

Abstract
Objectives: The 2015 Workshop of the Society for Hematopathology/European Association for Haematopathology aimed to review immunodeficiency-related lymphoproliferative disorders with plasmablastic and plasma cell differentiation.
Methods: The workshop panel reviewed human herpes virus 8 (HHV8)/Kaposi sarcoma herpesvirus (KSHV)–associated lesions and other lesions exhibiting plasma cell differentiation, including plasmablastic proliferations with features of myeloma/plasmacytoma, plasmablastic neoplasms presenting in extranodal sites and effusion-based lymphomas, and rendered a consensus diagnosis.
Results: The spectrum of HHV8/KSHV-associated proliferations ranged from multicentric Castleman disease (MCD) to MCD with plasmablastic aggregates to HHV8+ diffuse large B-cell lymphoma and germinotrophic lymphoproliferative disorder. Comparisons across effusion-based lymphomas with and without HHV8/KSHV and plasmablastic lymphomas in immunodeficient and immunocompetent patients were discussed.
Conclusions: The presence or absence of HHV8/KSHV is a defining feature in disorders associated with Castleman disease, although their differential diagnosis and recognition of progression may be challenging. Plasmablastic proliferations overlap with myeloma/plasmacytoma as well as extranodal and effusion-based lymphomas. The involvement of Epstein-Barr virus is typically variable.
Keywords: HHV8, KSHV, Multicentric Castleman disease, Germinotropic lymphoproliferative disorder, Plasmablastic lymphoma, Primary effusion lymphoma
Upon completion of this activity you will be able to:
recognize the salient histologic features of human herpesvirus 8 (HHV8)–associated multicentric Castleman disease.
list the differential diagnosis of HHV8+ lymphoid lesions and specify the information needed to identify each entity.
provide a differential diagnosis of plasmablastic lesions, incorporating pathologic features (morphology, immunophenotype, genetics) and also viral content, clinical setting, and clinical presentation.
The ASCP is accredited by the Accreditation Council for Continuing Medical Education to provide continuing medical education for physicians. The ASCP designates this journal-based CME activity for a maximum of 1 AMA PRA Category 1 Credit™ per article. Physicians should claim only the credit commensurate with the extent of their participation in the activity. This activity qualifies as an American Board of Pathology Maintenance of Certification Part II Self-Assessment Module.
The authors of this article and the planning committee members and staff have no relevant financial relationships with commercial interests to disclose.
Exam is located at www.ascp.org/ajcpcme.
Drs Amy Chadburn and Jonathan Said chaired session 3 of the workshop on human herpes virus 8 (HHV8)/Kaposi sarcoma herpesvirus (KSHV)–positive lymphoproliferative disorders and the spectrum of plasmablastic and plasma cell neoplasms in the immunodeficiency setting, and they functioned as the co-lead authors of this article. This session discussed the spectrum of lesions with plasmablastic differentiation, including KSHV/HHV8-associated lymphoproliferative disorders, plasmablastic lymphomas, and a number of morphologically similar lesions. These proliferations are highly associated with infection by γ-herpes viruses, possibly more so than the other categories included in this workshop. The term plasmablast refers to a cell that retains proliferative capacity together with an almost fully mature plasma cell phenotype and the transcriptional profile of plasma cells with expression of PR domain zinc finger protein 1 (PDRM1)/Blimp1 and X-box binding protein 1 (XBP1).1,2 Morphologically, these lesions are characterized by medium to large cells with relatively abundant cytoplasm and cytologic features of immunoblasts, plasmablasts, and plasmacytoid cells and may be difficult to separate based on morphology alone. Thus, these lesions highlight the need to perform a multiparameter investigation, including clinical information, laboratory studies, and viral status in addition to morphology, immunophenotype, and as needed genetic studies as part of their evaluation.
The spectrum of plasmablastic/plasmacytoid neoplasms in the setting of immunodeficiency includes plasmablastic lymphoma, which can exhibit a spectrum of morphology, as well as plasmacytoma, plasma cell myeloma, and plasma cell leukemia.3 While “classic” examples of these lesions are relatively straightforward to diagnose, many cases exhibit overlapping features that can cause confusion. For example, the lesions that are grouped under the term plasmablastic lymphoma are quite heterogeneous with differences that may be related to the immunodeficiency state of the patient (human immunodeficiency virus [HIV] vs posttransplant lymphoproliferative disorder [PTLD], for example). Separation of plasmablastic lymphoma and blastic myeloma can be difficult even with adequate clinical information. Furthermore, lymphomas with plasmacytic/plasmablastic differentiation can present as malignant effusions, causing confusion with the HHV8+ primary effusion lymphomas (PELs). Several cases are highlighted in this report to illustrate the characteristics of these lesions in the different immunodeficiency settings and their differential with other lesions with plasma cell/plasmacytic differentiation, including PEL.
HHV8-related lymphoproliferative disorders include germinotrophic lymphoproliferative disorder, multicentric Castleman disease (MCD), HHV8+ diffuse large B-cell lymphoma not otherwise specified (HHV8+ DLBCL NOS), and PEL/extracavitary PEL.4 While most HHV8-infected individuals have an underlying cause for immunosuppression, such as HIV, the rare condition, germinotropic lymphoproliferative disorder (GLPD), preferentially occurs in immunocompetent hosts.5 These lesions, like most extracavitary PELs (EC-PELs), not only are HHV8+ but also are Epstein-Barr virus (EBV) positive and composed of cells that may resemble those seen in EC-PEL. The differential immunophenotypic and genotypic findings of these entities will be discussed based on the cases submitted to the workshop. Plasmablastic differentiation is also a feature of many HHV8-positive lymphoid proliferations. HHV8 may infect naive B cells (CD19+, CD20+, IgM+, IgD+, CD27–) or IgM memory B cells (CD19+, CD20+, IgM+, ID+/–, CD27+). Latently infected cells under the influence of viral interleukin (IL)–6 and other factors have the ability to transform into B cells with a plasmablastic appearance that variably expresses intracellular and surface immunoglobulin. Plasmablasts are invariably present in cases of HHV8+ MCD. Cases submitted to this workshop are illustrated in this report to discuss criteria for the diagnosis of HHV8+ MCD, the significance of plasmablastic aggregates in HHV8+ MCD, and the criteria for progression of MCD to HHV8+ DLBCL NOS.
Key points include recognition of HHV8+ MCD and differential from other forms of lymphoid hyperplasia. Plasmablasts may be increased in cases of MCD showing progression, with extension outside of the germinal center. Differential includes HHV8+ DLBCL NOS, a newly defined World Health Organization (WHO) entity in which there is destruction of nodal or splenic architecture. GLPD can be a challenging diagnosis, although follicles are easily identified and the plasmablasts are also positive for EBV. Cases of PEL and EC-PEL have unique histologic and immunophenotypic characteristics illustrated in cases from the workshop. The spectrum of HHV8+ lymphoid proliferation is expanding with reports of cases showing unique or overlapping features, suggesting that HHV8 staining may be valuable in the assessment of lymphoproliferative disorders in the immunodeficiency setting.
HHV8+ Lymphoproliferative Disorders
MCD
Castleman disease was initially described in 1956 by Castleman et al,6 who reported on 13 cases of localized mediastinal lymphoid proliferations in asymptomatic patients. Since that time, it has been recognized that there are different morphologic variants (hyaline vascular, plasma cell/plasmablastic, and mixed or transitional) as well as clinical forms (unicentric and multicentric) classified under the broad clinicopathologic syndrome termed Castleman disease. The multicentric form, which usually exhibits morphologic features of the plasma cell/plasmablastic variant, is associated with constitutional symptoms, laboratory abnormalities, and widespread (ie, multicentric) lymphadenopathy often with hepatosplenomegaly.7‐10 Previously, MCD was classified into lesions that occurred in HIV+ and HIV– individuals. However, it has become increasingly recognized that separation based on HHV8+ status is clinically and biologically more relevant.10,11 HHV8– MCD, also known as idiopathic MCD, may have a variety of etiologies, but too few cases were submitted to the workshop for further evaluation and discussion. HHV8+ MCD is commonly seen in HIV-infected patients but can occur in the absence of HIV infection.12 MCD includes a group of systemic conditions in which there is a proliferation of lymphocytes, plasma cells, and vessels in response to the overproduction of cytokines and growth factors, particularly human interleukin-6 (huIL-6), a homologue of which is produced by HHV8 (viral IL-6 [vIL-6]). A number of other viral encoded genes and microRNAs contribute to the pathogenesis of HHV8+ MCD, including viral FLICE inhibitory protein, viral G protein–coupled receptor, viral macrophage inflammatory protein (vMIP)–I, vMIP-II, and kaposin B, among others. In addition, elevations of a number of human cytokines, including human IL-6, IL-10, and, to a lesser extent, tumor necrosis factor α and IL-1β are also seen in HHV8+ MCD.13 Disease symptoms correlate with serum levels of huIL-6, vIL-6, IL-10, and HHV8 viral load, while treatment with anti–IL-6 or anti–IL-6 receptor antibodies can result in amelioration of symptoms, highlighting the pathogenetic relationship between HHV8+ MCD and cytokines, particularly IL-6.10,14‐18,19‐21
Seventeen cases of HHV8+ MCD were submitted to the workshop from 12 HIV+, three HIV–, and two unknown HIV status patients (male, n = 15; female, n = 2). As distinct criteria for the diagnosis of HHV8+ MCD have not been fully defined, these cases were closely examined for features that would be helpful in their identification based on morphology and immunophenotypic markers. Although there was some variation in morphology, most cases showed variably involuted follicles that exhibited some degree of hyalinization, prominent vascular proliferation, sheets of interfollicular plasma cells, and plasmablasts that were located primarily in the mantle cell zones and/or abutting the germinal centers (Image 1 and cases SH2015-250 and SHS2015-287). Additional cases not presented but relevant to this discussion include the other cases of HHV8+ MCD cases submitted to the workshop. In some instances, the plasmablasts, which are medium to large cells with vesicular nuclei and amphophilic cytoplasm, form larger collections invading or replacing the germinal centers, abutting the germinal centers in the mantle zone and/or seen randomly in the interfollicular area. These plasmablastic aggregates were previously termed microlymphomas (see below).14,22,23
Image 1.

