very high sensitivity for the diagnosis of ARVC. A diagnostic test for ARVC based on reduced intercalated disc plakoglobin immunofluorescence in endomyocardial biopsies has been previously proposed40 and critiqued41,42 but has not been adopted in practice. Our serum blood test provides a more practical and accurate alternative.
The identification of an autoantibody associated with ARVC may open new therapeutic avenues, similar to those proposed for autoimmune pemphigus vulgaris.25 Immunosuppression is often effective for disorders resulting from autoimmunity and is usually the standard to which other therapies are compared. Newer therapies include co-stimulation blockade, regulatory T-cell therapy, antigen-specific immunotherapy, and manipulation of the interleukin-2 pathway.43 Each of these may provide an avenue for therapy in ARVC, once studies of the mechanism of autoimmunity have been completed. Of particular interest is a recent report of reengineered chimeric antigen receptor T-cells to target autoimmunity specifically, without off-target immuno-suppression.44 This technique was applied successfully to treat a murine model of pemphigus vulgaris due to anti-desmoglein-3 antibodies, and may hold promise for an effective targeted therapy in ARVC. Finally, a ‘negative vaccination’ approach exposing at-risk subjects to the DSG2 antigen early in life might provide a specific and potentially safe approach to preventing autoimmunity in ARVC, similar to therapies under study for Type 1 diabetes.45
Take home figure.
(Left panel) Mutations within the cardiomyocyte desmosomal complex result in release of desmoglein-2 into the circulation, stimulating an autoimmune response. (Right panel) Anti-desmoglein-2 antibodies result in distortion of desmoglein-2 and failure to crosslink.
Limitations
Our study evaluated a limited number of commercial control sera from ostensibly normal subjects for which we had limited clinical information, as well as a limited number of control sera from two other specific forms of cardiomyopathy: hypertrophic cardiomyopathy and dilated cardiomyopathy. Other cardiomyopathic processes including myocarditis and sarcoidosis should be assessed to determine if anti-DSG2 antibodies will also be detected in these disorders. Viral myocarditis has been associated with ARVC in some patients as an acquired cause of ARVC disease.37 We have not yet assessed these disorders to further assess the specificity of our test.
For this initial study, we have not tested individuals in whom the disease has yet to manifest, and therefore cannot characterize the ability of this test to detect silent mutation carriers at this time.
Conclusion
Our discovery will enable the development of a new serological test for ARVC that can easily be adapted to clinical diagnosis and the prediction of disease. The assay is negative in normal control individuals and also negative in two other forms of cardiomyopathy, but further assessment is required among other myocardial diseases to determine its specificity to ARVC. Further research is required to elucidate the pathophysiology of this autoimmune response, its sensitivity in predicting disease risk or hot phases of disease, and its utility in directing or monitoring new therapies for ARVC.
Supplementary Material
Acknowledgements
The authors are grateful to Stephen W. Scherer, Ronald D. Cohn, and Michael W. Salter for manuscript review, to technician Yanting Wang for technical assistance, to the many patients who participated as subjects for this study and to the owners, breeders, and veterinarians contributing samples to the Boxer dog studies.
Funding
This study was funded by an innovation grant from the Labatt Family Heart Centre to R.M.H. and also was made possible by materials and data from a Canadian Institutes of Health Research Team Grant (2009 to 2014) to R.M.H. The Caitlyn Elizabeth Morris Memorial Foundation, Alex Corrance Memorial Foundation and Meredith Cartwright, LLB also provided funding support. Further studies are also being funded y a HSBC Bank Canada Catalyst Research Grant from The Hospital for Sick Children. T.T.K. and K.M. are supported by Carter Heart Rhythm Fellowships. The corresponding author has submitted a patent application covering this work. The Zurich ARVC Program is supported by grants from the Georg und Bertha Schwyzer-Winiker Foundation, the Baugarten Foundation, and the Swiss National Science Foundation.
Conflict of interest: M.J.A. is a consultant for Audentes Therapeutics, Boston Scientific, Gilead Sciences, Invitae, Medtronic, MyoKardia, and St. Jude Medical. M.J.A. and Mayo Clinic have an equity/royalty relationship with AliveCor, Blue Ox Health, and StemoniX. However, none of these entities had any involvement with this study. All other authors have declared no conflict of interest.
Footnotes
This paper was guest edited by Anthony N. DeMaria, MD, University of California La Jolla, USA, ademaria@ucsd.edu.
