

 Unravelling the molecular basis of thalidomide embryotoxicity, which is remarkably species-specific, is challenging in view of its low toxicity in the mature animal.
Unravelling the molecular basis of thalidomide embryotoxicity, which is remarkably species-specific, is challenging in view of its low toxicity in the mature animal.
Abstract
Unravelling the molecular basis of thalidomide embryotoxicity, which is remarkably species–specific, is challenging in view of its low toxicity in the mature animal. Employing data derived solely from proven sensitive primate species or susceptible strains of rabbit, the structure–activity relationship of over 50 compounds which are, arguably, congeners of thalidomide has been reviewed. The molecular requirement for ‘thalidomide-type’ teratogenicity was highly structure dependent. Both the phthalimide and glutarimide groups were essential for embryopathic activity, although minor substitutions in either or both rings could be tolerated without a loss of toxicity. An α-linkage between the two cyclic structures was essential; a β-link resulted in a complete loss of embryopathic activity. Crucially, this α-configuration provided a centre of asymmetry enabling the existence of stereoisomers. The thalidomide molecule is not a static entity and under physiological conditions it undergoes a number of intra- and inter-molecular reactions. Besides irreversible hydrolysis, its keto–enol tautomerism, base-assisted proton transfer and glutarimide ring rotation lead to rapid interconversion of the thalidomide enantiomers. These enantiomers form equilibria between themselves and also between both homochiral and heterochiral dimers. It is proposed that the more energetically favourable and stable heterochiral dimer of thalidomide is an active agent that possesses the structural features of the paired nucleotides of the double-stranded DNA. Its capacity to enter into hydrogen bonding interactions affects DNA expression in a chaotic manner without causing permanent mutations. This disruption may well be concentrated at nucleotide sites known to be involved in specific promoter regions of the genome.
1. Introduction
The mechanism(s) whereby thalidomide exerts teratogenic effects in human pregnancy and in certain animal species remains enigmatic. Some 50 years after the original tragic events, it is still one of the most challenging problems in reproductive toxicology; put at its simplest, how could a relatively uncomplicated molecule of low acute toxicity elicit such specific embryopathic effects at critical periods of embryogenesis? Over the years there have been two types of approach in attempting to unravel the possible mechanisms underlying the toxicity of thalidomide.
The first has used a variety of biochemical, immunological and molecular modelling techniques and the outcome of these have been reviewed critically at various times.1–4 Most recent evidence suggests that thalidomide prevents inter alia angiogenic outgrowth thereby interfering with early limb development.5,6 Furthermore, this effect appears to be a property of the intact drug, racemic thalidomide, as its metabolites including its hydrolysis products seem to be inactive in this respect.7 The second approach has been through an evaluation of the structural requirements for thalidomide-type teratogenic effects based on the results of testing structural analogues in appropriate sensitive species such as various strains of rabbit and primate. In total about 50–60 structurally-related compounds have been evaluated in this manner over the years. Such studies had two objectives: (a) the results might provide an insight into embryotoxic mechanism and (b) the definition of structural requirements for teratogenicity could conceivably help in the design of future therapeutic agents, so as to avoid this appalling toxicity. A review of this nature was published several decades ago.1
The present paper provides an overview of the structural relationships for ‘thalidomide-type’ of teratogenicity; on the basis of this overview a novel hypothesis is proposed which takes into account the fact that racemic (dl) thalidomide is most satisfactorily considered as a dimeric heterochiral assembly structure rather than as simple monomers as is usually depicted. For the purpose of this overview it is necessary to begin with a review of two related aspects, namely (a) the unique features of the chemistry of dl-thalidomide and (b) the choice of species used for evaluating the embryotoxicity of various thalidomide congeners.
2. Chemistry of thalidomide
Although of relatively simple chemical structure, the physicochemical properties of thalidomide are surprisingly complex. The thalidomide molecule consists of two heterocyclic ring structures linked together, namely the N-position of the phthalimide ring joined to the alpha or 3′-position of the glutarimide ring. The alpha (3′) position on the glutarimide ring is chiral hence thalidomide can exist in three forms; the optically active d and l enantiomers and the optically inactive dl racemic form. Crystalline racemic thalidomide can exist in different allelomorphic forms probably reflecting the various solvents and conditions used for crystallisation.8,9 Furthermore, X-ray evidence indicates that dl-thalidomide in the crystalline state exists as a dimeric racemic compound formed by hydrogen bonding interactions between the d and l enantiomers.9–11 Similar situations have been found with 4-bromothalidomide.12,13 Theoretical calculations of hydrogen bond energies have indicated that the component heterochiral dimer is a more stable entity than the homochiral dimer.14 More recent evidence suggests that racemic thalidomide can exist also in aqueous solution and under physiological conditions as a racemic dimeric compound (a heterochiral assembly) rather than the simple monomeric structure.15 Thus, when aqueous solutions of equal parts of d- and l-thalidomide are mixed together there separates on standing crystals of dl-thalidomide with physical properties different from those of its isomers. It seems probable therefore that in aqueous solution racemic dl-thalidomide exists in equilibrium with the individual d and l enantiomers. This is quite reasonable as the molecular structure of thalidomide carries numerous centres allowing powerful hydrogen bonding interactions to occur (Fig. 1).15
Fig. 1. Thalidomide heterochiral assembly. Proposed structure of a dimeric heterochiral assembly for dl-thalidomide showing potential sites for interconnecting hydrogen bonds.

A second feature of the thalidomide molecule is that the two enantiomers can rapidly racemise. Enantiomerization pathways via keto–enol tautomerism, base-catalysed proton transfer and subsequent rotation of the glutarimide ring have been proposed.16–18 Mean rates of chiral inversion in blood at 37 °C (in vitro) of 0.31 h–1 and 0.30 h–1 and in human subjects (in vivo) of 0.17 h–1 and 0.12 h–1 have been measured for the d to l and l to d inversions, respectively.19 These authors concluded that this rapid racemisation would offset any theoretical differences in toxicity or therapeutic properties between the two isomers when administered in vivo. A number of studies, pharmacological, toxicological and embryopathic have been undertaken but the interpretation of the results can be confounded by this problem of rapid racemisation and isomer interconversion.
A third issue is its inherent instability. The thalidomide molecule contains 4 amide bonds each of which is susceptible to hydrolysis at pH values of 6 and above. At the physiological pH of 7.4, hydrolysis is rapid and can result in the formation of 12 hydrolysis products.20 Some early experiments on the pharmacological properties of thalidomide were conducted on the basis of dissolving the compound in sodium hydroxide, in which it is readily soluble; unfortunately such solutions would have been devoid of any structurally intact thalidomide.21
3. Testing for thalidomide teratogenicity
It is generally recognised that probably the most reliable way of testing for ‘thalidomide-type’ teratogenicity is by means of in vivo animal models although this is now ethically undesirable. Some in vitro models have been employed but their use is effectively restricted to the investigation of mechanisms of embryotoxicity. There is firstly the issue of which animal species would most closely emulate the situation seen in human pregnancy. Initially rodent species such as rats and mice were chosen, mainly for reasons of convenience and practicality, but these proved to be remarkably resistant to the teratogenic effects of thalidomide. In these two species one sees marked embryolethality (embryonic resorption) rather than evident teratogenic effects and early studies performed in these two species have led to a number of misleading conclusions. The most widespread misconception arising from testing of thalidomide in pregnant rats was that while the l-isomer of thalidomide was teratogenic the d-isomer was inactive in this respect.22 The d-isomer was viewed as the therapeutically effective entity and it was claimed that if this enantiomerically pure isomer had been marketed rather than the racemic form then the human tragedy of teratogenicity would have been avoided. This popular claim has now been discredited on the basis of the rapid isomerisation and interconversion of the two enantiomers as well as the fact that both isomers are teratogenic in vivo in the susceptible New Zealand White rabbit.23,24 Developing chick embryos were widely used also in early testing but they too have limitations as a model. The chick embryo has been proved vulnerable to a variety of chemicals tested including many solvent materials and the results cannot with confidence be extrapolated to humans.
The results of developmental toxicity studies with thalidomide have been reviewed and the underlying differences in species sensitivities highlighted.25,26 Some such as the hamster and Guinea pig appear to be relatively resistant and it is generally agreed and accepted that the species that most closely parallel human responses to thalidomide, but certainly not identical with, are several strains of rabbit (Himalayan and New Zealand White) and the few non-human primates that have been evaluated. The latter, as might be expected, appear to provide the closest parallel to thalidomide teratogenicity in humans. In early primate studies the use of large Old World monkey species posed several significant practical problems. Many were already scarce and endangered, most carried only a single embryo/foetus and large facilities were required to accommodate an adequate population of pregnant females to allow for meaningful testing. The use of smaller New World monkeys such as the marmoset (Callithrix jacchus) proved more popular for teratogenicity testing as they had several advantages over their Old World counterparts. They could be bred in large numbers in captivity, they did not have a breeding season, pregnant females carried twins and even triplets, they were easier to handle and manage and were very sensitive to the teratogenic effects of thalidomide. However, despite this the rabbit has been the major species employed in the teratogenic evaluation of the various thalidomide congeners. In this survey of structure–activity relationship for ‘thalidomide-type’ teratogenicity of some 56 compounds, consideration has been given to findings employing the rabbit and non-human primate species as the suitability of these species above all others has been endorsed by numerous workers.25,26
4. Embryopathic effects
It should be acknowledged at the outset that many of the studies were conducted with small numbers of animals which may be seen to limit the ‘power’ of the investigation. Indeed, most were undertaken prior to the development of officially recognised protocols for the conduct of reproductive procedures but nevertheless the findings remain. That is all we have at present and are likely to have in the foreseeable future.
Table 1 shows the structures of some 12 compounds including thalidomide itself which have been shown beyond reasonable doubt to have teratogenic properties in pregnant rabbits and/or monkey species.23,27–41 It should be noted that the actual number is 8 compounds because 4 of these are enantiomers. Thalidomide and its structural analogue EM12 both carry an asymmetric chiral centre and all three forms (dl, d, l) have been shown to be teratogenic. Inspection of the active structures show that all compounds retain the common feature of a phthalimide ring function linked to the glutarimide ring at the alpha (3′) position so effectively determining a centre of asymmetry for these compounds.
Table 1. Compounds shown to elicit thalidomide-like embryopathic effects in the rabbit or non-human primate species.
| Name | Systematic name | Structure | Test species and references |
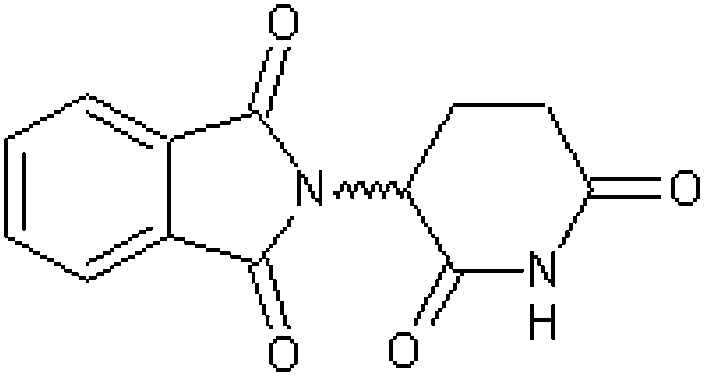
| dl-Thalidomide (dl-α-phthalimidoglutarimide) [1] | 2-(2,6-Dioxopiperidin-3-yl)-1H-isoindole-1,3(2H)-dione |
|
Cynomolgus monkey27 |
| Rhesus monkey28 | |||
| Baboon29 | |||
| Marmoset30 | |||
| Rabbit31,32 | |||
| d-Thalidomide (d-α-phthalimidoglutarimide) [2] | 2-[(3S)-2,6-dioxopiperidin-3-yl]-1H-isoindole-1,3(2H)-dione |