Morphologic features characteristic of human herpes virus 8–positive multicentric Castleman disease (courtesy of Drs Soderquist, Chadburn, and Bagg; Dr Goodlad; and Drs Pandey, Post, and Alapat). A, SH2015-0287. Prominent vascular proliferation (arrow) and interfollicular polytypic plasma cells (arrowhead) are shown (H&E, x10). B, SH2015-0237. Variably involuted, hyalinized follicles (arrow) and interfollicular polytypic plasma cells (arrowhead) are shown (H&E, x40). C, SH2015-0079. Variably involuted, hyalinized follicles (black arrow), penetrating vessels (white arrow), and a plasmablast (arrowhead) are shown (H&E, x40). D, SH2015-0287. A plasmablast aggregate (arrow) is shown (H&E, x20).
Summation of the immunophenotypic studies done on the 17 HHV8+ MCD cases (not all markers done on all cases; Image 2 and Image 3) showed that the plasmablasts were the HHV8-infected cells based on expression of latent nuclear antigen (LANA; ORF73; LNA-1), which shows a dot-like nuclear staining pattern (Image 2, upper left and middle images). Where tested, a proportion of the LANA-positive cells were also positive for vIL-6. These LANA-positive plasmablastic cells expressed cytoplasmic IgM λ immunoglobulin (Image 2, upper right image and lower images) and were uniformly multiple myeloma oncogene 1 (MUM1) positive, and between 33% and 50% of the cases were variably positive for CD20 and/or CD79a (Image 3, upper images). However, all cases tested were paired box protein 5 (PAX5); also known as B-cell-specific activator protein (BSAP), CD10, and B-cell CLL/lymphoma 6 (BCL6) negative. Immunostaining for follicular dendritic cell (FDC) markers, CD21 and CD23, showed variable patterns with respect to the follicle structure; however, a number of cases contained follicles with concentric, tight, collapsed, or small FDC meshworks. The large numbers of plasma cells in the interfollicular area were CD138+ and polytypic based on light chain expression (Image 3, bottom images). Although rarely reported in the literature,24 none of the cases submitted contained LANA-positive plasmablasts that were also positive for EBV based on in situ hybridization.
Image 2.

The human herpes virus 8–positive plasmablasts are best identified based on latent nuclear antigen (LANA) positivity (A [SH2015-0264] and B [SH2015-0079]). Notice the dot positivity in the nucleus (A). The LANA-positive plasmablasts are preferentially IgM positive (C [SH2015-0079]). Double immunohistochemistry for LANA (brown, D and E) and κ light chain (red, D) and λ light chain (red, E) shows that the LANA positive cells lack κ light chain expression (left side, D) but are preferentially λ light chain positive (left side, E). Note that the LANA negative plasma cells are polytypic (right side, D and E). (A, immunohistochemistry, x40; B, immunohistochemistry; C, immunohistochemistry; D, double immunohistochemistry, x40; E, double immunohistochemistry; x40; courtesy of Drs Monaghan, Kumar, and Luu; Dr Goodlad; and Drs Pizzi, Wu, Williams, Furman, Liu, and Chadburn.)
Image 3.

The human herpes virus 8–positive (HHV8+) plasmablasts are variably positive for CD20 (A [SH2015-264]) and CD79a (B [SH2015-264]). The interfollicular CD138+ plasma cells are often numerous (C [SH2015-228]) but in contrast to the HHV8+ plasmablasts are polytypic based on cytoplasmic κ (D [SH2015-264]) and λ (E [SH2015-264]) light chain expression (courtesy of Drs Monaghan, Kumar, and Luu and Drs Oak, Tan, and Fong).
MCD With Plasmablastic Aggregates
One of the characteristic features of HHV8+ MCD as described above are the HHV8-infected plasmablasts, which are present in the lymph nodes and spleens of these cases. Although these cells are usually seen in the mantle zones, as the disease progresses, the plasmablasts increase in number and may coalesce to form variably sized aggregates both within and outside of the germinal centers (case SH2015-287). Immunostaining with antibodies to the HHV8 latent nuclear antigen, LANA, highlights the plasmablast aggregates in this case, which are also positive for vIL-6, IgM, and λ immunoglobulin light chain. Although these aggregates exhibit λ light chain restriction, they are, by polymerase chain reaction (PCR) analysis, polyclonal or oligoclonal with unmutated immunoglobulin genes.
Diagnostic difficulties arise when the plasmablasts in HHV8+ MCD coalesce to from aggregates (previously called microlymphomas) as illustrated by several of the cases submitted to the workshop, including case SH2015-287. This was a 57-year-old HIV+ male patient who developed lymphadenopathy in the chest, abdomen, and pelvis. Although the histologic sections from a cervical lymph node showed features of HHV8+ MCD, there were aggregates of plasmablasts in the follicles and the interfollicular zones outside the germinal centers. Of interest in this case was the phenotypic diversity of the plasmablasts based on location in the lymph node with expression of CD138 by the interfollicular plasmablasts but not by the follicle-associated HHV8+ cells. The significance of the clusters of plasmablasts seen in this case is uncertain but should not be considered diagnostic of malignancy. However, progression to a monoclonal proliferation and frank lymphoma may occur. By contrast, HHV8+ DLBCL (see below) differs from microlymphoma in that there are sheets of clonal malignant cells effacing the tissue architecture. It is also of interest that a number of the plasmablasts in this case were found to be dim p53 and dim c-MYC protein positive. Two reports in the literature have also described the presence of p53-positive plasmablasts in HHV8+ MCD; in both of these cases, the patients had aggressive disease.25,26 In the case submitted to the workshop, the patient was treated with chemotherapy and subsequently went into remission, and the significance of p53 and MYC expression is uncertain.
HHV8+ DLBCL NOS
The incidence of non-Hodgkin lymphoma in patients with MCD is reported to be about 15 times that expected in the general HIV+ population.14 More recently, it has been suggested that treatment of MCD with rituximab reduces the risk of lymphoma.27,28 HHV8+ DLBCL NOS usually arises in a background of HHV8+ MCD in patients who are also HIV positive.22,29 Development of overt lymphoma is characterized by sheets and confluent clusters of plasmablasts and destruction of normal architecture. Lymph nodes, spleen, or extranodal sites may be involved. The proliferation is usually monomorphic with the malignant cells resembling plasmablasts or immunoblasts. The malignant cells are monoclonal and can express λ or κ light chains but usually the former. In some cases, a leukemic component is present.14,22 The neoplastic cells are variably positive for B-cell markers, including CD20; usually express markers of more terminal B-cell differentiation, such as MUM1; but are often negative for CD138 and are negative for EBV. In comparison with other plasmablastic lymphomas that have class-switched immunoglobulin genes, in HHV8+ DLBCL, there are no immunoglobulin somatic hypermutations. DLBCL arising in HHV8+ MCD commonly has an immunoblastic or plasmablastic appearance and should be differentiated from cases of PEL and EC-PEL discussed below, which may occur as well in patients with MCD. Although both PEL and EC-PEL may arise in patients with HHV8+ MCD, these are considered separate entities, and there is currently no definitive evidence of evolution of MCD to PEL/EC-PEL.30 The phenotype of the malignant cells in PEL differs from the plasmablasts in MCD in that they are usually negative for pan–B-cell antigens and positive for plasma cell and activation markers, including interferon regulatory factor 4 (IRF4)/MUM1. Unlike the HHV8+ cells in MCD and in HHV8+ DLBCL NOS, neoplastic PEL/EC-PEL cells are positive for EBV and have hypermutated genes and immunoglobulin gene rearrangements.
Case SH2015-250 is a case of HHV8+ DLBCL NOS arising in HHV8+ MCD and illustrates features of progression from MCD to lymphoma. This is a 37-year-old HIV+ male patient who developed widespread lymphadenopathy and B symptoms over the course of 2 years, while his HHV8 viral load increased to 1,200 copies/mL. An axillary lymph node revealed typical features of HHV8+ MCD. Clusters of plasmablasts were present, which were positive for IgM λ. The patient remained in remission for 2 years following rituximab therapy but then relapsed with progression to HHV8+ DLBCL NOS.
Evidence suggests that DLBCL in the setting of HHV8+ MCD arises from the plasmablastic cells, which are present either as individual blasts or as intra- or perifollicular clusters. In case SH2015-250, there was progression of HHV8+ MCD to MCD with plasmablastic aggregates (plasmablastic microlymphoma in WHO 2008)31 and finally to HHV8+ DLBCL. The term microlymphomas refers to aggregates of plasmablasts with moderate amounts of dense amphophilic cytoplasm, vesicular nuclei, and one to two prominent nucleoli, usually adjacent to lymphoid follicles. The aggregates of plasmablasts are typically negative for CD138 and variably positive for CD20 and CD79a. IRF4/MUM1 are positive, and despite lack of clonal restriction at the DNA level, they are positive for IgM λ and lack κ light chains. The plasmablasts are strongly positive for HHV8 LANA and in addition may express vIL-6. The term microlymphoma has been eliminated from the 2016 revision of the WHO since these are not clonal neoplasms and do not necessarily progress to lymphoma. In some cases, the blasts continue to proliferate, progressing to large aggregates, which grow independent of follicles until they efface the normal architecture meeting criteria for a diagnosis of lymphoma. The DLBCL cells resemble the plasmablasts in MCD in exhibiting variable positivity for B-cell markers and usually failing to express CD79a. They are variably positive and often negative for plasma cell markers, including CD38 and CD138, and lack activation markers such as CD30 but are IRF4/MUM1+.32 They are EBV– and express IgM with light chain restriction, which is λ in MCD but may be λ or, infrequently, κ in the lymphomas. The individual plasmablasts in MCD are polyclonal, while the plasmablastic aggregates may be poly- or oligoclonal or may contain weak dominant clones. Development of frank lymphoma is characterized by clonal immunoglobulin gene rearrangements. It is interesting to note, however, that in this case, the plasmablasts in the plasmablast aggregates were found to contain a MYC rearrangement based on fluorescence in situ hybridization (FISH) analysis, indicative of clonality and which may portend progression to frank lymphoma. Figure 1 (courtesy of Dr John Goodlad) illustrates the progression of MCD with individual blasts to larger aggregates (previously called microlymphomas) and finally HHV8+ DLBCL.
