

Summary
In this manuscript, we report the role of LTA4H in skin cancer development. LTA4H regulates cell cycle at the G0/G1 phase by negatively regulating p27 ubiquitination, which is associated with decreased phosphorylation of CDK2 and inhibition of the CDK2/cyclin E complex.
Abstract
Leukotriene A4 hydrolase (LTA4H), a bifunctional zinc metallo-enzyme, is reportedly overexpressed in several human cancers. Our group has focused on LTA4H as a potential target for cancer prevention and/or therapy. In the present study, we report that LTA4H is a key regulator of cell cycle at the G0/G1 phase acting by negatively regulating p27 expression in skin cancer. We found that LTA4H is overexpressed in human skin cancer tissue. Knocking out LTA4H significantly reduced skin cancer development in the 7,12-dimethylbenz(a)anthracene (DMBA)-initiated/12-O-tetradecanoylphorbol-13-acetate (TPA)-promoted two-stage skin cancer mouse model. LTA4H depletion dramatically decreased anchorage-dependent and -independent skin cancer cell growth by inducing cell cycle arrest at the G0/G1 phase. Moreover, our findings showed that depletion of LTA4H enhanced p27 protein stability, which was associated with decreased phosphorylation of CDK2 at Thr160 and inhibition of the CDK2/cyclin E complex, resulting in down-regulated p27 ubiquitination. These findings indicate that LTA4H is critical for skin carcinogenesis and is an important mediator of cell cycle and the data begin to clarify the mechanisms of LTA4H’s role in cancer development.
Introduction
Leukotriene A4 hydrolase (LTA4H; EC 3.3.2.6) is known as a bifunctional zinc metallo-enzyme exhibiting anion-dependent aminopeptidase activity in addition to its epoxide hydrolase activity (1,2). As an epoxide hydrolase, it converts the unstable allelic epoxide leukotriene A4 (LTA4) to leukotriene B4 (LTB4), which is a potent inducer of neutrophil, macrophage, and T lymphocyte chemotaxis in human diseases (3–10). As an aminopeptidase, LTA4H degrades the N-terminus of peptides. The aminopeptidase activity of LTA4H is generally assumed to be involved in the processing of peptides related to inflammation and host defense (11) and may be involved in human cancer (12–14). We have discovered that resveratrol (3,5,4’-trihydroxy-trans-stilbene) and [6]-gingerol directly bind to LTA4H and inhibit its enzyme activity, resulting in suppression of cancer development in vitro and in vivo (15,16). However, the molecular mechanism as to how LTA4H mediates cancer development remains elusive.
Cyclin-dependent kinase inhibitor 1B (CDKN1B, p27Kip1) is encoded by the CDKN1B gene (17) and was originally identified as a protein preventing the activation of the CDK2/cyclin E or CDK4/cyclin D complex, and thus controls the transition from the G1 phase into the S phase of the cell cycle (18–21). Inactivation of p27 is generally accomplished post-transcriptionally by the oncogenic activation of various pathways, including receptor tyrosine kinases (RTKs), phosphatidylinositol 3-kinase (PI3-K), Src or Ras-mitogen activated protein kinases (MAPKs). These pathways accelerate the proteolysis of the p27 protein and allow cancer cells to undergo rapid division and uncontrolled proliferation. The absence or reduction of p27 protein expression is also reported to be associated with a poor prognosis in several human cancers (22–26). Therefore, p27 is considered to be a tumor suppressor.
In the present study, we discovered that LTA4H is a key modulator of cell cycle through its negative effect on p27 expression. We found that LTA4H is overexpressed in human skin cancer tissues and knockout of LTA4H reduced skin cancer development in an in vivo mouse skin cancer model. Moreover, our findings showed that depleting LTA4H protein expression enhanced p27 protein stability by suppressing phosphorylation of CDK2 at Thr160 and inhibiting the formation of the CDK2/cyclin E complex, resulting in down-regulation of p27 ubiquitination. These findings reveal a novel mechanism of LTA4H-mediated cell cycle regulation through p27 ubiquitination and regulation of skin carcinogenesis.
Materials and methods
Cell culture, plasmids, antibodies and reagents
A431, SCC-12, SCC-13 and HaCaT cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM)/10% fetal bovine serum (FBS) with antibiotics. JB6 P+ cells were cultured in Eagle’s Minimum Essential Medium (MEM)/5% FBS with antibiotics. HEK 293T cells were cultured in DMEM/10% FBS. All cell lines were purchased from American Type Culture Collection (ATCC, Manassas, VA) and cytogenetically tested and authenticated before the cells were frozen. Each vial of frozen cells was thawed and maintained for a maximum of 8 weeks. The Xpress-LTA4H, HA-Ubiquitin (HA-Ub), pcDNA3, and pcDNA4A plasmids were purchased from Addgene (Cambridge, MA). Anti-LTA4H for Western blotting and anti-BLT1 were obtained from Cayman Chemicals (Ann Arbor, MI). Anti-LTA4H for immunohistochemistry, anti-p27, anti-p-p27 (Thr187), anti-Ub-HRP, anti-CDK2, and anti-cyclin E), anti-HA-tag (C29F4), anti-SKP2, and anti-pCDK2 (T160) were obtained from Cell Signaling (Danvers, MA). Anti-Xpress was from Life Technologies (Carlsbad, CA). Anti-HA-HRP (16B12) was purchased from Roche (Basel, Switzerland).
RNA interference
The lentiviral expression vectors (PLKO.1-shLTA4H and PLKO.1-shBLT1) and packaging vectors (pMD2.0G and psPAX) were purchased from OpenBioSystems (Huntsville, AL). The sequences were as follows: LTA4H shRNA#1: CGGCCCTTA TTCAAGGATCTT; LTA4H shRNA#2: GCCTCCCATAAAGCCCAATTA; LTA4H shR NA#3: CCCTGCTACCTGATTGCTTTA; LTA4H shRNA#4: CCTTCTGTGAAATTAA CCTAT; LTA4H shRNA#5: CGCAATTCCTTTGGCGCTAAA; BLT1 shRNA#1: CGCA CTTTCCTCCTGGCAGAA; BLT1 shRNA#2: TCTCAAGTTAAACGAACTGAA; BL T1 shRNA#3: CGCACAGTAGTGCCCTGGAAA; BLT1 shRNA#4: GCGCTCTGTCACT GCCCTGAT; and BLT1 shRNA#5: GTTCATCTCTCTGCTGGCTAT. Lentivirus shRNAs were constructed using the protocol shown on the OpenBioSystems website.