|
Rabbit23 |
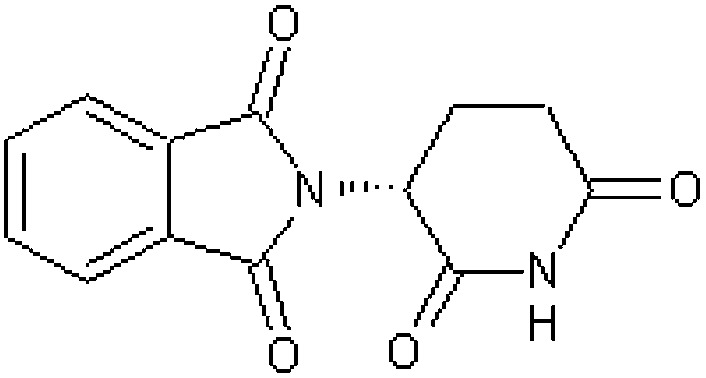
| l-Thalidomide (l-α-phthalimidoglutarimide) [3] | 2-[(3R)-2,6-Dioxopiperidin-3-yl]-1H-isoindole-1,3(2H)-dione |
|
Rabbit23 |
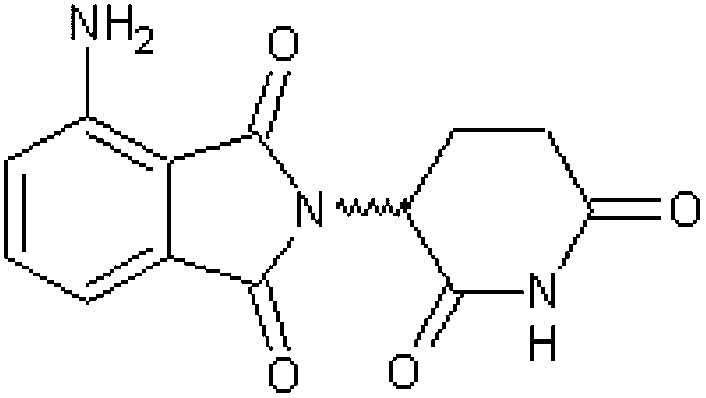
| dl-3-Aminothalidomide (dl-3-amino-α-phthalimidoglutarimide) [4] | 4-Amino-2-(2,6-dioxopiperidin-3-yl)-1H-isoindole-1,3(2H)-dione |
|
Rabbit33 |
| dl-3-Nitrothalidomide (dl-3-nitro-α-phthalimidoglutarimide) [5] | 2-(2,6-Dioxopiperidin-3-yl)-4-nitro-1H-isoindole-1,3(2H)-dione |

|
Rabbit33,34 |
| 3-Methylthalidomide (α-phthalimido-3-methylglutarimide) [6] | 2-(3-Methyl-2,6-dioxopiperidin-3-yl)-1H-isoindole-1,3(2H)-dione |

|
Marmoset35 |
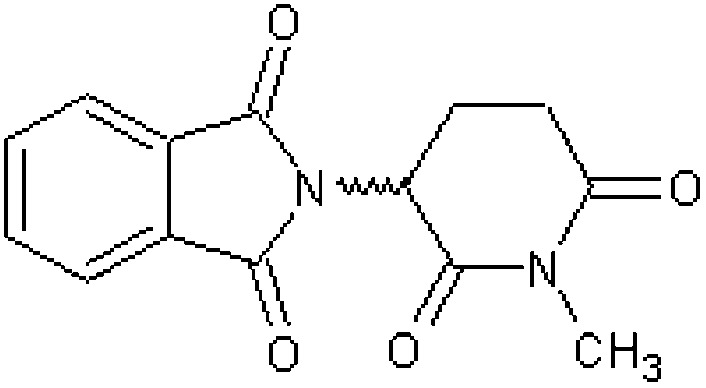
| N-Methylthalidomide (α-phthalimido-N-methylglutarimide) [7] | 2-(1-Methyl-2,6-dioxopiperidin-3-yl)-1H-isoindole-1,3(2H)-dione |
|
Rabbit36 |
| Thalidomide analogue EM12 (2-(2,6-dioxopiperidine-3-yl)-phthalimide) [8] | 3-(1-Oxo-1,3-dihydro-2H-isoindol-2-yl)piperidine-2,6-dione |

|
Rhesus monkey38 |
| Marmoset39,40 | |||
| Rabbit37,38 | |||
| Thalidomide analogue EM12 S-(–)-enantiomer [9] | (3S)-3-(1-Oxo-1,3-dihydro-2H-isoindol-2-yl)piperidine-2,6-dione |

|
Marmoset39,41 |
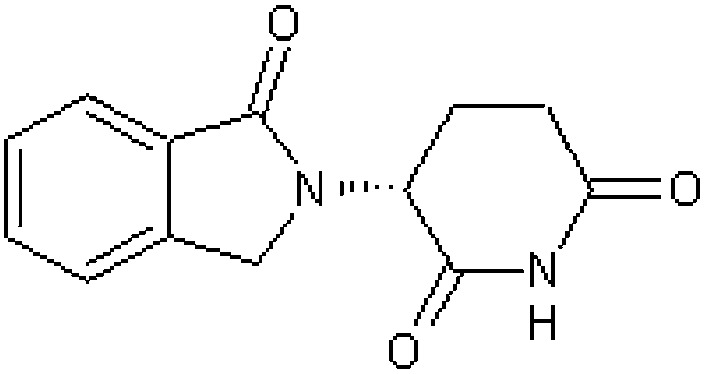
| Thalidomide analogue EM12 R-(+)-enantiomer [10] | (3R)-3-(1-Oxo-1,3-dihydro-2H-isoindol-2-yl)piperidine-2,6-dione |
|
Marmoset39,41 |
| Thalidomide analogue EM136 [11] | 3-(1,3-Dioxo-1,3-dihydro-2H-isoindol-2yl)piperidine-2-one |