Figure 1.

Cartoon illustrating progression of human herpes virus 8–positive (HHV8+) multicentric Castleman disease to HHV8+ diffuse large B-cell lymphoma not otherwise specified (courtesy of Dr John Goodlad).
GLPD
GLPD occurs in HIV– immunocompetent individuals and is characterized by HHV8+ large atypical cells with immunoblastic, anaplastic, or plasmablastic features, which infiltrate and replace germinal centers as illustrated by cases SH2015-27 and -358. The atypical cells may extend into the interfollicular regions, causing confusion with large cell lymphoma. The plasmablastic cells are usually negative for CD20 and CD138 but positive for IRF4/MUM1 with monotypic κ or λ light chain expression, although the two cases submitted to the workshop (cases SH2015-27 and 358) either did not express monotypic light chains or lacked immunoglobulin expression. In addition to expression of LANA, the atypical cells are positive for EBV-encoded small RNAs (EBER) and vIL-6 (cases SH2015-27 and 358). Although the cells often express monotypic immunoglobulin, they have a poly- or oligoclonal pattern of immunoglobulin gene rearrangements. In addition to somatic mutations, there is intraclonal variation in the rearranged immunoglobulin genes, suggesting origin from germinal center B cells.5 Although cases of GLPD usually respond to treatment, there are rare reported cases that progressed to high-grade HHV8+ EBV+ lymphoma.33 Similarly, there are reports of unique cases intermediate between HHV8+ lymphoma and GLPD,34,35 indicating that there may be a wider spectrum of HHV8+ lymphoid proliferations than generally described.
Case SH2015-358 was from a 60-year-old immunocompetent patient whose lymph node biopsy specimen showed the typical features of GLPD in 2013 and again in 2014. The patient had an indolent clinical course with persistence of lymphadenopathy. Cytogenetic studies from the 2014 biopsy specimen demonstrated trisomy 3 in two of 20 cells, which was not considered diagnostic of a malignant process in this case. Although DNA analysis for B-cell clonality was not performed, monotypia was not identified by immunophenotypic studies, in contrast to what is usually reported in the literature. The patient refused treatment and nearly a year after the initial diagnosis appears to have stable lymphadenopathy without significant symptomology, a clinical course that is consistent with GLPD.
The differential of GLPD includes EC-PEL (see below; Image 4 and Table 1). GLPD is localized to lymph nodes, which generally show retention of overlying architecture. Unlike PEL, GLPD arises from germinal center B cells, and the proliferating cells tend to be positive for immunoglobulin κ or λ light chains. However, caution is advised as the number of studied GLPD cases in the literature is small, and exceptions, as exemplified by cases SH2015-27 and -358, may occur. As noted above, there are exceptional cases with features intermediate between GLPD and HHV8+ lymphoma.34
Image 4.
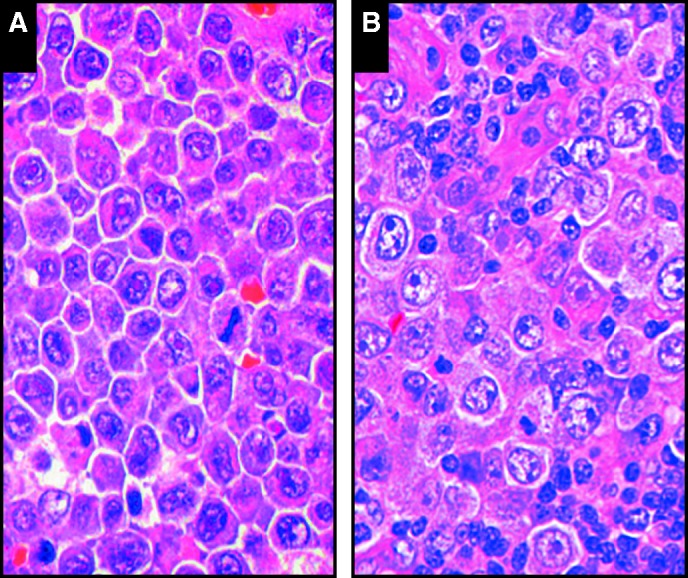
Morphologic similarities are seen when comparing the large immunoblastic/plasmablastic cells in extracavitary primary effusion lymphoma (A [SH2015-0309]) with those seen in germinotropic lymphoproliferative disorder (B [SH2015-0027]; courtesy of Dr Bhavsar and Drs Zhang, Vahdat, and Sokol).
Table 1.
GLPD and EC-PEL: Comparison of the Clinical and Immunophenotypic Features of the Submitted Workshop Casesa
| Characteristic | GLPD (n = 2) | EC-PEL (n = 15) |
|---|---|---|
| Age, mean, y | 72 | 51 |
| Sex | M = 1; F = 1 | M = 15; F = 0 |
| HIV | Yes = 0; no = 2 | Yes = 13; no = 2b |
| Presenting location; specimen site | LN = 2 | LN = 10c; GI = 4c; skin = 1; BM = 1 |
| CD45 | ND | 10/11d |
| CD20 | 0/2 | 2/13d |
| CD79A | 0/2 | 4/12d |
| Immunoglobulin | Noe = 1; κ/λ = 1 | Noe = 6; κ = 1; λ = 7; κ/λ = 1 |
| CD138 | 0/2 | 5/14d |
| CD38 | ND | 7/7 |
| MUM1 | 1/1 | 12/13 |
| CD30 | 1/2 | 7/13d |
| CD10 | 0/1 | 0/7 |
| EBER | 2/2 | 13/15 |
| HHV8 | 2/2 | 15/15 |
| T-cell antigen | 1/2f | 3/10f |
BM, bone marrow; EBER, Epstein-Barr virus–encoded small RNAs; EC-PEL, extracavitary primary effusion lymphoma; GI, gastrointestinal; GLPD, germinotropic lymphoproliferative disorder; HHV8, human herpes virus 8; HIV, human immunodeficiency virus; LN, lymph node; ND, not done.
Only first GLPD/EC-PEL diagnosis evaluated; includes SH2015 cases 003, 020, 027, 191, 238, 253, 262, 309, 326, 338, 358, 362, 402, 406, 425, 429, and 446; not all studies done in all cases. Values are presented as number or number/total number unless otherwise indicated.
1 = posttransplant, 1 = chemotherapy.
One patient had LN and GI involvement.
At least some cases showed partial, focal, and/or dim expression.
No = no light chain expression.
T-cell antigens expressed included partial CD3 for GLPD and CD3/CD4, partial CD2, and partial CD4 for EC-PEL.
PEL/EC-PEL
A DNA fragment was isolated from Kaposi sarcoma lesions using representational differential analysis in 1994 by Chang et al.36 This DNA fragment was from the Kaposi sarcoma herpesvirus (HHV8), which is now known to be etiologically related to the development of all forms of Kaposi sarcoma. Soon after the discovery of HHV8, Cesarman et al37 identified the virus in an unusual group of lymphomas seen preferentially in HIV+ individuals, which occurred primarily as effusions, lacked T- or B-cell lineage markers, but were composed of clonal B cells based on DNA analysis.38 These lesions were initially called body cavity–based lymphomas but are now known as primary effusion lymphomas. These cases constitute a unique entity, which tend to arise in severely immunosuppressed individuals with a history of AIDS but can occur in HIV– individuals as well.39 HIV+ patients with PEL are usually highly immunosuppressed with extremely low CD4 cell counts. However, there are a few reported cases where the CD4 count is high, and therefore a CD4 count more than 200 should not exclude the diagnosis.40 Approximately 10 years after the initial description of PEL, a solid form of the disease, known as EC-PEL, was described, which is essentially identical to classic PEL, except for the lack of an effusion.40‐42 These lesions, both classic PEL and EC-PEL, by definition, are HHV8+, and about 75% of the HIV+ cases are also EBV+, with the incidence of EBV coinfection lower in other patient populations.30,40,43 These neoplasms, although usually negative for B-cell lineage markers, such as CD19, CD20, PAX5, and surface immunoglobulin, and the germinal center markers CD10 and BCL6, express markers of terminal B-cell differentiation such as MUM1, Blimp1 (like the plasmablasts in MCD), CD138, and CD38 (in contrast to the plasmablasts in MCD) and are usually CD30 and epithelial membrane antigen (EMA) positive as well (see Table 1 for summation of the immunophenotype of the workshop cases).24,30,39,43 Evaluation of the immunoglobulin gene configuration shows the presence of somatic hypermutations in most cases.44,45 Genetic expression profiling studies have shown that PELs exhibit a profile between that of DLBCL and plasma cells.46,47 These immunophenotypic and genetic features indicate that PEL/EC-PELs are derived from terminally differentiated B cells that have gone through the germinal center process. Furthermore, in contrast to many other non-Hodgkin lymphomas, PEL/EC-PELs only rarely contain structural alterations in oncogenes or tumor suppressor genes,39 but cytogenetics usually shows a complex karyotype.48,49 Morphologically, the neoplastic cells, as with many of the other lesions in this section, are plasmablastic/immunmoblastic/anaplastic in appearance, as demonstrated by cases SH2015-309 and SH2015-446. Although PEL/EC-PELs are almost always of B-cell lineage, there are cases, including case SH2015-309, where T-cell antigens, such as CD3 and CD4, have been identified. Although very rare cases have been shown by DNA studies to contain clonal T cells,50‐52 most cases in the literature, as in case SH2015-309, consist of clonal B cells, based on immunoglobulin gene rearrangement studies, associated with a polyclonal T-cell population.53‐55 Thus, the T-cell antigen expression is aberrant in most PEL/EC-PELs and does not indicate T-cell origin, as shown by case SH2015-309.