Western blotting
Cells were harvested and disrupted with lysis buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1% sodium deoxycholate and 0.1% SDS). Whole cell lysates were subjected to SDS-PAGE and transferred to a polyvinylidene difluoride (PVDF) membrane. After blocking with 5% milk, the membrane was incubated with a specific primary antibody, and then protein bands were visualized using the ECL system after hybridization with a horseradish peroxidase-conjugated secondary antibody.
Immunohistochemistry
Human skin cancer tissues were kindly provided from Dr. G. Tim Bowden (University of Arizona Cancer Center, Tucson, AZ). The skin tissue array was purchased from US Biomax, Inc (Rockville, MD). All skin tissues were de-paraffinized in xylene and rehydrated in serial concentrations of alcohol. After boiling in 10 mM sodium citrate buffer (pH 6.0) for 12 min, the tissues were incubated with 5% H2O2 for 10 min and then blocked with 50% normal goat serum for 1 h followed by incubation with anti-LTA4H (1:100) at 4°C overnight. After incubation with a secondary antibody for 1 h, LTA4H expression was visualized with 3,3’-diaminobenzidine. Images were captured under a microscope and analyzed using the Image-Pro Premier software (v.9.0) program (Media Cybernetics, Rockville, MD).
Anchorage-independent cell growth assay
Cells were suspended in basal medium eagle (BME) containing 10% FBS and 0.33% agar and plated on solidified BME containing 10% FBS and 0.5% agar. After incubation for 5–14 days, the colonies were counted using the Image-Pro Plus Software (v.4) program (Media Cybernetics, Rockville, MD).
Animals
LTA4H knockout mice were obtained from Jackson Laboratory (Bar Harbor, ME) and 129SvEv mice, the wild-type control, were purchased from Taconic (Hudson, NY). Animals were acclimated for 1 week before the study and had free access to food and water. The animal study was conducted according to the guidelines approved by the University of Minnesota Institutional Animal Care and Use Committee (IACUC). The animals were housed in climate-controlled quarters with a 12-hour light/dark cycle.
Skin carcinogenesis was induced by a two-stage skin carcinogenesis procedure using 7,12-dimethylbenz[a]anthracene (DMBA) and 2-O-tetradecanoylphorbol-13-acetate (TPA). The backs of the animals were shaved at least once a week over the entire experimental period. All animals initially received topical applications of 200 nmol of DMBA in 150 µl of acetone. After 1 week, animals were treated with or without 4 nmol TPA in 200 µl of acetone twice weekly. Mice were weighed and tumors were measured with caliper once a week until week 22 or tumors reached 1 cm3 total volume, at which time mice were euthanized.
Cell proliferation assay
Cells were seeded in 96-well plates and then proliferation was determined using the CellTiter 96® AQueous Non-Radioactive Cell Proliferation Assay (Promega, Madison, WI) according to the manufacturer’s instructions.
Cell cycle analysis
Cells seeded were harvested, and fixed with ice-cold 70% ethanol at −20°C overnight. Cells were then washed twice with PBS followed by incubation with 200 µg/ml RNase A and 20 µg/ml propidium iodide in PBS at room temperature for 30 min in the dark. The samples were subjected to flow cytometry using the FACS Calibur flow cytometer and data were analyzed using ModFit LT (Verity Software House, Inc., Topsham, ME).
Quantitative PCR analysis
Cells were harvested and total RNA was purified with Trizol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. The cDNA was then synthesized using the Superscript III Reverse transcription kit (Invitrogen). The PCR reaction was performed using SYBR® Select Master Mix (Applied Biosystems, Foster City, CA) with the following primers: p27, sense 5′-AGCTGTCTCTGAAAGGGACATT-3′; antisense 5′-TTTGACTTGCATGAAGAGAAGC-3′; GAPDH, sense 5′-AGCCACATCGCT CAGACAC-3′; antisense 5′-TGGGATTTCCATTGATGACA-3′.
Ex vivo p27 ubiquitination assay
Cells were transfected with HA-Ub for 48 h and then incubated with 10 µM MG132 for 12 h. Whole cell lysates were pre-cleared with protein A/G agarose beads in NP-40 lysis buffer for 30 min at 4°C and then co-immunoprecipitated with 2 µg anti-p27 at 4°C overnight. After incubation with protein A/G agarose beads for 2 h at 4°C, the immunocomplexes were analyzed by Western blotting.
Co-immunoprecipitation assay
Whole cell lysates were pre-cleared with protein A/G agarose beads in NP-40 lysis buffer for 30 min at 4°C and then incubated with 2 µg antibody at 4°C overnight. After incubation with protein A/G agarose beads for 2 h at 4°C, the immunocomplexes were analyzed by Western blotting.
LTB4 production
Cells were seeded in 6-well plates and LTB4 production in the medium was quantified using the Leukotriene B4 EIA kit (Cayman Chemical) following the supplier’s instructions.
Statistical analysis
All quantitative data are expressed as means ± S.D. GraphPad PRISM software was used to evaluate statistical significance. One-way ANOVA was used in Figures 1D and 2E. Two-tailed unpaired t-tests were used for statistical analysis in other figures. A probability of P < 0.05 was used as the criterion for statistical significance.
Figure 1.

LTA4H is overexpressed in human cancer cells and tissues. Endogenous protein levels of LTA4H were analyzed by Western blotting in (A) human skin, (B) colon and (C) lung cancer cell lines. (D) LTA4H levels in human skin were analyzed by immunohistochemistry and the density score from each sample was determined (bottom panel, bar = 100 µm). Representative cases are shown (upper panels).
Figure 2.

LTA4H mediates cell transformation. (A) JB6 P+ or (B) HaCaT cells expressing the indicated shRNAs were grown in soft agar and then colonies were counted. (C) JB6 P+ or (D) HaCaT cells were transfected with the indicated constructs and grown in soft agar and then colonies were counted. For A–D, data are shown as mean values ± SD (*P < 0.05). (E) All animals (n = 25) were administered 200 nmol of DMBA and then treated with vehicle or 200 nmol of TPA twice a week for 21 weeks. Data are shown as mean values ± SEM and statistical significance was determined by one-way ANOVA (*P < 0.05).
Results
LTA4H is overexpressed in human cancers
LTA4H is reportedly overexpressed in several cancers, including colorectal, lung and esophageal (12–14). To determine the cancer tissue type on which to focus in this study, the protein levels of LTA4H were determined in several cancer cell lines. Interestingly, overexpression of LTA4H was observed in human skin squamous cell carcinoma (SCC), but not in normal skin cells or malignant melanoma cells (Figure 1A). We also observed overexpression of LTA4H in lung and colon cancer cell lines (Figure 1B and C). The expression levels of LTA4H were also examined in human skin cancer tissues, and our results indicated that LTA4H protein expression is significantly higher in actinic keratosis (AK) and SCC compared to normal human skin tissues (Figure 1D). Interestingly, the tissue array revealed that overexpression of LTA4H was not observed in human basal cell carcinoma (Supplementary Figure 1a). These results indicated that LTA4H might be a critical molecule in human skin carcinogenesis, but especially in SCC.