|
Rabbit37 |
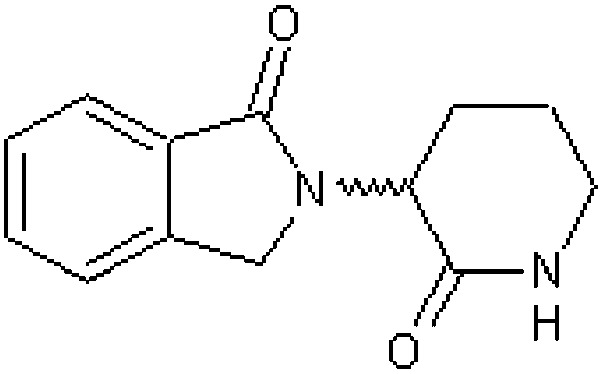
| Thalidomide analogue EM255 [12] | 3-(1-Oxo-1,3-dihydro-2H-isoindol-2-yl)piperidine-2-one |
|
Rabbit37 |
Further perusal of the structures reveals that some minor changes can be made without loss of teratogenic activity. Thus, aromatic substitution of the phthalimide function with an amino group (compound 4) or nitro group (compound 5) does not result in loss of teratogenic activity. The reduction of a single carbonyl to a methylene function affords EM12 (compound 8) which is of interest as this teratogen is considerably more active than thalidomide (10 times) presumably because it is more resistant to spontaneous hydrolysis at physiological pH.33 Similarly, modest structural changes to the glutarimide ring do not lead to loss of teratogenic activity. Thus, the N-methylglutarimide analogue of thalidomide (compound 7) is a positive teratogen in the rabbit although it needs to be acknowledged that this compound could undergo metabolic demethylation in the body to form thalidomide itself as an active metabolite. In a further analogue (compound 11) in which one of the glutarimido keto functions has been reduced to a methylene function (EM136) teratogenic activity in the rabbit is retained. Finally, EM255 (compound 12) in which modest structural changes in both ring structures have been made does not result in loss of activity. What emerges at this stage is that the implication for ‘thalidomide-type’ teratogenicity is the coupling of the two heterocyclic ring structures, namely, the phthalimido and glutarimido functions, linked at the alpha (3′) position to provide a centre of asymmetry. The importance of the alpha linkage and the chiral nature so afforded will become evident later as other structural analogues are considered. Also, one should note that limited structural changes in the two ring structures can be tolerated without loss of activity and indeed may be accompanied by enhanced activity (compound EM12).
Table 2 summarises the findings for the teratogenicity testing of various hydrolysis products of thalidomide (compounds 13–22).33,42,43 It has been pointed out previously that thalidomide is unstable at pH values above 6–7 and rapidly undergoes spontaneous hydrolysis in vivo to some 12 hydrolysis products.44,45 There is also some evidence that besides spontaneous hydrolysis there may be additional enzyme-assisted hydrolysis at the amide bonds. Some 10 of these hydrolysis products have been tested for embryotoxicity in the New Zealand White rabbit all with negative results. It has been suggested that because the hydrolysis products are polar in nature that they may not penetrate to the early blastocyst when orally administered to the pregnant mother but this has been shown not to be so.42,44 It therefore appears unlikely that one or more of the hydrolysis products of thalidomide are responsible for the teratogenicity and rather it is a function of the parent compound.
Table 2. Hydrolysis and other breakdown products of thalidomide.
| Name | Systematic name | Structure | Test species and references |
| 4-Phthalimidoglutaramic acid [13] | 5-Amino-4-(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)-5-oxopentanoic acid |

|
Rabbit33,42 |
| 2-Phthalimidoglutaramic acid [14] | 5-Amino-2-(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)-5-oxopentanoic acid |

|
Rabbit33,42,43 |
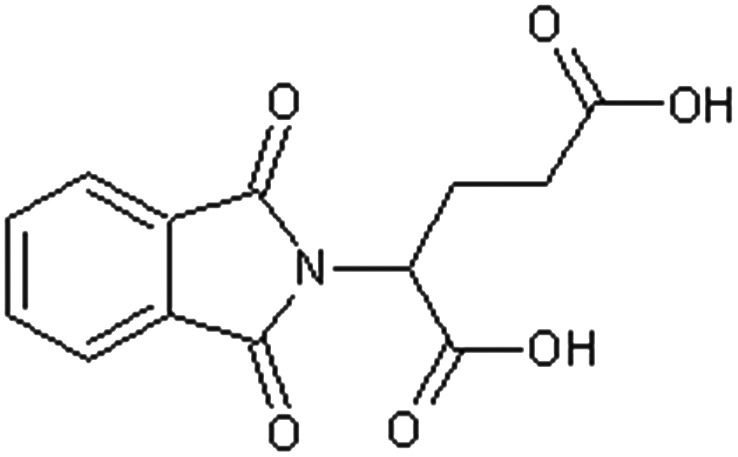
| 2-Phthalimidoglutaric acid [15] | 2-(1,3-Dioxo-1,3-dihydro-2H-isoindol-2-yl)pentanedioic acid |
|
Rabbit33,42,43 |
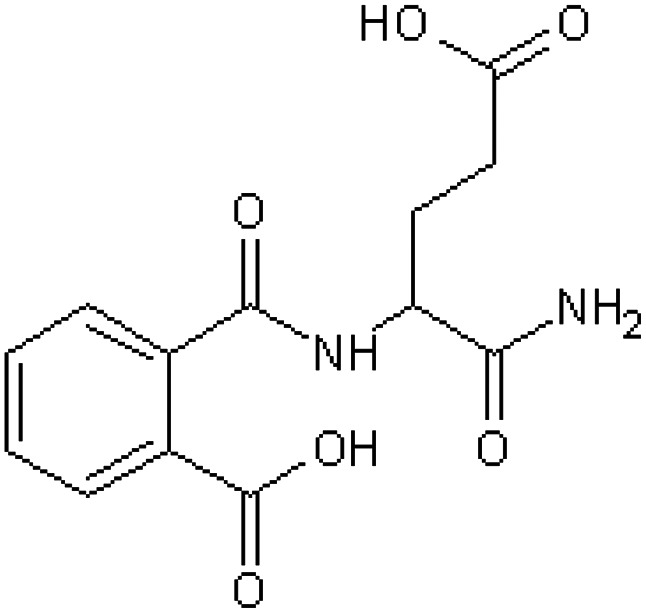
| 4-(O-Carboxybenzamido) glutaramic acid [16] | 2-[(1-Amino-4-carboxy-1-oxobutan-2-yl)carbamoyl] benzoic acid |
|
Rabbit33,42 |
| 2-(O-Carboxybenzamido) glutaramic acid [17] | 2-[(4-Amino-1-carboxy-4-oxobutyl)carbamoyl] benzoic acid |
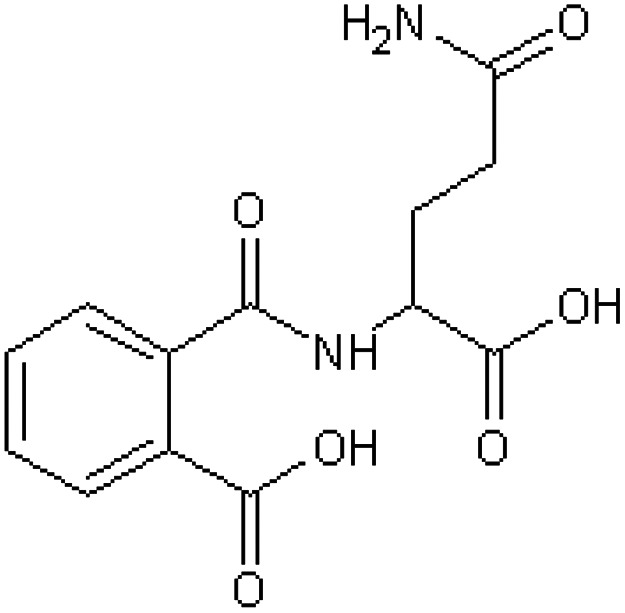
|
Rabbit33,42 |
| 2-(O-Carboxybenzamido) glutaric acid [18] | 2-[(2-Carboxybenzoyl) amino]pentanedioic acid |