A relatively large number of PELs/EC-PELs were submitted to the workshop (23 specimens from 18 patients). These cases were preferentially from HIV+ patients but also arose in HIV– individuals, including two who were posttransplant. These EC-PELs that occur in HIV– patients can lead to diagnostic difficulties as both EC-PELs (cases SH2015-309 and SH2015-446) and GLPDs (cases SH2015-27 and SH2015-358; Image 4) are composed of morphologically similar immunoblastic/plasmablastic/anaplastic-appearing cells. Although immunophenotypically, these two lesions are reportedly different, in that EC-PELs are classically CD138+ and only infrequently express cytoplasmic immunoglobulin30,56‐58 while GLPDs usually lack CD138 expression but are positive for cytoplasmic immunoglobulin,33,59 approximately two thirds of the EC-PELs submitted to the workshop were CD138–, and one of submitted GLPDs was immunoglobulin light chain negative, suggesting care must be taken when relying only on morphology and phenotype (Table 1). Genetically, however, the EC-PELs are monoclonal lesions while the GLPDs are polyclonal or oligoclonal,40,59 characteristics that are highlighted by the submitted cases (eg, GLPD in case SH2015-27 and EC-PEL in case SH2015-309).
There are several caveats to remember about PEL/EC-PEL:
Although PEL/EC-PELs occur preferentially in HIV+ individuals, they can also occur in other patients, including some who have no known cause for immunodeficiency.60‐63 This is highlighted by case SH2015-446 and case SH2015-295. In case SH2015-446, the patient was a 72-year-old man with mediastinal and iliac lymphadenopathy and no known cause of immunosuppression. Biopsy specimen of an iliac lymph node showed an EBER+, HHV8+, EMA+, and MUM1+ EC-PEL. The patient from case SH2015-295 was a 65-year-old bilateral lung transplant recipient who was taking azathioprine, cyclosporine, and prednisolone. Six months after transplant, he developed Kaposi sarcoma and later bilateral pleural effusions. Subsequent examination of the effusion showed neoplastic cells phenotypically and genetically similar to those of classic PEL in HIV+ patients.39
Not all PELs present as effusions in the pleural, pericardial, and abdominal cavities and can occur in other “cavities,” including in the space surrounding breast implants, in the cerebrospinal fluid, and in the peripheral blood, as shown by case SH2015-369 from a 59-year-old HIV+ positive man.40,63,64
Not all effusions are PELs. A variety of B-cell neoplasms (as described below) can occur as an effusion, many of which are morphologically similar to PEL.65‐67 Several non-PEL effusion or effusion-like lymphomas were presented at this workshop, including some in unusual sites (see cases SH2015-394, 300, 463, 324, 436, and 189). While PEL should be considered in the differential, the lack of HHV8 positivity clearly indicates that these lesions should be classified as another entity.
Effusion-Based DLBCL HHV8–
The term PEL is restricted to cavity-based lymphomas in which the neoplastic cells are infected with HHV8. Other lymphomas, which are HHV8–, can uncommonly present as neoplastic effusions and should not be confused with PEL. The term PEL-like lymphoma, which has been applied to these cases because of cytologic overlap, is confusing and is best avoided.68 Case SH2015-300 is an example of an HHV8– pleural-based lymphoma with cytology and phenotype resembling PEL but which was HHV8–. The patient was a 44-year-old woman taking highly active antiretroviral therapy (HAART) therapy for HIV who had an undetectable viral load. In addition to pleural effusions and ascites, she had multiple enlarged nodes on imaging. Axillary lymph node and bone marrow biopsy specimens were negative for lymphoma. Thoracentesis revealed large pleomorphic malignant cells with prominent nucleoli, amphophilic cytoplasm, and cytoplasmic vacuoles. Flow cytometry was positive for CD45 and CD38 but negative for B-cell markers and immunoglobulins. EBV EBER was positive, and HHV8 LANA was negative. There was a complex karyotype in addition to abnormalities involving MYC (t(8;14)(q24;q32) and immunoglobulin heavy locus (IGH)/MYC fusion by FISH) that are not seen in PEL.
Effusion-based DLBCL in the absence of HHV8 infection is characteristically seen in association with longstanding chronic inflammation and associated with EBV. Pyothorax-associated lymphoma (PAL) is considered the prototypic form but has unique characteristics.69 PAL was first described in Japan in patients with longstanding artificial pneumothorax and pyothorax after treatment for pulmonary tuberculosis. Similar lymphomas have been described with chronic inflammation from osteomyelitis, foreign material such as surgical mesh implants, and cardiac prosthetic valves, among others. Like cases of PAL, these lymphomas have a non–germinal center B-cell immunophenotype and EBV infection latency III, and they usually remain localized. The pathogenesis may be related to longstanding chronic inflammation in a confined compartmentalized space, resulting in localized immune impairment and proliferation of EBV-infected cells.70 Cytogenetically, these are complex neoplasms with frequent abnormalities involving MYC. DLBCL associated with chronic inflammation is frequently immunoblastic or anaplastic and related to DLBCL NOS rather than PEL.
In addition to chronic inflammation, similar lymphomas occur in patients with medical conditions leading to fluid overload, such as patients with liver disease and ascites. This is illustrated by case SH2015-394. This case was included in a series of HHV8– effusion-based lymphomas.71 The patient was a 45-year-old man with alcoholic cirrhosis, following an orthotopic liver transplant and increasing abdominal distention due to ascites. There were no mass lesions and the patient was negative for HIV but positive for hepatitis B and C. Cytology of the malignant cells revealed large pleomorphic cells with an immunoblastic or plasmablastic appearance and vacuolated amphophilic cytoplasm. In addition to expressing CD45 and CD138, the cells were positive for MYC by immunohistochemistry and EBV EBER. HHV8 was negative, as was expression of multiple B-cell markers, and flow cytometry revealed intracellular κ light chain restriction.
Unlike PEL and PAL, the EBV+ KSHV– effusion-based lymphomas are not well characterized and may be heterogeneous in their clinical and pathologic features, particularly as there is no defining genomic abnormality that has been implicated in the pathogenesis. In general, effusion-based B-cell lymphomas of this type tend to occur in older male patients (median 70 years) with frequent preexisting conditions leading to fluid overload states, particularly heart failure and cirrhosis. Unlike PEL, most cases express B-cell markers. Interestingly, there may be an association with chronic hepatitis C infection, which has been documented in up to 30% of cases. The cells tend to be pleomorphic and are frequently plasmablastic or immunoblastic, but cases with a Burkitt-like appearance have also been described.71 They have a complex karyotype with no recurrent chromosomal abnormalities, and prognosis is variable but generally more favorable than PEL. It is suggested that persistent antigenic stimulation by chronic inflammation and viral infection, including EBV and hepatitis C virus, triggers intraperitoneal B-cell expansion in an immune-sequestered site, leading to escape of immune surveillance and clonal expansion of neoplastic cells.72,73
Plasmablastic Lymphoma in Immunodeficiency and Nonimmunodeficiency Settings
Plasmablastic lymphoma is characterized by a proliferation of large malignant lymphoid cells resembling plasmablasts or immunoblasts. Important for the definition is the phenotype, generally negative for CD20 and positive for CD138, CD38, and IRF4/MUM1, indicating origin from a proliferating B cell that has switched to the plasma cell gene expression program. Plasmablastic lymphoma was first described in the oral cavity of HIV-infected individuals,74 and this variety is now known as the “oral mucosa type” of plasmablastic lymphoma. Although the oral cavity is the most common site at presentation, it may be seen at other sites.29 There may be different pathogenetic mechanisms contributing to the development of plasmablastic lymphomas in different immunologic settings.75 Other cases have more obvious plasmacytic differentiation and may resemble plasmablastic myeloma. The plasmablastic phenotype may also occur in other B-cell lymphomas such as anaplastic lymphoma receptor tryosine kinase–positive B-cell lymphoma, but these are not included in this category.76
Case SH2015-94 is an example of the oral cavity type of plasmablastic lymphoma. The patient was a 49-year-old HIV+ male patient who had a mass lesion of the gingiva. He was severely immunosuppressed with a CD4/CD8 ratio of 0.1 and an HIV viral load of 1,061 copies/mL. Biopsy specimen of the oral cavity mass revealed sheets of large immunoblastic cells with prominent nucleoli and basophilic cytoplasm. The phenotype was characteristically negative for B-cell markers, including CD19, CD20, and PAX5, but positive for the plasma cell marker CD138 and for IRF4/MUM1 Table 2. In this case, MYC was positive by immunohistochemistry in a large percentage of the cells, as was in situ hybridization for EBV EBER. There was no MYC translocation detected by break-apart FISH. HIV-related plasmablastic lymphoma usually presents in extranodal sites, with the majority in the oral cavity or jaw, but also in other locations such as the gastrointestinal tract, skin, and soft tissues.77 Lymph node involvement is less frequent. Clinical stage at presentation is most often stage III or IV, and bone marrow involvement is common. Improved survival has been reported in patients treated with combination chemotherapy and antiretroviral therapy.77 The patient described in case SH2015-94 has been in remission more than 5 years after his initial diagnosis. Favorable outcomes are associated with low stage at diagnosis, younger age group, oral location, and the absence of MYC/IgH rearrangements.
Table 2.
