

Abstract
Background
Progressive familial intrahepatic cholestasis (PFIC) is an autosomal recessive inherited disease that disrupts the genes for bile formation. Liver transplantation (LT) is the only effective treatment for PFIC patients with end-stage liver disease. We describe our experience in terms of clinical characteristics, complications, and outcome of LT for PFIC.
Case Report
The data of 5 pediatric PFIC patients recipients (3 PFIC1, 1 PFIC2, and 1 PFIC3) who received LT at our Liver Transplant Center from June 2013 to February 2017 were retrospectively analyzed. Four patients received liver transplantation from donation after cardiac death (DCD) donors. One patient received a living donor liver transplantation (LDLT). All the LT recipients received an immunosuppressive regimen of tacrolimus (FK 506) + methylprednisolone + mycophenolate mofetil (MMF). Diarrhea did not improve in 2 PFIC1 patients after LT, and they both developed steatohepatitis several months after LT. The other PFIC1 patient received ABO blood group incompatible LT and developed biliary complications and a severe Epstein-Barr virus infection; this patient underwent endoscopic retrograde cholangiopancreatography. She recovered after treatment with ganciclovir and reduction of tacrolimus dosage. The PFIC2 patient had abnormal liver function 19 months after LT, and recovered after administration of increased dosage of immunosuppressant agents. Liver function in the PFIC3 patient was normal during 2-year follow-up.
Conclusions
Liver transplantation is an effective treatment in PFIC patients. However, PFIC1 patients may develop aggravated diarrhea and steatohepatitis after LT. PFIC2 and PFIC3 patients have good outcomes after LT.
MeSH Keywords: Cholestasis, Intrahepatic; Diarrhea; Liver Transplantation
Background
Progressive familial intrahepatic cholestasis (PFIC) is an autosomal recessive inherited disease that disrupts the genes encoding protein transporters responsible for bile formation [1]. PFIC is classified into 3 types (PFIC 1-3): PFIC1 is caused by a mutation of the ATP8B1 gene encoding the FIC1 protein [2], PFIC2 is caused by a mutation of ABCB11 gene encoding the bile salt excretion protein (BSEP), and PFIC3 is due to a mutation of ABCB4 gene encoding the MDR3 protein [3].
The clinical manifestations in PFIC patients include progressive jaundice, pruritus, and growth retardation. Subsequently, patients develop liver cirrhosis and hepatic failure. Liver transplantation (LT) is the only effective treatment for PFIC at present [4].
We report 5 cases of PFIC who received LT at our center from June 2013 to February 2017, and their clinical characteristics and outcomes were analyzed.
Case Report
Since June 2013, 470 patients, including 290 pediatric patients, underwent LT at the Liver Transplant Section in Beijing Friendship Hospital. This is a retrospective study of 5 cases of these 290 pediatric liver transplant recipients for PFIC.
The 5 PFIC patients (3 with PFIC1, 1 with PFIC2, and 1 with PFIC3) consisted of 3 boys and 2 girls, and their age at liver transplantation ranged from 2 to 7 years old. Four patients received liver transplantation from donation after cardiac death (DCD) donors. One patient received Ditate. ABO blood groups were identical in 4 cases and incompatible in 1 case of PFIC1. All LT recipients received an immunosuppressive regimen of tacrolimus (FK 506) + methylprednisolone + mycophenolate mofetil (MMF). The regimen for ABO incompatibility included administration of rituximab before LT and gamma globulin after LT.
We analyzed the hospital records of these patients with regard to clinical characteristics, treatment, complications, outcome of LT, and impact of LT on growth. The study conformed to the ethics guidelines of the 1975 Declaration of Helsinki and was approved by Ethics Committee in Beijing Friendship Hospital.
Clinical characteristics of PFIC patients
Since June 2013, we have treated 5 pediatric patients diagnosed with PFIC. Three cases were PFIC1, 1 wasPFIC2, and 1 was PFIC3. Three were boys and the other 2 patients were girls. Their age at liver transplantation ranged from 2 to 7 years old. Clinical symptoms in the PFIC1 and PFIC2 patients were jaundice, pruritus, and growth retardation. The PFIC1 patients also had diarrhea. The PFIC3 patient had ascites and portal hypertension without severe jaundice or pruritus. Their Pediatric End-Stage Liver Disease Model (PELD) score ranged from 5 to 16. Their height S.D. scores ranged from −4.17 to 4.5, and weight S.D. scores ranged from −4.37 to 2.55 (Table 1).