LTA4H mediates cell transformation
To investigate the role of LTA4H in skin carcinogenesis, we first depleted LTA4H expression in mouse skin epidermal JB6 P+ and HaCaT cells (human skin keratinocytes) by using a lentiviral vector-based system with LTA4H specific short hairpin RNA (shRNA). We then examined epidermal growth factor (EGF)- or 12-O-tetradecanoylphorbol-13-acetate (TPA)-induced cell transformation. Depletion of LTA4H dramatically suppressed neoplastic transformation of both JB6 P+ and HaCaT cells (Figures 2A and B). Consistently, JB6 P+ and HaCaT cells overexpressing LTA4H exhibited enhanced neoplastic transformation (Figures 2C and D). The function of LTA4H in cancer development in vivo was also examined using the 7,12-dimethylbenz[a]anthracene (DMBA)/TPA-induced 2 stage skin carcinogenesis mouse model (Figure 2E). Both the number and volume of tumors were significantly less in LTA4H knockout mice compared with wild-type mice. We also examined the status of inflammation in the skin of these mice by measuring inflammatory cell infiltration and vascular dilatation (Supplementary Figure 1b). The results indicated that both wild-type and LTA4H knockout mice with no TPA treatment showed no signs of obvious inflammation. On the other hand, wild-type, but not LTA4H knockout mice, treated with TPA showed inflammatory cell infiltration and vascular dilatation in the dermis, which are indicated by black arrow heads (inflammatory cells) and white arrow heads (vascular dilatation; Supplementary Figure 1b). These results indicated that LTA4H is critical in skin cancer development.
LTA4H mediates cancer cell growth
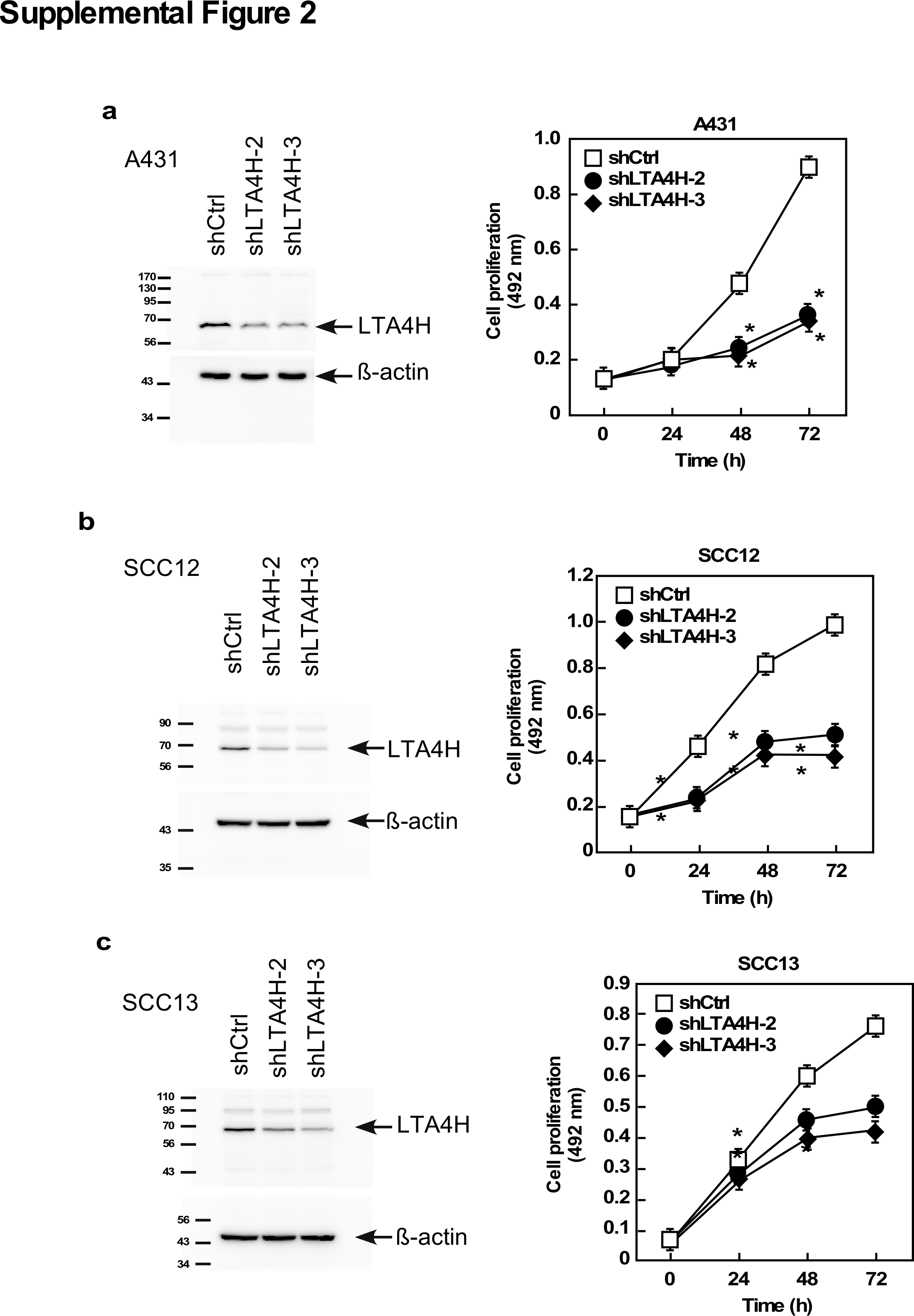
To determine the impact of LTA4H on cancer cell growth, LTA4H protein expression was depleted in three types of human SCC cell lines (A431, SCC12 and SCC13) using an LTA4H shRNA lentiviral vector. A431 cells were established from human skin epidermoid carcinomas and SCC12 and SCC13 cells are human facial skin squamous cell carcinoma cell lines. Depletion of LTA4H significantly suppressed growth of A431 human SCC (Supplementary Figure 2a), SCC12 (Supplementary Figure 2b) and SCC13 cell lines (Supplementary Figure 2c). These results suggest that LTA4H mediates skin cancer development by influencing cell growth.
