

Abstract
Structural interactions that enable electron transfer to cytochrome-P450 (CYP450) from its redox partner CYP450-reductase (CPR) are a vital prerequisite for its catalytic mechanism. Here, we report the first structural model for the membrane-bound functional complex to reveal interactions between the full-length CYP450 and a minimal domain of CPR. Our results suggest that anchorage of the proteins in a lipid bilayer is a minimal requirement for CYP450 catalytic function. Akin to cytochrome-b5 (cyt-b5), Arg125 on the C-helix of CYP450s is found to be important for effective electron transfer, thus supporting the competitive behavior of redox partners for CYP450s. We report a general approach to study protein-protein interactions combining the use of nanodiscs with NMR spectroscopy and SAXS. Linking structural details to the mechanism will help unravel the xenobiotic metabolism of diverse microsomal CYP450s in their native environment and facilitate the design of new drug entities.
Keywords: Cytochrome P450, Cytochrome P450 reductase, Nanodiscs, Membrane protein
Graphical Abstract
Layout 1:
Solving a structure of the cytochrome P450 (CYP450) complex with its redox partner is a vital prerequisite to gaze at the selective route of electron transfer. Here we report structural interactions of CYP450-redox partner complex anchored in lipid membrane as a minimal requirement for functionality (electron transfer). This study brings about a paradigm shift to unravel the drug/xenobiotic metabolism by diverse microsomal CYPs in their native membrane environment.
The marked ability of CYP450 for biosynthesis, interconversion and efficient metabolism of steroids, vitamins, fatty acids and drugs makes it a vital target to fight various diseases.[1–4] Its catalytic efficiency depends on its interaction with either its redox partner CPR or cyt-b5, which is a rate-limiting step involving electron transfer.[5] During its catalytic cycle, CYP450 sequentially receives two electrons. CPR must provide the first electron whereas the second may originate from either CPR or cyt-b5. With few exceptions[6, 7], most structural information about CYP450 and CPR are based on crystal structures of their respective truncated soluble domains.[8–16] Intriguingly, the crystal structure of the FMN (flavin mononucleotide) binding domain (FBD) of CPR which lacks the transmembrane domain (truncated-FBD, tr-FBD) is identical to the FBD segment in full-length CPR,[17, 18] albeit it lacks activity.[19] Therefore, membrane counterparts of these proteins need to be considered to study their catalytic interaction, which is achieved by the use of nanodiscs in this study. Furthermore, lipid membrane has also been considered to be a crucial prerequisite to facilitate the access of hydrophobic ligands to the active site of CYP450s.[20] Earlier work from our lab and other research groups have demonstrated the use of small peptide-based nanodiscs which have facilitated the successful investigation of structure and dynamics of membrane proteins by solution-[21–27] and solid-state[28] NMR experiments. Their unique ability to accommodate multiple proteins in a natively folded functional state inside the lipid bilayer allows for a better understanding of protein-protein interactions.[29] Additionally, detergent-free reconstitution of sensitive CYP450s and related proteins increases their stability from few days to weeks.[27] In this study, we extend this approach to study the interaction of CYP450 2B4 (CYP2B4) with its redox partner CPR. We demonstrate that our system allows for functional characterization of protein-protein interactions in the complex and provides novel insights into the mechanisms of electron transfer.