See page 3945 for the editorial comment on this article (doi: 10.1093/eurheartj/ehy410)
References
- 1. Thiene G, Nava A, Corrado D, Rossi L, Pennelli N.. Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med 1988;318:129–133. [DOI] [PubMed] [Google Scholar]
- 2. Tabib A, Loire R, Chalabreysse L, Meyronnet D, Miras A, Malicier D, Thivolet F, Chevalier P, Bouvagnet P.. Circumstances of death and gross and microscopic observations in a series of 200 cases of sudden death associated with arrhythmogenic right ventricular cardiomyopathy and/or dysplasia. Circulation 2003;108:3000–3005. [DOI] [PubMed] [Google Scholar]
- 3. Corrado D, Basso C, Schiavon M, Thiene G.. Screening for hypertrophic cardiomyopathy in young athletes. N Engl J Med 1998;339:364–369. [DOI] [PubMed] [Google Scholar]
- 4. Pilmer CM, Kirsh JA, Hildebrandt D, Krahn AD, Gow RM.. Sudden cardiac death in children and adolescents between 1 and 19 years of age. Heart Rhythm 2014;11:239–245. [DOI] [PubMed] [Google Scholar]
- 5. Basso C, Corrado D, Marcus FI, Nava A, Thiene G.. Arrhythmogenic right ventricular cardiomyopathy. Lancet 2009;373:1289–1300. [DOI] [PubMed] [Google Scholar]
- 6. McKenna WJ, Thiene G, Nava A, Fontaliran F, Blomstrom-Lundqvist C, Fontaine G, Camerini F.. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology. Br Heart J 1994;71:215–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, Calkins H, Corrado D, Cox MG, Daubert JP, Fontaine G, Gear K, Hauer R, Nava A, Picard MH, Protonotarios N, Saffitz JE, Sanborn DM, Steinberg JS, Tandri H, Thiene G, Towbin JA, Tsatsopoulou A, Wichter T, Zareba W.. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the Task Force Criteria. Eur Heart J 2010;31:806–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Corrado D, Wichter T, Link MS, Hauer R, Marchlinski F, Anastasakis A, Bauce B, Basso C, Brunckhorst C, Tsatsopoulou A, Tandri H, Paul M, Schmied C, Pelliccia A, Duru F, Protonotarios N, Estes NA 3rd, McKenna WJ, Thiene G, Marcus FI, Calkins H.. Treatment of arrhythmogenic right ventricular cardiomyopathy/dysplasia: an international task force consensus statement. Eur Heart J 2015;36:3227–3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Beffagna G, Occhi G, Nava A, Vitiello L, Ditadi A, Basso C, Bauce B, Carraro G, Thiene G, Towbin JA, Danieli GA, Rampazzo A.. Regulatory mutations in transforming growth factor-beta3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc Res 2005;65:366–373. [DOI] [PubMed] [Google Scholar]
- 10. Gerull B, Heuser A, Wichter T, Paul M, Basson CT, McDermott DA, Lerman BB, Markowitz SM, Ellinor PT, MacRae CA, Peters S, Grossmann KS, Drenckhahn J, Michely B, Sasse-Klaassen S, Birchmeier W, Dietz R, Breithardt G, Schulze-Bahr E, Thierfelder L.. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat Genet 2004;36:1162–1164. [DOI] [PubMed] [Google Scholar]
- 11. Merner ND, Hodgkinson KA, Haywood AF, Connors S, French VM, Drenckhahn JD, Kupprion C, Ramadanova K, Thierfelder L, McKenna W, Gallagher B, Morris-Larkin L, Bassett AS, Parfrey PS, Young TL.. Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am J Hum Genet 2008;82:809–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Otten E, Asimaki A, Maass A, van Langen IM, van der Wal A, de Jonge N, van den Berg MP, Saffitz JE, Wilde AA, Jongbloed JD, van Tintelen JP.. Desmin mutations as a cause of right ventricular heart failure affect the intercalated disks. Heart Rhythm 2010;7:1058–1064. [DOI] [PubMed] [Google Scholar]
- 13. Pilichou K, Nava A, Basso C, Beffagna G, Bauce B, Lorenzon A, Frigo G, Vettori A, Valente M, Towbin J, Thiene G, Danieli GA, Rampazzo A.. Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation 2006;113:1171–1179. [DOI] [PubMed] [Google Scholar]
