

Abstract
Glutaminase-1 (GLS1) is a mitochondrial enzyme found in endothelial cells (ECs) that metabolizes glutamine to glutamate and ammonia. Although glutaminolysis modulates the function of human umbilical vein ECs, it is not known whether these findings extend to human ECs beyond the fetal circulation. Furthermore, the molecular mechanism by which GLS1 regulates EC function is not defined. In this study, we show that the absence of glutamine in the culture media or the inhibition of GLS1 activity or expression blocked the proliferation and migration of ECs derived from the human umbilical vein, the human aorta, and the human microvasculature. GLS1 inhibition arrested ECs in the G0/G1 phase of the cell cycle and this was associated with a significant decline in cyclin A expression. Restoration of cyclin A expression via adenoviral-mediated gene transfer improved the proliferative, but not the migratory, response of GLS1-inhibited ECs. Glutamine deprivation or GLS1 inhibition also stimulated the production of reactive oxygen species and this was associated with a marked decline in heme oxygenase-1 (HO-1) expression. GLS1 inhibition also sensitized ECs to the cytotoxic effect of hydrogen peroxide and this was prevented by the overexpression of HO-1. In conclusion, the metabolism of glutamine by GLS1 promotes human EC proliferation, migration, and survival irrespective of the vascular source. While cyclin A contributes to the proliferative action of GLS1, HO-1 mediates its pro-survival effect. These results identify GLS1 as a promising therapeutic target in treating diseases associated with aberrant EC proliferation, migration, and viability.
Keywords: glutaminase, endothelial cells, proliferation, migration
Graphical Abstract:
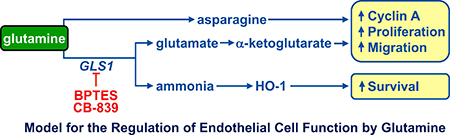
1. Introduction
Glutamine is the most abundant amino acid in the circulation and serves as a major carrier of nitrogen between organs. Although traditionally viewed as a non-essential amino acid, glutamine is indispensable during periods of rapid growth or in catabolic states such as trauma, critical illness, and sepsis [1]. Glutamine contributes to numerous metabolic and biosynthetic pathways [2–5]. It is a required nitrogen donor for the de novo synthesis of purines, pyrimidines, nucleotides, and hexosamine. Glutamine is predominantly metabolized to glutamate and ammonia by the mitochondrial enzyme glutaminase (GLS) [6]. Glutamate is subsequently converted by glutamate dehydrogenase and/or aminotransferases to α-ketoglutarate which feeds into the tricarboxylic acid (TCA) cycle for ATP production or as an anaplerotic source of carbon for the synthesis of non-essential amino acids and lipids, thereby fulfilling both the energetic and macromolecular requirements of cells. In addition, glutamate is used for the synthesis of glutathione which buffers the cell against oxidative stress.
Two distinct isoforms of GLS, GLS1 and GLS2, have been identified that exhibit distinct structural and kinetic properties and tissue specific expression profiles [7,8]. GLS2 expression is largely confined to the liver where it provides nitrogen for the urea cycle. In contrast, GLS1 is expressed in most tissues and plays a key role in regulating acid-base balance in the kidney and generating the excitatory neurotransmitter glutamate in the brain [9,10]. Considerable evidence indicates that the metabolism of glutamine by GLS1 plays a critical role in cancer [8]. Elevated levels of GLS1 have been detected in various cancer cells and tumors, and correlate with increased glutamine consumption and proliferation [11,12]. Significantly, glutamine deprivation or GLS1-silencing reduces tumor cell growth while pharmacological inhibition of GLS1 inhibits the growth of tumors both in culture and in mouse xenograft models [13,14]. In fact, several clinical trials are exploring the safety and efficacy of the GLS1-specific inhibitor CB-839 against a broad range of cancers.
Interestingly, GLS1 and its substrate glutamine have been shown to play a fundamental role in the immune system by regulating the activation and proliferation of T-lymphocytes [15–17]. Cell cycle synchronization studies revealed that GLS1 activity is required for G1/S phase transition and completion of S phase in human T-lymphocytes while metabolic flux studies using radiolabeled glutamine disclosed a proliferative pathway linking glutaminolysis to the biosynthesis of polyamines in these cells. GLS1 has also been identified in the heart where glutaminolysis has been coupled to maladaptive right ventricular hypertrophy in an animal model of pulmonary hypertension but to cardioprotection in a rodent model of ischemia-reperfusion injury [18,19]. Notably, abundant GLS1 activity has been reported in endothelial cells resulting in high rates of ammonia synthesis [20–22]. Moreover, recent studies demonstrate a critical role for glutaminolysis in regulating endothelial cell senescence, proliferation, redox potential, and energy balance [23–25]. In addition, the selective loss of GLS1 in endothelial cells results in impaired vessel sprouting in mouse models of physiologic or pathologic angiogenesis, suggesting a critical role for this enzyme in blood vessel formation [24,25].
Although glutaminolysis has been shown to modulate the function of human umbilical vein endothelial cells (HUVEC) [23–25], it is not known whether these findings extend to human endothelial cells beyond the fetal circulation. In addition, the molecular mechanism by which GLS1 regulates endothelial cell function is not fully defined. These are important issues as defects in endothelial cell function precipitate the development of vascular disease and a large number of clinical complications in several organ systems. Moreover, a better understanding of factors that regulate endothelial cell function may lead to the development of novel therapeutic approaches in treating vascular disease. Accordingly, the present study investigated the role of glutamine in various human endothelial cells. We now show that the metabolism of glutamine by GLS1 stimulates the proliferation, migration, and survival of human endothelial cells and is critical for redox homeostasis in these cells. This is observed in human endothelial cells derived from venous and arterial blood vessels as well as the microcirculation. Furthermore, we identified cyclin A and heme oxygenase-1 (HO-1) as important downstream targets of GLS1 that modulate the proliferative and pro-survival actions of this enzyme.
2. Materials and Methods
2.1. Reagents
Gelatin, sodium dodecyl sulfate (SDS), dithiothreitol, NaCl, EDTA, heparin, trichloroacetic acid, trypan blue, propidium iodide, RNase, cesium chloride, M199 medium, dimethyl-α-ketoglutarate, aspartate, hydrogen peroxide, glutamine, dialyzed fetal bovine serum, trypsin, 6-diazo-5-oxo-L-norleucine (DON), mercaptoethanol, streptomycin, penicillin, bis-2-(5-phenylacetomido-1,3,4-thiadiazol-2-yl)ethyl sulfide (BPTES), glutamate, ammonium chloride, and diethylamine NONOate (DEA-NO), K2HPO4, ATP, NAD, hydrazine, bovine liver glutamate dehydrogenase, and asparagine were from Sigma-Aldrich (St. Louis, MO). Endothelial cell growth factor was from Becton Dickinson Biosciences (Bedford, MA). Rainbow molecular weight markers were from GE Healthcare (Piscataway, NJ). Lipofectamine and 5-(and-6)-chloromethyl-2,7-dichlorodihydrofluorescein diacetate acetyl ester (CM-H2DCFDA) were from Life Technologies Corporation (Carlsbad, CA). CB-839 and NG-nitro-L-arginine methyl ester (L-NAME), were from Selleckchem (Houston, TX). Antibodies against GLS1, cyclin D1, cyclin E, cyclin A, p21, p27, p53, and β-actin were from Santa Cruz Biotechnology (Santa Cruz, CA), an antibody against phospho-retinoblastoma protein was from Cell Signaling Technologies (Beverley, MA), and an antibody against HO-1 was from Assay Designs (Ann Arbor, MI). [3H]Thymidine (20 Ci/mmol) was from Perkin Elmer (Boston, MA).