|
Rabbit42 |
| α-Aminoglutarimide [19] | 3-Aminopiperidine-2,6-dione |

|
Rabbit33,42 |
| l-Glutamine [20] | (2S)-2,5-Diamino-5-oxopentanoic acid |

|
Rabbit33,42 |
| dl-Glutamine [21] | 2,5-Diamino-5-oxopentanoic acid |

|
Rabbit33,42 |
| Phthalic acid [22] | Benzene-1,2-dicarboxylic acid |
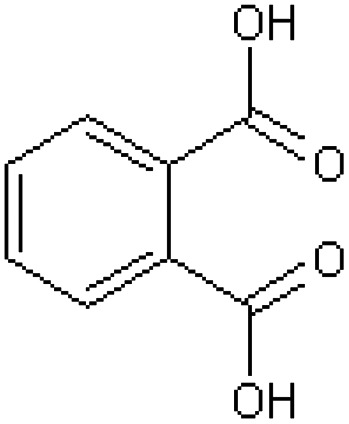
|
Rabbit33,42 |
Table 3 (compounds 23 and 24) illustrates the findings for the positional isomers of thalidomide and its highly teratogenic analogue (EM12).46 For these two compounds the point of attachment of the phthalimido function to the glutarimide ring has been moved from the alpha to the beta position to create positional isomers of the parent compounds and thereby removing the centre of asymmetry and their chiral nature. Remarkably, both compounds are devoid of embryotoxicity when tested in pregnant rabbits and these investigators emphasised that the point of attachment of the two ring structures was crucial for some reason for embryopathic activity, but this observation appears to have been overlooked.46
Table 3. Non-teratogenic thalidomide analogues; positional isomers; alteration from α- to β-configuration.
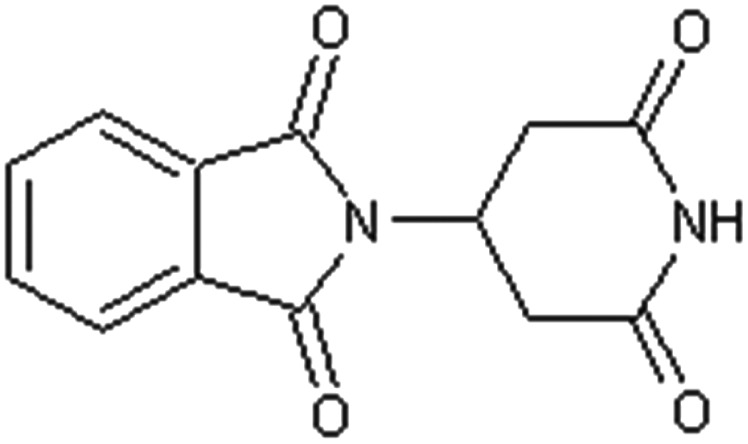

Table 4 indicates 8 structural analogues of thalidomide (compounds 25–32) in which the phthalimide ring has been replaced by various alternative ring structures such as benzothiazole (compounds 25–27) or succinimido (compound 32) all of which proved to be inactive as teratogens in the pregnant rabbit.33,34,36,37,46–49
Table 4. Non-teratogenic analogues of thalidomide; replacement of the phthalimide ring by alternative ring structures with intact glutarimide function.
| Name | Systematic name | Structure | Test species and references |
| 3-(1,3-Dihydro-1-oxo-2H-isoindol-2-yl)-2,6-dioxopiperidine [25] | 3-(1,1-Dioxido-1,2-benzothiazol-2(3H)-yl)piperidine-2,6-dione |

|
Rabbit46 |
| 2-(3-Oxo-1,2-benzisothiazolin-2-yl)glutarimide-S,S-dioxide [26] | 3-(1,1-Dioxido-3-oxo-1,2-benzothiazol-2(3H)-yl)piperidine-2,6-dione |

|
Rabbit47 |
| EM8 3-(2,3-Dihydro-1,1-dioxido-3-oxo-1,2-benzoisothiazol-2-yl)-2,6-dioxopiperidine [27] | 3-(1,1-Dioxido-3-oxo-1,2-benzothiazol-2(3H)-yl)piperidine-2,6-dione |

|
Rabbit37,46 |
| 2-(4-Oxo-3,4-dihydroquinazolin-3-yl)-glutarimide [28] | 3-(4-Oxoquinazolin-3(4H)-yl)piperidine-2,6-dione |
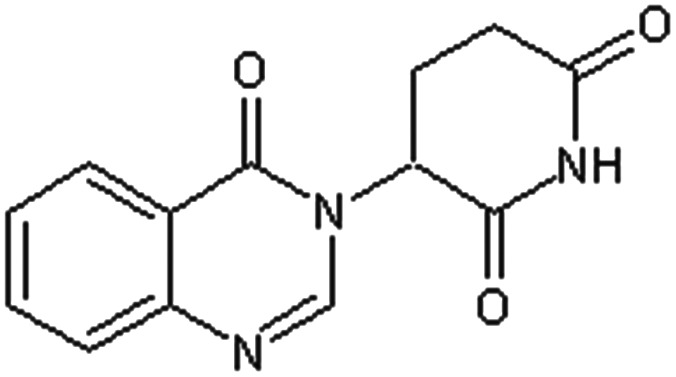
|
Rabbit47 |
| 2-(Hexahydrophthalimido)-glutarimide [29] | 2-(2,6-Dioxopiperidin-3-yl)hexahydro-1H-isoindole-1,3(2H)-dione |

|
Rabbit33,36 |
| 3-(1,4-Endoxocyclohexan-2-exo-3-exo-dicarboximido)-piperidin-2,6-dione [30] | 4-(2,6-Dioxopiperidin-3-yl)-10-oxa-4-azatricyclo[5.2.1.02,6]decane-3,5-dione |

|
Rabbit48,49 |
| 3-(1,4-Endomethylene-cyclohexane-2,3-endo-cis-dicarboximido)-piperidin-2,6-dione [31] | 4-(2,6-Dioxopiperidin-3-yl)-4-azatricyclo[5.2.1.02,6]decane-3,5-dione |

|
Rabbit48,49 |
| α-Succinimidoglutarimide [32] | 3-(2,5-Dioxopyrrolidin-1-yl)piperidine-2,6-dione |

|
Rabbit34 |
Table 5 shows 16 compounds (compounds 33–48) in which the phthalimide ring has been left intact with structural modification or replacement of the glutarimido ring.34,43,47,50–52 All compounds were inactive as teratogens in the pregnant rabbit and two of these chemicals are used as pesticides (compounds 36, 37). The negative findings for N-hydroxythalidomide (compound 43) and N-methoxythalidomide (compound 44) were a little surprising considering they possess only minor additions to the glutarimido ring, but these observations might be explained if they were to undergo a rapid conjugation with glucuronic acid, the latter after demethylation.52
Table 5. Non-teratogenic structural thalidomide analogues; phthalimide ring intact with structural modification or replacement of the glutarimide ring.
| Name | Systematic name | Structure | Test species and references |
| Phthalimidophthalimide [33] | 2,2′-Biisoindole-1,1′,3,3′-tetrone |

|
Marmoset50 |
| 2-Phthalimido-dl-succinimide [34] | 2-(2,5-Dioxopyrrolidin-3-yl)-1H-isoindole-1,3(2H)-dione |

|
Rabbit43 |
| N-Phthaloyl-dl-alanylamide [35] | 2-(1,3-Dioxo-1,3-dihydro-2H-isoindol-2-yl)propanamide |
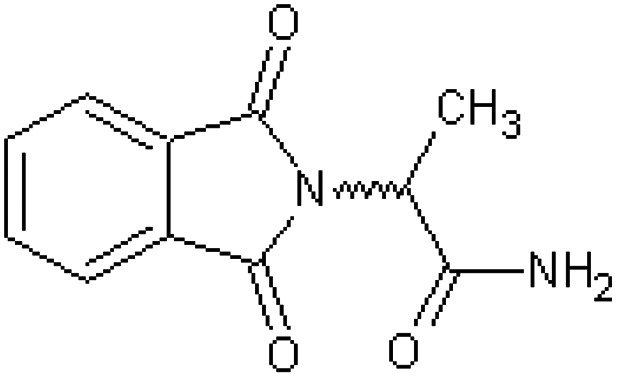
|
Rabbit43 |
| Imidan O,O-dimethyl-S-phthalimidomethylphosphorodithiolate [36] | S-[(1,3-Dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl] O,O-dimethyl phosphorodithioate |

|
Rabbit51 |
| Phaltan N-(trichloromethylthio)-phthalimide [37] | 2-[(Trichloromethyl)sulfanyl]-1H-isoindole-1,3(2H)-dione |
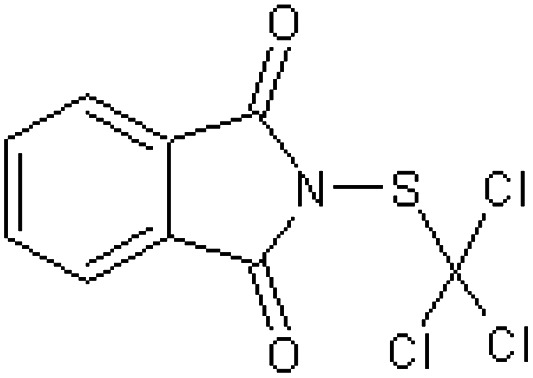
|
Rabbit51 |
| N-Phenylphthalimide [38] | 2-Phenyl-1H-isoindole-1,3(2H)-dione |
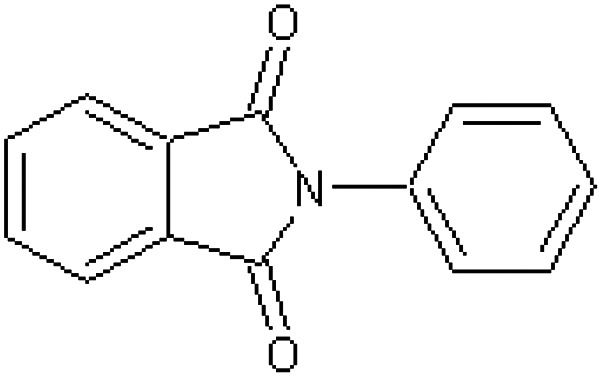
|
Rabbit34 |
| 2-Phthalimidoglutaric acid anhydride [39] | 2-(2,6-Dioxotetrahydro-2H-pyran-3-yl)-1H-isoindole-1,3(2H)-dione |

