

Current antiepileptic drugs cannot prevent the onset of epilepsy or its progression. Pauletti et al. show that antioxidant drugs in medical use administered early post-injury block spontaneous seizure progression and neurodegeneration in an epilepsy model, and rescue cognitive deficits. This intervention may be considered for patients exposed to potential epileptogenic insults.
Keywords: cognitive deficit, neuronal cell loss, HMGB1, spontaneous seizures, neuroinflammation
Abstract
Epilepsy therapy is based on antiseizure drugs that treat the symptom, seizures, rather than the disease and are ineffective in up to 30% of patients. There are no treatments for modifying the disease—preventing seizure onset, reducing severity or improving prognosis. Among the potential molecular targets for attaining these unmet therapeutic needs, we focused on oxidative stress since it is a pathophysiological process commonly occurring in experimental epileptogenesis and observed in human epilepsy. Using a rat model of acquired epilepsy induced by electrical status epilepticus, we show that oxidative stress occurs in both neurons and astrocytes during epileptogenesis, as assessed by measuring biochemical and histological markers. This evidence was validated in the hippocampus of humans who died following status epilepticus. Oxidative stress was reduced in animals undergoing epileptogenesis by a transient treatment with N-acetylcysteine and sulforaphane, which act to increase glutathione levels through complementary mechanisms. These antioxidant drugs are already used in humans for other therapeutic indications. This drug combination transiently administered for 2 weeks during epileptogenesis inhibited oxidative stress more efficiently than either drug alone. The drug combination significantly delayed the onset of epilepsy, blocked disease progression between 2 and 5 months post-status epilepticus and drastically reduced the frequency of spontaneous seizures measured at 5 months without modifying the average seizure duration or the incidence of epilepsy in animals. Treatment also decreased hippocampal neuron loss and rescued cognitive deficits. Oxidative stress during epileptogenesis was associated with de novo brain and blood generation of disulfide high mobility group box 1 (HMGB1), a neuroinflammatory molecule implicated in seizure mechanisms. Drug-induced reduction of oxidative stress prevented disulfide HMGB1 generation, thus highlighting a potential novel mechanism contributing to therapeutic effects. Our data show that targeting oxidative stress with clinically used drugs for a limited time window starting early after injury significantly improves long-term disease outcomes. This intervention may be considered for patients exposed to potential epileptogenic insults.
Introduction
Epilepsy is a brain disorder affecting over 50 million people worldwide and is associated with increased mortality, significant co-morbidities, unique stigmatization of affected individuals, and high societal cost. Current antiepileptic drugs mainly provide symptomatic relief from seizures, have multiple adverse effects, and fail to control seizures in up to 30% of people (Kwan et al., 2010). Moreover, there are no drugs that prevent disease onset or its progression. New antiseizure treatments for epilepsy are unlikely to bridge this treatment gap. The next generation of drugs should potentially be able to delay or prevent the onset of epilepsy in susceptible individuals (anti-epileptogenesis) or to halt or reverse its progression and/or improve the neuropathology and the associated comorbidities (disease-modifying) (Varvel et al., 2015). To develop such drugs, we need to understand the pathological processes occurring during epileptogenesis in the brain of people exposed to injuries, or with an established diagnosis of epilepsy. Epileptogenesis is a complex multifactorial process, including both the development of an epilepsy condition and its progression after the condition is established (Pitkanen and Engel, 2014). It is characterized by various modifications in the brain, including neuroinflammation and oxidative stress. These two phenomena are rapidly and persistently induced in the injured brain; moreover, they are functionally interconnected and reinforce each other (Hsieh and Yang, 2013; Rowley and Patel, 2013; Vezzani et al., 2015). Notably, neuroinflammation occurs in human epilepsy and peripheral markers of oxidative stress are increased in people with epilepsy (Aronica and Crino, 2011; Vezzani et al., 2011a; Rowley and Patel, 2013; Cardenas-Rodriguez et al., 2014; Puttachary et al., 2015). They both contribute to neuropathology and behavioural deficits and play a role in determining seizure threshold in animal models (Rowley and Patel, 2013; Vezzani et al., 2013; Pearson et al., 2015).
