

Abstract
Motivation
High-parameter single-cell technologies can reveal novel cell populations of interest, but studying or validating these populations using lower-parameter methods remains challenging.
Results
Here, we present GateFinder, an algorithm that enriches high-dimensional cell types with simple, stepwise polygon gates requiring only two markers at a time. A series of case studies of complex cell types illustrates how simplified enrichment strategies can enable more efficient assays, reveal novel biomarkers and clarify underlying biology.
Availability and implementation
The GateFinder algorithm is implemented as a free and open-source package for BioConductor: https://nalab.stanford.edu/gatefinder.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Modern single-cell analysis platforms can measure up to 42 anti-body-based markers per cell in normal and malignant tissues (Han et al., 2015; Inoue et al., 2016). Such high-parameter cytometry data enables the identification of novel cell populations defined by numerous, unexpected or unintuitive combinations of markers. A host of computational tools are available to facilitate clustering of cell phenotypes in n-dimensional cytometry data (Saeys et al., 2016; Weber and Robinson, 2016), but human interpretation of these clusters can be challenging due to biologically irrelevant or redundant markers and the vast number of possible marker combinations.
The computational method introduced here, termed GateFinder, constructs simple phenotypic signatures for target cell populations identified through high-dimensional single-cell cytometry. By inspecting all possible two-dimensional spaces and prioritizing the most informative markers, GateFinder provides a series of polygon filters (‘gates’) that best discriminate the target cell population from all other cells. The practical uses for this concise visual description of a target population include (i) designing fluorescence-activated cell sorting strategies for physical isolation of cells, (ii) communicating novel cell types in figures for publication, (iii) creating purpose-built assays for large-scale studies, or (iv) facilitating assignment into cell ontogenies (Bakken et al., 2017).
2 Materials and methods
Multi-parameter cytometry data can be conceptualized as a cloud of points in n-dimensional space. Selecting cells from this high-parameter cloud using a low-dimensional filter, or gate, is fundamentally imperfect and entails a tradeoff in terms of purity (i.e. the proportion of the captured cells that were desired by the researcher) and yield (i.e. the proportion of desired cells that were successfully captured). The goal of the GateFinder algorithm is to produce a series of two-dimensional polygon gates that best discriminates the target cells from non-target cells according to the user’s desired balance of purity and yield.
The typical GateFinder workflow begins with the researcher selecting a target cell population of interest such as a manually gated sub-population or the output of a clustering algorithm (Fig. 1A, left panel). Depending on the goals of the analysis, the parameter(s) that were originally used to define the target population may be withheld from the GateFinder algorithm. Briefly, the algorithm takes a randomly selected sub-sample of the complete high-dimensional dataset and projects it in all possible two-dimensional scatter plots (i.e. 861 plots for a 42-parameter dataset; Fig. 1A, second panel; see Supplementary Material for details). For each scatter plot of target cells, a bootstrapped outlier-detection test is used to eliminate outliers based on a user-defined threshold. Next, a non-convex polygon gate is constructed around the remaining target cells on each scatter plot, which reflects the type of gates manually drawn by researchers. The enrichment of target cells achieved by each gate is quantified by the F-measure statistic (i.e. the harmonic mean of purity and yield). The gate that achieves the best enrichment of the target cells is selected as the first gate in the overall gating strategy, and the markers defining that gate are excluded from subsequent rounds of the algorithm (Fig. 1A, third panel). These steps are then repeated on the cells within the gate, attempting to further enrich for the target cell population using only the remaining markers. This process is repeated until all markers have been exhausted, with each marker being used exactly once. Finally, to produce a robust and reproducible gating strategy (Fig. 1A, right panel), the entire gate-finding sequence is repeated several times (five times by default) using unique random sub-samples of the original data to avoid sub-optimal solutions. In cases where exploration of all sub-spaces is not feasible, GateFinder uses a supervised feature selection algorithm to limit the search to the most relevant markers (see Supplementary Material for details).
Fig. 1.
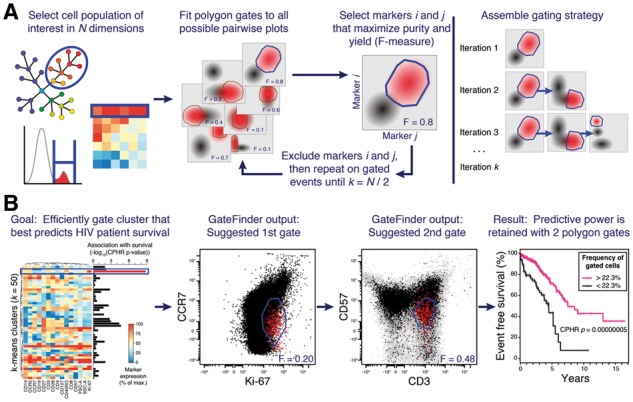
GateFinder produces optimal serial gating strategies for high-dimensional single-cell populations. (A) The researcher selects a target cell population using any number of dimensions (left panel). The algorithm searches all possible pairwise plots for the convex polygon gate that best segregates the target population (red) from the other cells in the dataset (black; center panels). To assemble a serial gating strategy, the search is repeated on the selected cells, ignoring markers used by earlier gates, until no markers are remaining (right panel). (B) In this example, the target population was defined as the k-means cluster that best predicted survival in 15-parameter polychromatic cytometry data an HIV cohort (Cluster 3; left panel, blue rectangle). Association with survival was quantified by Cox proportional hazards ratio. The first two gates from the serial gating strategy produced by GateFinder (center panels) clarified the phenotype of Cluster 3 as CCR7dim Ki-67dim CD57dim CD3+. The association with clinical outcome was nearly as strong in the cell population captured by the first two gates as it was in the 15-dimensional target population, Cluster 3 (right panel)
3 Results