Table 1.
Demographics and characteristics of patients.
| Case No. | Primary disease | Age at transplantation (y) | Sex | Clinical symptoms | PELD score | Height S.D. score | Weight S.D. score |
|---|---|---|---|---|---|---|---|
| 1 | PFIC1 | 4 | Female | Jaundice, pruritus, diarrhea | 16 | −3.72 | −4.03 |
| 2 | PFIC1 | 3 | Female | Jaundice, pruritus, diarrhea | 12 | −4.17 | −4.37 |
| 3 | PFIC2 | 5 | Male | Jaundice, pruritus | 5 | −1.11 | −0.59 |
| 4 | PFIC3 | 7 | Male | Ascites | 6 | 4.5 | 2.55 |
| 5 | PFIC1 | 2 | Male | Jaundice, diarrhea | 8 | −1.9 | −1.52 |
Liver transplantation in PFIC patients
Four patients received liver transplantation from DCD donors. One patient received LDLT. Blood loss during surgery was 150–300 mL. The cold ischemia time ranged from 110 min to 550 min, and the retrieval warm ischemia time was approximately 3–5 min. The ABO blood groups were identical in 4 cases and incompatible in 1 case of PFIC1. Our regimen for ABO incompatibility included administration of rituximab (375 mg/m2) before LT and gamma globulin (400 mg/kg daily) for 7–10 days after LT. Methylprednisolone was given at 10 mg/kg during surgery. All recipients received an immunosuppressive regimen of tacrolimus (FK 506) + methylprednisolone + mycophenolate mofetil (MMF) after LT.
The trough level of tacrolimus was maintained at 8–10 ng/mL for 6 months after LT, and then 5–8 ng/mL for the next 6 months. Methylprednisolone was stopped after 3 months and MMF was discontinued after 1 year. Length of hospital stay was 19–54 days (Table 2).
Table 2.
Clinical findings in PFIC patients with liver transplantation.
| Case No. | Donor type | ABO blood group | Operation time | Blood loss (mL) | Cold Ischemia time (min) | Retrieval warm Ischemia time (min) | Immunosuppressants during LT/post-LT | Trough level of tacrolimus (ng/mL) within 6 m/after one year | Hospital stay (d) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | DCD | Compatible | 2014/9/2 | 150 | 420 | 5 | Methylprednisolone (10 mg/kg)/FK 506 + methylprednisolone + MMF | 8–10/5–8 | 25 |
| 2 | DCD | Incompatible | 2014/7/25 | 150 | 480 | 5 | Methylprednisolone (10 mg/kg)/FK 506 + methylprednisolone + MMF | 8–10/5–8 | 54 |
| 3 | DCD | Compatible | 2014/9/23 | 200 | 370 | 5 | Methylprednisolone (10 mg/kg)/FK 506 + methylprednisolone + MMF | 8–10/5–8 | 28 |
| 4 | DCD | Compatible | 2016/1/14 | 300 | 550 | 5 | Methylprednisolone (10 mg/kg)/FK 506 + methylprednisolone + MMF | 8–10/5–8 | 19 |
| 5 | LDLT | Compatible | 2017/2/23 | 230 | 110 | 3 | Methylprednisolone (10 mg/kg)/FK 506 + methylprednisolone + MMF | 7–10/4–7 | 21 |
Complications in PFIC patients after LT and treatment
Case 1 was a PFIC1 patient who recovered well after surgery and was followed up after discharge. However, she had aggravated diarrhea after LT. Following treatment with loperamide and cholestyramine, diarrhea improved. Her liver function was abnormal with increased transaminase (ALT 90 IU/L, AST 74 IU/L) at 7 months after LT. Liver biopsy was performed and showed steatohepatitis with 90% hepatocyte steatosis (Figure 1). The patient was treated with Essentiale (a commercial preparation of essential phospholipids). At 19 months after LT, ALT was 41 IU/L and AST was 55 IU/L. Liver biopsy was repeated and showed 70% hepatocyte steatosis (Figure 2). The latest review results showed that liver function was normal, but liver biopsy showed nonalcoholic steatohepatitis with 66% hepatocyte steatosis and hepatic fibrosis (F3) (Figures 3, 4).