|
Rabbit34 |
| 2-Phthalimidopyridine [40] | 2-(Pyridin-3-yl)-1H-isoindole-1,3(2H)-dione |
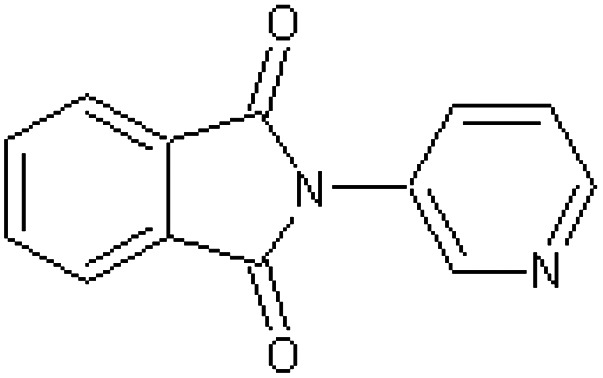
|
Rabbit34 |
| N-Butylphthalimide [41] | 2-Butyl-1H-isoindole-1,3(2H)-dione |

|
Rabbit34 |
| Phthalimide [42] | 1H-Isoindole-1,3(2H)-dione |
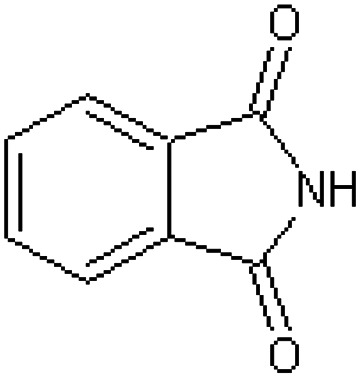
|
Rabbit34 |
| N-Hydroxythalidomide [43] | 2-(1-Hydroxy-2,6-dioxopiperidin-3-yl)-1H-isoindole-1,3(2H)-dione |
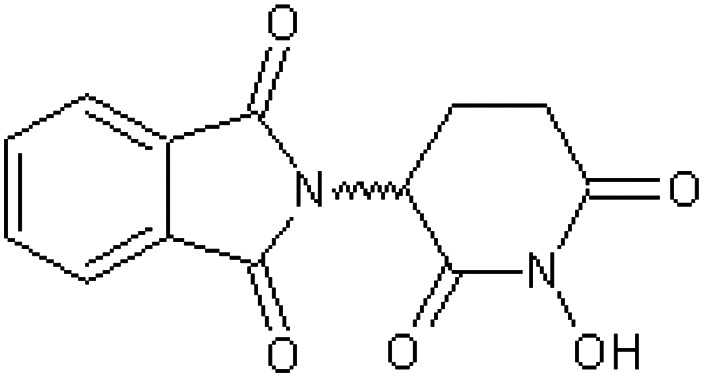
|
Rabbit52 |
| N-Methoxythalidomide [44] | 2-(1-Methoxy-2,6-dioxopiperidin-3-yl)-1H-isoindole-1,3(2H)-dione |
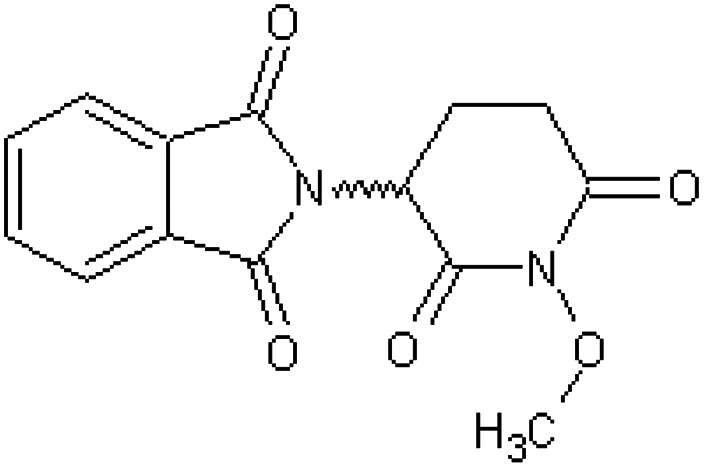
|
Rabbit52 |
| 2-Phthalimidoacetamide [45] | 2-(1,3-Dioxo-1,3-dihydro-2H-isoindol-2-yl)acetamide |
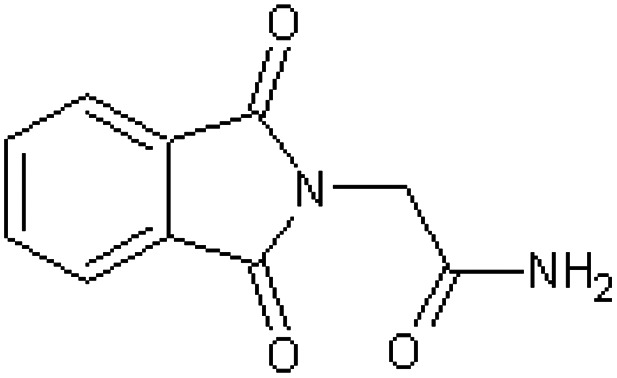
|
Rabbit34,47 |
| 4-Phthalimidobutyramide [46] | 4-(1,3-Dioxo-1,3-dihydro-2H-isoindol-2-yl)butanamide |

|
Rabbit34 |
| 2-Phthalimidobutyramide [47] | 2-(1,3-Dioxo-1,3-dihydro-2H-isoindol-2-yl)butanamide |
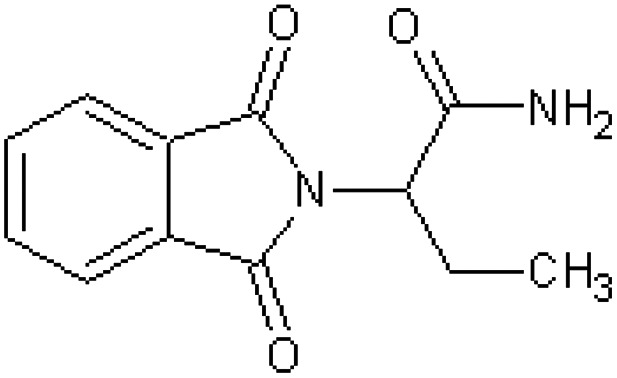
|
Rabbit47 |
| 2-Phthalimidoglutaric diamide [48] | 2-(1,3-Dioxo-1,3-dihydro-2H-isoindol-2-yl)pentanediamide |
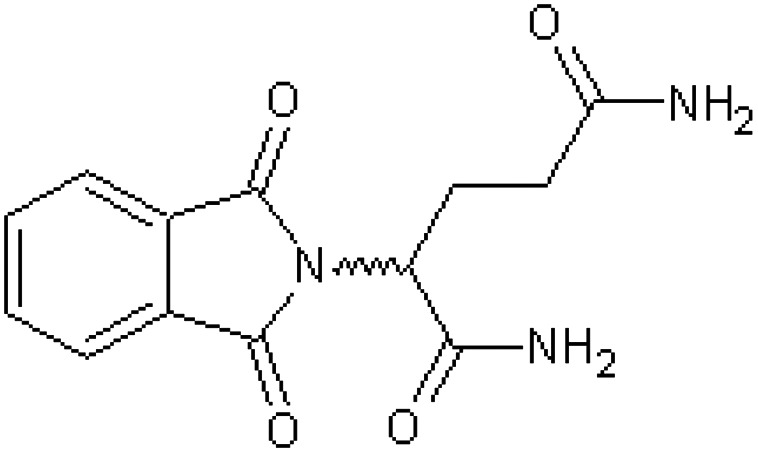
|
Rabbit52 |
Table 6 affords a compilation of the findings for the teratogenic testing of 11 compounds (compounds 49–59) which are arguably structural analogues of thalidomide where both the phthalimido and glutarimido functions have been replaced.37,46,47,51,53,54 All compounds appeared to be non-teratogenic in sensitive species such as the rabbit and certain non-human primates. Several of these compounds involved replacement of the carbonyl function in the phthalidimido group by a sulphone group (compounds 49–51, 56, 57). Others include compounds where the phthalimido group has been replaced by an oxoquinazoline function (compounds 53–55).
Table 6. Non-teratogenic structural analogues of thalidomide; compounds in which both phthalimide and glutarimide functions have been replaced.
| Name | Systematic name | Structure | Test species and references |
| 2-(3-Oxo-1,2-benzisothiazolin-2-yl)succinimide-S,S-dioxide [49] | 3-(1,1-Dioxido-3-oxo-1,2-benzothiazol-2(3H)-yl)pyrrolidine-2,5-dione |