- 14. Rampazzo A, Nava A, Malacrida S, Beffagna G, Bauce B, Rossi V, Zimbello R, Simionati B, Basso C, Thiene G, Towbin JA, Danieli GA.. Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet 2002;71:1200–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Syrris P, Ward D, Evans A, Asimaki A, Gandjbakhch E, Sen-Chowdhry S, McKenna WJ.. Arrhythmogenic right ventricular dysplasia/cardiomyopathy associated with mutations in the desmosomal gene desmocollin-2. Am J Hum Genet 2006;79:978–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tiso N, Stephan DA, Nava A, Bagattin A, Devaney JM, Stanchi F, Larderet G, Brahmbhatt B, Brown K, Bauce B, Muriago M, Basso C, Thiene G, Danieli GA, Rampazzo A.. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum Mol Genet 2001;10:189–194. [DOI] [PubMed] [Google Scholar]
- 17. van Hengel J, Calore M, Bauce B, Dazzo E, Mazzotti E, De Bortoli M, Lorenzon A, Li Mura IE, Beffagna G, Rigato I, Vleeschouwers M, Tyberghein K, Hulpiau P, van Hamme E, Zaglia T, Corrado D, Basso C, Thiene G, Daliento L, Nava A, van Roy F, Rampazzo A.. Mutations in the area composita protein alphaT-catenin are associated with arrhythmogenic right ventricular cardiomyopathy. Eur Heart J 2013;34:201–210. [DOI] [PubMed] [Google Scholar]
- 18. Asimaki A, Syrris P, Wichter T, Matthias P, Saffitz JE, McKenna WJ.. A novel dominant mutation in plakoglobin causes arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet Nov 2007;81:964–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mayosi BM, Fish M, Shaboodien G, Mastantuono E, Kraus S, Wieland T, Kotta MC, Chin A, Laing N, Ntusi NB, Chong M, Horsfall C, Pimstone SN, Gentilini D, Parati G, Strom TM, Meitinger T, Pare G, Schwartz PJ, Crotti L.. Identification of cadherin 2 (CDH2) mutations in arrhythmogenic right ventricular cardiomyopathy. Circ Cardiovasc Genet 2017;10:e001605.. [DOI] [PubMed] [Google Scholar]
- 20. James CA, Bhonsale A, Tichnell C, Murray B, Russell SD, Tandri H, Tedford RJ, Judge DP, Calkins H.. Exercise increases age-related penetrance and arrhythmic risk in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. J Am Coll Cardiol 2013;62:1290–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Garcia-Gras E, Lombardi R, Giocondo MJ, Willerson JT, Schneider MD, Khoury DS, Marian AJ.. Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J Clin Invest 2006;116:2012–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fidler LM, Wilson GJ, Liu F, Cui X, Scherer SW, Taylor GP, Hamilton RM.. Abnormal connexin43 in arrhythmogenic right ventricular cardiomyopathy caused by plakophilin-2 mutations. J Cell Mol Med 2009;13:4219–4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Campuzano O, Alcalde M, Iglesias A, Barahona-Dussault C, Sarquella-Brugada G, Benito B, Arzamendi D, Flores J, Leung TK, Talajic M, Oliva A, Brugada R.. Arrhythmogenic right ventricular cardiomyopathy: severe structural alterations are associated with inflammation. J Clin Pathol 2012;65:1077–1083. [DOI] [PubMed] [Google Scholar]
- 24. Saguner AM, Brunckhorst C, Duru F.. Arrhythmogenic ventricular cardiomyopathy: a paradigm shift from right to biventricular disease. World J Cardiol 2014;6:154–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Koga H, Tsuruta D, Ohyama B, Ishii N, Hamada T, Ohata C, Furumura M, Hashimoto T.. Desmoglein 3, its pathogenecity and a possibility for therapeutic target in pemphigus vulgaris. Expert Opin Ther Targets 2013;17:293–306. [DOI] [PubMed] [Google Scholar]
- 26. Chatterjee-Chakraborty M, Chatterjee D.. Artificial rearing inhibits apoptotic cell death through action on pro-apoptotic signaling molecules during brain development: replacement licking partially reverses these effects. Brain Res 2010;1348:10–20. [DOI] [PubMed] [Google Scholar]
- 27. Basso C, Fox PR, Meurs KM, Towbin JA, Spier AW, Calabrese F, Maron BJ, Thiene G.. Arrhythmogenic right ventricular cardiomyopathy causing sudden cardiac death in boxer dogs: a new animal model of human disease. Circulation 2004;109:1180–1185. [DOI] [PubMed] [Google Scholar]
- 28. Chatterjee D, Chatterjee-Chakraborty M, Rees S, Cauchi J, de Medeiros CB, Fleming AS.. Maternal isolation alters the expression of neural proteins during development: ′Stroking′ stimulation reverses these effects. Brain Res 2007;1158:11–27. [DOI] [PubMed] [Google Scholar]