Combining the use of peptide (4F) - lipid (DMPC) nanodiscs and full-length proteins with high-resolution solution NMR spectroscopy, we set out to structurally characterize the FBD as well as the complex formed between native CYP2B4 and FBD, each containing their N-terminal transmembrane (TM) domain (fl-CYP2B4 and fl-FBD, respectively), taking their functionally vital membrane-supported binding nature into account. The individually purified proteins (Figure S1) were incubated with nanodiscs in a stepwise manner, in order to form a functional complex suitable for structural analyses. Indeed, nanodiscs efficiently monomerize fl-FBD from its otherwise aggregated state in solution, resulting in 1H-15N-TROSY-HSQC NMR spectra featuring remarkable resolution of a well-folded protein (Figure 1a). The 1H-15N-TROSY-HSQC of the fl-FBD in nanodiscs shows significant resemblance to tr-FBD in solution published previously,[30] indicating that the overall fold of the soluble domain is not affected by the presence of the TM domain nor the membrane. A similar dynamic structural property has been observed for other membrane-anchored proteins such as Bcl-xL and cyt-b5.[31, 32] However, we observe approximately 55 additional peaks for fl-FBD, accounting for the N-terminal transmembrane domain, and several chemical shift perturbations towards the N-terminus of the truncated protein, indicative of protein reconstitution in the nanodisc. The full-length sample shows significant stability in nanodiscs (Insert, Figure 1a), facilitating acquisition of 3D NMR spectra. The enhanced signal dispersion and signal-to-noise ratio enabled sequential assignment of 83% of FBD residues, including a large portion of the N-terminal domain (Figure S2a). A three-dimensional structural model for the (soluble domain of) membrane-anchored fl-FBD in lipid bilayer was generated using NMR based chemical shifts and the CS-ROSETTA server from the Biological Magnetic Resonance Data Bank[33] (Figure S3). The canonical fold of fl-FBD in nanodiscs was verified by prediction of secondary structural elements calculated from chemical shifts, which are highly comparable to those reported in the literature[34] (Figs. 1b and S2b). Superimposed X-ray and the solution structural model of FBD show that the overall Rossmann fold (alternating β-strand with α-helical segments) of FBD is well conserved (Figure S3). Structural investigation of FBD has to-date been limited to NMR spectroscopic and crystallographic studies of its truncated soluble domain. The restrictions of this approach are demonstrated when overlaying NMR and X-ray structures of FBD (Figure S3), which result in relatively large r.m.s.d values (~3.0 Ǻ).
Figure 1:
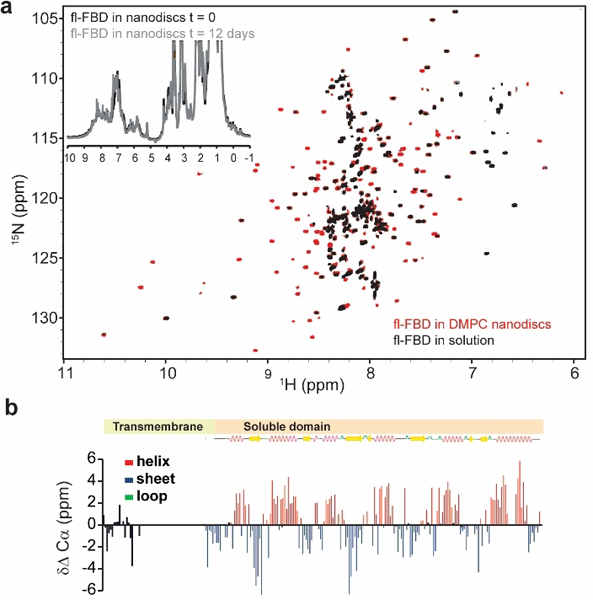
(a) Overlapped 2D 1H-15N TROSY-HSQC NMR spectra of fl-FBD reconstituted in nanodiscs (red) and fl-FBD in solution (grey). The insert shows 1D 1H NMR spectra of fl-FBD in nanodiscs recorded immediately (black) and after 12 days (grey), demonstrating the stability of the sample. (b) Secondary structural elements based on the comparison of experimentally measured Cα chemical shift values to the random coil Cα chemical shift values (ΔCα).