|
Rabbit47 |
| [50] | 2-(1,1-Dioxido-3-oxo-1,2-benzothiazol-2(3H)-yl)acetamide |

|
Rabbit47 |
| [51] | 2-(1,1-Dioxido-3-oxo-1,2-benzothiazol-2(3H)-yl)butanamide |
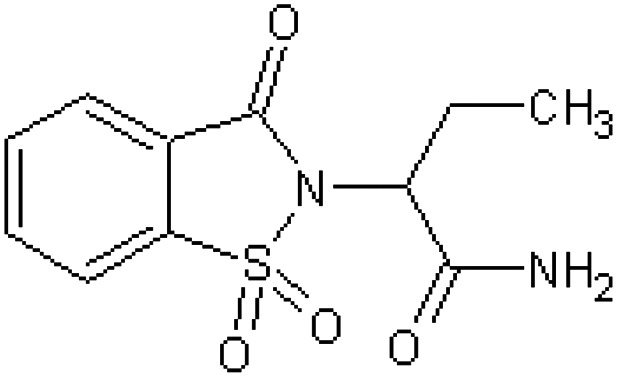
|
Rabbit47 |
| [52] | 2-(1-Oxo-1,3-dihydro-2H-isoindol-2-yl)acetamide |

|
Rabbit47 |
| 4-Oxo-3(4H)-quinazolineacetamide [53] | 2-(4-Oxoquinazolin-3(4H)-yl)acetamide |