2.2. Cell Culture
HUVEC, human aortic endothelial cells (HAEC), and human dermal microvascular endothelial cells (HMEC) were purchased from Lonza Incorporated (Allendale, NJ). HAEC and HUVEC were serially cultured on gelatin-coated dishes in M199 medium supplemented with 20% dialyzed fetal bovine serum, 50 μg/ml endothelial cell growth factor, 90 μg/ml heparin, and 100 U/ml of penicillin and streptomycin while HMEC were grown on gelatin-coated plates in EGM-2 medium (Lonza Incorporated, Allendale, NJ) augmented with 10% serum, 5 ng/ml epidermal growth factor, 1 mg/ml hydrocortisone, and 100 U/ml of penicillin and streptomycin, as we previously described [26]. Unless otherwise indicated endothelial cells were cultured in the presence of 2 mM L-glutamine. All cells were incubated at 37°C in a humidified 95% air-5% CO2 atmosphere. HUVEC were used between passage 4 and 12, and HAEC and HMEC between passages 4 and 8.
2.3. Small Interference RNA (siRNA) and Adenovirus Infection Protocol
Gene expression was silenced using siRNAs targeting GLS1. The experimental and control, non-targeting siRNAs were obtained from Dharmicon (Lafayette, CO) and delivered to endothelial cells (100nM) using lipofectamine. A replication-defective adenovirus expressing a cyclin A (AdCyclin A) was generated by cloning the cyclin A cDNA into the pAACMVpLpA vector and cotransfecting HEK-293 cells (American Type Culture Collection, Manassas, VA) with the pJM17 plasmid [27]. Virus from cytopathic HEK-293 cells were harvested 48 hours after virus infection, purified by gradient cesium chloride centrifugation, and viral titers determined by plaque assay. Gene transfer of HO-1 was achieved by infecting endothelial cells with a replication-defective adenovirus expressing HO-1 (AdHO-1), as we previously reported [28]. Subconfluent endothelial cells were infected with AdCyclin A, AdHO-1, or an adenovirus expressing green fluorescent protein (AdGFP) at a multiplicity of infection of 50.
2.4. Cell Proliferation and DNA Synthesis
Cells were plated onto 6-well plates at a density of 2 × 104 cells/well. The following day, cells were washed and incubated in the presence and absence of various test compounds. At selected times, cells were dissociated from the plate with trypsin and counted in a Beckman Z1 Coulter Counter (Beckman Coulter Incorporated, Brea, CA). Cell proliferation was also monitored by measuring the incorporation of radiolabeled thymidine into DNA [26]. For these experiments, cells were pulsed with [3H]thymidine for 4 hours, washed with PBS, and fixed with trichloracetic acid (10%). DNA was then solubilized with 0.2% SDS/0.2N NaOH and radioactivity quantified by scintillation spectroscopy [26].
2.5. Cell Viability
Cell viability was determined by trypan blue exclusion staining, as we previously described [28].
2.6. Cell Cycle Analysis
Cell cycle progression was determined by flow-activated cell sorting, as we previously reported [29,30]. Cells were plated onto 10cm dishes, grown until 80% confluent, and then exposed to various test compounds for 24 hours. Cells were then collected, fixed in ethanol (70%), and incubated with propidium iodide (50μg/ml) and RNase A (100μg/ml) for 1 hour at room temperature. DNA fluorescence was measured in a Beckman Coulter CyAN™ ADP Cytometer (Brea, CA) and the fraction of cells in each phase of the cell cycle determined using FlowJo™ software (FlowJO LLC, Ashland, OR).
2.7. Cell Migration
Cell migration was monitored using a scratch wound assay. Cells were seeded onto 6-well plates and incubated until confluent. Using a sterile 200μl pipette tip, the cell layer was scratched in each well to generate a cleared line. Cell monolayers were photographed immediately and 20 hours after scratch injury with a digital camera (Q-Imaging, QICAM; Hitschfel Instruments, Incorporated, St. Louis, MO) and the cell-free area determined by planimetry.
2.8. Western Blotting
Cells were collected in electrophoresis buffer (125mM Tris [pH 6.8], 12.5% glycerol, 2% SDS, and trace bromophenol blue) and proteins separated by SDS-polyacrylamide gel electrophoresis under reducing conditions. Following electrophoretic transfer to nitrocellulose membranes, membranes were blocked with PBS and nonfat milk (5%) and then incubated with antibodies against cyclin D1 (1:200), cyclin E (1:100), cyclin A (1:500), p27 (1:250), p21 (1:250), p53 (1:200), phospho-retinoblastoma protein (1:100), HO-1 (1:1500), GLS1 (1:500), or β-actin (1:200). Membranes were washed in PBS, incubated with horseradish peroxidase-conjugated goat anti-rabbit or rabbit anti-goat antibodies and developed with commercial chemoluminescence reagents (Amersham, Arlington Heights, IL). Blots were then stripped of antibodies at 50°C for 30 minutes using a stripping solution (10% SDS, and 100mM mercaptoethanol in 62.5mM Tris buffer, pH 6.8) before reprobing with other antibodies. Protein expression was quantified by densitometry and normalized with respect to β-actin.
2.9. Real-Time PCR
Total RNA was extracted with TRIzol reagent (Life Technologies, Grand Island, NY) and transcribed to cDNA using the iScript cDNA Synthesis Kit (Bio-Rad Laboratories, Hercules, CA) according to the manufacturer’s instructions. Real-time PCR was performed with SYBR Green Supermix in a SYBER Green Cycler iQ 5RT-PCR detection system (Bio-Rad Laboratories, Hercules, CA). The primer sequences were as follows: cyclin A, forward 5’-CAGAAAACCATTGGTCCCTC-3’ and reverse 5’-CACTCACTGGCTTTTCATCTTC-3’, and 18S RNA forward 5”-TCAAGAACGAAAGTCGGAGG-3’ and reverse 5’-GGACATCTAAGGGCATCACA-3’. Transcript levels were quantified using the ΔΔCT method and normalized to 18S rRNA, as we previously described [22].
2.10. Cyclin A Promoter Assay
Cyclin A promoter activity was examined using a 471-bp (266/205) fragment of the human cyclin A 5’-flanking region that was inserted into the promoterless luciferase reporter plasmid pGL2-basic, as previously described [30]. Transfection efficiency was determined by introducing a plasmid encoding renilla luciferase (Promega, Madison, WI). Cells were transfected with plasmids (2μg) using lipofectamine (Invitrogen Corporation, Carlsbad, CA), incubated for 24 hours, and then exposed to GLS1 inhibitors for 24 hours. Firefly luciferase activity was measured in a Glomax luminometer (Promega, Madison, WI), and normalized to renilla luciferase.