Figure 1.
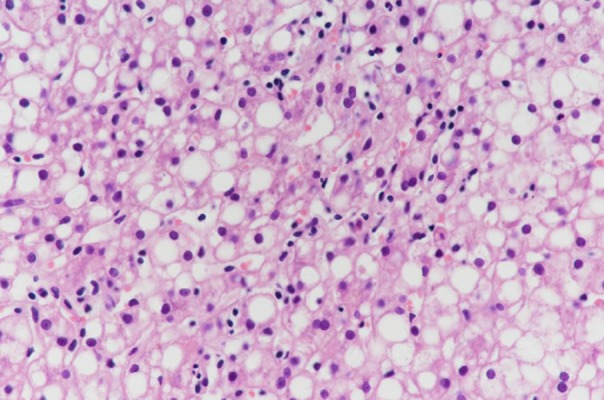
Liver biopsy in Case 1 at 7th month after LT. Liver biopsy showed steatohepatitis with 90% hepatocyte steatosis.
Figure 2.
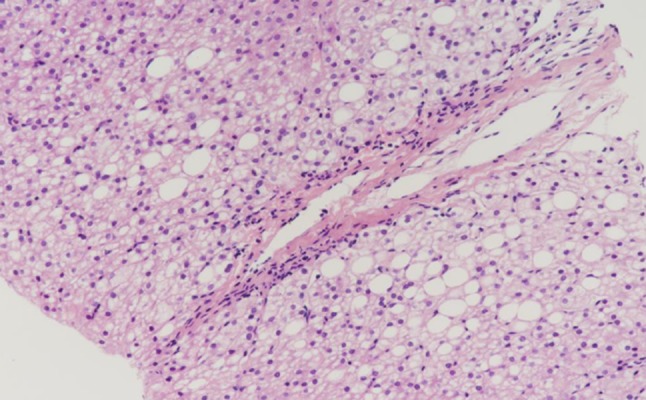
Liver biopsy in Case 1 at 19th month after LT. Liver biopsy showed 70% hepatocyte steatosis.
Figure 3.

Liver biopsy in Case 1 at 31th month after LT. Liver biopsy showed hepatocyte steatosis and hepatic fibrosis (F3).
Figure 4.

Post-LT liver function in Case 1. The patient underwent liver biopsy three times due to steatohepatitis. Following treatment with Essentiale, her liver did not significantly improve.
Case 2 was a PFIC1 patient with ABO blood group incompatibility. Her blood group was A and the donor’s blood group was AB. Anti-B IgM was 1: 32 and anti-B IgG was 1: 64. The patient received rituximab (375 mg/m2) before LT and gamma globulin (400 mg/kg daily) for 7 days after LT. On the 12th day after LT, ultrasound showed that left and right branches of the hepatic artery were undetectable and there was a low-echo area in the left liver lobe (Figure 5). Contrast-enhanced ultrasonography was performed, showing that the intrahepatic branches of the hepatic artery were normal, but she still received anticoagulant therapy. She developed biliary complications at 10 months and suffered repeated biliary infections and progressive jaundice (TB 196 mmol/L, DB 108 mmol/L). Ultrasound showed biloma formation in the liver and multiple intrahepatic biliary dilatations. She received percutaneous transhepatic cholangio-drainage (PTCD) followed by endoscopic retrograde cholangiopancreatography (ERCP) (Figures 6, 7). After these treatments, TB gradually decreased to 38 mmol/L and DB decreased to 19 mmol/L. At 2 years after LT, the recipient had a severe Epstein-Barr virus (EBV) infection with high fever and enlarged lymph nodes. EBV-DNA copies were 5×106/mL. Post-transplant lymphoproliferative disease was suspected, and lymph node biopsy of the left axilla was performed. The results showed lymphadenitis accompanied by EBV infection (Figure 8). Ganciclovir was administered and tacrolimus dosage was reduced. Following this treatment, she recovered and was discharged from the hospital.
Figure 5.