|
Rabbit47 |
| 2-(4-Oxo-3,4-dihydroquinazolin-3-yl)-succinimide [54] | 3-(4-Oxoquinazolin-3(4H)-yl)pyrrolidine-2,5-dione |
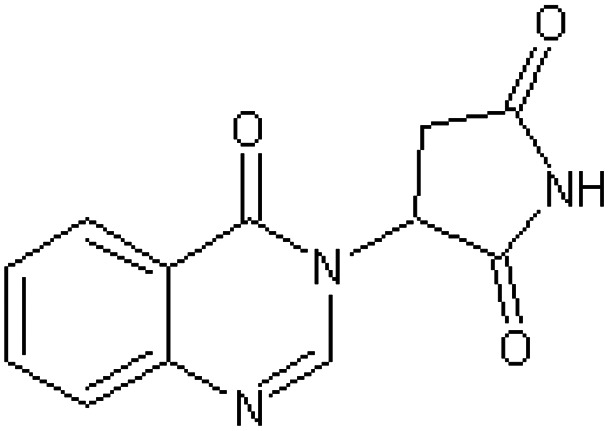
|
Rabbit47 |
| α-Ethyl-4-oxo-3(4H)-quinazolineacetamide [55] | 2-(4-Oxoquinazolin-3(4H)-yl)butanamide |
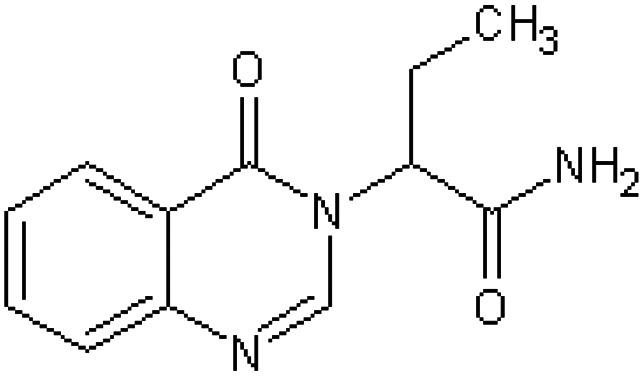
|
Rabbit47 |
| CG3033 supidimide 3-(2,3-dihydro-1,1-dioxido-3-oxo-1,2-benzisothiazol-2-yl)-2-oxopiperidine [56] | 2-(2-Oxopiperidin-3-yl)-1,2-benzothiazol-3(2H)-one 1,1-dioxide |
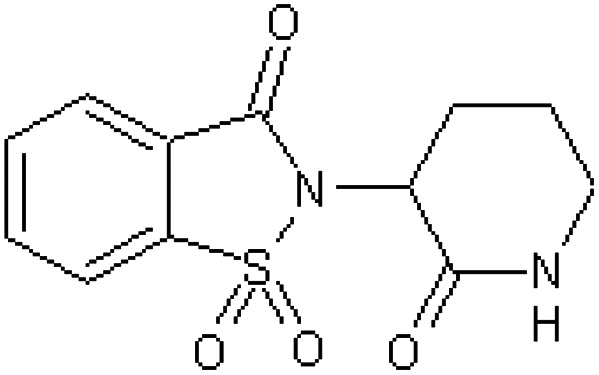
|
Baboon37,54 |
| Rabbit46 | |||
| EM240 [57] | 2-[(2-Oxopiperidin-3-yl)sulfamoyl]benzoic acid |
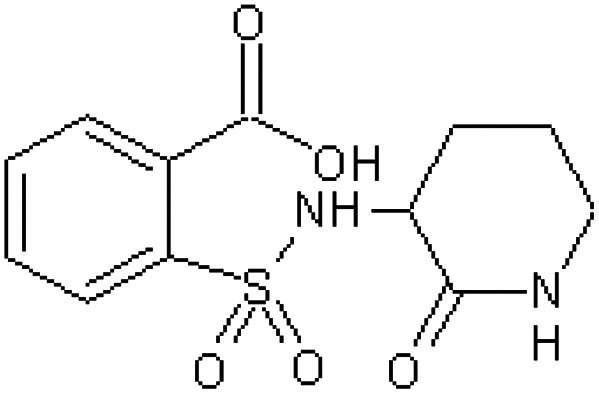
|
Rhesus monkey53 |
| Phthalamudine 3-(4′-chloro-3′-sulphmoyl)-3-hydroxy-phthalimidine [58] | 2-Chloro-5-(1-hydroxy-3-oxo-2,3-dihydro-1H-isoindol-1-yl)benzenesulfonamide |
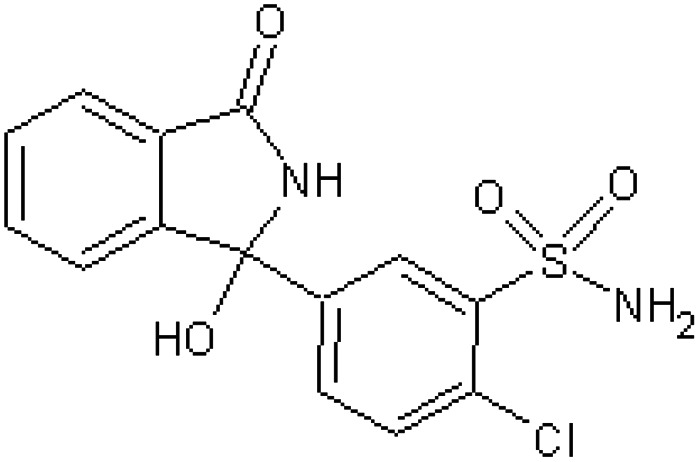
|
Rabbit51 |
| Captan N-(trichloromethylthio) –Δ4-tetrahydrophthalimide [59] | 2-[(Trichloromethyl)sulfanyl]-3a,4,7,7a-tetrahydro-1H-isoindole-1,3(2H)-dione |
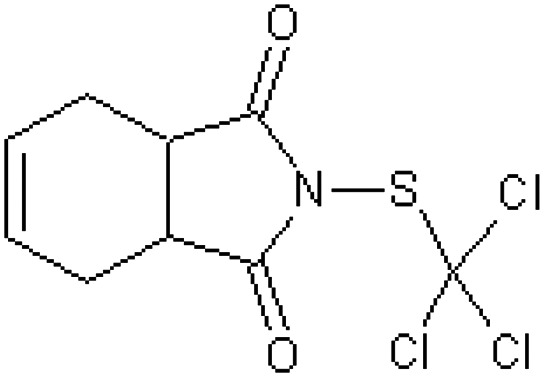
|
Rabbit51 |
5. Discussion
The most striking defect caused by thalidomide is that of amelia or meromelia (total or partial absence of extremities) and it was an unprecedented rise in the frequency of this relatively unusual type of anomaly that enabled the causal relationship with thalidomide intake to be discovered. However, the effects of thalidomide are not that specific and in addition to an absence or gross deformity of the long bones other problems embraced within the ‘thalidomide syndrome’, include facial haemangiomas, damage to the ears and eyes, vertebral column irregularities, genital malformations and abnormalities of internal organs including the heart, kidney and gastrointestinal tract. Indeed, almost any organ or tissue may be affected by thalidomide.55 Despite this extensive list of teratogenic effects, thalidomide is not a mutagen and its induced defects are not passed on to the next generation.56,57 Its interference with embryological development appears transitory and limited to the time of uterine exposure with no permanent and irreversible effects on the nucleic acids being evident. Interestingly, in regenerating rat liver (following partial hepatectomy) thalidomide treatment did not interfere with the de novo synthesis of DNA or RNA.58 This is the enigma presented.
From the data tendered in this overview several important points become evident. Both optical isomers of thalidomide are active as teratogens although the experimental results are difficult to interpret because of rapid racemisation and interconversion. The hydrolysis products of thalidomide are inactive as teratogens suggesting embryopathic activity is due to the parent molecule. The basic α-phthalimidoglutarimide arrangement appears essential for the teratogenic properties although minor modifications may be made without loss of activity. In the case of thalidomide and its active congener, EM12, the positional relationship of the phthalimide and glutarimide functions appears to be of critical importance with only the α-configuration being active. One consequence of this α-linkage is that it endows a point of asymmetry, a chiral centre, within the thalidomide and EM12 molecules. These considerations imply that the structural features for ‘thalidomide type’ of embryopathy are really quite conservative and essentially involve intact phthalimido and glutarimido functions linked crucially through an α-linkage.46
The furtive search for the underlying mechanism of thalidomide embryopathy has led to the proposal of many (over 30) potential possibilities, several of which have now been shown to be incorrect.59–63 Curently favoured mechanisms include inhibition of angiogenesis, obstructing chondrogenesis, interference with the synthesis, binding or function of growth factors, transmembrane receptors and mediator complexes, oxidative stress and redox misregulation (redox sensitive transcription factors) and affecting DNA replication and transcription events.5,64 With regards to the latter options, a detailed examination has indicated that intercalation of a single thalidomide molecule within the nucleic acid polymer may be strained and not an ‘easy fit’.5 Also, no permanent association may be undertaken as thalidomide does not induce mutagenesis and the ensuing defects are not hereditary. However, what appears to have been overlooked is that it is only a chiral molecule which may form a heterochiral dimer that is active in this respect.
The idea that thalidomide may intercalate within, or attach to, the DNA helix is not new and it had been noted that its structure resembled that of purine–pyrimidine base pairs.12,65,66 It has been shown to have an affinity for areas that are rich in guanine and cytosine residues, particularly the GC-boxes (GGGCGG) around GC-promoter sites. This intercalation with G-rich promoter regions disrupts transcription, interfering with transcription factor binding, but its association is insufficiently strong to result in permanent intercalation or an enduring disruption that leads to mutation.5,67
To date, it has been assumed that racemic thalidomide exists in biological systems as a collection of simple monomeric molecules whereas actually the active structure may be a dimeric heterochiral assembly associated by extensive intermolecular hydrogen bonding. For potential homochiral assemblies relatively unfavourable hydrogen-bonding abilities exist as donor and receptor groups are on opposite faces of the molecule.15 The α-linkage alone joining the two rings permits the existence of the two stereoisomers and hence the heterochiral assembly. When thalidomide is present at high concentration after dosing could such a hetero-dimer form and interfere with the normal functioning of the nuclear machinery before dissociating back into its component enantiomers?
The thalidomide molecule is not passive; in a biological environment it may be viewed as a dynamic ‘restless’ molecule continuously undergoing intramolecular and intermolecular rearrangements as well as hydrolysis. The individual isomers undergo rapid interconversion through keto–enol tautomerism and are in equilibrium with heterochiral dimer assemblies which incessantly form and then dissociate. Major additions to the ring structures or interference with the rings themselves extinguish these activities. It is suggested that once the thalidomide molecules have gained access to the embryonic DNA, it is these constant molecular events that principally involve the heterochiral thalidomide dimer possessing the structural features of the paired nucleotides of the double-stranded DNA, that subtly interfere at many places within the DNA molecule which is being transcripted extensively during this critical time. This subtle interference with gene activities leads to the host of malformations subsequently experienced following thalidomide exposure.† Hence, thalidomide may have a unique mechanism of action, acting as a general ‘chaotic disorganiser’ of embryonic DNA expression, thereby frustrating attempts to uncover a single and discrete mechanism of action but may help to explain why so many different mechanistic proposals have been forwarded.55,68
A recent comprehensive review, whilst commenting upon the spectrum of disorders that thalidomide may induce within individuals, remarked, ‘how and why the drug caused such a range and variability in damage remains unclear, but likely includes individual differences in metabolism and clearance of the drug, as well [as] genetic and environmental factors’.55 This sums up the frustratingly ambivalent position that still exists over half a century later.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgments
The authors are indebted to Professor John Lindon for the construction of the figure and tables.
Footnotes
†To avoid confusion when consulting virtually all previous thalidomide literature concerning teratogenicity, the Fischer–Rosanoff d/l configurational descriptors have been retained for thalidomide instead of adopting the more recent Cahn–Ingold–Prelog R/S nomenclature.
References
- Jönsson M. A. Acta Pharm. Suec. 1972;9:521–542. [PubMed] [Google Scholar]
- Jönsson M. A. Acta Pharm. Suec. 1972;9:543–562. [PubMed] [Google Scholar]
- Eriksson T., Björkman S., Höglund P. Eur. J. Clin. Pharmacol. 2001;57:365–376. doi: 10.1007/s002280100320. [DOI] [PubMed] [Google Scholar]
- Stephens T. D., Fillmore B. J. Teratology. 2000;61:189–195. doi: 10.1002/(SICI)1096-9926(200003)61:3<189::AID-TERA6>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Stephens T. D., Bunde C. J., Fillmore B. J. Biochem. Pharmacol. 2000;59:1489–1499. doi: 10.1016/s0006-2952(99)00388-3. [DOI] [PubMed] [Google Scholar]