LTA4H mediates the G0/G1 cell cycle phase through p27 proteasome degradation
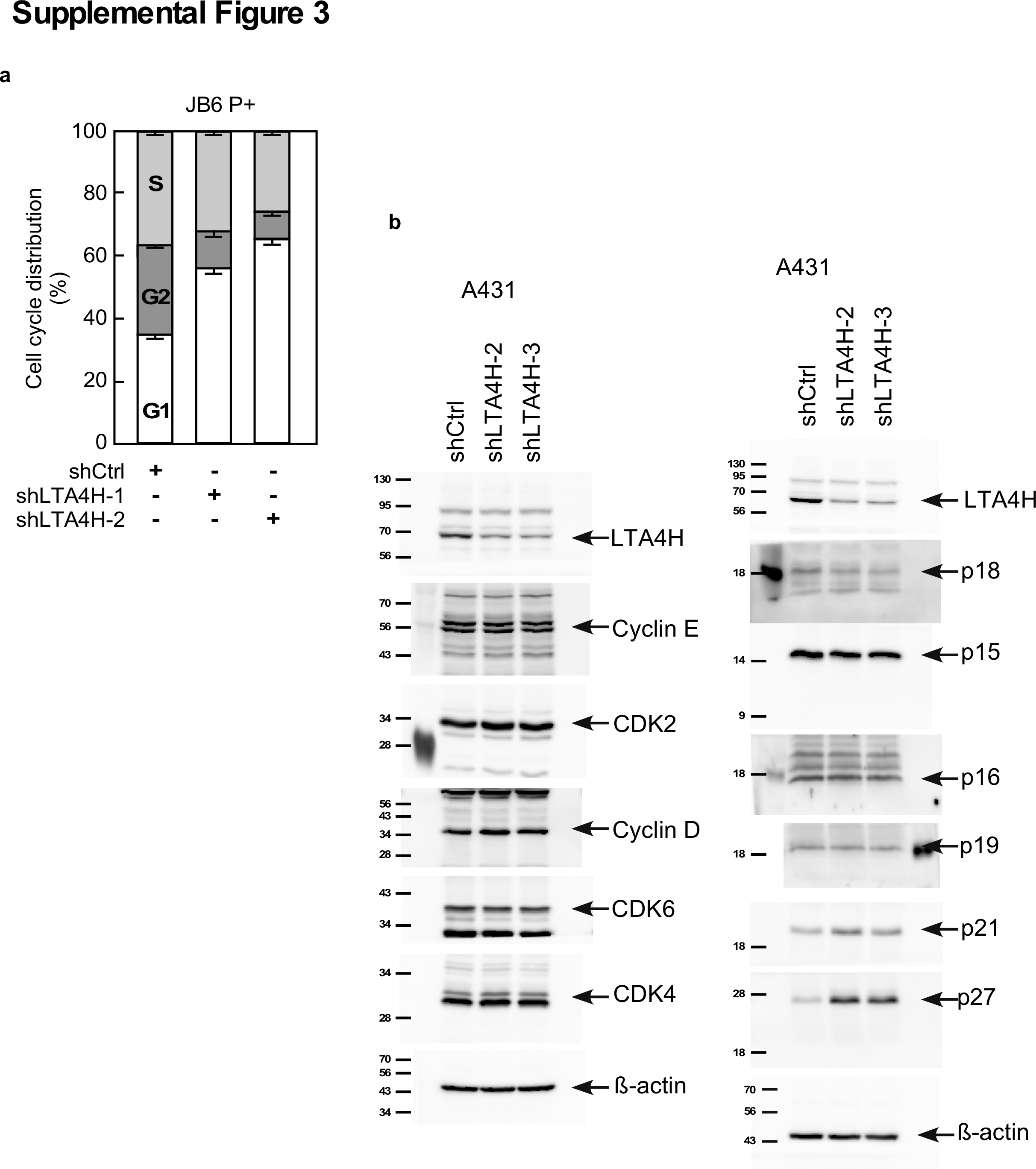
Because our results showed that LTA4H is implicated in cell growth, cell cycle distribution was determined in LTA4H-depleted cells. Knockdown of LTA4H strongly induced G0/G1 phase arrest in A431 cells (Figure 3A) as well as in JB6 P+ cells (Supplementary Figure 3a). Protein levels of cyclins, cyclin-dependent kinases (CDKs) and CDK inhibitors were determined in LTA4H-depleted cells. Among the 11 proteins examined, p27 expression was obviously increased by knocking down LTA4H (Supplementary Figure 3b). Enhanced expression of p27 by depletion of LTA4H was also observed in SCC12 and SCC13 cells (Figure 3B). On the other hand, p27 mRNA levels were not affected by depletion of LTA4H expression (Figure 3C), indicating that LTA4H-mediated p27 expression does not involve transcription. We then examined p27 protein stability in LTA4H-depleted cells and found that the p27 protein was significantly stabilized in LTA4H-depleted cells compared with sh-control cells (Figure 3D). Additionally, induction of p27 protein expression by LTA4H depletion was not observed in cells treated with the proteasome inhibitor MG132 (Figure 3E). These results indicate that LTA4H mediates p27 expression through the proteasomal degradation pathway.
Figure 3.

LTA4H mediates p27 stability. (A) A431 cells were infected with the indicated shRNAs for 48 h and then cell distribution was analyzed by flow cytometry. (B) A431, SCC12, or SCC13 cancer cells were infected with the indicated shRNAs for 48 h and then whole cell lysates were analyzed by Western blotting to detect LTA4H or p27 protein expression. (C) A431 cells were infected with the indicated shRNAs for 48 h and then mRNA levels were analyzed by quantitative PCR (qPCR). Relative mRNA levels were normalized against gapdh. Data are shown as mean values ± S.D. (D) A431 cells expressing the indicated shRNAs were treated with 20 µg/ml cycloheximide (CHX) and harvested at the indicated time points. Whole cells lysates were analyzed by Western blot to detect LTA4H and p27. Relative protein levels were normalized against ß-actin and data are shown as mean values ± S.D. (*P < 0.05). (E) A431 cells expressing the indicated shRNAs were incubated with or without MG132 (10 µM) for 12 h. Whole cell lysates were analyzed by Western blotting as indicated.