Plasmablastic Lymphoma: Summation of Workshop Findingsa
| Category | HIV+ | HIV– | Posttransplant | Transformed |
|---|---|---|---|---|
| % of cases | 45 | 13 | 28 | 4 |
| M:F, No. | 4:1 | 2:1 | 3:4 | 1 |
| Age, median, y | 45 | 76 | 40 | 62 |
| No. 1 site | Analb | Nasal | GI/effusionb | Gingiva |
| No. 2 site | Various sitesc | GI | Various sitesc | NA |
| CD20 | 0/10 | 0/3 | 0/7 | 0/1 |
| CD79ad | 5/8 | 2/3 | 3/6 | 1/1 |
| MUM1 | 6/7 | 2/2 | 5/5 | 1/1 |
| CD138d | 7/9 | 3/3 | 7/7 | 1/1 |
| EBER | 9/10 | 3/3 | 4/7 | 0/1 |
| Ki-67, mean (range), % | >90% (80->95%) | ∼80% (60->90%) | 90% (70-100%) | >90% |
| MYC-R | 6/6 | ND | 1/2 | ND |
EBER, Epstein-Barr virus–encoded small RNAs; GI, gastrointestinal; HIV, human immunodeficiency virus; NA, not applicable; ND, not done.
Summation of submissions: SH2015-009, 032, 037, 094, 098, 108, 118, 142, 161, 189, 231, 300, 306, 394, 411, 412, 430, 436, 463, 473, and 482; iatrogenic associated (SH2015-005), congenital associated (SH2015-256), and indeterminate plasmablastic lymphoma vs plasma cell neoplasm (ie, SH2015-266) not included in this table. Values are presented as number/total number unless otherwise indicated. Findings are based on information provided and studies reportedly done by the submitting authors; not all studies were done in all workshop cases.
Only two cases each.
One case per anatomic site.
Variable expression in some cases.
EBV is positive in most cases of plasmablastic lymphoma, particularly in patients with AIDS and other forms of immunosuppression. The latency pattern in patients with HIV is usually type I with expression of EBER, EBV nuclear antigen 1, negative for EBV nuclear antigen 2 and EBV latent membrane protein 1, while latency pattern III may be seen in the posttransplantation setting.75,78 MYC translocations are seen in over 50% of cases and may be important for the pathogenesis. The genotype is usually complex with rearranged immunoglobulin genes among other abnormalities.
Plasmablastic lymphoma in the non-HIV-infected population has a more variable appearance and clinical presentation.75 These may have nodal presentation and different degrees of plasmacytic differentiation and are less frequently associated with EBV.79 Case SH2015-108 is from a 28-year-old male patient treated with an allogeneic stem cell transplant for lymphoblastic leukemia. Six years after the transplant, he developed a soft tissue mass in the right knee with infiltration into the tibia. Biopsy specimen revealed sheets of plasmablasts with oval nuclei, prominent nucleoli, and amphophilic cytoplasm. Immunohistochemical stains for B-cell markers were negative, but the malignant cells were strongly positive for CD138, IRF4/MUM1, and MYC. EBV EBER was negative, as has been described in 25% to 40% of plasmablastic lymphomas posttransplant.75 FISH with break-apart probe showed evidence of a MYC translocation plus extra copies of MYC. Molecular analysis demonstrated two distinct clonal B-cell populations of donor origin.
Plasmablastic lymphomas occurring in the setting of iatrogenic immunosuppression, including following transplantation, is characterized by phenotypic evidence of plasmacytic differentiation with variable loss of B-cell markers, including CD20 and PAX5, and expression of plasma cell markers, including CD38, CD138, IRF4/MUM1, VS38c, Blimp1, and XBP-1. Cytoplasmic immunoglobulin is variably present in about half or the cases.75 MYC and p53 are commonly overexpressed, and MYC translocations are seen in 50% to 70% of the cases. EBV positivity is seen in 50% to 80% of cases, usually latency type I, rarely type III.
Case SH2015-32 illustrates the difficulties in differential diagnosis between plasmablastic lymphoma and myeloma, a problem that was also recognized in a previous workshop.80 The patient was a 34-year-old HIV+ woman with a scalp mass and lytic lesions in the right humerus. Bone marrow biopsy specimen revealed sheets of plasmacytoid blasts with large nuclei, prominent often central nucleoli, and plasmacytoid cytoplasm. Cells were positive for CD138 and MYC by immunohistochemistry, as well as EBV EBER by in situ hybridization (ISH). Ki-67 revealed a high proliferation rate. Serologic studies revealed high titers of EBV (1,800 IU/mL) and HIV copies (51,084 copies/mL). Cytogenetic studies with a myeloma panel did not detect additional aberrations, including absence of hyperdiploidy, cyclin D1/IgH rearrangement, 13q deletion, or deletion of TP53. After treatment with combination chemotherapy, autologous stem cell transplant, and HAART, she developed a soft tissue mass in the left arm, which also revealed plasmablastic cells on fine-needle aspirate. Cells were positive for EBV EBER by ISH, and FISH revealed a MYC/IgH rearrangement. In this case, separation from myeloma was particularly problematic because of the presence of lytic bone lesions.
Features favoring plasmablastic lymphoma in this case included the background of immunodeficiency, the presence of EBV, the morphologic features resembling B immunoblasts, and the high Ki-67 proliferation rate. Also, cytogenetic studies were negative for myeloma-related abnormalities and positive for MYC rearrangements. Staining for CD56 and cyclin D1, more frequent in myeloma than plasmablastic lymphoma, was absent in this case. The proliferation rate as determined by staining for Ki-67 is generally low in myeloma in comparison with plasmablastic lymphoma.81 Clinical findings may be required to differentiate between myeloma and plasmablastic lymphoma, particularly in the bone marrow. With the exception of HIV+ patients and those with other forms of immunodeficiency, late-stage disease with bone marrow involvement is uncommon in plasmablastic lymphoma. Paraproteins may present in association with plasmablastic lymphoma, but this occurs in a minority of cases. MYC translocation has been associated with tumor progression in myeloma, and dysregulation of MYC may be a common genetic mechanism that imparts plasmablastic morphology and aggressive clinical course to B-cell neoplasms at a later stage of differentiation.82
Plasma Cell Neoplasms in the Immunodeficiency State, Including Plasmacytoma, Myeloma, and Plasma Cell Leukemia
Plasma Cell Neoplasms
Myeloma and other mature plasma cell neoplasms are uncommonly associated with immunosuppression. The risk of plasma cell neoplasms following transplant is estimated at 1.8 times that of the general population,83 and plasmacytomas comprise about 4% of cases of PTLD. Association with EBV is also variable (38%-78%) but less common than in most other forms of PTLD.84 As with other types of PTLD, the risk is greater in patients who are EBV seronegative prior to transplantation. Other risk factors include older age at transplantation.83,85 Plasma cell neoplasms usually occur late, 3 to 8 years following transplantation. Plasmacytomas are usually extranodal and may involve the skin or other sites, including the transplanted organ such as the kidney or heart in cardiac transplants. In addition to reduction of immunosuppression, treatment options include local therapy (radiation and surgery), chemotherapy regimens similar to those used in myeloma, and newer agents such as lenalinomide.86 However, these lesions in the post–solid organ transplant setting have been found to be associated with a relatively good clinical outcome.87 Case SH2015-209 was a 51-year-old male patient who received an unrelated donor bone marrow transplant following treatment for acute monoblastic leukemia. Within 2 months, he developed inguinal lymphadenopathy and was diagnosed with polymorphic B-cell lymphoproliferative disorder, EBV+, posttransplantation. One week after this biopsy, he developed plasma cell leukemia characterized by a WBC count of 13.4 × 103/μL and 20% plasma cells. In addition to serum free light chains and Bence Jones protein, serum protein electrophoresis revealed an IgA λ M-protein of 4.5 g/dL. Bone marrow biopsy specimen revealed 50% plasma cells, including sheets positive for CD138 and λ light chains. This case is unusual in occurring soon after transplantation and rapid progression to plasma cell leukemia. A similar case of a patient who developed plasma cell leukemia was recently described in a cardiac transplant patient less than 1 month following an allograft.3 Even with clinical information, it may not be possible to differentiate between plasmacytoma and myeloma in this setting. Myeloma cases may more frequently express CD56 and CD117 and are less frequently positive for EBV, but exceptions may be encountered.
Effusion-Based Plasmablastic Lymphoma, HHV8–
Plasmablastic lymphoma, HHV8– causing an effusion is a rare entity, while primary effusion-based plasmablastic lymphomas have been infrequently described in the literature, even when using a wide definition of effusion.65,79,88‐96 Several cases exhibiting plasmablastic morphology but occurring in fluids were submitted to the workshop, including two cases discussed above (cases SH2015-300 and 394). These latter cases occurred in the posttransplant setting as did case SH2015-463, a patient who had undergone both a kidney and pancreas transplants. In this case, the patient developed subcutaneous nodules 4 years after her most recent transplant, which on biopsy specimen were composed of EBV+, monotypic IgA κ-positive lymphoplasmacytic cells. A bone marrow at the time showed scattered EBV+ cells but no large plasma cell infiltrate. A year later, after a good response to treatment, a breast nodule was biopsied that showed sheets of EBV+, IgA κ-positive plasma cells, this time consistent with a plasmacytoma. Two years later, the patient developed progressive dyspnea and had bilateral pleural effusions but no lymphadenopathy or mass lesions. Evaluation of the effusion showed pleomorphic, plasmablastic-appearing cells consistent with plasmablastic lymphoma. The cells were EBV+ and cytoplasmic κ light chain restricted, which by PCR analysis contained clonal B-cell rearrangements identical to the previous breast nodule. Plasmablastic PTLDs, like plasmacytoma-like PTLDs, are rare, accounting for 4% to 6% of all plasmablastic lesions.78 However, progression from plasmacytoma to plasmablastic lymphoma is extremely unusual and only rarely reported in the literature. A recent case report of this occurring in an immunocompetent patient97 showed that the progression was associated with increased numbers of EBV+ cells, an increased proliferation rate, and increased c-MYC protein expression, the latter two findings similar to what was seen in this case. In addition, in case SH2015-463, p53 expression, not seen in the plasmacytoma, was identified in the plasmablastic lesion. Thus, the acquisition of additional genetic abnormalities in this EBV+ lesion most likely resulted in a more aggressive disease process.81
Conclusions
This session addressed the broad spectrum of cases of immunodeficiency-related lymphoproliferative disorders with plasmablastic differentiation that were submitted to the workshop. The entities discussed ranged from plasmablastic proliferations that overlapped with myeloma/plasmacytoma to plasmablastic neoplasms presenting in extranodal sites and as malignant effusions. EBV was variably involved in the different immunodeficiency settings while characteristically absent in others (eg, HHV8+ DLBCL NOS). Proliferations associated with HHV8 were highlighted and difficulties in the differential diagnosis discussed. A summary of the key features is found in Table 3.