2.11. GLS Activity
GLS activity was determined using a two-step assay where glutamine is first hydrolyzed to glutamate and then glutamate dehydrogenase used to catalyze the oxidative deamination of glutamate to form α-ketoglutarate and NADH, which is followed by absorbance spectroscopy [31]. Briefly, cell lysates were incubated in reaction buffer I (50mM Tris [pH 8.6], 0.5mM EDTA, 20mM glutamine, and 100mM K2HPO4) for one hour at 37°C and the reaction stopped by adding 3N HCl and placing samples on an ice-bath for 5 minutes. An aliquot of the quenched reaction mixture was then add to reaction buffer II (150Mm Tris [pH 9.4], 0.35mM ATP, 1.7mM NAD, 1% hydrazine, and 2.0 U glutamate dehydrogenase), incubated at room temperature for 45 minutes, and the formation of NADH monitored by absorbance spectroscopy at 340nm.
2.12. Determination of Intracellular Glutamate
Glutamate levels were determined in cell lysates using an enzymatic assay kit according to the manufacturer’s instructions (Sigma-Aldrich, St. Louis, MO). Samples were incubated in a glutamate enzyme mix containing a glutamate developer for 30 minutes at 37°C and absorbance measured at 450nm with a μQuant spectrophotometer (Bio-Tek Instruments, Winooski, VT). Concentrations were determined relative to a standard curve obtained using aqueous solutions of glutamate.
2.13. Measurement of Intracellular ROS
Intracellular ROS production was monitored using the cell-permeable probe CM-H2DCFDA, as we previously described [26,32]. Cells were incubated with the dye (10μM) for 30 minutes at 37°C, washed with PBS, and fluorescence quantified by microplate fluorimetry with excitation at 485nm and emission at 530nm. Mean values from each treatment were expressed relative to control cells.
2.14. Statistics
Results are presented as mean ± SD. Statistical analyses were performed with the use of a Student’s two-tailed t-test when comparing two groups and with a one-way analysis of variance with the Holm-Sidak post-hoc test when multiple treatment groups were compared. A value of P < 0.05 was considered statistically significant.
3. Results
The proliferation of HUVECs was dependent on the presence of extracellular glutamine. Cell proliferation was absent in HUVECs cultured in glutamine-free media (Figure 1A). However, increasing the concentration of glutamine in the culture media evoked a marked concentration-dependent rise in cell proliferation (Figure 1B). Incubation of HUVECs in the absence of glutamine in the culture media nearly abolished DNA synthesis and this was also observed in HAECs and HMECs (Figure 1C). Removal of glutamine from the culture media also retarded the migration of all three types of endothelial cells (Figure 1D). Since glutamine is largely metabolized by GLS1 to glutamate in vascular endothelium [20,21], we investigated the role of this enzyme on endothelial cell function. HUVECs grown in glutamine-free media have low levels of intracellular glutamate (Figure 2A). However, addition of glutamine in the culture media for 24 hours elevated intracellular glutamate concentrations by approximately 6-fold. The increase in glutamate concentration following glutamine supplementation was largely eliminated by treating HUVECs with the non-selective GLS inhibitor DON [33] or the GLS1-specific inhibitors BPTES and CB-839 [34,35], confirming their efficacy in blocking GLS1 activity. Notably, treatment of HUVECs with the GLS1 inhibitors blocked the proliferative response to glutamine in a concentration-dependent manner (Figure 2B, C, and D). In addition, the GLS1 inhibitors dramatically reduced DNA synthesis in HUVECs, HAECs, and HMECs grown in glutamine-replete media (Figure 3A, B, and C). The anti-proliferative effect of these agents was not related to any change in cell viability, as assessed by trypan blue exclusion (Figure 3D). Incubation of HUVEC with DON, BPTES, or CB-839 also blocked the migration of HUVEC grown in glutamine-containing media in a concentration-dependent fashion (Figure 4A, B, and C). Similar anti-migratory effects were attained by the GLS1 inhibitors in HAECs and HMECs (Figure 4D and E).
1.

Glutamine (Gln) stimulated the proliferation and migration of human endothelial cells. (A) Gln stimulated the proliferation of HUVECs in a time-dependent manner. (B) Gln stimulated the proliferation of HUVECs in a concentration-dependent manner. HUVECs were incubated in the absence and presence of different concentrations of Gln for three days. (C) Gln deprivation for three days inhibited DNA synthesis in HUVECs, HAECs, and HMECs. (D) Gln deprivation for three days inhibited the migration of HUVECs, HAECs, and HMECs. Results are means ± SD (n=4–6). *Statistically significant effect of Gln addition or deprivation.
2.

GLS1 inhibition blocked glutamine (Gln)-mediated increases in intracellular glutamate and proliferation of HUVECs. (A) Gln-mediated increases in intracellular glutamate were inhibited by GLS1 inhibitors. HUVECs were incubated with Gln in the presence or absence of DON (20μM), BPTES (20μM), or CB-839 (20μM) for 24 hours. (B) DON inhibited the proliferation of HUVECs in a concentration-dependent manner. (C) BPTES inhibited the proliferation of HUVECs in a concentration-dependent manner. (D) CB-839 inhibited the proliferation of HUVECs in a concentration-dependent manner. For the proliferation experiments, HUVEC were incubated in the presence or absence of GLS1 inhibitors for three days. Results are means ± SD (n=6).
*Statistically significant effect of GLS1 inhibitors. +Statistically significant effect of Gln
3.

GLS1 inhibition blocked DNA synthesis in human endothelial cells. (A-C) GLS1 inhibitors blocked DNA synthesis in HUVECs, HAECs, and HMECs. (D) GLS1 inhibition had no effect on endothelial cell viability. HUVECs were treated with DON (20μM), BPTES (20μM), or CB-839 (20μM) for 24 hours prior to DNA synthesis or viability measurements. Results are means ± SD (n=4–6).
*Statistically significant effect of GLS1 inhibitors.
4.

GLS1 inhibition blocked the migration of human endothelial cells. (A) DON inhibited the migration of HUVECs. (B) BPTES inhibited the migration of HUVECs. (C) CB-839 inhibited the migration of HUVECs. (D) DON (20μM) or BPTES (20μM) inhibited the migration of HAECs. (E) DON (20μM) or BPTES (20μM) inhibited the migration of HMECs. Cells were treated with the GLS1 inhibitors for 24 hours prior to the migration assay. Results are means ± SD (n=4–6). *Statistically significant effect of GLS1 inhibitors.
To complement the pharmacological studies a siRNA approach was used to silence GLS1 expression. Transfection of HUVEC with GLS1 siRNA markedly inhibited the expression of GLS1 protein by ◻70% while the non-targeting oligonucleotide had no effect (Figure 5A). Moreover, silencing GLS1 expression blocked the proliferation and migration of HUVEC by glutamine (Figure 5B and C).
5.