LTA4H mediates p27 stability through LTB4/BLT1 pathway
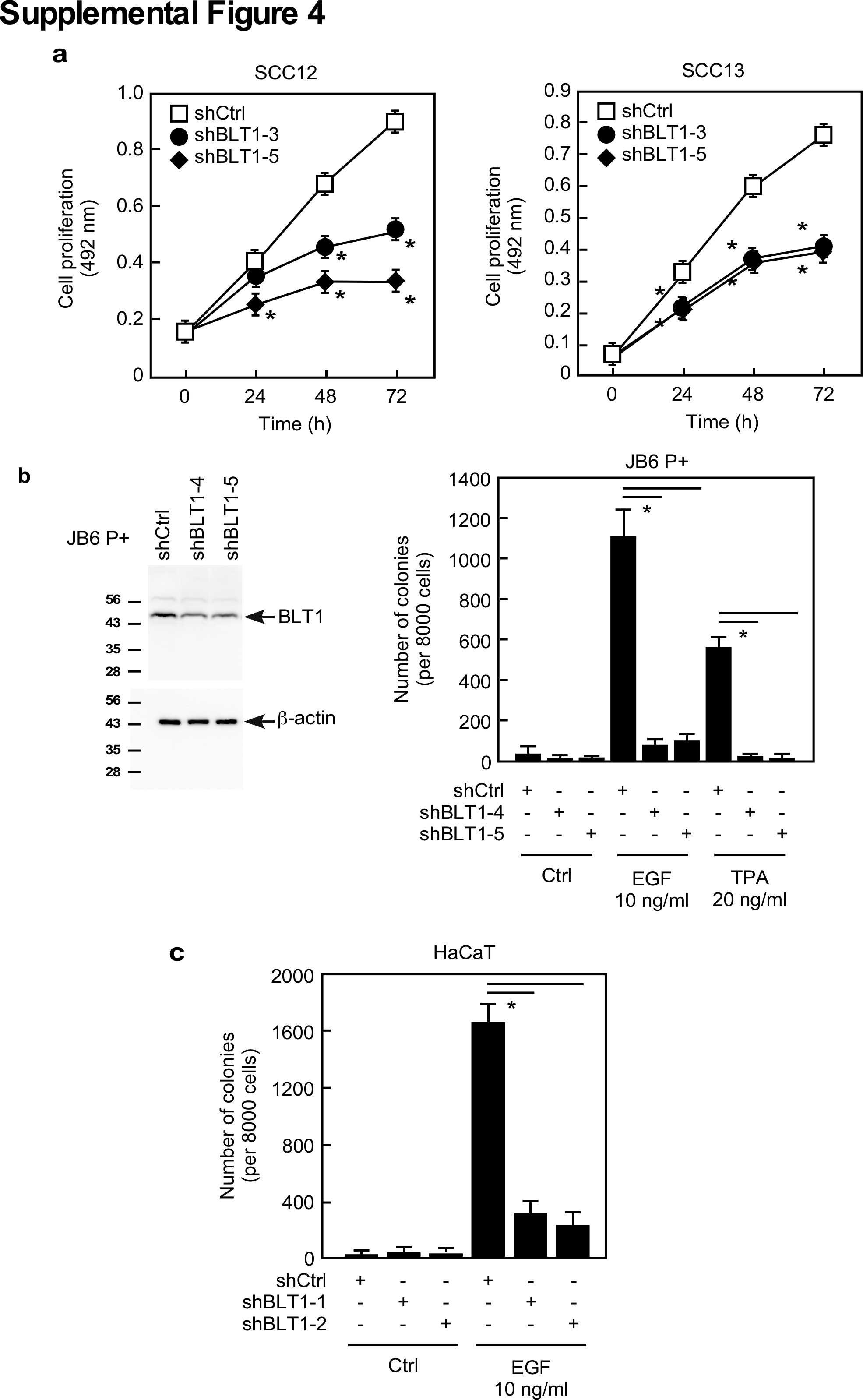
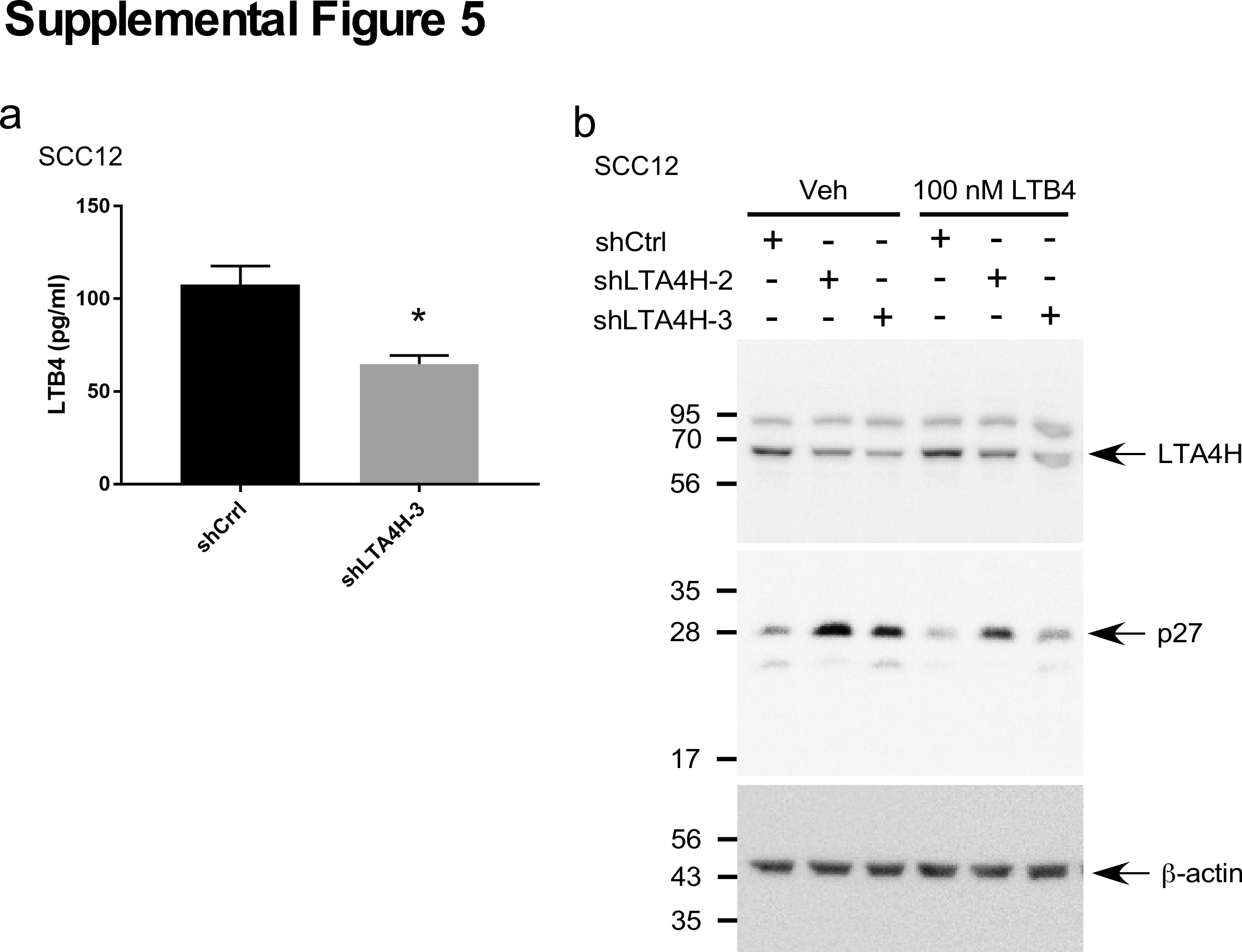
The leukotriene B4 receptor 1 (BLT1, LTB4R) is a G protein-coupled seven-transmembrane receptor that has high affinity towards leukotriene B4 (LTB4). LTB4 is an enzyme product of LTA4H and is known to increase mRNA and protein expression of BLT1 (27). We therefore questioned whether the LTB4/BLT1 pathway is implicated in LTA4H mediation of p27 expression. Our results showed that knockdown of BLT1 dramatically decreased anchorage-independent growth of A431 cells (Figure 4A). Suppression of anchorage-dependent cell growth by depletion of BLT1 was also observed in A431 cells (Figure 4B) as well as in SCC12 and SCC13 cells (Supplementary Figure 4a). Knockdown of BLT1 also caused G1 arrest (Figure 4C) and EGF- or TPA-induced cell transformation was reduced by BLT1 depletion in JB6 P+ and HaCaT cells (Supplementary Figures 4b, c). Knocking down BLT1 dramatically increased p27 expression (Figure 4D), but no significant change in p27 mRNA levels was observed (Figure 4E). Results also showed that depletion of BLT1 increased p27 protein stability (Figure 4F), but enhancement of p27 expression by BLT1 knock down was not observed in cells treated with MG132 (Figure 4G). We also measured LTB4 production in LTA4H-depleted cells, and found that knocking down LTA4H significantly reduced LTB4 production (Supplementary Figure 5a). Moreover, the induction of p27 protein expression by LTA4H depletion was not completely blocked but markedly reduced by LTB4 treatment (Supplementary Figure 5b). These results strongly suggest that LTA4H mediation of p27 expression occurs through the LTB4/BLT1 pathway.