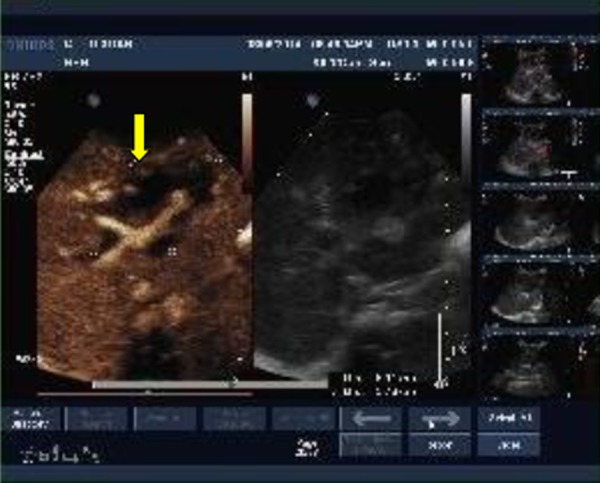
Ultrasound revealed a 6.5×3.9 cm low-echo area (see arrow) in the left liver lobe on day 12 after LT in Case 2.
Figure 6.
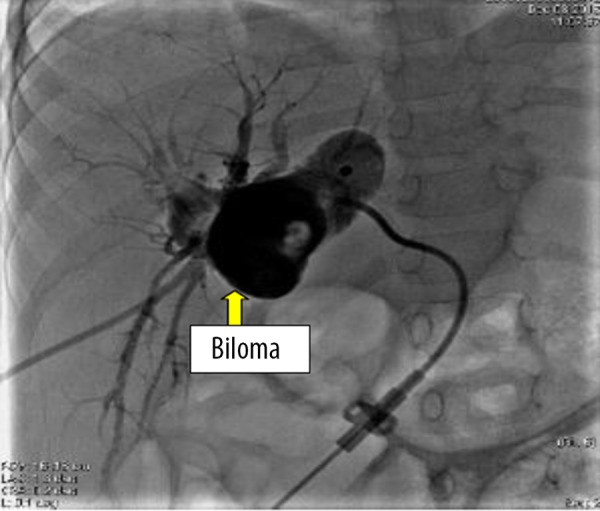
In case 2, biliary cholangiography showed biloma formation in the liver and multiple intrahepatic biliary dilatations, and biliary drainage was carried out.
Figure 7.

In case 2, ERCP showed multiple intrahepatic biliary dilatations, and a stent was implanted.
Figure 8.

Biopsy of lymph nodes in the left axilia showed lymhadenitis accompanied by EBV infection with EBER(+) in case 2.
Case 3 was a PFIC2 patient. The patient recovered well after LT and was discharged from the hospital. He was followed up regularly. At 19 months after LT, his liver function appeared abnormal (ALT 513 IU/L, AST 654 IU/L, TB 18 mmol/L, ALP 254 IU/L, GGT 248 IU/L). B ultrasound of the liver was normal. Immunosuppressants, including methylprednisolone and MMF, were administered again. Tacrolimus trough level was 6.5 ng/mL. Thus, tacrolimus was increased from 1 mg to 1.5 mg every 12 h and tacrolimus trough level was maintained at 7–8 ng/mL. Liver function subsequently recovered (Figure 9).
Figure 9.

Liver function was abnormal at 19th month after LT in case 3 and recovered after adjustment of immunosuppressant.
Case 4 was a PFIC3 patient. He recovered rapidly after LT and was discharged on day 19. Liver function was normal at 1-year follow-up, without complications.
Case 5 was a PFIC1 patient. The patient recovered well after LDLT and liver function was almost normal. He had recurrent diarrhea at 2 months after LDLT. According to the experience of case 1, a protocol liver biopsy was performed at 4 months after LT. The result showed steatohepatitis with 50% hepatocyte steatosis. The patient was treated with Essentiale (Table 3). At 10 months after LT, he had another liver biopsy. The result showed steatohepatitis with 20–30% hepatocyte steatosis, which was better than before (Figure 10).
Table 3.
Complications in PFIC patients after LT and treatment.
| Case No. | Complications | Occurrence time | Treatment |
|---|---|---|---|
| 1 | Diarrhea | 1st month | Loperamide, cholestyramine |
| Steatohepatitis | 7th month | Essentiale | |
|
| |||
| 2 | Biliary complications | 10th month | PTCD and ERCP |
| EBV infection | 2 years | Ganciclovir and tacrolimus reduction | |
|
| |||
| 3 | Liver function abnormal | 19th month | Increase in immunosuppressants |
|
| |||
| 4 | None | None | None |
|
| |||
| 5 | Diarrhea | 2nd month | Dioctahedral smectite |
| Steatohepatitis | 4th month | Essentiale | |
Figure 10.