GLS1 knockdown inhibited the proliferation and migration of HUVEC. (A) GLS1 siRNA inhibited HUVEC GLS1 protein expression. (B) GLS1 siRNA inhibited HUVEC DNA synthesis. (C) GLS1 siRNA inhibited HUVEC migration. Cells were transfected with GLS1 siRNA (0.1μM) or non-targeting (NT) siRNA (0.1μM) for 48 hours prior to the proliferation or migration assay. Results are means ± SD (n=4). *Statistically significant effect of GLS1 silencing.
Given the importance of endothelial nitric oxide (NO) synthase (eNOS) in the circulation, we also explored the role of this enzyme in regulating endothelial cell function. Treatment of HUVEC with a high concentration of the eNOS inhibitor L-NAME (5mM) [36] significantly inhibited endothelial cell proliferation and migration (Figure 6A and B). This high concentration of L-NAME was previously reported to completely block basal NO release by HUVEC [37]. Consistent with the high degree of GLS1 protein expression, abundant GLS activity was detected in HUVEC, and this was significantly attenuated by the GLS1 inhibitor, BPTES (Figure 6C). In contrast, exogenous administration of the NO donor, DEA-NO, had no effect on endothelial GLS activity.
6.

Effect of eNOS on the proliferation, migration, and activity of GLS activity in HUVEC. (A) Effect of L-NAME on endothelial cell proliferation. Cells were incubated in the presence or absence of L-NAME (0–5mM) for three days. (B) Effect of L-NAME on endothelial cell migration. Cells were incubated in the presence or absence of L-NAME (0–5mM) for 24 hours. (C) Effect of BPTES and DEA-NO on GLS Activity. Cells were incubated in the presence or absence of BPTES (20μM) or DEA-NO (20μM) for 24 hours. Results are means ± SD (n=4–6). *Statistically significant effect of L-NAME or BPTES.
In other studies, we determined which of the glutamine metabolites contributes to the proliferative and migratory action of this amino acid. Supplementing the culture media with ammonia, given as ammonium chloride, or glutamate failed to restore the proliferation or migration of glutamine-deprived HUVECs (Figure 7A and B). In contrast, the addition of cell-permeable dimethyl-α-ketoglutarate resulted in a modest, but significant, rescue of endothelial cell growth and migration. Since, glutamine contributes to the intracellular pool of aspartate and asparagine [25], the role of these amino acids was also explored. While aspartate failed to rescue endothelial cell function, supplementation of the culture media with asparagine significantly increased HUVEC proliferation and migration. Moreover, the combined addition of dimethyl-α-ketoglutarate and asparagine largely restored the proliferative and migratory capacity of glutamine-deprived endothelial cells, demonstrating a critical role for these two glutamine metabolites in promoting endothelial cell growth and motility.
7.

Effect of glutamine (Gln) metabolites on the proliferation and migration of Gln-deprived HUVECs. A. α-Ketoglutarate (α-KG) and asparagine (Asp) are required for endothelial cell proliferation in Gln-depleted conditions. B. α-KG and Asp are required for endothelial cell migration in Gln-depleted conditions. Cells were grown in Gln (2mM)-replete or -deprived conditions with or without supplementation of glutamate (Glu;2Mm), ammonium chloride (NH4Cl;2mM), dimethyl-α-KG (DMKG;2mM), aspartate (Asp;100μM), and/or asparagine (Asn;100μM) for three days prior to performing the proliferation or migration assay. Results are means ± SD (n=5–6). *Statistically significant effect of Gln deprivation. +Statistically significant effect of DMKG and/or Asn supplementation.
Subsequently, we investigated the role of GLS1 on endothelial cell cycle progression. Administration of DON or BPTES arrested HUVECs in the G0/G1 phase of the cell cycle, as demonstrated by an increase in the percentage of cells in G0/G1 with a corresponding decline in the fraction of cells in S phase and G2/M (Figure 8A). Cell cycle arrest by the GLS1 inhibitors was associated with a striking reduction in the expression of cyclin A and phosphorylation of pRb with minimal changes in the expression of cyclin D1, cyclin E, p53, or the cyclin-dependent kinase inhibitors, p21 and p27 (Figure 8B and C). Both GLS1 inhibitors also reduced cyclin A mRNA expression and cyclin A promoter activity (Figure 8D and E). Given the potent and specific inhibition of cyclin A by the GLS1 inhibitors, we tested whether restoration of cyclin A levels would negate the effect of these inhibitors on cell growth and migration. Infection of HUVECs with AdCyclin A fully restored cyclin A protein expression in the presence of BPTES and partially rescued endothelial cell growth in the GLS1-inhibited cells (Figure 9A and B). In contrast, AdCyclin A had no effect on HUVEC migration (Figure 9C).
8.

GLS1 inhibition blocked cell cycle progression and cyclin A expression by HUVEC. (A) Effect of DON (20μM) or BPTES (20μM) exposure for 24 hours on the distribution of HUVECs in the cell cycle. (B) Effect of DON (20μM) or BPTES (20μM) exposure for 24 hours on cell cycle protein expression and phosphorylation determined by western blotting. (C) Effect of DON (20μM) or BPTES (20μM) exposure for 24 hours on cyclin A mRNA expression. (D) Effect of DON (20μM) or BPTES (20μM) exposure for 24 hours on cyclin A promoter activity. Results are means ± SD (n=4). *Statistically significant effect of GLS1 inhibitors.
9.

Cyclin A contributed to the anti-proliferative effect of GLS1 inhibition in HUVEC. (A) AdCyclin A increased cyclin A protein levels in GLS1-inhibited HUVEC. (B) AdCyclin A increased DNA synthesis in GLS1-inhibited HUVEC. (C) AdCyclin A had no effect on migration in GLS1-inhibited HUVEC. Cells were infected with AdCyclin A (50 MOI) or AdGFP (50 MOI) for 24 hours and then incubated in the presence or absence of BPTES (10μM) for another 24 hours prior to performing the proliferation or migration assay. Results are means ± SD (n=3–4). *Statistically significant effect of AdCyclin A. +Statistically significant effect of BPTES.
Finally, glutamine-deprivation resulted in a significant increase in ROS production in HUVECs, HAECs, and HMECs (Figure 10A). The GLS1 inhibitors DON, BPTES, and CB-839 also elevated oxidative stress in HUVECs (Figure 10B). The increase in oxidative stress following GLS1 inhibition was paralleled by a decline in HO-1 expression (Figure 10C). Moreover, BPTES-treated HUVECs were sensitive to hydrogen peroxide-induced cell death (Figure 10D). However, upregulation of HO-1 expression following the infection of HUVEC with AdHO-1 blocked hydrogen peroxide-mediated death in GLS1-inhibited endothelial cells (Figure 10C and D).
10.

GLS1 preserved oxidative balance and viability of human endothelial cells. (A) Glutamine (Gln) deprivation for 24 hours increased ROS production in HUVECs, HAECs, and HMECs. (B) GLS1 inhibition increased ROS production in HUVECs. Cells were treated with DON (20μM), BPTES (20μM), or CB-839 (20μM) for 24 hours. (C) BPTES (20μM) exposure for 24 hours decreased HO-1 protein levels in HUVECs. (D) AdHO-1 increased survival of GLS1-inhibited HUVECs exposed to ROS. Cells were infected with AdCyclin A (50 MOI) or AdGFP (50 MOI) for 24 hours, incubated in the presence or absence of BPTES (20μM) for another 24 hours, and then exposed to hydrogen peroxide (250μM) for 4 hours. Results are means ± SD (n=4).