In order to study the native protein-protein interactions occurring between the two redox partners, a functional complex was established in a membrane environment (Figure 2). Successful reconstitution of fl-FBD and fl-CYP2B4 peptide-based nanodiscs is supported by dynamic light scattering (DLS) and size exclusion chromatography (SEC) measurements (Figure 2a-b). At the optimized 1:1.5 w/w peptide:lipid ratio, the obtained empty peptide nanodiscs feature hydrodynamic radii of 4.45±0.40 nm in DLS measurements, which is in excellent agreement with the radius of gyration determined from small-angle X-ray scattering (SAXS) data (4.49±0.07 nm) (Figure S4). Despite the significant molecular weight of nanodiscs (~124.5 kDa), the hydrodynamic radii of aggregated protein assemblies are reduced upon insertion into the lipid bilayer. Incubation of fl-CYP2B4 followed by fl-FBD resulted in a stepwise increase of the constructs, indicating successful incorporation of both proteins into the membrane (Figure 2a-b). The relative ratio of reconstituted full-length proteins in the CYP2B4-FBD complex was estimated to be 0.95 using UV-Vis spectroscopy (Figure S5). Correct orientation of the two proteins in the complex is supported by SAXS measurements (Figure 2c-d). The linear Guinier region from experimental scattering curves of empty and protein-loaded nanodiscs demonstrate sample uniformity (Figure S4a-b). The maximum dimension (Dmax) of nanodiscs and the membrane reconstituted redox complex (fl-CYP2B4-fl-FBD) was calculated using pair-distance distribution function with GNOM module in PRIMUS.[35] The Dmax was found to increase from 114 to 197.64 Å, which is attributed to the association of the soluble domains of the fl-CYP2B4-fl-FBD complex with the nanodiscs (Figure S4c-d). An ab initio model of the redox-CYP450 complex anchored in lipid nanodiscs (reddish brown) was reconstructed using DAMMIN/DAMMIF module in Primus from ATSAS package[35] (Figure 2d). The bell-shaped curve of nanodisc anchoring fl-CYP450-fl-FBD complex in normalized Kratky plot demonstrates the presence of a well-folded redox-complex in membrane (Figure S4f).
Figure 2: Reconstitution of the fl-FBD and fl-CYP2B4 redox complex in peptide-based nanodiscs.
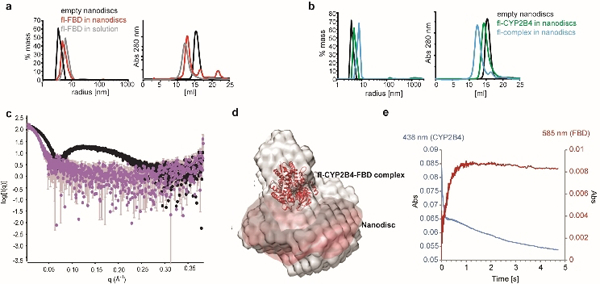
(a) DLS (left) and SEC measurements (right) of empty nanodiscs (black), fl-FBD incubated with nanodiscs (red) and fl-FBD in solution (grey) demonstrate successful reconstitution of fl-FBD in nanodiscs. (b) fl-CYP2B4 reconstituted in nanodiscs (green), the complex between fl-FBD and fl-CYP2B4 reconstituted in nanodiscs (blue), and empty nanodiscs (black). (c) Overlapped experimental scattering curves of empty (black) and fl-CYP2B4-fl-FBD complex containing nanodiscs (purple) from SAXS data. (d) An ab initio reconstruction of the fl-FBD-fl-CYP2B4 complex anchored in lipid nanodiscs (reddish brown) using the DAMMIN/DAMMIF module in Primus from ATSAS package.[35] A low resolution molecular envelop is superimposed with docked structure of the fl-FBD-fl-CYP2B4 complex. (e) Transfer of the second electron from hydroquinone FBD to oxyferrous CYP2B4 under anaerobic conditions can be monitored by the increase of absorbance at 585 nm (FBD hydroquinone to semiquinone transfer, red), as well as the decrease at 438 nm (CYP2B4 reduction of oxyferrous state, blue).