- Knobloch J., Shaughnessy J. D., Rüther U. FASEB J. 2007;21:1410–1421. doi: 10.1096/fj.06-7603com. [DOI] [PubMed] [Google Scholar]
- Therapontos C., Erskine L., Gardner E. R., Figg W. D., Vargesson N. Proc. Natl. Acad. Sci. U. S. A. 2009;106:8573–8578. doi: 10.1073/pnas.0901505106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carini J. P., Pavei C., Silva A. P. C., Machado G., Mexias A. S., Pereira V. P., Fialho S. L., Mayorga P. Int. J. Pharm. 2009;372:17–23. doi: 10.1016/j.ijpharm.2008.12.034. [DOI] [PubMed] [Google Scholar]
- Reepmeyer J. C., Rhodes M. O., Cox D. C., Silverton J. V. J. Chem. Soc., Perkin Trans. 2. 1994:2063–2067. [Google Scholar]
- Allen F. H., Trotter J. J. Chem. Soc. D. 1970:778. [Google Scholar]
- Allen F. H., Trotter J. J. Chem. Soc. B. 1971:1073–1079. [Google Scholar]
- Furberg S., Petersen C. S. Acta Chem. Scand. 1965;19:253–254. doi: 10.3891/acta.chem.scand.19-1266. [DOI] [PubMed] [Google Scholar]
- Peterson C. S. Acta Chem. Scand. 1969;23:2389–2402. [Google Scholar]
- Suzuki T., Tanaka M., Shiro M., Shibata N., Osaka T., Asahi T. Phase Transitions. 2010;83:223–234. [Google Scholar]
- Hague D. E., Idle J. R., Mitchell S. C., Smith R. L. Xenobiotica. 2011;41:837–843. doi: 10.3109/00498254.2011.590547. [DOI] [PubMed] [Google Scholar]
- Reist M., Carrupt P. A., Testa B. Chem. Res. Toxicol. 1998;11:1521–1528. doi: 10.1021/tx9801817. [DOI] [PubMed] [Google Scholar]
- Schoetz G., Trapp O., Schurig V. Electrophoresis. 2001;22:3185–3190. doi: 10.1002/1522-2683(200109)22:15<3185::AID-ELPS3185>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Tian C., Xiu P., Meng Y., Zhao W., Wng Z., Zhou R. Chem. – Eur. J. 2012;18:14305–14313. doi: 10.1002/chem.201202651. [DOI] [PubMed] [Google Scholar]
- Eriksson T., Björkman S., Roth B., Fyge A., Höglund P. Chirality. 1995;7:44–52. doi: 10.1002/chir.530070109. [DOI] [PubMed] [Google Scholar]
- Schumacher H., Smith R. L., Williams R. T. Br. J. Pharmacol. Chemother. 1965;25:324–337. doi: 10.1111/j.1476-5381.1965.tb02053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams R. T., Schumacher H., Fabro S. and Smith R. L., The chemistry and metabolism of thalidomide, in Embryopathic Activity of Drugs: Biological Council Symposium, ed. J. M. Robson, E. M. Sullivan and R. L. Smith, J. & A. Churchill, London, 1965, pp. 167–182. [Google Scholar]
- Blaschke G., Kraft H. P., Fickentscher K., Köhler F. Arzneim.-Forsch./Drug Res. 1979;29:1640–1642. [PubMed] [Google Scholar]
- Fabro S., Smith R. L., Williams R. T. Nature. 1967;215:296. doi: 10.1038/215296a0. [DOI] [PubMed] [Google Scholar]
- Shibata N., Yamamoto T., Toru T. Top. Heterocycl. Chem. 2007;8:73–97. [Google Scholar]
- Schardein J. L., Schwetz B. A., Kenel M. F. Environ. Health Perspect. 1985;61:55–67. doi: 10.1289/ehp.856155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schardein J. L. and Macina O. T., Human Developmental Toxicants: Aspects of Toxicology and Chemistry, Taylor & Francis, Florida, 2007, pp. 127–141. [Google Scholar]
- Delahunt C. S., Lassen L. J. Science. 1964;146:1300–1305. doi: 10.1126/science.146.3649.1300. [DOI] [PubMed] [Google Scholar]
- Wilson J. G., Gavan A. J. Anat. Rec. 1967;158:99–109. doi: 10.1002/ar.1091580111. [DOI] [PubMed] [Google Scholar]
- Hendrickx A. G., Axelrod L. R., Clayborn L. D. Nature. 1966;210:958–959. doi: 10.1038/210958a0. [DOI] [PubMed] [Google Scholar]
- Poswillo D. E., Hamilton W. J., Sopher D. Nature. 1972;239:460–462. doi: 10.1038/239460a0. [DOI] [PubMed] [Google Scholar]
- Somers G. S. Lancet. 1962;1(7235):912–913. doi: 10.1016/s0140-6736(62)91943-8. [DOI] [PubMed] [Google Scholar]
- Fabro S., Smith R. L. J. Pathol. Bacteriol. 1966;91:511–519. doi: 10.1002/path.1700910227. [DOI] [PubMed] [Google Scholar]
- Smith R. L., Fabro S., Schumacher H. and Williams R. T., Studies on the relationship between the chemical structure and embryotoxic activity of thalidomide and related compounds, in Embryopathic Activity of Drugs: Biological Council Symposium, ed. J. M. Robson, E. M. Sullivan and R. L. Smith, J. & A. Churchill, London, 1965, pp. 194–209. [Google Scholar]
- Fabro S., Schumacher H., Smith R. L., Williams R. T. Life Sci. 1964;3:987–992. doi: 10.1016/0024-3205(64)90109-2. [DOI] [PubMed] [Google Scholar]
- Heger W., Sames K., Merker H. J., Frankus E., Graudums I., Neubert D., Teratology, 1986, 34 , 415 , (abstract) . [Google Scholar]
- Wuest H. M., Sigg E. B., Frâtta I. Life Sci. 1964;3:721–724. doi: 10.1016/0024-3205(64)90025-6. [DOI] [PubMed] [Google Scholar]
- Helm F. C., Frankus E., Friderichs E., Graudums I., Flohé L. Arzneim.-Forsch. 1981;31:941–949. [PubMed] [Google Scholar]
- Schumacher H. J., Terapane J., Jordan R. L., Wilson J. G. Teratology. 1972;5:233–240. doi: 10.1002/tera.1420050213. [DOI] [PubMed] [Google Scholar]
- Heger W., Schmahl H. J., Klug S., Felies A., Nau H., Merker H. J., Neubert D. Teratog., Carcinog., Mutagen. 1994;14:115–122. doi: 10.1002/tcm.1770140303. [DOI] [PubMed] [Google Scholar]
- Merker H. J., Heger W., Sames K., Stürge H., Neubert D. Arch. Toxicol. 1988;61:165–179. doi: 10.1007/BF00316631. [DOI] [PubMed] [Google Scholar]
- Heger W., Klug S., Schmahl H. J., Nau H., Merker H. J., Neubert D. Arch. Toxicol. 1988;62:205–208. doi: 10.1007/BF00570141. [DOI] [PubMed] [Google Scholar]
- Fabro S., Schumacher H., Smith R. L., Stagg B. R., Williams R. T. Br. J. Pharmacol. 1965;25:352–362. doi: 10.1111/j.1476-5381.1965.tb02055.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuest H. M., Frâtta I., Sigg E. B. Life Sci. 1966;5:393–396. doi: 10.1016/0024-3205(66)90151-2. [DOI] [PubMed] [Google Scholar]
- Fabro S., Smith R. L., Williams R. T. Biochem. J. 1967;104:570–574. doi: 10.1042/bj1040570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabro S., Smith R. L., Williams R. T. Biochem. J. 1967;104:565–569. doi: 10.1042/bj1040565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helm C. F., Frankus E., Teratology, 1982, 25 , 47A , (abstract) . [Google Scholar]
- Jönsson N. A., Mikiver L., Selberg U. Acta Pharm. Suec. 1972;9:431–446. [PubMed] [Google Scholar]
- Stockinger L., Koch H. Arzneim.-Forsch. 1969;19:167–169. [PubMed] [Google Scholar]
- Koch H., Stockinger L. Arzneim.-Forsch. 1971;21(Nr12):2022–2024. [PubMed] [Google Scholar]
- Klug S., Felies A., Stürje H., Nogueira A. C., Neubert R., Frankus E. Arch. Toxicol. 1994;68:203–205. doi: 10.1007/s002040050055. [DOI] [PubMed] [Google Scholar]
- Fabro S., Smith R. L., Williams R. T. Food Cosmet. Toxicol. 1966;3:587–590. doi: 10.1016/s0015-6264(65)80205-x. [DOI] [PubMed] [Google Scholar]
- Wuest H. M., Fox R. R., Crary D. D. Experientia. 1968;24:993–994. doi: 10.1007/BF02138700. [DOI] [PubMed] [Google Scholar]
- Scott W. J., Wilson J. G., Helm F. C. Teratology. 1980;22:183–185. doi: 10.1002/tera.1420220207. [DOI] [PubMed] [Google Scholar]
- Hendrickx A. G., Helm F. C. Teratology. 1980;22:179–182. doi: 10.1002/tera.1420220206. [DOI] [PubMed] [Google Scholar]
- Vargesson N. Birth Defects Res., Part C. 2015;105(2):140–156. doi: 10.1002/bdrc.21096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashby J., Tinwell H. Br. Med. J. 2002;325:1245. doi: 10.1136/bmj.325.7374.1245/a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strömland K., Philipson E., Anderson-Gronland M. Teratology. 2002;66:115–121. doi: 10.1002/tera.10083. [DOI] [PubMed] [Google Scholar]
- Fabro S., Session 6; Discussion section, in Embryopathic Activity of Drugs: Biological Council Symposium, ed. J. M. Robson, E. M. Sullivan and R. L. Smith, J. & A. Churchill, London, 1965, p. 229. [Google Scholar]
- Stephens T. D. Teratology. 1988;35:229–239. doi: 10.1002/tera.1420380307. [DOI] [PubMed] [Google Scholar]
- Hansen J. M., Harris C. Antioxid. Redox Signaling. 2004;6:1–14. doi: 10.1089/152308604771978291. [DOI] [PubMed] [Google Scholar]
- Vargesson N. BioEssays. 2009;31:1327–1336. doi: 10.1002/bies.200900103. [DOI] [PubMed] [Google Scholar]
- Vargesson N. ISRN Develop. Biol. 2013:241016. doi: 10.1155/2013/241016. [DOI] [Google Scholar]
- Knobloch J., Jungck D., Cock A. Curr. Mol. Med. 2017;17:108–117. doi: 10.2174/1566524017666170331162315. [DOI] [PubMed] [Google Scholar]
- Kin J. H., Sciall A. R. Toxicol. Sci. 2011;122:1–6. doi: 10.1093/toxsci/kfr088. [DOI] [PubMed] [Google Scholar]
- Koch H. P., Czelka M. J. Z. Naturforsch. 1986;C41:1057–1061. [PubMed] [Google Scholar]
- Furberg S. Acta Chem. Scand. 1965;19:1266–1267. doi: 10.3891/acta.chem.scand.19-1266. [DOI] [PubMed] [Google Scholar]
- Drucker L., Uziel O., Tohami T., Shapiro H., Radnay J., Yarkoni S., Lahav M., Lishner M. Mol. Pharmacol. 2003;64:415–420. doi: 10.1124/mol.64.2.415. [DOI] [PubMed] [Google Scholar]
- Patil D., Patwardhan B. and Kumbhae K., Why and how drugs fail, in Innovative Approaches in Drug Discovery. Ethnopharmacology, Systems Biology and Holistic Targeting, ed. B. Patwardhanand, R. Chaguturu, Academic Press, London, 2017, pp. 23–46. [Google Scholar]