*Statistically significant effect of Gln deprivation or GLS1-inhibition.
+Statistically significant effect of AdHO-1.
4. Discussion
In the present study, we identified the metabolism of glutamine by GLS1 as an essential driver of endothelial cell proliferation, migration, and survival. In addition, GLS1 functions as a key regulator of oxidative status. This was observed in endothelial cells derived from various vascular sources and likely represents a generalized response evoked by GLS1 in these cells. In addition, we determined that cyclin A and HO-1 are critical downstream targets of glutamine metabolism that modulate endothelial cell growth and survival, respectively. These results further illustrate the importance of amino acid metabolism in regulating endothelial cell function and suggest that therapeutic strategies targeting GLS1 may be a promising approach in treating diseases associated with abnormal endothelial cell proliferation, migration, and viability.
The current study found that glutamine is required for DNA synthesis and proliferation by endothelial cells. The proliferative effect of glutamine on endothelial cells is concentration-dependent and reliant on GLS1 activity since it is prevented by three distinct pharmacological inhibitors of GLS1 or by the siRNA-mediated knockdown of GLS1. Notably, inhibition of endothelial cell growth by the GLS1 inhibitors occurs in the absence of cell death, demonstrating that they function as cytostatic rather than cytotoxic agents. Our finding that glutamine stimulates the proliferation of HUVECs via the GLS1 pathway is consistent with two recent reports in these cells [24,25]. Moreover, we extend this work to show that GLS1 also fuels the growth of human endothelial cells derived from the arterial and microvascular circulation. Interestingly, GLS1 also stimulates the growth of murine endothelial cells as deletion of the GLS1 gene in these cells mitigates their proliferative response both in vivo and in vitro [24,25]. Thus, GLS1 may function in a universal fashion to promote the proliferation of the vascular endothelium.
Endothelial cell proliferation requires the navigation of cells through distinct phases of the cell cycle where DNA synthesis and mitosis occur. Analysis of cell cycle distribution indicated that GLS1 is required for endothelial cells to progress into S phase where DNA synthesis arises as GLS1 inhibitors arrest endothelial cells in the G0/G1 phase of the cell cycle. In addition, blockade of cell cycle progression by GLS1 inhibitors is associated with a pronounced decrease in the phosphorylation of retinoblastoma protein and with a selective reduction in the expression of cyclin A, independent of any change in the expression of other cyclins or the cyclin-dependent kinase inhibitors. The hypophosphorylation of retinoblastoma protein observed following GLS1 inhibition maintains the protein in its growth-suppressive state which represses the transcription of genes, including cyclin A, that are required for DNA synthesis [38]. Interestingly, cyclin A contributes to the phosphorylation and deactivation of retinoblastoma protein via its association with cyclin-dependent kinase 2, highlighting important reciprocal regulatory interactions between these two proteins that govern cell cycle progression. Importantly, we show that adenoviral-mediated restoration of cyclin A expression in GSL1-inhibited endothelial cells increases DNA synthesis in these cells, demonstrating that cyclin A is a key downstream target of the GLS1 inhibitors. However, complete restoration of cyclin A levels does not fully rescue DNA synthesis, suggesting that other factors may also contribute to the anti-proliferative action of the GLS1 inhibitors. In this respect, inhibitors of glutaminolysis have been shown to block cell growth by downregulating mTOR1 activity in lung squamous cell carcinoma [39]. Since the breakdown of glutamine by GLS1 provides for the synthesis of TCA-intermediates for protein and lipid production, GLS1 inhibitors may also curb endothelial cell growth by limiting the availability of essential glutamine metabolites required to fuel the TCA cycle [3–5]. In support of this concept, we found that replenishment of the TCA cycle via the administration of cell permeable dimethyl α-ketoglutarate increases the proliferation of glutamine-deprived endothelial cells. Moreover, co-administration of dimethyl α-ketoglutarate with the glutamine derivative asparagine largely restores the proliferation of glutamine-starved endothelial cells. Aside from providing additional nitrogen for biomass synthesis, asparagine promotes cell growth by activating signaling pathways involved in protein and nucleotide synthesis [40]. Our findings are consistent with previous work showing that the combination of these two glutamine metabolites rescues glutamine-dependent phenotypes in endothelial cells as well as in cancer cells [24,25,40].
The role of glutamine in endothelial cell migration is controversial. We found that glutamine stimulates the migration of endothelial cells derived from the venous, arterial, or microvascular circulation. Here again, the effect of glutamine is dependent on GLS1 since inhibition of GLS1 activity or expression antagonizes the migratory action of the amino acid. This is in-line with the study by Huang et al. [25] which found that HUVEC migration was dependent on extracellular glutamine and the presence of GLS1. However, it contrasts with another study which failed to show an effect of glutamine on HUVEC migration [24]. The cause for these variable findings is not known but may be related to differences in culture conditions, duration of glutamine deprivation, and/or the concentration of GLS1 inhibitors between studies. Notably, the dependence of endothelial cell migration on glutamine is not as severe as proliferation. Substantial migration is observed in glutamine-deprived or GLS1-inhibited endothelial cells whereas DNA synthesis is practically abolished under these conditions.
The precise mechanism by which GLS1 inhibition attenuates endothelial cell migration is unclear but it does not involve loss of cyclin A expression as restoration of cyclin A levels in GLS1-inhibited endothelial cells has no effect on their movement. Glutamine has been demonstrated to facilitate the spreading and migration of human melanoma cell lines by inducing specific integrin expression and actin cytoskeleton remodeling [41]. In addition, the catabolism of glutamine to glutamate may favor cell migration. High extracellular concentrations of glutamate stimulate glioma cell migration and augments the invasion and migration of pancreatic cancer cells via AMPA receptor activation and K-ras-MAPK signaling [42,43]. However, we found that glutamate fails to stimulate endothelial cell migration. This may reflect the poor permeability of glutamate and/or the lack of functional glutamate receptors on endothelial cells [44,45]. Limited uptake and receptor deficiency may also explain the inability of glutamate to increase the proliferation of endothelial cells grown in a glutamine-free environment. Interestingly, the combined addition of α-ketoglutarate and asparagine is able to largely restore endothelial cell migration in the absence of glutamine. This latter finding is consistent with recent work showing that asparagine stimulates the migration of colorectal cells [46], and further illustrates the importance of these glutamine metabolites in regulating endothelial cell phenotype.