To unambiguously ensure catalytic activity of the membrane-embedded complex, we used stopped-flow data to monitor the reduction of oxyferrous to ferric fl-CYP2B4 upon electron transfer from FMN under anaerobic conditions (Figs 2e and S6). An increase in absorbance at 585 nm (FBD hydroquinone to semiquinone) and a decrease at 438 nm (CYP2B4 reduction of oxyferrous state) were observed as a result of the second electron transfer from hydroquinone fl-FBD to oxyferrous fl-CYP2B4. In the full-length CYP2B4-FBD complex, FBD oxidizes rapidly (3.7 s−1), which is attributed to electron transfer from FBD to CYP2B4 (Figure S6). The kinetic traces of the individual proteins in nanodiscs are shown in figure S6, along with the rates of oxidation of fl-CYP2B4 (0.09 s−1) and fl-FBD (1.5 s−1). In the presence of FBD, CYP2B4 oxidizes more rapidly (~19 s−1 and 0.33 s−1) due to reduction of oxyferrous CYP2B4 by FBD; the rapid electron transfer (~19 s−1) process needs further investigation for a complete understanding of the process. After receiving an electron CYP450 undergoes catalysis and returns to the ferric protein. The slower oxidation of CYP2B4 in the complex than FBD is expected because it has been shown that catalysis by CYP2B4 proceeds via a long-lived hydroperoxo intermediate in the presence of reductase.[5]
Mapping the interacting hot-spot region of the functionally active full-length redox complex (CYP2B4-FBD) significantly contributes to the design and development of novel drug molecules. The successful reconstitution of the uniformly 15N-labeled FBD and unlabeled CYP2B4 complex in nanodiscs enabled us to probe the protein-protein binding interface by NMR (Figure 3). Notably, the sample resulted in a well-dispersed spectrum featuring resolved peaks. The Rossmann fold of FBD is unaffected by the presence of fl-CYP2B4, yet, the protein-protein interaction is reflected in the spectrum (Figure 3b). The FMN stabilizing anionic loop of FBD interacts with the cationic surface on the proximal side of CYP2B4. Small, yet relevant, chemical shift perturbations (CSPs) were observed for fl-FBD in the complex, in particular in the linker region (S67-V70) and soluble domain (T88, Y117, L119, G143, K176, and H180) (Figs 3c and S7a). Contact induced conformational dynamics for residues Y117-L119 of fl-FBD was observed, although they do not seem to be directly involved in the interaction. Upon binding to fl-CYP2B4, an overall intensity decrease of fl-FBD resonances was observed (Fig. 3d and S7b), indicating the complex formation and interaction between the proteins. Lipid-protein interactions have also been reported to play important roles in amyloid aggregation.[36, 37] Most of the residues in the globular domain are affected by an exchange in an intermediate timescale. Addition of the solvent paramagnetic relaxation enhancement (sPRE) agent [Gd(DTPA-BMA)] provides further identification of residues buried in the interacting interface (Figs 3e and S7c). Residues in the protein-protein interacting interface of the redox complex will not be quenched by the sPRE agent, as they are no longer solvent accessible. The difference in relative intensities between fl-FBD and the fl-complex in the absence and presence of the sPRE agent (ΔQuenching) demonstrates which residues are recovered in the complex. The interacting interfacial residues of fl-FBD, T88 and G143, as well as residues 174–180 are protected from quenching in sPRE experiments. Several residues recovered from sPRE effects coincide with regions of the protein undergoing chemical shift perturbations, especially the linker, as well as residues surrounding M137 and G143.
Figure 3: Probing hot spots for redox complex formation.
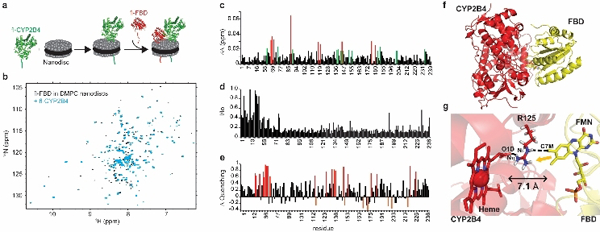
(a) Schematic representation of complex formation. (b) 1H-15N TROSY-HSQC of 15N fl-FBD in peptide-based nanodiscs in the absence (black) and presence (blue) of fl-CYP2B4. (c) CSPs of fl-FBD shifted by 1 (green) and 2 (red) standard deviation of the mean upon binding to fl-CYP2B4. (d) Line-broadening of fl-FBD resonances upon binding to fl-CYP2B4. (e) sPRE data displaying the recovery of fl-FBD resonances by 1 standard deviation (red), which no longer undergo quenching once in the complex. (f) Structural model of globular domain of full-length FBD-CYP2B4 complex derived using HADDOCK simulations.[38] (g) The shortest edge-to-edge distance between FMN and heme was calculated using HARLEM.[43] An yellow arrow marks the probable pathway of electron transfer.