Table 3.
Summary Table: HHV8/KSHV-Positive Lymphoproliferative Disorders and the Spectrum of Plasmablastic and Plasma Cell Neoplasms
| HHV8+ MCD |
| Castleman disease is classified according to the presence or absence of HHV8+ |
| HHV8+ MCD is a lymphoid, histiocytic, and plasma cell proliferation related to cytokine production, particularly viral interleukin 6 |
| Polyclonal plasmablasts with IgM λ light chain restriction are characteristically present in HHV8+ MCD |
| Some cases are associated with plasmablastic aggregates (previously called microlymphomas), which, despite light chain restriction, are poly- or oligoclonal |
| HHV8+ DLBCL |
| Usually but not always related to MCD |
| In some cases, there may be evolution from MCD with plasmablastic aggregates to sheets of plasmablasts with tissue destruction and monoclonal expression of light chains |
| Lymphomas express markers of terminal B-cell differentiation but are negative for EBV |
| HHV8+ GLPD |
| Occurs in immunocompetent patients, EBV+, and may be monotypic κ or λ but polyclonal or oligoclonal (compared with EC-PEL, which is clonal) |
| PEL and PEL/EC-PEL |
| PELs are negative for B-cell markers, but immunoglobulin gene rearrangements are positive, indicating a B-cell genotype |
| Usually but not always associated with HIV infection |
| Not all PELs present as effusions, and cases have been reported in the space surrounding breast implants, CSF, and peripheral blood |
| PELs are HHV8+ by definition, and EBV is present in about 75% of cases, particularly those associated with HIV |
| PELs express markers of terminal B-cell differentiation (MUM1, Blimp1) |
| Not all effusions are PELs, and other HHV8– lymphomas can present as effusions |
| Effusion-based DLBCL HHV8– |
| May be seen with longstanding chronic inflammation and associated with EBV as in cases of PAL |
| Similar lymphomas occur in patients with medical conditions leading to fluid overload such as liver disease and ascites |
| Unlike cases of PEL, most express B-cell markers |
| Plasma cell neoplasms associated with immunodeficiency |
| Spectrum ranges from marginal zone–like lymphoma to plasmacytoma to plasmablastic lymphoma and plasma cell leukemia, sometimes in the same patient |
| Plasmablastic lymphomas differ in different settings (immunocompetent vs HIV vs PTLD) |
| Clinical features are important in making the correct diagnosis |
| EBV and MYC are variably involved in different immunodeficiency settings |
| CD56, CD117, and myeloma FISH are helpful in separating blastic myeloma from plasmablastic lymphoma |
| PL in the immunodeficiency and nonimmunodeficiency setting |
| Phenotype is important for the definition, generally negative for CD20 and positive for CD138, CD38, and IRF4/MUM1 |
| Although the oral cavity is a common site of presentation, particularly in association with HIV, PL may occur at other sites |
| In the non-HIV population, plasmablastic lymphoma has a more variable appearance and clinical presentation |
| Cases with marked plasmacytic differentiation may resemble plasmablastic myeloma and be difficult to differentiate, even with clinical information |
| EBV is positive in most cases, particularly in patients with AIDS, and MYC translocations are seen in over 50% of cases |
CSF, cerebrospinal fluid; DLBCL, diffuse large B-cell lymphoma; EBV, Epstein-Barr virus; EC-PEL, extracavitary primary effusion lymphoma; FISH, fluorescence in situ hybridization; GLPD, germinotropic lymphoproliferative disorder; HHV8, human herpes virus 8; HIV, human immunodeficiency virus; IRF4, interferon regulatory factor 4; MCD, multicentric Castleman disease; PAL, pyothorax-associated lymphoma; PEL, primary effusion lymphoma; PL, plasmablastic lymphoma; PTLD, posttransplant lymphoproliferative disorder.
Acknowledgments
Acknowledgment: The authors are grateful to the submitters of cases discussed in this section of the workshop.
References
- 1. Tarte K, Zhan F, De Vos J, et al. Gene expression profiling of plasma cells and plasmablasts: toward a better understanding of the late stages of B-cell differentiation. Blood. 2003;102:592-600. [DOI] [PubMed] [Google Scholar]
- 2. Montes-Moreno S, Montalban C, Piris MA. Large B-cell lymphomas with plasmablastic differentiation: a biological and therapeutic challenge. Leuk Lymphoma. 2012;53:185-194. [DOI] [PubMed] [Google Scholar]
- 3. Ramia S, Guy J. An unusual posttransplant EBV-associated lymphoproliferative disorder exhibiting plasmacytic features in leukemic phase. Blood. 2015;125:1672. [DOI] [PubMed] [Google Scholar]
- 4. Carbone A, Cesarman E, Spina M, et al. HIV-associated lymphomas and gamma-herpesviruses. Blood. 2009;113:1213-1224. [DOI] [PubMed] [Google Scholar]
- 5. Du MQ, Diss TC, Liu H, et al. KSHV- and EBV-associated germinotropic lymphoproliferative disorder. Blood. 2002;100:3415-3418. [DOI] [PubMed] [Google Scholar]
- 6. Castleman B, Iverson L, Menendez VP. Localized mediastinal lymphnode hyperplasia resembling thymoma. Cancer. 1956;9:822-830. [DOI] [PubMed] [Google Scholar]
- 7. Frizzera G, Peterson BA, Bayrd ED, et al. A systemic lymphoproliferative disorder with morphologic features of Castleman's disease: clinical findings and clinicopathologic correlations in 15 patients. J Clin Oncol. 1985;3:1202-1216. [DOI] [PubMed] [Google Scholar]
- 8. Talat N, Schulte KM. Castleman's disease: systematic analysis of 416 patients from the literature. Oncologist. 2011;16:1316-1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cronin DM, Warnke RA. Castleman disease: an update on classification and the spectrum of associated lesions. Adv Anat Pathol. 2009;16:236-246. [DOI] [PubMed] [Google Scholar]
- 10. Dossier A, Meignin V, Fieschi C, et al. Human herpesvirus 8-related Castleman disease in the absence of HIV infection. Clin Infect Dis. 2013;56:833-842. [DOI] [PubMed] [Google Scholar]
- 11. Fajgenbaum DC, Liu A, Ruth J, et al. HHV-8-negative, idiopathic multicentric Castleman Disease (iMCD): a description of clinical features and therapeutic options through a systematic literature review. Blood. 2014;124:4861. [Google Scholar]
- 12. Soulier J, Grollet L, Oksenhendler E, et al. Kaposi's sarcoma–associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood. 1995;86:1276-1280. [PubMed] [Google Scholar]
- 13. Hengge UR, Ruzicka T, Tyring SK, et al. Update on Kaposi's sarcoma and other HHV8 associated diseases. Part 2: pathogenesis, Castleman's disease, and pleural effusion lymphoma. Lancet Infect Dis. 2002;2:344-352. [DOI] [PubMed] [Google Scholar]
- 14. Oksenhendler E, Boulanger E, Galicier L, et al. High incidence of Kaposi sarcoma–associated herpesvirus-related non-Hodgkin lymphoma in patients with HIV infection and multicentric Castleman disease. Blood. 2002;99:2331-2336. [DOI] [PubMed] [Google Scholar]
- 15. Aoki Y, Tosato G, Fonville TW, et al. Serum viral interleukin-6 in AIDS-related multicentric Castleman disease. Blood. 2001;97:2526-2527. [DOI] [PubMed] [Google Scholar]
- 16. Polizzotto MN, Uldrick TS, Wang V, et al. Human and viral interleukin-6 and other cytokines in Kaposi sarcoma herpesvirus-associated multicentric Castleman disease. Blood. 2013;122:4189-4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sakakibara S, Tosato G. Contribution of viral mimics of cellular genes to KSHV infection and disease. Viruses. 2014;6:3472-3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bhutani M, Polizzotto MN, Uldrick TS, et al. Kaposi sarcoma–associated herpesvirus-associated malignancies: epidemiology, pathogenesis, and advances in treatment. Semin Oncol. 2015;42:223-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. van Rhee F, Stone K, Szmania S, et al. Castleman disease in the 21st century: an update on diagnosis, assessment, and therapy. Clin Adv Hematol Oncol. 2010;8:486-498. [PubMed] [Google Scholar]
- 20. Matsuyama M, Suzuki T, Tsuboi H, et al. Anti-interleukin-6 receptor antibody (tocilizumab) treatment of multicentric Castleman's disease. Intern Med. 2007;46:771-774. [DOI] [PubMed] [Google Scholar]
- 21. van Rhee F, Rothman M, Ho KF, et al. Patient-reported outcomes for multicentric Castleman's disease in a randomized, placebo-controlled study of siltuximab. Patient. 2015;8:207-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dupin N, Diss TL, Kellam P, et al. HHV-8 is associated with a plasmablastic variant of Castleman disease that is linked to HHV-8-positive plasmablastic lymphoma. Blood. 2000;95:1406-1412. [PubMed] [Google Scholar]
- 23. Du MQ, Liu H, Diss TC, et al. Kaposi sarcoma-associated herpesvirus infects monotypic (IgM lambda) but polyclonal naive B cells in Castleman disease and associated lymphoproliferative disorders. Blood. 2001;97:2130-2136. [DOI] [PubMed] [Google Scholar]