Our study also confirmed previous work demonstrating a role for eNOS-derived NO in stimulating endothelial cell proliferation and migration [37,47,48]. Incubation of HUVEC with the eNOS inhibitor, L-NAME, attenuates endothelial cell mitogenesis and motogenesis. However, the magnitude of growth-inhibition following pharmacological blockade of eNOS is substantially less than that observed with the GLS1 inhibitors, indicating that GLS1 is the more potent driver of endothelial cell proliferation. Current knowledge regarding the interaction between eNOS and GLS1 is limited. Although these enzymes occupy distinct intracellular compartments, with eNOS largely expressed in the plasma membrane while GLS1 is localized to the mitochondria, channeling of substrate between these two enzymes may occur. An early report [49] identified a GLS-initiated pathway for arginine synthesis from glutamine in macrophages and monocytes. This study found that the addition of glutamine to macrophages evokes a significant increase in NO production whereas pharmacological inhibition of GLS activity decreases NO synthesis. In contrast, glutamine inhibits the generation of arginine and NO by cultured endothelial cells [50,51]. However, the inhibitory effect of glutamine on NO synthesis does not appear to involve GLS1 as GLS1 inhibition has no effect on eNOS expression, NO synthesis as measured by endothelium-dependent vasodilation, and the sensitivity of blood vessels to NO [25]. Finally, we show that the eNOS product NO has no effect on endothelial GLS activity, suggesting limited cross-talk between these two enzymes in vascular endothelium.
We also found that glutamine plays a critical role in maintaining redox balance in endothelial cells. Glutamine deprivation or GLS1 inhibition evokes a significant increase in oxidative stress in endothelial cells. This is observed in all three types of human endothelial cells tested irrespective of their vascular source and likely reflects the loss in cellular glutathione under these conditions [25]. In addition, we discovered that GLS1-inhibited endothelial cells are more sensitive to the toxic effect of ROS. Increases in the sensitivity to chemotherapy drugs and cytotoxic stimuli following GLS1 inhibition has also been reported in various tumor and immune cells, indicating an important role for this enzyme in preserving cell viability [52–55]. Interestingly, the increased susceptibility to ROS in GLS1-inhibited endothelial cells is paralleled by a decline in the levels of HO-1. This finding is consistent with our recent work demonstrating that glutamine-derived ammonia is a major driver of HO-1 expression in endothelial cells [22]. The lower rate of ammonia synthesis by endothelial cells following GLS1 inhibition likely accounts for the decrease in HO-1 expression. Given the well-established protective role of HO-1 in endothelial cells [28,32,46,57], we examined if restoration of HO-1 expression in BPTES-treated cells protects against ROS-mediated injury. In fact, adenoviral-mediated gene transfer of HO-1 prevents the toxic effect of ROS in GLS1-inhibited endothelial cells, establishing a key role for the GLS1-HO-1 signaling axis in promoting endothelial cell survival. The protection afforded by HO-1 is likely mediated via the release of carbon monoxide and bilirubin from heme as both of these HO-1 products protect endothelial cells against oxidative injury [58].
The finding that GLS1 promotes the proliferation, migration, and survival of endothelial cells is of pharmacological significance given that these processes contribute to numerous pathological disorders including cancer, atherosclerosis, and retinopathy [59–61]. The dependence of cancer cells on glutamine metabolism has made GLS1 an attractive therapeutic target [62]. GLS1 inhibitors have shown promise in preclinical models of cancer and CB-839 is currently the subject of several clinical trials. Aside from direct inhibitory actions on tumor cells, the ability of GLS1 inhibitors to block endothelial cell growth and migration may compromise the formation of new blood vessels that are required to nourish growing tumors and further contribute to their antineoplastic action. Support for this notion is provided from recent work demonstrating that pharmacological blockade of GLS1 by CB-839 reduces angiogenesis in postnatal and oxygen-induced mouse models of retinopathy [24,25]. However, whether GLS1 contributes to angiogenesis by other stimuli or in other organs remains to be established. Given that GLS activity is elevated in diabetes [63,64], the use of GLS1 inhibitors may provide a novel approach in treating diabetic retinopathy. Finally, neovascularization in atherosclerotic lesions plays a major role in plaque growth by increasing the influx of lipoproteins and leukocytes into the lesion [65]. Furthermore, as newly formed vessels within the plaque are fragile and leaky they contribute to intraplaque hemorrhage and plaque instability. Thus, drugs targeting GLS1 may also diminish angiogenesis-driven atherosclerotic plaque development and promote the reduction and stability of atherosclerotic lesions.
On a cautionary note, our studies employed cultured human endothelial cells which do not fully simulate the complex natural environment of these cells within blood vessels and the complex biochemical and biophysical interactions that occur in vivo. In addition, cell passage can lead to endothelial cell dedifferentiation and diminished production of important anti-inflammatory and anti-thrombotic molecules such as nitric oxide. In particular, Lange et al. [66] found that eNOS expression is downregulated in rodent coronary microvascular endothelial cells following culture while studies from our laboratory [67] noted a progressive loss of NO activity in HUVEC with increasing cell passage. Thus, the use of cultured HUVEC may have contributed to the muted response to L-NAME observed in our study. Interestingly, increased glutamine consumption and GLS1 expression has been observed in highly passaged, senescent endothelial cells [23]. However, consistent with our in vitro findings, GLS1 expression is detected in the vascular endothelium of murine blood vessels [24,25]. Moreover, genetic deletion of GLS1 in endothelial cells of newborn mice impairs endothelial cell proliferation [24,25] and migration [25] leading to poorly branched and stunted vascular networks in a model of retinal angiogenesis, lending credence to our results in cultured endothelial cells.
In conclusion, the present study demonstrates that the metabolism of glutamine by GLS1 plays a fundamental role in regulating human endothelial cell biology. GLS1 stimulates the proliferation, migration, and survival of endothelial cells, and maintains oxidative balance in these cells. The actions of GLS1 are widespread and occur in endothelial cells derived from the venous, arterial, and microvascular circulation. While cyclin A plays a key role in transducing the proliferative effect of GLS1, HO-1 mediates the cytoprotective action of GLS1. Collectively, these studies provide novel insight into the molecular mechanisms by which glutamine modulates endothelial function, and further supports GLS1 as a therapeutic target in ameliorating diseases dependent on aberrant endothelial cell proliferation, migration, and viability.