Figure 4.

The effect of BLT1 on p27 is similar to LTA4H. (A) A431 cells expressing the indicated shRNAs were grown in soft agar and then colonies counted. Data are shown as mean values ± S.D. (*P < 0.05). (B) A431 cells expressing the indicated shRNAs were plated and then formazan production was determined at the indicated time points by MTS assay. Data are shown as mean values ± SD (*P < 0.05). (C) A431 cells were infected with the indicated shRNAs for 48 h and then cell cycle distribution was analyzed by flow cytometry. (D) Cells were infected with the indicated shRNAs for 48 h and then whole cell lysates were analyzed by Western blotting. (E) A431 cells were infected with the indicated shRNAs for 48 h and then mRNA levels were analyzed by qPCR. Relative mRNA levels were normalized against gapdh. Data are shown as mean values ± SD. (F) A431 cells expressing the indicated shRNAs were treated with 20 µg/ml CHX and harvested at the indicated time points. Whole cells lysates were analyzed by Western blotting. Relative protein levels were normalized against ß-actin and data are shown as mean values ± SD (*P < 0.05). (G) A431 cells expressing the indicated shRNAs were incubated with or without MG132 (10 µM) for 12 h. Whole cell lysates were analyzed by Western blotting as indicated.
LTA4H and BLT1 mediate phosphorylation of p27 at Thr187 and affects p27 ubiquitination
Our results indicate that LTA4H and BLT1 mediate p27 stability through the proteasome degradation pathway. Protein levels of p27 are mainly regulated through degradation by ubiquitin-dependent proteolysis (28). We therefore conducted an ex vivo ubiquitination assay (Figures 5A and B). The results showed that depletion of LTA4H decreased the ubiquitination of p27 and knocking down BLT1 had a similar effect. Consistently, overexpression of LTA4H dramatically enhanced p27 ubiquitination compared with cells expressing control shRNA (Figure 5C). The most well-known E3 ligase to ubiquitinate p27 is S-phase kinase-associated protein 2 (Skp2) (29,30). However, no significant changes were observed in Skp2 protein levels in LTA4H- or BLT1-depleted cells (Figure 5D). Interestingly, phosphorylation of Thr187 was obviously down-regulated by LTA4H or BLT1 depletion, whereas no difference was observed in Thr198 phosphorylation (Figure 5D). These results clearly indicate that LTA4H and BLT1 mediate p27 ubiquitination through phosphorylation of p27 at Thr187.
Figure 5.

LTA4H and BLT1 mediate phosphorylation of p27 at Thr187 affecting p27 ubiquitination. (A and B) A431 cells expressing control or LTA4H or BLT1 shRNA were transfected with the indicated constructs for 48 h and then incubated with MG132 (10 µM) for an additional 12 h. Whole cell lysates were co-immunoprecipitated with a p27 antibody followed by Western blotting with anti-HA or anti-p27. (C) A431 cells were transfected with the indicated constructs for 48 h and then incubated with MG132 (10 µM) for an additional 12 h. Whole cell lysates were co-immunoprecipitated with a p27 antibody followed by Western blotting with anti-HA or anti-p27. (D) A431 cells were infected with the indicated shRNAs for 48 h and then whole cell lysates were analyzed by Western blotting as indicated.
LTA4H and BLT1 influence the formation of the CDK2/cyclin E complex and the phosphorylation of CDK2 at Thr160
Phosphorylation of p27 at Thr187 by the CDK2/cyclin E complex is a prerequisite for p27 ubiquitination and degradation (31), and phosphorylation of CDK2 at Thr160 is required for its kinase activity (32). We therefore determined whether the CDK2/cyclin E complex or phosphorylation levels of CDK2 are influenced by LTA4H or BLT1. Our results showed that depletion of LTA4H or BLT1 dramatically decreased the interaction between CDK2 and cyclin E (Figure 6A and B). The phosphorylation levels of CDK2 (Thr160) were dramatically decreased in cells expressing shLTA4H or shBLT1 (Figure 6C), indicating that depletion of LTA4H or BLT1 results in inhibition of CDK2 activity.
Figure 6.

LTA4H and BLT1 influence the formation of the CDK2/cyclin E complex and the phosphorylation of CDK2 at Thr160. (A and B) A431 cells were infected with the indicated shRNAs for 48 h. Whole cell lysates were co-immunoprecipitated with anti-cyclin E followed by Western blotting with anti-CDK2 or anti-cyclin E. (C) A431 cells were infected with the indicated shRNAs for 48 h and then whole cell lysates were analyzed by Western blotting. (D) A working model of the mediation of p27 by LTA4H and/or BLT1.