- 29. Liu J, Siragam V, Gong Z, Chen J, Fridman MD, Leung C, Lu Z, Ru C, Xie S, Luo J, Hamilton RM, Sun Y.. Robotic adherent cell injection for characterizing cell-cell communication. IEEE Trans Bio-Med Eng 2015;62:119–125. [DOI] [PubMed] [Google Scholar]
- 30. Meyer RA, Laird DW, Revel JP, Johnson RG.. Inhibition of gap junction and adherens junction assembly by connexin and A-CAM antibodies. J Cell Biol 1992;119:179–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Brasch J, Harrison OJ, Honig B, Shapiro L.. Thinking outside the cell: how cadherins drive adhesion. Trends Cell Biol 2012;22:299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Harrison OJ, Brasch J, Lasso G, Katsamba PS, Ahlsen G, Honig B, Shapiro L.. Structural basis of adhesive binding by desmocollins and desmogleins. Proc Natl Acad Sci USA 2016;113:7160–7165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vreeker A, van Stuijvenberg L, Hund TJ, Mohler PJ, Nikkels PG, van Veen TA.. Assembly of the cardiac intercalated disk during pre- and postnatal development of the human heart. PLoS One 2014;9:e94722.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kessler EL, Nikkels PG, van Veen TA.. Disturbed desmoglein-2 in the intercalated disc of pediatric patients with dilated cardiomyopathy. Hum Pathol Jul 2017;67:101–108. [DOI] [PubMed] [Google Scholar]
- 35. Tizard IR. Autoimmunity: general principles In: Tizard IR, ed. Veterinarry Immunology. 9th ed.St. Louis, MO: Elsevier Saunders; 2013, pp. 400–407. [Google Scholar]
- 36. Caforio AL, Mahon NG, McKenna WJ.. Clinical implications of anti-cardiac immunity in dilated cardiomyopathy. Ernst Schering Res Found Workshop 2006;169–193. [DOI] [PubMed] [Google Scholar]
- 37. Caforio AL, Zachara E, Vinci A, Re F, Baratta P, Tona F, Thiene G, Iliceto S, Tondo C, McKenna WJ. Serum organ-specific anti-heart autoantibodies in arrhythmogenic right ventricular cardiomyopathy patients and relatives: evidence for autoimmune involvement in a genetically-determined myocarditis. Circulation 2008;118:S948. [Google Scholar]
- 38. Caforio AL, Zachara E, Re F, Baratta P, Bagato F, Thiene G, Iliceto S, Syrris P, McKenna WJ. Serum anti-intercalated disk autoantibodies in arrhythmogenic right ventricular cardiomyopathy are autoimmune markers associated with pathogenic desmosomal mutations. Circulation 2011;124:A13404. [Google Scholar]
- 39. Ponten F, Jirstrom K, Uhlen M.. The human protein atlas—a tool for pathology. J Pathol 2008;216:387–393. [DOI] [PubMed] [Google Scholar]
- 40. Asimaki A, Tandri H, Huang H, Halushka MK, Gautam S, Basso C, Thiene G, Tsatsopoulou A, Protonotarios N, McKenna WJ, Calkins H, Saffitz JE.. A new diagnostic test for arrhythmogenic right ventricular cardiomyopathy. N Engl J Med 2009;360:1075–1084. [DOI] [PubMed] [Google Scholar]
- 41. Kohn MA, Newman TB.. Arrhythmogenic right ventricular cardiomyopathy. N Engl J Med 2009;360:2784–2785; author reply 2785–2786. [PubMed] [Google Scholar]
- 42. Tavora F, Creswell N, Burke AP.. Arrhythmogenic right ventricular cardiomyopathy. N Engl J Med 2009;360:2784; author reply 2785–2786. [DOI] [PubMed] [Google Scholar]
- 43. Rosenblum MD, Gratz IK, Paw JS, Abbas AK.. Treating human autoimmunity: current practice and future prospects. Sci Transl Med 2012;4:125sr1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ellebrecht CT, Bhoj VG, Nace A, Choi EJ, Mao X, Cho MJ, Di Zenzo G, Lanzavecchia A, Seykora JT, Cotsarelis G, Milone MC, Payne AS.. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science 2016;353:179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Harrison LC, Wentworth JM, Zhang Y, Bandala-Sanchez E, Bohmer RM, Neale AM, Stone NL, Naselli G, Bosco JJ, Auyeung P, Rashidi M, Augstein P, Morahan G.. Antigen-based vaccination and prevention of type 1 diabetes. Curr Diab Rep 2013;13:616–623. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.