In the arena of clinical research, GateFinder has great potential to translate a complex phenotypic signature identified during the discovery phase into a concise, readily interpretable, low-cost assay suitable for clinical validation. As an example, we re-analyzed a public 15-parameter digital optical flow cytometry dataset containing measurements of peripheral blood from HIV-positive patients (n = 466; Ganesan et al., 2010). A version of this dataset was recently used as a benchmark to evaluate automated flow cytometry analysis algorithms (FlowCAP-IV; Aghaeepour et al., 2016). Unsupervised k-means clustering of this dataset (k = 50) identified a cell population that was strongly associated with survival (i.e. days from first detection of HIV until either progression to AIDS or death). The 15-dimensional definition of this population, termed ‘Cluster 3’ (Fig. 1B), would be challenging to implement as a fluorescent flow cytometry assay in a clinical pathology environment due to the high number of simultaneous fluorescent channels required. Visual inspection of the heatmap (Fig. 1B, left panel) reveals that Cluster 3 is a type of memory T cell (CD45RO+ Ki-67+ CD3+ CCR5+ CD14dim), but it is unclear which markers should be prioritized when distilling this 15-dimensional phenotype down to a practical signature for a clinical assay. Toward this end, GateFinder analysis was performed on a composite data file representing 10 000 cells randomly sub-sampled from each patient, with the cells from Cluster 3 configured as the target population. The first two gates in the GateFinder-computed gating strategy identified the cells from Cluster 3 with 38% purity and 67% yield (Supplementary Fig. S1 and Fig. 1B, center panels). Notably, the gating strategy suggested an unintuitive but potent combination of three markers with intermediate expression (i.e. CCR7, CD57 and Ki-67), and only one marker with high expression (i.e. CD3). When the complete dataset was re-analyzed using only these first two gates, the strong association with patient survival was retained (Supplementary Fig. S2 and Fig. 1B, right panel). At least two gates were necessary, as predictive power was lost when only one gate was used (Supplementary Figs S3 and S4). The co-expression of CD57 and Ki-67, markers of replicative senescence and recent cell cycle activity, respectively, points toward a model that these cells have recently divided and have since committed to senescence. In this example, the GateFinder algorithm provided a rapid and unbiased method of simplifying a 15-parameter signature into a 4-parameter signature that could be more practically translated to a clinical setting, as well as provided a mechanistic hypothesis to guide further investigation. Two additional examples are provided in Supplementary Material.
4 Conclusions
GateFinder uses a combination of supervised feature-selection, heuristic local search and bootstrapping to address several related challenges in cytometry analysis: The identification of surrogate phenotypes, the design of efficient follow-up experiments and distilling mechanistic insights from high-dimensional signatures. This flexible automated tool can accelerate the analysis pipeline for high-dimensional single-cell experiments.
Funding
During this work, N.A. was supported by an Ann Schreiber Mentored Investigator Award from the Ovarian Cancer Research Fund (OCRF 292495), a Canadian Institute of Health Research (CIHR) Postdoctoral Fellowship (CIHR 321510), and an International Society for Advancement of Cytometry Scholarship. E.F.S. was supported by a Damon Runyon Cancer Research Foundation Postdoctoral Fellowship (DRG 2190-14). Additional support was provided by the March of Dimes Prematurity Research Center at Stanford University, the Gates Foundations, and the Department of Anesthesiology, Perioperative and Pain Medicine, Stanford University. D.J.K. was supported by a Vanier Scholarship from CIHR. G.K.F. was supported by the Stanford Bio-X graduate research fellowship and the US National Institute of Health (T32GM007276). This work was supported by grants (to the Nolan lab) from the NIH (0158GKB065, 1R01CA130826, 5U54CA143907, HHSN272200700038C, N01-HV-00242, 41000411217, 5-24927, P01 CA034233-22A1, P01 CA034233-22A1, PN2EY018228, RFA CA 09-009, RFA CA 09-011, U19 AI057229 and U54CA149145), the California Institute for Regenerative Medicine (DR1-01477 and RB2-01592), the European Commission (HEALTH.2010.1.2-1), the US FDA (HHSF223201210194C: BAA-12-00118), and the Entertainment Industry Foundation. This work was also supported by grants to the Weiss lab from the NIH (1R33CA183692-01) and The Cure Starts Now Foundation and a Terry Fox Foundation Program Project grant (TFF 122869) to the Eaves lab.
Conflict of Interest: none declared.
Supplementary Material
References
- Aghaeepour N. et al. (2016) A benchmark for evaluation of algorithms for identification of cellular correlates of clinical outcomes. Cytometry A, 89, 16–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakken T. et al. (2017) Cell type discovery and representation in the era of high-content single cell phenotyping. BMC Bioinformatics, 18, 559.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganesan A. et al. (2010) Immunologic and virologic events in early HIV infection predict subsequent rate of progression. J. Infect. Dis., 201, 272–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han L. et al. (2015) Single‐cell mass cytometry reveals intracellular survival/proliferative signaling in FLT3‐ITD‐mutated AML stem/progenitor cells. Cytometry A, 87, 346–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue S. et al. (2016) Mutant IDH1 downregulates ATM and alters DNA repair and sensitivity to DNA damage independent of TET2. Cancer Cell, 30, 337–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeys Y. et al. (2016) Computational flow cytometry: helping to make sense of high-dimensional immunology data. Nat. Rev. Immunol., 16, 449–462. [DOI] [PubMed] [Google Scholar]
- Weber L.M., Robinson M.D. (2016) Comparison of clustering methods for high‐dimensional single‐cell flow and mass cytometry data. Cytometry A, 89, 1084–1096. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.