Acknowledgements
This work was supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health under award number R01HL59976 and by the American Heart Association Grant 15GRNT25250015.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lacey JM, Wilmore DW. Is glutamine a conditionally essential amino acid? Nutr Rev 48:297–309, 1990. [DOI] [PubMed] [Google Scholar]
- 2.Curi R, Newholme P, Procopio J, Langranha C, Gorjao R, Pithon-Curi TC. Glutamine, gene expression and cell function. Front Biosci 2007;12:344–357. [DOI] [PubMed] [Google Scholar]
- 3.Young VR, Ajami AM. Glutamine: the emperor or his clothes. J Nutr 2001;131:2449S2459S. [DOI] [PubMed] [Google Scholar]
- 4.DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, et al. Beyond aerobic glycolysis: transformed cells can exceed the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci USA 2007;104:19345–19350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DeBarardinis RJ, Cheng T. Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010;29:313–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Curthoys NP, Watford M. Regulation of glutaminase activity and glutamine metabolism. Annu Rev Nutr 1995;15:133–159. [DOI] [PubMed] [Google Scholar]
- 7.Aledo JC, Gomez-Fabre PM, Olalla L, Marquez J. Identification of two human glutaminase loci and tissue-specific expression of the two related genes. Mamm Genome 2002;9:157–166. [DOI] [PubMed] [Google Scholar]
- 8.Mates J, Segura JA, Martin-Rufian M, Campos-Sandoval JA, Alonso FJ, Marquez J. Glutaminase isozymes as key regulators in metabolic and oxidative stress against cancer. Curr Mol Med 2012;12:1–21. [DOI] [PubMed] [Google Scholar]
- 9.Curthoys NP. Role of mitochondrial glutaminase in rat renal glutamine metabolism. J Nutr 2001;131:2491S–2497S. [DOI] [PubMed] [Google Scholar]
- 10.Chaudhary FA, Reimer RJ, Edwards RH. The glutamine commute: take the N line and transfer to the A. J Cell Biol 2002;157:349–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Szeliga M, Orbara-Michlewska M. Glutamine in neoplastic cells: focus on the expression and roles of glutaminases. Neurochem Int 2009;55:71–75. [DOI] [PubMed] [Google Scholar]
- 12.Knox WE, Horowitz MI, Friedell GH. The proportionality of glutaminase content to growth rate and morphology of rat neoplasms. Cancer Res 1969;29:669–680. [PubMed] [Google Scholar]
- 13.Ahluwalia GS, Grem JL, Hao Z, Cooney DA. Metabolism and action of amino acid analog anti-cancer agents. Pharmacol Ther 1990;46:243–271. [DOI] [PubMed] [Google Scholar]
- 14.Shelton LM, Huysentruyt LC, Seyfried TN. Glutamine targeting inhibits systemic metastasis in the VM-M3 murine tumor model. Int J Cancer 2010;127:2478–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, et al. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J Immunol 2010;185:1037–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor Myc controls metabolic reprogramming upon T-lymphocyte activation. Immunity 2011;35:871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Colombo SL, Palacios-Callender M, Frakich N, De Leon J, Schmitt CA, Boorn L, et al. Anaphase promoting complex/cyclosome-cdh1 coordinates glycolysis and glutaminolysis with transition to S-phase in human T lymphocytes. Proc Natl Acad Sci USA 2010;107:18868–18873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Piao L, Fang Y-H, Parikh K, Ryan JJ, Toth PT, Archer SL.Cardiac glutaminolysis: a maladaptive cancer metabolism pathway in the right ventricle in pulmonary hypertension. J Mol Med 2013;91:1185–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu F, Li Y, Liu G. MicroRNA-200c exacerbates the ischemia/reperfusion injury of heart through targeting the glutaminase (GLS)-mediated glutamine metabolism. Eur Rev Med Pharmacol Sci 2017;21:3282–3289. [PubMed] [Google Scholar]
- 20.Leighton B, Curi R, Hussein A, Newsholme EA. Maximum activities of some key enzymes of glycolysis, glutaminolysis, Krebs cycle and fatty acid utilization in bovine pulmonary endothelial cells. FEBS Lett 1987:225:93–96. [DOI] [PubMed] [Google Scholar]
- 21.Wu G, Haynes TE, Li H, Meininger CJ. Glutamine metabolism in endothelial cells: ornithine synthesis from glutamine via pyrroline-5-carboxylate synthase. Comp Biochem Physiol Part A 2000;126:115–123. [DOI] [PubMed] [Google Scholar]
- 22.Liu XM, Peyton KJ, Durante W. Ammonia promotes endothelial cell survival via the heme oxygenase-1-mediated release of carbon monoxide. Free Radic Biol Med 2017;102:37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Unterluggauer H, Mazurek S, Lener B, Hutter E, Eigenbrodt E,Zwersche W, et al. Premature senescence of human endothelial cells induced by inhibition of glutaminase. Biogerentology 2008;9:247–259. [DOI] [PubMed] [Google Scholar]
- 24.Kim B, Li J, Jang C, Arany Z. Glutamine fuels proliferation but not migration of endothelial cells. EMBO J 2017;36:2321–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang H, Vandekeere S, Kalucka J, Bierhansl L, Zecchin A, Bruning U, et al. Role of glutamine and interlinked asparagine metabolism in vessel formation. EMBO J 2017;36:2334–2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu XM, Durante Z, Peyton KJ, Durante W. Heme oxygenase-1-derived bilirubin counteracts HIV protease inhibitor-mediated endothelial cell dysfunction. Free Radic Biol Med 2016;94:218–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang H, Jiang X, Yang F, Chapman GB, Durante W, Sibinga NE, et al. Cyclin A transcriptional suppression is the major mechanism mediating homocysteine-induced endothelial cell growth inhibition. Blood 2002;99:939–945. [PMC free article] [PubMed] [Google Scholar]
- 28.Liu XM, Peyton KJ, Durante W. Physiological cyclic strain promotes endothelial cell survival via the induction of heme oxygenase-1. Am J Physiol Heart Circ Physiol 2013; 304:H1634–H1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peyton KJ, Liu XM, Yu Y, Yates B, Durante W. Activation of AMP-activated protein kinase inhibits the proliferation of human endothelial cells. J Pharmacol Exp Ther 2012;342:827–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jamaluddin Md S, Chen I, Yang F, Jiang X, Jan M, Liu XM, Schafer AI, Durante W, Yang X, Wang H. Homocysteine inhibits endothelial cell growth via DNA hypomethylation of the cyclin A gene. Blood 2007;110:3648–3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Curthoys NP, Lowry OH. The distribution of glutaminase isoenzymes in the various structures of the nephron in normal, acidotic, and alkalotic rat kidney. J Biol Chem 1973;248:162–168. [PubMed] [Google Scholar]
- 32.Liu XM, Peyton KJ, Shebib AR, Wang H, Durante W. Compound C stimulates heme oxygenase-1 gene expression via the Nrf2-ARE pathway to preserve human endothelial cell survival. Biochem Pharmacol 2011;82:371–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Willis RC, Seegmiller JE. The inhibition by diazo-5-oxo-l-norleucine of glutamine catabolism of the cultured human lymphoblast. J Cell Physiol 1977;93:375–382. [DOI] [PubMed] [Google Scholar]
- 34.Robinson MM, McBryant SJ, Tsukamoto T, Rojas C, Ferraris DV, Hamilton SK, et al. Novel mechanism of inhibition of rat kidney-type glutaminase by bis-2-(5-phenylacetomido-1,3,4-thiadiazol-2-yl)ethyl sulfide (BPTES). Biochem J 2007;406:407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gross ML, Demo SD, Dennison JB, Chen L, Chernov-Rogan T, Goyal B, et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol Cancer Ther 2014;13:1–12. [DOI] [PubMed] [Google Scholar]