A structural model of the full-length protein complex (CYP2B4-FBD) anchored in the membrane was derived using HADDOCK 2.2.[38] NMR based CSPs and differential line broadening data were used as proximity restraints to guide the docking simulation. HADDOCK involves rigid-body docking, followed by molecular dynamics simulations that allow selected amino acid side chains, as well as parts of the backbone, to move freely to improve the complementarity and electrostatic interactions at the interface. The active ambiguous restraints for CYP2B4 were obtained from published mutagenesis data[39] (Table 1). However, the interfacial residues on the CPR side were selected from CSPs and differential line broadening data using fl-FBD. HADDOCK simulation resulted in 183 complex structures which covered ~91.5% of total structures based on energy statistics and better Z-scores (Table 2). An energetically minimized structure of the complex shows that the binding surface of CYP2B4 covers mostly charged or hydrophobic residues, including R133, F135, M137 and K139, spanning the C-D loop along with some residues on other loops on the proximal surface of heme of CYP2B4. As shown in Figure 3f-g, both prosthetic groups are nearly perpendicular to each other (~118.3° angle) with a shortest edge-to-edge distance of 7.1 Å and it is within the 14.0 Å limit predicted for electron transfer to occur.[40] The relative orientation of two cofactors in CPR (ΔTGEE) and rat HO-1 complex[41] is similar to our proposed model. Moreover, the crystal structure of bacterial CYPBM3 complex revealed the distance between heme and FMN to be ~18 Å along with a similar orientation to our proposed model.[42] Both studies support the validity of our biologically active complex (CYP2B4-FBD) in lipid bilayer.
In addition, we predicted the electron transfer pathway in the membrane-embedded protein complex using HARLEM.[43] The guanidinium group of R125 on the C-helix of CYP2B4 acts as a bridge to transfer an electron from FMN to the D-propionate of the heme (Figure 3g). It should be noted that the structural model for the truncated proteins complex is also feasible in solution[44], possibly due to non-specific electrostatic interaction, but lacks functional intent.[19]
The structural perspective of different CYP450 isoforms reveals a common binding surface for CPR. Thus amino acids critical for maintaining the structural conformation and biological function are often under evolutionary constraints and evolve slowly. As expected, ConSurf analysis [45, 46] of CYP2B4 showed that the majority of conserved residues are located on the proximal side of the protein (Figure S8). Evolutionary conservational analysis also reveals that R125 is one of the most conserved residues on the CYP2B4 proximal surface, which is well supported by earlier published reports on CYPcam, CYP241 and CYP2B4.[47–49] This ubiquitous presence of R125 in CYP450s may be responsible for the competitive nature of redox partners for binding CYP450s.
In summary, we successfully report the first atomic-resolution structural characterization of a minimal and fully functional CYP450-FBD complex anchored in lipid membrane. In addition to unambiguously proving the functional nature of the complex, we depict the binding interface and propose an electron transfer pathway via R125. The use of a minimal CPR domain (fl-FBD) imparts vital insights into the electron transfer process and can facilitate a better understanding of drug metabolism towards the design of therapeutics. While a simple model lipid bilayer is utilized in this study to overcome numerous challenges posed by the large-size of the membrane-bound functional complex, investigations of the roles of membrane composition[50] and raft domain[51] that have recently been shown to stabilize CYP450, would be important to obtain further insights into the mechanism of electron transfer process and metabolism by CYP450.
Supplementary Material
Acknowledgements