Liver biopsy in case 5 at 10th month after L. Liver biopsy showed steatohepatitis with 20–30% hepatocyte steatosis and ballooning degeneration (see arrow).
Outcome and the impact of LT on growth
All patients were alive during the follow-up period. At the end of the first year, their height S.D. scores ranged from −2.56 to 4.61, and weight S.D. scores ranged from −2.14 to 2.78 (Table 4). Both height and weight S.D. scores improved from baseline.
Table 4.
Outcome and growth after LT.
| Case No. | Height S.D. score before LT | Height S.D. score at the 1st year | Weight S.D. score before LT | Weight S.D. score at the 1st year | Outcome |
|---|---|---|---|---|---|
| 1 | −3.72 | −2.56 | −4.03 | −2.09 | Alive |
| 2 | −4.17 | −1.89 | −4.37 | −2.14 | Alive |
| 3 | −1.11 | 2.43 | −0.59 | 2.31 | Alive |
| 4 | 4.5 | 4.61 | 2.55 | 2.78 | Alive |
| 5 | −1.9 | −1.7 | −1.52 | −1.2 | Alive |
Discussion
Liver transplantation is the only effective treatment at present for PFIC patients with end-stage liver disease. The indications for LT include severe pruritus, significant growth retardation, liver cirrhosis, and liver failure. PFIC patients usually have growth retardation and metabolic bone disease, mainly related to impaired vitamin D and calcium absorption due to reduced bile secretion. Aydogdu [5] reported a significant improvement at 6 months and 12 months after transplantation in terms of weight and height S.D. compared to baseline at the time of transplantation.
In the present study, the growth development of all the cases was improved compared to before. Cases 1, 2, and 5 were PFIC1 and suffered growth retardation before LT. After LT, growth retardation improved, but the catch-up growth of 2 of the 3 patients was affected by recurrent diarrhea and biliary complications.
Unlike PFIC2, the timing of LT in PFIC1 is controversial. This is because LT may aggravate diarrhea and affect catch-up growth. The main cause is thought to be expression of the FIC1 gene in various organs, including the liver, pancreas, small intestine, and kidney, and a higher expression level is found in the small intestine than in the liver. Bile acid secretion may be increased in the small intestine after LT and watery diarrhea can lead to metabolic acidosis. Bile acid resin and loperamide may improve the symptoms [6]. In our study, case 1 and 5 showed aggravated diarrhea after LT. Mycophenolate mofetil was stopped and the patients recovered after loperamide and dioctahedral smectite administration. Case 2 was also a PFIC1 patient, but she did not develop aggravated diarrhea after LT.
Hori et al. [7,8] reported on 14 patients who underwent living donor liver transplantation (LDLT) for PFIC (11 cases of PFIC1 and 3 cases of PFIC2). Three of the 11 PFIC1 recipients died, while all 3 PFIC2 recipients survived. Eight of the 11 PFIC1 recipients developed steatosis after LDLT. Nine of these 11 PFIC1 recipients developed fibrosis after LDLT. In contrast to the PFIC1 recipients, PFIC2 recipients did not develop steatosis or fibrosis after LDLT. In our study, 2 cases of PFIC1 developed steatohepatitis within 1 year after LT. Long-term follow-up of these recipients is required to monitor for fibrosis.
PFIC2 patients with some subtypes are prone to recurrences of liver diseases [9]. Some patients have a BSEP protein antibody to the donor liver [10]. In some cases, these antibodies have resulted in recurrent graft failure [11]. When allo-antibodies are detected, increased immunosuppression and the initiation of plasmapheresis/molecular adsorbent recirculating system therapies are shown to improve cholestatic episodes after transplantation in some of these PFIC2 patients[11]. The use of rituximab has also been reported to improve symptoms[12]. In our present study, the PFIC2 recipient showed fluctuations in liver function 19 months after LT. He recovered following an increase in immunosuppressants. The PFIC3 recipient recovered well without complications.