- 24. Chadburn A, Hyjek EM, Tam W, et al. Immunophenotypic analysis of the Kaposi sarcoma herpesvirus (KSHV; HHV-8)–infected B cells in HIV+ multicentric Castleman disease (MCD). Histopathology. 2008;53:513-524. [DOI] [PubMed] [Google Scholar]
- 25. Dargent JL, Lespagnard L, Sirtaine N, et al. Plasmablastic microlymphoma occurring in human herpesvirus 8 (HHV-8)–positive multicentric Castleman's disease and featuring a follicular growth pattern. APMIS. 2007;115:869-874. [DOI] [PubMed] [Google Scholar]
- 26. Li CF, Ye H, Liu H, et al. Fatal HHV-8-associated hemophagocytic syndrome in an HIV-negative immunocompetent patient with plasmablastic variant of multicentric Castleman disease (plasmablastic microlymphoma). Am J Surg Pathol. 2006;30:123-127. [DOI] [PubMed] [Google Scholar]
- 27. Gerard L, Michot JM, Burcheri S, et al. Rituximab decreases the risk of lymphoma in patients with HIV-associated multicentric Castleman disease. Blood. 2012;119:2228-2233. [DOI] [PubMed] [Google Scholar]
- 28. Hoffmann C, Schmid H, Muller M, et al. Improved outcome with rituximab in patients with HIV-associated multicentric Castleman disease. Blood. 2011;118:3499-3503. [DOI] [PubMed] [Google Scholar]
- 29. Dong HY, Scadden DT, de Leval L, et al. Plasmablastic lymphoma in HIV-positive patients: an aggressive Epstein-Barr virus–associated extramedullary plasmacytic neoplasm. Am J Surg Pathol. 2005;29:1633-1641. [DOI] [PubMed] [Google Scholar]
- 30. Pan ZG, Zhang QY, Lu ZB, et al. Extracavitary KSHV-associated large B-cell lymphoma: a distinct entity or a subtype of primary effusion lymphoma? Study of 9 cases and review of an additional 43 cases. Am J Surg Pathol. 2012;36:1129-1140. [DOI] [PubMed] [Google Scholar]
- 31. Isaacson PG, Campo E. Large B-cell lymphoma arising in HHV8-associated multicentric Castleman disease In: Swerdlow SH, Campo E, Harris NL, et al. , eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed.Lyon, France: IARC; 2008:258-259. [Google Scholar]
- 32. Said J, Isaacson P, Campo E, Harris NL, HHV8+ DLBCL NOS In: Swerdlow SH, Campo E, Harris NL, et al. , eds. WHO Classification of Tumours of Haematoploietic and Lymphoid Tissue. Lyon, France: IARC. In press. [Google Scholar]
- 33. Courville EL, Sohani AR, Hasserjian RP, et al. Diverse clinicopathologic features in human herpesvirus 8–associated lymphomas lead to diagnostic problems. Am J Clin Pathol. 2014;142:816-829. [DOI] [PubMed] [Google Scholar]
- 34. Seliem RM, Griffith RC, Harris NL, et al. HHV-8+, EBV+ multicentric plasmablastic microlymphoma in an HIV+ man: the spectrum of HHV-8+ lymphoproliferative disorders expands. Am J Surg Pathol. 2007;31:1439-1445. [DOI] [PubMed] [Google Scholar]
- 35. Papoudou-Bai A, Hatzimichael E, Kyriazopoulou L, et al. Rare variants in the spectrum of human herpesvirus 8/Epstein-Barr virus-copositive lymphoproliferations. Hum Pathol. 2015;46:1566-1571. [DOI] [PubMed] [Google Scholar]
- 36. Chang Y, Cesarman E, Pessin MS, et al. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science. 1994;266:1865-1869. [DOI] [PubMed] [Google Scholar]
- 37. Cesarman E, Chang Y, Moore PS, et al. Kaposi's sarcoma–associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med. 1995;332:1186-1191. [DOI] [PubMed] [Google Scholar]
- 38. Walts AE, Shintaku IP, Said JW. Diagnosis of malignant lymphoma in effusions from patients with AIDS by gene rearrangement. Am J Clin Pathol. 1990;94:170-175. [DOI] [PubMed] [Google Scholar]
- 39. Nador RG, Cesarman E, Chadburn A, et al. Primary effusion lymphoma: a distinct clinicopathologic entity associated with the Kaposi's sarcoma–associated herpes virus. Blood. 1996;88:645-656. [PubMed] [Google Scholar]
- 40. Chadburn A, Hyjek E, Mathew S, et al. KSHV-positive solid lymphomas represent an extra-cavitary variant of primary effusion lymphoma. Am J Surg Pathol. 2004;28:1401-1416. [DOI] [PubMed] [Google Scholar]
- 41. DePond W, Said JW, Tasaka T, et al. Kaposi's sarcoma–associated herpesvirus and human herpesvirus 8 (KSHV/HHV8)-associated lymphoma of the bowel: report of two cases in HIV-positive men with secondary effusion lymphomas. Am J Surg Pathol. 1997;21:719-724. [DOI] [PubMed] [Google Scholar]
- 42. Carbone A, Gloghini A, Vaccher E, et al. KSHV/HHV-8 associated lymph node based lymphomas in HIV seronegative subjects: report of two cases with anaplastic large cell morphology and plasmablastic immunophenotype. J Clin Pathol. 2005;58:1039-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li J, Zhao S, Wang J, et al. CD20-negative diffuse large B cell lymphoma: a comprehensive analysis of 695 cases. Tumour Biol. 2016;37:3619-3637. [DOI] [PubMed] [Google Scholar]
- 44. Hamoudi R, Diss TC, Oksenhendler E, et al. Distinct cellular origins of primary effusion lymphoma with and without EBV infection. Leuk Res. 2004;28:333-338. [DOI] [PubMed] [Google Scholar]
- 45. Matolcsy A, Nador RG, Cesarman E, et al. Immunoglobulin VH gene mutational analysis suggests that primary effusion lymphomas derive from different stages of B cell maturation. Am J Pathol. 1998;153:1609-1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Klein U, Gloghini A, Gaidano G, et al. Gene expression profile analysis of AIDS-related primary effusion lymphoma (PEL) suggests a plasmablastic derivation and identifies PEL-specific transcripts. Blood. 2003;101:4115-4121. [DOI] [PubMed] [Google Scholar]
- 47. Jenner RG, Maillard K, Cattini N, et al. Kaposi's sarcoma–associated herpesvirus-infected primary effusion lymphoma has a plasma cell gene expression profile. Proc Natl Acad Sci U S A. 2003;100:10399-10404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Guillet S, Gerard L, Meignin V, et al. Classic and extracavitary primary effusion lymphoma in 51 HIV-infected patients from a single institution. Am J Hematol. 2016;91:233-237. [DOI] [PubMed] [Google Scholar]
- 49. Wilson KS, McKenna RW, Kroft SH, et al. Primary effusion lymphomas exhibit complex and recurrent cytogenetic abnormalities. Br J Haematol. 2002;116:113-121. [DOI] [PubMed] [Google Scholar]
- 50. Lechapt-Zalcman E, Challine D, Delfau-Larue MH, et al. Association of primary pleural effusion lymphoma of T-cell origin and human herpesvirus 8 in a human immunodeficiency virus-seronegative man. Arch Pathol Lab Med. 2001;125:1246-1248. [DOI] [PubMed] [Google Scholar]
- 51. Kalogeraki A, Haniotis V, Karvelas-Kalogerakis M, et al. Primary effusion lymphoma with aberrant T-cell phenotype in an iatrogenically immunosuppressed renal transplant male: cytologic diagnosis in peritoneal fluid. Diagn Cytopathol. 2015;43:144-148. [DOI] [PubMed] [Google Scholar]
- 52. Coupland SE, Charlotte F, Mansour G, et al. HHV-8-associated T-cell lymphoma in a lymph node with concurrent peritoneal effusion in an HIV-positive man. Am J Surg Pathol. 2005;29:647-652. [DOI] [PubMed] [Google Scholar]
- 53. Said JW, Shintaku IP, Asou H, et al. Herpesvirus 8 inclusions in primary effusion lymphoma: report of a unique case with T-cell phenotype. Arch Pathol Lab Med. 1999;123:257-260. [DOI] [PubMed] [Google Scholar]
- 54. Juskevicius D, Dietsche T, Lorber T, et al. Extracavitary primary effusion lymphoma: clinical, morphological, phenotypic and cytogenetic characterization using nuclei enrichment technique. Histopathology. 2014;65:693-706. [DOI] [PubMed] [Google Scholar]
- 55. Brimo F, Michel RP, Khetani K, et al. Primary effusion lymphoma: a series of 4 cases and review of the literature with emphasis on cytomorphologic and immunocytochemical differential diagnosis. Cancer. 2007;111:224-233. [DOI] [PubMed] [Google Scholar]
- 56. Liao G, Cai J, Yue C, et al. Extracavitary/solid variant of primary effusion lymphoma presenting as a gastric mass. Exp Mol Pathol. 2015;99:445-448. [DOI] [PubMed] [Google Scholar]
- 57. Costes V, Faumont N, Cesarman E, et al. Human herpesvirus-8-associated lymphoma of the bowel in human immunodeficiency virus–positive patients without history of primary effusion lymphoma. Hum Pathol. 2002;33:846-849. [DOI] [PubMed] [Google Scholar]
- 58. Mate JL, Navarro JT, Ariza A, et al. Oral solid form of primary effusion lymphoma mimicking plasmablastic lymphoma. Hum Pathol. 2004;35:632-635. [DOI] [PubMed] [Google Scholar]
- 59. D'Antonio A, Boscaino A, Addesso M, et al. KSHV- and EBV-associated germinotropic lymphoproliferative disorder: a rare lymphoproliferative disease of HIV patient with plasmablastic morphology, indolent course and favourable response to therapy. Leuk Lymphoma. 2007;48:1444-1447. [DOI] [PubMed] [Google Scholar]