This research was supported by funds from NIH (GM084018 to A.R.). The authors thank Dr. Thirupathi Ravula for help with nanodiscs, Dr. Vivekanandan with the Biophysics NMR facility, and the staff at BioCAT Beamline at Argonne National Laboratory for help with SAXS experiments.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].Shen AL, O’Leary KA, Kasper CB, J Biol Chem 2002, 277, 6536–6541. [DOI] [PubMed] [Google Scholar]
- [2].Nebert DW, Russell DW, Lancet 2002, 360, 1155–1162. [DOI] [PubMed] [Google Scholar]
- [3].Orlando L, Schiavone P, Fedele P, Calvani N, Nacci A, Rizzo P, Marino A, D’Amico M, Sponziello F, Mazzoni E, Cinefra M, Fazio N, Maiello E, Silvestris N, Colucci G, Cinieri S, Cancer Treat Rev 2010, 36 Suppl 3, S67–71. [DOI] [PubMed] [Google Scholar]
- [4].O’Donnell A, Judson I, Dowsett M, Raynaud F, Dearnaley D, Mason M, Harland S, Robbins A, Halbert G, Nutley B, Jarman M, Br J Cancer 2004, 90, 2317–2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Pearl NM, Wilcoxen J, Im S, Kunz R, Darty J, Britt RD, Ragsdale SW, Waskell L, Biochemistry 2016, 55, 6558–6567. [DOI] [PubMed] [Google Scholar]
- [6].Monk BC, Tomasiak TM, Keniya MV, Huschmann FU, Tyndall JD, O’Connell JD 3rd, Cannon RD, McDonald JG, Rodriguez A, Finer-Moore JS, Stroud RM, Proc Natl Acad Sci U S A 2014, 111, 3865–3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ghosh D, Griswold J, Erman M, Pangborn W, Nature 2009, 457, 219–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hamdane D, Xia C, Im SC, Zhang H, Kim JJ, Waskell L, J Biol Chem 2009, 284, 11374–11384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ellis J, Gutierrez A, Barsukov IL, Huang WC, Grossmann JG, Roberts GC, J Biol Chem 2009, 284, 36628–36637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Poulos TL, Finzel BC, Howard AJ, J Mol Biol 1987, 195, 687–700. [DOI] [PubMed] [Google Scholar]
- [11].Scott EE, He YA, Wester MR, White MA, Chin CC, Halpert JR, Johnson EF, Stout CD, Proc Natl Acad Sci U S A 2003, 100, 13196–13201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Yano JK, Hsu MH, Griffin KJ, Stout CD, Johnson EF, Nat Struct Mol Biol 2005, 12, 822–823. [DOI] [PubMed] [Google Scholar]
- [13].Mast N, White MA, Bjorkhem I, Johnson EF, Stout CD, Pikuleva IA, Proc Natl Acad Sci U S A 2008, 105, 9546–9551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zhang A, Zhang T, Hall EA, Hutchinson S, Cryle MJ, Wong LL, Zhou W, Bell SG, Mol Biosyst 2015, 11, 869–881. [DOI] [PubMed] [Google Scholar]
- [15].Petrunak EM, DeVore NM, Porubsky PR, Scott EE, J Biol Chem 2014, 289, 32952–32964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Scott EE, White MA, He YA, Johnson EF, Stout CD, Halpert JR, J Biol Chem 2004, 279, 27294–27301. [DOI] [PubMed] [Google Scholar]
- [17].Pandey AV, Fluck CE, Pharmacol Ther 2013, 138, 229–254. [DOI] [PubMed] [Google Scholar]
- [18].Zhao Q, Modi S, Smith G, Paine M, McDonagh PD, Wolf CR, Tew D, Lian LY, Roberts GC, Driessen HP, Protein Sci 1999, 8, 298–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Black SD, French JS, Williams CH Jr., Coon MJ, Biochem Biophys Res Commun 1979, 91, 1528–1535. [DOI] [PubMed] [Google Scholar]
- [20].Baylon JL, Lenov IL, Sligar SG, Tajkhorshid E, J Am Chem Soc 2013, 135, 8542–8551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hagn F, Etzkorn M, Raschle T, Wagner G, J Am Chem Soc 2013, 135, 1919–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Etzkorn M, Raschle T, Hagn F, Gelev V, Rice AJ, Walz T, Wagner G, Structure 2013, 21, 394–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Puthenveetil R, Vinogradova O, Proteins 2013, 81, 1222–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bibow S, Carneiro MG, Sabo TM, Schwiegk C, Becker S, Riek R, Lee D, Protein Sci 2014, 23, 851–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Fox DA, Larsson P, Lo RH, Kroncke BM, Kasson PM, Columbus L, J Am Chem Soc 2014, 136, 9938–9946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ding Y, Fujimoto LM, Yao Y, Plano GV, Marassi FM, Biochim Biophys Acta 2015, 1848, 712–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zhang M, Huang R, Ackermann R, Im SC, Waskell L, Schwendeman A, Ramamoorthy A, Angew Chem Int Ed Engl 2016, 55, 4497–4499. [DOI] [PubMed] [Google Scholar]
