

Abstract
Motivation
Co-localization of trait associated SNPs for specific transcription-factor binding sites or regulatory regions in the genome can yield profound insight into underlying causal mechanisms. Analysis is complicated because the truly causal SNPs are generally unknown and can be either SNPs reported in GWAS studies or other proxy SNPs in their linkage disequilibrium. Hence, a comprehensive pipeline for SNP co-localization analysis that utilizes all relevant information about both the genotyped SNPs and their proxies is needed.
Results
We developed an R package snpEnrichR for SNP co-localization analysis. The software integrates different tools for random SNP generation and genome co-localization analysis to automatize and help users to create custom SNP co-localization analysis. We show via an example that including proxy SNPs in SNP co-localization analysis enhances the sensitivity of co-localization detection.
Availability and implementation
The software is available at https://github.com/kartiek/snpEnrichR.
1 Introduction
Assessing co-localization of SNPs on given genomic regions requires an empirical hypothesis test. For a given population, SNPs have several quantifiable properties, such as allele frequency, the number of SNPs in linkage disequilibrium (LD), distance to nearest gene and gene density, which can be used to draw random sets of SNPs that have similar characteristics as the original SNP set. Such an empirical randomization approach provides a calibrated null distribution for co-localization analysis.
Genome-wide association studies have successfully linked SNPs to various traits. So-called tag-SNPs are generally consireded as proxies for causal SNPs. Because it is difficult to pinpoint the actual causal SNPs to a phenotype, taking other SNPs in their LD into account may enhance the sensitivity of the co-localization analysis.
2 Materials and methods
R package snpEnrichR facilitates SNP co-localization analysis by computing required statistics and integrates to several existing tools to enable efficient and automated data management for the analysis. The package consists of five main functions: (i) getSNPs retrieves trait associated SNPs directly from the NHGRI-EBI GWAS Catalog (MacArthur et al., 2017). Alternatively, user can manually provide custom SNP lists. (ii) clumpSNPs detects linked SNPs in a list, removes the correlated SNPs, and returns a list of (decorrelated) tag-SNPs. Removing correlated SNPs from a SNP list is needed to avoid biases in random SNP set generation. (iii) submitSNPsnap connects to SNPsnap server (Pers et al., 2015) and sends a retrieval request to generate a specified number of randomly sampled SNP sets. Each set consists of randomly sampled SNPs that have similar properties as the list of (decorrelated) tag-SNPs. (iv) findProxies expands a list of SNPs with all linked SNPs within a genomic distance d and above a correlation level r2 that are set by the user. (v) analyzeEnrichment computes the overlap between the genomic regions and each of the randomly sampled SNP sets that are extended to contain all SNPs that are in LD. These overlap scores form an empirical null distribution for the hypothesis test, and the empirical P-value is computed the standard way by counting the number of times randomly sampled SNP sets have at least as many overlaps with the genomic regions as the original input SNP set (which is also extended with LD SNPs). Empirical P-values are computed for all input SNP lists (e.g. different diseases) separately and the obtained P-values are corrected for multiple testing by the Benjamini–Hochberg method providing false discovery rate (FDR) values.
The functions can be easily used as the basis of SNP co-localization analysis pipeline. External tools are required only to liftover different genomic builds to correspond to each other, such as, e.g. GWAS catalog uses build GRCh38 whereas SNPsnap relies on GRCh37. Due to the dependency of an external server and the resulting time lag in random SNP set generation, we suggest that pipeline should be run in two phases. snpEnrichR requires R packages RSelenium, readr, dplyr, httr, utils, parallel, rtracklayer and GenomicFeatures, and external software PLINK version 1.9 (Chang et al., 2015).
2.1 Input files
snpEnrichR requires three user-specified data sources: (i) a list of genomic regions, (ii) a list of trait associated SNPs, (iii) a processed version of 1000 Genomes Project phase 3 SNP data for the studied population in a format supported by PLINK, i.e. a sample information file (.bed), a binary biallelic genotype table (.bim) and an extended set variant information file (.fam) (The 1000 Genomes Project Consortium, 2015). In our analyses, 1000 genomes data is annotated based on the genome coordinates, long indels and duplicate variants have been removed, and the data is filtered with the same quality control criteria used by SNPsnap, i.e. minimum minor allele frequency is 0.01, Hardy–Weinberg equilibrium test’s P-value is 10–6 and maximum missing genotype rate is 0.1. Note that in snpEnrichR the SNP files can be directly accessed from NHGRI-EBI GWAS Catalog database and 1000 genomes data is preprocessed into PLINK compatible format for convenience. All data is mapped into human genome assembly hg19 and represented in one-based coordinate system.