- 36.Rees DD, Palmer RM, Schulz R, Hodson SF, Moncada S. Characterization of three inhibitors of endothelial nitric oxide synthase in vitro and in vivo. Br J Pharmacol 1990;101:746–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Papapetropoulos A, Garcia-Cardena G, Madri JA, Sessa WC. Nitric oxide production contributes to the angiogenic properties of vascular endothelial growth factor in human endothelial cells. J Clin Invest 1997;100:3131–3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weinberg RA. The retinoblastoma protein and cell cycle control. Cell 1995;81:323–330. [DOI] [PubMed] [Google Scholar]
- 39.Ye X, Zhou Q, Matsumoto Y, Moriyama M, Kageyama S, Komatsu M, et al. Inhibition of glutaminolysis inhibits cell growth via down-regulating Mtor1 signaling in lung squamous cell carcinoma. Anticancer Res 2016;36:6021–6029. [DOI] [PubMed] [Google Scholar]
- 40.Krall AS, XU S, Graeber TG, Brass D, Christofk HR Asparagine promotes cancer cell proliferation through the use as an amino acid exchange factor. Nat Commun 7:11457, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fu YM, Xhang H, Ding M, Li YQ, Fu X, Y ZS et al. Specific amino acid restriction inhibits attachment and spreading of human melanoma via modulation of the integrin/focal adhesion kinase pathway and actin cytoskeleton remodeling. Clin Exp Metastasis 2004;21:587–598. [DOI] [PubMed] [Google Scholar]
- 42.Ishiuchi S, Tsuzuki K, Yoshida Y, Yamada N, Hagimura N, Okado H, et al. Blockage of Ca(2+)-permeable AMPA receptors suppresses migration and induces apoptosis in human glioblastoma cells. Nat Med 2002;8:971–978. [DOI] [PubMed] [Google Scholar]
- 43.Hemer A, Sauliunaite D, Michalski CW, Erkan M, De Oliveira T, Abiatari I, et al. Glutamate increases pancreatic cancer cell invasion and migration via AMPA receptor activation and Kras-MAPK signaling. Int J Cancer 2011;129:2349–2359. [DOI] [PubMed] [Google Scholar]
- 44.Schachter D, Sang JC. Regional differentiation in the rat aorta for a novel signaling pathway: leucine to glutamate. Am J.Physiol Heart Circ Physiol. 1997;273:H1484–H1492. [DOI] [PubMed] [Google Scholar]
- 45.Morley P, Small DL, Murray CL, Mealing GA, Poulter MO, Durkin JP, et al. Evidence that functional glutamate receptors are not expressed on rat or human cerebromicrovascular endothelial cells. J Cereb Blood Flow Metab 1998;18:396–406. [DOI] [PubMed] [Google Scholar]
- 46.Wong CC, Qian Y, Li X, Xu J, Kang W, Tong JH, et al. SLC25A22 promotes proliferation and survival of colorectal cancer cells with KRAS mutations and xenograft tumor progression in mice via intracellular synthesis of aspartate. Gastroenterology 2016;151:945–960. [DOI] [PubMed] [Google Scholar]
- 47.Ouchi N, Oshima Y, Ohashi K, Higuchi A, Ikegami C, Izumiya Y, et al. Follistatin-like-1, a secreted muscle protein, promotes endothelial cell function and revascularization in ischemic tissue through a nitric oxide synthase-dependent mechanism. J Biol Chem 2008;283:32802–32811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ziche M, Morbidelli L, Masini E, Amerini S, Granger HJ, Maggi CA, et al. Nitric oxide mediates angiogenesis in vivo and endothelial cell growth and migration in vitro promoted by substance P. J Clin Invest 1994;94:2036–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Murphy C, Newsholme P. Importance of glutamine metabolism in murine macrophages and human monocytes to L-arginine biosynthesis and rates of nitrite or urea production. Clin Sci 1998;95:397–407. [PubMed] [Google Scholar]
- 50.Sessa WC, Hecker M, Mitchell JA, Vane JR. The metabolism of L-arginine and its significance for the biosynthesis of endothelium-derived relaxing factor: L-Glutamine inhibits the generation of L-arginine by cultured endothelial cells. Proc Natl Acad Sci USA 1990;87:8607–8611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu G, Haynes TE, Li H, Yan W, Meininger CJ. Glutamine metabolism to glucosamine is necessary for glutamine inhibition of endothelial nitric oxide synthesis. Biochem J 2001;353:245–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Herranz D, Ambesi-Impiobato A, Sudderth J, Sanchez-Martin M, Belver L, Tosello V, et al. Metabolic reprogramming induces resistance to anti-NOTCH1 therapies in T cell acute lymphoblastic leukemia. Nat Med 2015;21:1182–111189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab 2012;15:110–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Simpson NE, Tryndyak VP, Pogribna M, Beland FA, Pogribny IP. Modifying metabolically sensitive histone marks by inhibiting glutamine metabolism affects gene expression and alters cancer cell phenotype. Epigenetics 2012;7:1413–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tanaka K, Sasayama T, Irino Y, Takata K, Nagashima H, Satoh N, et al. Compensatory glutamine metabolism promotes glioblastoma resistance to mTOR inhibitor treatment. J Clin Invest 2015;125:1591–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brouard S, Otterbein LE, Anrather J, Tobiasch E, Bach FH, Choi AM, et al. Carbon monoxide generated by heme oxygenase-1 suppresses endothelial cell apoptosis. J Exp Med 2000;192:1015–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen S, Segal M, Agarwal A. “Lumen digestion” technique for isolation of aortic endothelial cells from heme oxygenase-1 knockout mice. Biotechniques 2010;37:84–89. [DOI] [PubMed] [Google Scholar]
- 58.Wei Y, Liu XM, Peyton KJ, Wang H, Johnson FK, Johnson RA, et al. Hypochlorous acid-induced heme oxygenase-1 gene expression promotes human endothelial cell survival. Am J Physiol Cell Physiol 2009;297:C907–C915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Folkman J Angiogenesis. Ann Rev Med 2006;57:1–18. [DOI] [PubMed] [Google Scholar]
- 60.Sluimer JC, Daemen MJ. Novel concepts in atherogenesis: angiogenesis and hypoxia in atherosclerosis. J Pathol 2009;218:7–29. [DOI] [PubMed] [Google Scholar]
- 61.Simo R, Carrasco E, Garcia-Ramirez M, Hernandez C. Angiogenic and antiangiogenic factors in proliferative diabetic retinopathy. Curr Diabetes Rev 2006;2:71–98. [DOI] [PubMed] [Google Scholar]
- 62.Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer 2016;16:619–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Watford M, Smith EM, Erbling EJ. The regulation of phosphate-activated glutaminase activity and glutamine metabolism in the streptozotocin-diabetic rat. Biochem J 1984;224:207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Squires SA, Ewart HS, McCarthy C Brosnan ME, Brosnan JT Regulation of hepatic glutaminase in the streptozotocin-induced diabetic rat. Diabetes 1997;46:1945–1949. [DOI] [PubMed] [Google Scholar]
- 65.Camere C, Pucelle M, Negre-Salvayre A, Salvayre R. Angiogenesis in the atherosclerotic plaque. Redox Biology 2017;12:18–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lang D, Bell JP, Bayraktutan U, Small GR, Shah AM, Lewis MJ. Phenotypic changes in rat and guinea pig coronary microvascular endothelium after culture: loss of nitric oxide synthase activity. Cardiovasc Res 1999;42:794–804. [DOI] [PubMed] [Google Scholar]
- 67.Durante W, Kroll ME, Vanhoutte PM, Schafer AI. Endothelium-derived relaxing factor inhibits thrombin-induced platelet aggregation by inhibiting platelet phospholipase C. Blood 1992;79:110–116. [PubMed] [Google Scholar]