- [28].Mors K, Roos C, Scholz F, Wachtveitl J, Dotsch V, Bernhard F, Glaubitz C, Biochim Biophys Acta 2013, 1828, 1222–1229. [DOI] [PubMed] [Google Scholar]
- [29].Ravula T, Barnaba C, Mahajan M, Anantharamaiah GM, Im SC, Waskell L, Ramamoorthy A, Chem Commun (Camb) 2017, 53, 12798–12801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Huang R, Zhang M, Rwere F, Waskell L, Ramamoorthy A, J Biol Chem 2015, 290, 4843–4855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Aisenbrey C, Sudheendra US, Ridley H, Bertani P, Marquette A, Nedelkina S, Lakey JH, Bechinger B, Eur Biophys J 2007, 37, 71–80. [DOI] [PubMed] [Google Scholar]
- [32].Durr UH, Yamamoto K, Im SC, Waskell L, Ramamoorthy A, J Am Chem Soc 2007, 129, 6670–6671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Shen Y, Vernon R, Baker D, Bax A, J Biomol NMR 2009, 43, 63–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Rwere F, Xia C, Im S, Haque MM, Stuehr DJ, Waskell L, Kim JJ, J Biol Chem 2016, 291, 14639–14661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Franke D, Petoukhov MV, Konarev PV, Panjkovich A, Tuukkanen A, Mertens HDT, Kikhney AG, Hajizadeh NR, Franklin JM, Jeffries CM, Svergun DI, J Appl Crystallogr 2017, 50, 1212–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Michalek M, Salnikov ES, Werten S, Bechinger B, Biochemistry 2013, 52, 847–858. [DOI] [PubMed] [Google Scholar]
- [37].Brender JR, Salamekh S, Ramamoorthy A, Acc Chem Res 2012, 45, 454–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].van Zundert GCP, Rodrigues J, Trellet M, Schmitz C, Kastritis PL, Karaca E, Melquiond ASJ, van Dijk M, de Vries SJ, Bonvin A, J Mol Biol 2016, 428, 720–725. [DOI] [PubMed] [Google Scholar]
- [39].Bridges A, Gruenke L, Chang YT, Vakser IA, Loew G, Waskell L, J Biol Chem 1998, 273, 17036–17049. [DOI] [PubMed] [Google Scholar]
- [40].Page C, Current Opinion in Chemical Biology 2003, 7, 551–556. [DOI] [PubMed] [Google Scholar]
- [41].Sugishima M, Sato H, Higashimoto Y, Harada J, Wada K, Fukuyama K, Noguchi M, Proc Natl Acad Sci U S A 2014, 111, 2524–2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Sevrioukova IF, Li H, Zhang H, Peterson JA, Poulos TL, Proc Natl Acad Sci U S A 1999, 96, 1863–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kurnikov IV, HARLEM molecular modeling package, 2000. [Google Scholar]
- [44].Estrada DF, Laurence JS, Scott EE, J Biol Chem 2016, 291, 3990–4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Glaser F, Pupko T, Paz I, Bell RE, Bechor-Shental D, Martz E, Ben-Tal N, Bioinformatics 2003, 19, 163–164. [DOI] [PubMed] [Google Scholar]
- [46].Landau M, Mayrose I, Rosenberg Y, Glaser F, Martz E, Pupko T, Ben-Tal N, Nucleic Acids Res 2005, 33, W299–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Nakamura K, Horiuchi T, Yasukochi T, Sekimizu K, Hara T, Sagara Y, Biochim Biophys Acta 1994, 1207, 40–48. [DOI] [PubMed] [Google Scholar]
- [48].Schlingmann KP, Kaufmann M, Weber S, Irwin A, Goos C, John U, Misselwitz J, Klaus G, Kuwertz-Broking E, Fehrenbach H, Wingen AM, Guran T, Hoenderop JG, Bindels RJ, Prosser DE, Jones G, Konrad M, N Engl J Med 2011, 365, 410–421. [DOI] [PubMed] [Google Scholar]
- [49].Ahuja S, Jahr N, Im SC, Vivekanandan S, Popovych N, Le Clair SV, Huang R, Soong R, Xu J, Yamamoto K, Nanga RP, Bridges A, Waskell L, Ramamoorthy A, J Biol Chem 2013, 288, 22080–22095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Barnaba C, Gentry K, Sumangala N, Ramamoorthy A, F1000Res 2017, 6, 662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Barnaba C, Sahoo BR, Ravula T, Medina-Meza IG, Im SC, Anantharamaiah GM, Waskell L, Ramamoorthy A, Angew Chem Int Ed Engl 2018, 57, 3391–3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.