- 60. Boulanger E, Hermine O, Fermand JP, et al. Human herpesvirus 8 (HHV-8)–associated peritoneal primary effusion lymphoma (PEL) in two HIV-negative elderly patients. Am J Hematol. 2004;76:88-91. [DOI] [PubMed] [Google Scholar]
- 61. Jones D, Ballestas ME, Kaye KM, et al. Primary-effusion lymphoma and Kaposi's sarcoma in a cardiac-transplant recipient. N Engl J Med. 1998;339:444-449. [DOI] [PubMed] [Google Scholar]
- 62. Regnier-Rosencher E, Barrou B, Marcelin AG, et al. Primary effusion lymphoma in two kidney transplant recipients [in French]. Ann Dermatol Venereol. 2010;137:285-289. [DOI] [PubMed] [Google Scholar]
- 63. Said JW, Tasaka T, Takeuchi S, et al. Primary effusion lymphoma in women: report of two cases of Kaposi's sarcoma herpes virus–associated effusion-based lymphoma in human immunodeficiency virus-negative women. Blood. 1996;88:3124-3128. [PubMed] [Google Scholar]
- 64. Crane GM, Xian RR, Burns KH, et al. Primary effusion lymphoma presenting as a cutaneous intravascular lymphoma. J Cutan Pathol. 2014;41:928-935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Friis A, Akerlund B, Christensson B, et al. Epstein Barr virus DNA analysis in blood predicts disease progression in a rare case of plasmablastic lymphoma with effusion. Infect Agent Cancer. 2013;8:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Cesarman E, Nador RG, Aozasa K, et al. Kaposi's sarcoma–associated herpesvirus in non-AIDS related lymphomas occurring in body cavities. Am J Pathol. 1996;149:53-57. [PMC free article] [PubMed] [Google Scholar]
- 67. Kim Y, Park CJ, Roh J, et al. Current concepts in primary effusion lymphoma and other effusion-based lymphomas. Korean J Pathol. 2014;48:81-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wu W, Youm W, Rezk SA, et al. Human herpesvirus 8–unrelated primary effusion lymphoma-like lymphoma: report of a rare case and review of 54 cases in the literature. Am J Clin Pathol. 2013;140:258-273. [DOI] [PubMed] [Google Scholar]
- 69. Fukayama M, Hayashi Y, Ooba T, et al. Pyothorax-associated lymphoma: development of Epstein-Barr virus-associated lymphoma within the inflammatory cavity. Pathol Int. 1995;45:825-831. [DOI] [PubMed] [Google Scholar]
- 70. Copie-Bergman C, Niedobitek G, Mangham DC, et al. Epstein-Barr virus in B-cell lymphomas associated with chronic suppurative inflammation. J Pathol. 1997;183:287-292. [DOI] [PubMed] [Google Scholar]
- 71. Alexanian S, Said J, Lones M, et al. KSHV/HHV8-negative effusion-based lymphoma, a distinct entity associated with fluid overload states. Am J Surg Pathol. 2013;37:241-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ascoli V, Lo Coco F, Artini M, et al. Primary effusion Burkitt's lymphoma with t(8;22) in a patient with hepatitis C virus–related cirrhosis. Hum Pathol. 1997;28:101-104. [DOI] [PubMed] [Google Scholar]
- 73. Franzin F, Efremov DG, Pozzato G, et al. Clonal B-cell expansions in peripheral blood of HCV-infected patients. Br J Haematol. 1995;90:548-552. [DOI] [PubMed] [Google Scholar]
- 74. Delecluse HJ, Anagnostopoulos I, Dallenbach F, et al. Plasmablastic lymphomas of the oral cavity: a new entity associated with the human immunodeficiency virus infection. Blood. 1997;89:1413-1420. [PubMed] [Google Scholar]
- 75. Morscio J, Dierickx D, Nijs J, et al. Clinicopathologic comparison of plasmablastic lymphoma in HIV-positive, immunocompetent, and posttransplant patients: single-center series of 25 cases and meta-analysis of 277 reported cases. Am J Surg Pathol. 2014;38:875-886. [DOI] [PubMed] [Google Scholar]
- 76. Montes-Moreno S, Montalban C, Piris MA. Large B-cell lymphomas with plasmablastic differentiation: a biological and therapeutic challenge. Leuk Lymphoma. 2012;53:185-194. [DOI] [PubMed] [Google Scholar]
- 77. Castillo JJ, Bibas M, Miranda RN. The biology and treatment of plasmablastic lymphoma. Blood. 2015;125:2323-2330. [DOI] [PubMed] [Google Scholar]
- 78. Zimmermann H, Oschlies I, Fink S, et al. Plasmablastic posttransplant lymphoma: cytogenetic aberrations and lack of Epstein-Barr virus association linked with poor outcome in the prospective German posttransplant lymphoproliferative disorder registry. Transplantation. 2012;93:543-550. [DOI] [PubMed] [Google Scholar]
- 79. Colomo L, Loong F, Rives S, et al. Diffuse large B-cell lymphomas with plasmablastic differentiation represent a heterogeneous group of disease entities. Am J Surg Pathol. 2004;28:736-747. [DOI] [PubMed] [Google Scholar]
- 80. Lorsbach RB, Hsi ED, Dogan A, et al. Plasma cell myeloma and related neoplasms. Am J Clin Pathol. 2011;136:168-182. [DOI] [PubMed] [Google Scholar]
- 81. Moller HE, Preiss BS, Pedersen P, et al. Clinicopathological features of plasmablastic multiple myeloma: a population-based cohort. APMIS. 2015;123:652-658. [DOI] [PubMed] [Google Scholar]
- 82. Taddesse-Heath L, Meloni-Ehrig A, Scheerle J, et al. Plasmablastic lymphoma with MYC translocation: evidence for a common pathway in the generation of plasmablastic features. Mod Pathol. 2010;23:991-999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Engels EA, Clarke CA, Pfeiffer RM, et al. Plasma cell neoplasms in US solid organ transplant recipients. Am J Transplant. 2013;13:1523-1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Trappe R, Zimmermann H, Fink S, et al. Plasmacytoma-like post-transplant lymphoproliferative disorder, a rare subtype of monomorphic B-cell post-transplant lymphoproliferation, is associated with a favorable outcome in localized as well as in advanced disease: a prospective analysis of 8 cases. Haematologica. 2011;96:1067-1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Caillard S, Agodoa LY, Bohen EM, et al. Myeloma, Hodgkin disease, and lymphoid leukemia after renal transplantation: characteristics, risk factors and prognosis. Transplantation. 2006;81:888-895. [DOI] [PubMed] [Google Scholar]
- 86. Sanchez Quintana A, Rull PR, Atienza JB, et al. Renal transplant in plasma cell dyscrasias with lenalidomide treatment after autologous stem cell transplantation. Nephrology (Carlton). 2013;18:641-643. [DOI] [PubMed] [Google Scholar]
- 87. Trappe RU, Choquet S, Dierickx D, et al. International prognostic index, type of transplant and response to rituximab are key parameters to tailor treatment in adults with CD20-positive B cell PTLD: clues from the PTLD-1 trial. Am J Transplant. 2015;15:1091-1100. [DOI] [PubMed] [Google Scholar]
- 88. Cooper AR, Burack WR, Allerton JP. A case of Kaposi sarcoma–associated herpesvirus/human herpesvirus 8-unrelated but Epstein-Barr virus-positive primary effusion lymphoma-like lymphoma in the setting of human immunodeficiency virus and hepatitis C virus infection. Leuk Lymphoma. 2010;51:2303-2305. [DOI] [PubMed] [Google Scholar]
- 89. Loghavi S, Alayed K, Aladily TN, et al. Stage, age, and EBV status impact outcomes of plasmablastic lymphoma patients: a clinicopathologic analysis of 61 patients. J Hematol Oncol. 2015;8:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Romero M, Gonzalez-Fontal GR, Saavedra C, et al. Primary CNS plasmablastic lymphoma in an HIV/EBV negative patient: a case report. Diagn Cytopathol. 2016;44:61-65. [DOI] [PubMed] [Google Scholar]
- 91. Valera A, Balague O, Colomo L, et al. IG/MYC rearrangements are the main cytogenetic alteration in plasmablastic lymphomas. Am J Surg Pathol. 2010;34:1686-1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Qing X, Sun N, Chang E, et al. Plasmablastic lymphoma may occur as a high-grade transformation from plasmacytoma. Exp Mol Pathol. 2011;90:85-90. [DOI] [PubMed] [Google Scholar]
- 93. Qing X, Sun N, French SW, et al. Oral and extraoral plasmablastic lymphoma: similarities and differences in clinicopathologic characteristics. Am J Clin Pathol. 2011;135:977-979. [DOI] [PubMed] [Google Scholar]
- 94. Mathews MS, Bota DA, Kim RC, et al. Primary leptomeningeal plasmablastic lymphoma. J Neurooncol. 2011;104:835-838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Dorwal P, Sachdev R, Mishra P, et al. Extraoral plasmablastic lymphoma detected using ascitic fluid cytology and flow cytometry: a case report with a review of the literature. Acta Cytol. 2014;58:309-317. [DOI] [PubMed] [Google Scholar]
- 96. Urrego PA, Smethurst M, Fowkes M, et al. Primary CNS plasmablastic lymphoma: report of a case with CSF cytology, flow cytometry, radiology, histological correlation, and review of the literature. Diagn Cytopathol. 2011;39:616-620. [DOI] [PubMed] [Google Scholar]
- 97. Ambrosio MR, De Falco G, Gozzetti A, et al. Plasmablastic transformation of a pre-existing plasmacytoma: a possible role for reactivation of Epstein Barr virus infection. Haematologica. 2014;99:e235-e237. [DOI] [PMC free article] [PubMed] [Google Scholar]