Conclusions
LT is currently the only effective treatment for end-stage liver diseases due to PFIC, although LT may worsen the extra-hepatic manifestations in PFIC1 recipients. More attention should be paid to the management of immunosuppressants in PFIC2 recipients because of its relapsing nature. Further studies are necessary find ways to prolong long-term survival and reduce long-term complications in PFIC patients.
Footnotes
Source of support: This study was supported by the Capital Special Program for Health Research and Development (No. 2016-1-2021)
Conflict of interests
None.
References
- 1.Nakanishi Y, Saxena R. Pathophysiology and diseases of the proximal pathways of the biliary system. Arch Pathol Lab Med. 2015;139:858–66. doi: 10.5858/arpa.2014-0229-RA. [DOI] [PubMed] [Google Scholar]
- 2.van Mil SW, Klomp LW, Bull LN, Houwen RH. FIC1 disease: A spectrum of intrahepatic cholestatic disorders. Semin Liver Dis. 2001;21:535–44. doi: 10.1055/s-2001-19034. [DOI] [PubMed] [Google Scholar]
- 3.de Vree JM, Jacquemin E, Sturm E, et al. Mutations in the MDR3 gene cause progressive familial intrahepatic cholestasis. Proc Natl Acad Sci USA. 1998;95:282–87. doi: 10.1073/pnas.95.1.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mehl A, Bohorquez H, Serrano M-S, et al. Liver transplantation and the management of progressive familial intrahepatic cholestasis in children. World J Transplant. 2016;6(2):278–90. doi: 10.5500/wjt.v6.i2.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aydogdu S, Cakir M, Arikan C, et al. Liver transplantation for progressive familial intrahepatic cholestasis: Clinical and histopathological findings, outcome and impact on growth. Pediatr Transplant. 2007;11(6):634–40. doi: 10.1111/j.1399-3046.2007.00722.x. [DOI] [PubMed] [Google Scholar]
- 6.Lykavieris P, van Mil S, Cresteil D, et al. Progressive familial intrahepatic cholestasis type 1 and extrahepatic features: No catch-up of stature growth, exacerbation of diarrhea, and appearance of liver steatosis after liver transplantation. J Hepatol. 2003;39(3):447–52. doi: 10.1016/s0168-8278(03)00286-1. [DOI] [PubMed] [Google Scholar]
- 7.Hori T, Egawa H, Miyagawa-Hayashino A, et al. Living-donor liver transplantation for progressive familial intrahepatic cholestasis. World J Surg. 2011;35(2):393–402. doi: 10.1007/s00268-010-0869-6. [DOI] [PubMed] [Google Scholar]
- 8.Hori T, Egawa H, Takada Y, et al. Progressive familial intrahepatic cholestasis: A single-center experience of living-donor liver transplantation during two decades in Japan. Clin Transplant. 2011;25(5):776–85. doi: 10.1111/j.1399-0012.2010.01368.x. [DOI] [PubMed] [Google Scholar]
- 9.Maggiore G, Gonzales E, Sciveres M, et al. Relapsing features of bile salt export pump deficiency after liver transplantation in two patients with progressive familial intrahepatic cholestasis type 2. J Hepatol. 2010;53:981–86. doi: 10.1016/j.jhep.2010.05.025. [DOI] [PubMed] [Google Scholar]
- 10.Stindt J, Kluge S, Dröge C, et al. Bile salt export pump-reactive antibodies form a polyclonal, multiinhibitory response in antibody-induced bile salt export pump deficiency. Hepatology. 2016;63:524–37. doi: 10.1002/hep.28311. [DOI] [PubMed] [Google Scholar]
- 11.Ketel V, Burdelski M, Vojnisek Z, et al. De novo bile salt transporter antibodies as a possible cause of recurrent graft failure after liver transplantation: A novel mechanism of cholestasis. Hepatology. 2009;50:510–17. doi: 10.1002/hep.23083. [DOI] [PubMed] [Google Scholar]
- 12.Lin HC, Alvarez L, Laroche G, et al. Rituximab as therapy for the recurrence of bile salt export pump deficiency after liver transplantation. Liver Transpl. 2013;19:1403–10. doi: 10.1002/lt.23754. [DOI] [PubMed] [Google Scholar]