Overall, our study demonstrates that LTA4H regulates phosphorylation of CDK2 at Thr160 and the formation of the CDK2/cyclin E complex, which in turn induces p27 ubiquitination consequently influencing the G0/G1 cell cycle phase (Figure 6D). These findings show for the first time that LTA4H is a key mediator of cell cycle and provides a mechanism explaining the role of LTA4H in cancer development.
Discussion
Our previous studies showed that inhibition of LTA4H enzyme activity by the natural compounds, resveratrol or [6]-gingerol, led to reduced cancer incidence and suppressed cancer growth (15,16). However, the fundamental role of LTA4H in carcinogenesis and the molecular mechanisms of LTA4H-mediated cancer development are not fully understood. In the present study, we provided evidence showing that LTA4H is critical for skin carcinogenesis and is a key mediator of cell cycle progression by its negative regulation of p27 stability.
Because LTA4H is a bifunctional enzyme with aminopeptidase and epoxide hydrolase activities, we questioned the identity of the enzyme activity that is implicated in LTA4H-mediated p27 ubiquitination. As an aminopeptidase, LTA4H degrades the N-terminus of peptides but no physiological substrate has been identified (33,34). In 2010, Snelgrove et al. (35) identified the neutrophil chemoattractant, Pro-Gly-Pro (PGP), as a physiological substrate of LTA4H. PGP is a biomarker for chronic obstructive pulmonary disease (COPD), and is implicated in neutrophil persistence in the lung. However, no critical report has shown a relationship between LTA4H and cancer. In contrast, several reports have shown that the hydrolase function of LTA4H is implicated in cancer development (36–39). The hydrolase function of LTA4H specifically catalyzes the rate-limiting step in the conversion of LTA4 to LTB4, which is known as one of the most potent chemoattractants and activators of neutrophils. BLT1 is a G protein-coupled seven-transmembrane receptor that has a high affinity towards LTB4 and is also known to increase the mRNA and protein expression of BLT1 (27). Our results showed that BLT1 depletion had similar effects on p27 regulation as did the depletion of LTA4H (Figures 4–6). We thus concluded that LTA4H mediates p27 expression.
In this study, we discovered that LTA4H and BLT1 mediate p27 ubiquitination by regulating CDK2 activity and formation of the CDK2/cyclin E complex. CDK2 is a serine/threonine protein kinase and its activation is essential for the initiation of DNA synthesis and cell cycle progression at the G1/S phase (40–42). Our results indicated that depletion of LTA4H or BLT1 decreased CDK2 phosphorylation at Thr160 (Figure 6C). Phosphorylation of CDK2 at Thr160 is essential to regulate CDK2 kinase activity (32), and is mediated by CDK-activating kinase (CAK), a complex of CDK7 and cyclin H (30). Moreover, CDK2 is localized in the cytoplasm of quiescent mammalian cells and then is translocated to the nucleus after forming a complex with cyclin E to become activated by CAK and Cdc25 (43). Further experiments such as determination of CAK expression and localization of the CDK2/cyclin E complex might be required to understand the detailed mechanisms of LTA4H/BLT1-mediated p27 regulation.
In conclusion, our results indicated that depletion of LTA4H induces cell cycle arrest at the G0/G1 phase, and knockout of LTA4H significantly reduced skin cancer development in an in vivo skin cancer mouse model. Moreover, we discovered that depleting LTA4H enhanced p27 protein stability by suppressing phosphorylation of CDK2 at Thr160 and inhibiting the formation of the CDK2/cyclin E complex, resulting in down-regulation of p27 ubiquitination. These findings reveal a new mechanism for the role of LTA4H in cancer development. Collectively, these findings support the idea that inhibiting LTA4H epoxide hydrolase activity is a promising strategy for skin cancer prevention.
Supplementary Material
Supplementary data are available at Carcinogenesis online.
Funding
This work was supported by The Hormel Foundation, AgStar, and National Institutes of Health grants CA196639, CA187027, CA166011 and CA027502.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We are grateful to Dr. G. Tim Bowden (University of Arizona Cancer Center, Tucson, AZ) for providing human skin tissues.
Conflict of Interest Statement: None declared.
Abbreviations
- LTA4H
leukotriene A4 hydrolase
- RTKs
receptor tyrosine kinases
- MAPKs
mitogen activated protein kinases
- PVDF
polyvinylidene difluoride
References
- 1. Thunnissen M.M., et al. (2002) Crystal structures of leukotriene A4 hydrolase in complex with captopril and two competitive tight-binding inhibitors. FASEB J., 16, 1648–1650. [DOI] [PubMed] [Google Scholar]
- 2. Andersson B., et al. (2003) Crystallization and X-ray diffraction data analysis of leukotriene A4 hydrolase from Saccharomyces cerevisiae. Acta Crystallogr. D. Biol. Crystallogr., 59, 1093–1095. [DOI] [PubMed] [Google Scholar]
- 3. Shim Y.M., et al. (2010) Role of LTB₄ in the pathogenesis of elastase-induced murine pulmonary emphysema. Am. J. Physiol. Lung Cell. Mol. Physiol., 299, L749–L759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stockley R.A., et al. (2002) The effect of augmentation therapy on bronchial inflammation in alpha1-antitrypsin deficiency. Am. J. Respir. Crit. Care Med., 165, 1494–1498. [DOI] [PubMed] [Google Scholar]
- 5. Hubbard R.C., et al. (1991) Neutrophil accumulation in the lung in alpha 1-antitrypsin deficiency. Spontaneous release of leukotriene B4 by alveolar macrophages. J. Clin. Invest., 88, 891–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rios-Santos F., et al. (2003) A critical role of leukotriene B4 in neutrophil migration to infectious focus in cecal ligaton and puncture sepsis. Shock, 19, 61–65. [DOI] [PubMed] [Google Scholar]
- 7. Pace E., et al. (2004) LTB4 is present in exudative pleural effusions and contributes actively to neutrophil recruitment in the inflamed pleural space. Clin. Exp. Immunol., 135, 519–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Marian E., et al. (2006) Up-regulated membrane and nuclear leukotriene B4 receptors in COPD. Chest, 129, 1523–1530. [DOI] [PubMed] [Google Scholar]