3 Example use case
To illustrate the utility and features of the tool, we applied it for studying SNP co-localization in transcription factor STAT6 binding sites in human CD4+ T cells during early Th2 cell differentiation (Elo et al., 2010). We downloaded STAT6 binding sites from Gene Transcription Regulation Database (GTRD) which hosts transcription factor binding sites identified by ChIP-seq experiments (Yevshin et al., 2017). The data consisted of STAT6 binding sites from five samples (EXP000514,…, EXP000518) of one biological replicate. After merging overlapping bindings sites, there are 15340 binding sites. The median length of the binding sites is 421. We fetched tag-SNPs of 11 immune-related and three non-immune related diseases/traits in European ancestry from NHGRI-EBI Catalog, and we removed tag-SNPs from HLA region and converted the coordinates into hg19 assembly. We used the snpEnrichR analysis pipeline with LD block parameters (d = 100 kb and r2 = 0.8) and used 1000 randomly generated SNPs sets when computing empirical P-values.
We used the tool to implement two analyses. The first pipeline computes the standard co-localizations using only the tag-SNPs whereas the second considers the proxy SNPs as well. Figure 1 shows that including proxy SNPs enhances the sensitivity of co-localization analysis. When considering the tag-SNPs only, two of the immune-related trait specific SNP co-localizations were detected. Whereas, five additional traits were identified as significantly enriched at STAT6 binding sites when proxy SNPs were taken into account. In addition, the inclusion of the proxy SNPs did not cause artificial co-localization signal for non-immune related traits where the tag-SNPs did not co-localize with STAT6 binding sites.
Fig. 1.
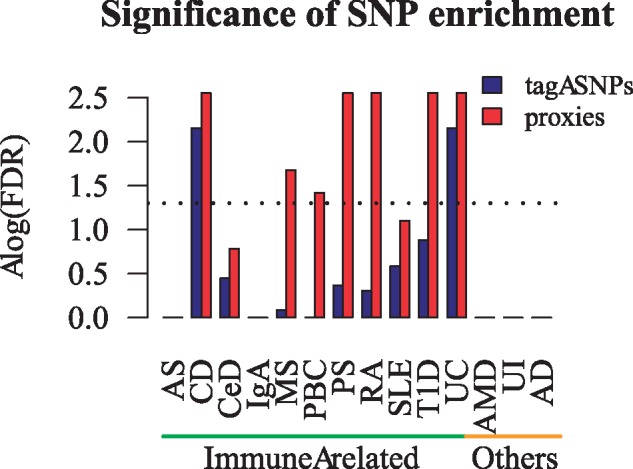
Co-localization results for SNPs from 11 immune-related and 3 non-immune-related diseases in STAT6 binding sites in human CD4+ T cells during early differentiation. Dashed line corresponds to the corrected P-value (FDR) of 0.05
4 Discussion and conclusion
We have implemented R package snpEnrichR to facilitate automated SNP co-localization analysis. The tool provides all major functionalities needed in co-localization analysis: an interface to fetch trait specific SNPs, detection and filtering tool for clumped SNPs, access to a web server that uses the best practises in generating random SNP sets that maintain characteristics of a given input SNP set, and the computation of proxy SNPs as well as co-localization tests. snpEnrichR; R package also enables flexible and easy integration to related analyses a user may have. Additional examples of this approach were recently reported (Tripathi et al., 2017; Ullah et al., 2018).
Funding
This work has been supported by the Academy of Finland [Centre of Excellence in Molecular Systems Immunology and Physiology Research (2012-2017) grant 250114; as well as the project 292832]. R.L. was supported by the Academy of Finland (AoF) grants 292335, 294337, 292482, 31444 and by grants from the JDRF, the Sigrid Jusélius Foundation and the Finnish Cancer Foundation.
Conflict of Interest: none declared.
References
- Chang C.C. et al. (2015) Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience, 4, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elo L.L. et al. (2010) Genome-wide profiling of interleukin-4 and STAT6 transcription factor regulation of human Th2 cell programming. Immunity, 32, 852–862. [DOI] [PubMed] [Google Scholar]
- MacArthur J. et al. (2017) The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res., 45, D896–D901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pers T.H. et al. (2015) SNPsnap: a Web-based tool for identification and annotation of matched SNPs. Bioinformatics, 31, 418–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The 1000 Genomes Project Consortium (2015) A global reference for human genetic variation. Nature, 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi S.K. et al. (2017) Genome-wide analysis of STAT3 mediated transcription during early human Th17 cell differentiation. Cell Rep., 19, 1888–1901. [DOI] [PubMed] [Google Scholar]
- Ullah U. et al. (2018) Transcriptional Repressor HIC1 Contributes to Suppressive Function of Human Induced Regulatory T Cells. Cell Rep., 22, 2094–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yevshin I.S. et al. (2017) GTRD: a database of transcription factor binding sites identified by ChIP-seq experiments. Nucleic Acids Res., 45, D61–D67. [DOI] [PMC free article] [PubMed] [Google Scholar]