- 9. Young R.E., et al. (2007) Role of neutrophil elastase in LTB4-induced neutrophil transmigration in vivo assessed with a specific inhibitor and neutrophil elastase deficient mice. Br. J. Pharmacol., 151, 628–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Grespan R., et al. (2008) CXCR2-specific chemokines mediate leukotriene B4-dependent recruitment of neutrophils to inflamed joints in mice with antigen-induced arthritis. Arthritis Rheum., 58, 2030–2040. [DOI] [PubMed] [Google Scholar]
- 11. Haeggström J.Z. (2004) Leukotriene A4 hydrolase/aminopeptidase, the gatekeeper of chemotactic leukotriene B4 biosynthesis. J. Biol. Chem., 279, 50639–50642. [DOI] [PubMed] [Google Scholar]
- 12. Chen X., et al. (2003) Leukotriene A4 hydrolase in rat and human esophageal adenocarcinomas and inhibitory effects of bestatin. J. Natl. Cancer Inst., 95, 1053–1061. [DOI] [PubMed] [Google Scholar]
- 13. Chen X., et al. (2004) Leukotriene A4 hydrolase as a target for cancer prevention and therapy. Curr. Cancer Drug Targets, 4, 267–283. [DOI] [PubMed] [Google Scholar]
- 14. Wang Q., et al. (2002) Expression of endoplasmic reticulum molecular chaperon GRP94 in human lung cancer tissues and its clinical significance. Chin. Med. J. (Engl)., 115, 1615–1619. [PubMed] [Google Scholar]
- 15. Jeong C.H., et al. (2009) [6]-Gingerol suppresses colon cancer growth by targeting leukotriene A4 hydrolase. Cancer Res., 69, 5584–5591. [DOI] [PubMed] [Google Scholar]
- 16. Oi N., et al. (2010) Resveratrol, a red wine polyphenol, suppresses pancreatic cancer by inhibiting leukotriene A₄hydrolase. Cancer Res., 70, 9755–9764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hershko D.D., et al. (2006) Prognostic role of p27Kip1 deregulation in colorectal cancer. Cancer, 107, 668–675. [DOI] [PubMed] [Google Scholar]
- 18. Polyak K., et al. (1994) p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev., 8, 9–22. [DOI] [PubMed] [Google Scholar]
- 19. Polyak K., et al. (1994) Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell, 78, 59–66. [DOI] [PubMed] [Google Scholar]
- 20. Slingerland J.M., et al. (1994) A novel inhibitor of cyclin-Cdk activity detected in transforming growth factor beta-arrested epithelial cells. Mol. Cell. Biol., 14, 3683–3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Toyoshima H., et al. (1994) p27, a novel inhibitor of G1 cyclin-Cdk protein kinase activity, is related to p21. Cell, 78, 67–74. [DOI] [PubMed] [Google Scholar]
- 22. Shapira M., et al. (2005) The prognostic impact of the ubiquitin ligase subunits Skp2 and Cks1 in colorectal carcinoma. Cancer, 103, 1336–1346. [DOI] [PubMed] [Google Scholar]
- 23. He W., et al. (2012) A crosstalk imbalance between p27(Kip1) and its interacting molecules enhances breast carcinogenesis. Cancer Biother. Radiopharm., 27, 399–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tian Y.F., et al. (2013) SKP2 overexpression is associated with a poor prognosis of rectal cancer treated with chemoradiotherapy and represents a therapeutic target with high potential. Tumour Biol., 34, 1107–1117. [DOI] [PubMed] [Google Scholar]
- 25. Hershko D.D. (2008) Oncogenic properties and prognostic implications of the ubiquitin ligase Skp2 in cancer. Cancer, 112, 1415–1424. [DOI] [PubMed] [Google Scholar]
- 26. Timmerbeul I., et al. (2006) Testing the importance of p27 degradation by the SCFskp2 pathway in murine models of lung and colon cancer. Proc. Natl. Acad. Sci. USA, 103, 14009–14014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yokomizo T., et al. (1997) A G-protein-coupled receptor for leukotriene B4 that mediates chemotaxis. Nature, 387, 620–624. [DOI] [PubMed] [Google Scholar]
- 28. Pagano M., et al. (1995) Role of the ubiquitin-proteasome pathway in regulating abundance of the cyclin-dependent kinase inhibitor p27. Science, 269, 682–685. [DOI] [PubMed] [Google Scholar]
- 29. Carrano A.C., et al. (1999) SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat. Cell Biol., 1, 193–199. [DOI] [PubMed] [Google Scholar]
- 30. Sutterlüty H., et al. (1999) p45SKP2 promotes p27Kip1 degradation and induces S phase in quiescent cells. Nat. Cell Biol., 1, 207–214. [DOI] [PubMed] [Google Scholar]
- 31. Sheaff R.J., et al. (1997) Cyclin E-CDK2 is a regulator of p27Kip1. Genes Dev., 11, 1464–1478. [DOI] [PubMed] [Google Scholar]
- 32. Gu Y., et al. (1992) Cell cycle regulation of CDK2 activity by phosphorylation of Thr160 and Tyr15. EMBO J., 11, 3995–4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Haeggström J.Z., et al. (1990) Leukotriene A4 hydrolase: a zinc metalloenzyme. Biochem. Biophys. Res. Commun., 172, 965–970. [DOI] [PubMed] [Google Scholar]
- 34. Orning L., et al. (1994) The bifunctional enzyme leukotriene-A4 hydrolase is an arginine aminopeptidase of high efficiency and specificity. J. Biol. Chem., 269, 11269–11273. [PubMed] [Google Scholar]
- 35. Snelgrove R.J., et al. (2010) A critical role for LTA4H in limiting chronic pulmonary neutrophilic inflammation. Science, 330, 90–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tong W.G., et al. (2005) LTB4 stimulates growth of human pancreatic cancer cells via MAPK and PI-3 kinase pathways. Biochem. Biophys. Res. Commun., 335, 949–956. [DOI] [PubMed] [Google Scholar]
- 37. Ihara A., et al. (2007) Blockade of leukotriene B4 signaling pathway induces apoptosis and suppresses cell proliferation in colon cancer. J. Pharmacol. Sci., 103, 24–32. [DOI] [PubMed] [Google Scholar]
- 38. el-Hakim I.E., et al. (1990) Leukotriene B4 and oral cancer. Br. J. Oral Maxillofac. Surg., 28, 155–159. [DOI] [PubMed] [Google Scholar]
- 39. Yang P., et al. (2008) Zyflamend reduces LTB4 formation and prevents oral carcinogenesis in a 7,12-dimethylbenz[alpha]anthracene (DMBA)-induced hamster cheek pouch model. Carcinogenesis, 29, 2182–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chi Y., et al. (2008) Identification of CDK2 substrates in human cell lysates. Genome Biol., 9, R149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dynlacht B.D., et al. (1997) Specific regulation of E2F family members by cyclin-dependent kinases. Mol. Cell. Biol., 17, 3867–3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. DeGregori J. (2004) The Rb network. J. Cell Sci., 117, 3411–3413. [DOI] [PubMed] [Google Scholar]
- 43. Keenan S.M., et al. (2001) Cyclin-dependent kinase 2 nucleocytoplasmic translocation is regulated by extracellular regulated kinase. J. Biol. Chem., 276, 22404–22409. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.