A potential critical point of intersection between oxidative stress and neuroinflammation is the generation of the disulfide isoform of high mobility group box 1 (HMGB1), a protein crucially involved in the molecular cascade that contributes to seizure mechanisms (Vezzani et al., 2011b). HMGB1 is a ubiquitous non-histone chromatin-binding protein that regulates gene transcription under physiological conditions. Following cell injury or pathophysiological cell activation, HMGB1 is hyper-acetylated at two nuclear localization sequences, a step required for its translocation to the cytoplasm from where it can be released extracellularly upon inflammasome activation (Yang et al., 2012; Lu et al., 2013). Extracellular HMGB1 can be partially oxidized by formation of the disulfide bond between C23 and C45 (i.e. disulfide HMGB1); this isoform has pro-inflammatory, neuromodulatory and ictogenic activities by activating toll-like receptor 4 (TLR4) (Venereau et al., 2012; Yang et al., 2012; Balosso et al., 2014). As reactive oxygen species (ROS) promote the stabilization of HMGB1 in its disulfide isoform (Yang et al., 2012), there is a vicious cycle that links ROS production, the generation of the pathologic HMGB1 isoform and neuroinflammation, thus representing a potential mechanism of epileptogenesis.
Previous approaches to targeting oxidative stress with small molecules in animal models of epilepsy have met with some success in reducing neuronal damage following prolonged seizures but have notably failed to show a convincing effect on the development of epilepsy (Rong et al., 1999; Liang et al., 2000; Barros et al., 2007; Rowley and Patel, 2013; Kovac et al., 2014; Pearson et al., 2015). We hypothesized that this is because the treatments given are either too short-lived or do not have a sufficiently rapid effect. We have overcome this hurdle by using a combination of two antioxidant drugs with complementary mechanism of action, N-acetylcysteine (NAC) acting as an acute antioxidant (Sun, 2010) and sulforaphane (SFN) increasing the longer-term endogenous antioxidant system (Houghton et al., 2013). We show that this combination reduces oxidative stress in brain and blood during epileptogenesis in a rat model of acquired epilepsy induced by status epilepticus, and significantly improves pathological outcomes by providing blockade of spontaneous seizure progression, reduction of cell loss and rescue of comorbidities. Notably, although treatment was transiently given during epileptogenesis starting early after status epilepticus onset, it mediates long-term therapeutic effects.
Moroever, we provide the first evidence that links seizure-induced oxidative stress to the brain generation of the ictogenic and pathologic neuroinflammatory molecule disulfide HMGB1, and therapeutic outcomes of antioxidant drugs are associated with abrogation of this molecule in brain and blood. HMGB1 may therefore contribute to the pathogenic consequences of oxidative stress, and could act as a biomarker to determine efficacy of treatment.
Since we show that oxidative stress occurs in the human brain after status epilepticus and in chronic epilepsy, antioxidant interventions may represent a potential therapeutic strategy for improving disease prognosis in patients exposed to epileptogenic injuries.
Materials and methods
Animals
Adult male Sprague-Dawley rats (225–250 g; Charles-River) were housed at constant temperature (23 ± 1°C) and relative humidity (60 ± 5%) with free access to food and water and a fixed 12-h light/dark cycle. In compliance with the ARRIVE guidelines, procedures involving animals and their care were conducted in conformity with the institutional guidelines that are in compliance with national (D.L. n.26, G.U. March 4, 2014) and international laws and policies (EEC Council Directive 86/609, OJ L 358, 1, December 12, 1987; Guide for the Care and Use of Laboratory Animals, U.S. National Research Council, 1996).
Electrical status epilepticus
Rats were implanted under 1.5% isoflurane anaesthesia with two bipolar Teflon™-insulated stainless-steel depth electrodes placed bilaterally into the temporal pole of the hippocampus (from bregma, mm: AP −4.7; L ± 5.0; −5.0 below dura) (Paxinos and Watson, 2005). Two screw electrodes were positioned over the nasal sinus and the cerebellum, and used as ground and reference electrodes, respectively. Electrodes were connected to a multipin socket and secured to the skull by acrylic dental cement. After surgical procedures, rats were treated locally with cicatrene powder (neomycin; bacitracin; glycine; l-cysteine; dl-threonin) and injected with ampicillin (100 mg/kg, subcutaneous) for 4 days to prevent infections. Rats were allowed to recover from surgery in their home cage for 10 days. Before electrical stimulation, EEG baseline hippocampal activity was recorded in freely-moving rats for 24 h. Then, rats were unilaterally stimulated (50 Hz, 400 μA peak-to-peak, 1 ms biphasic square waves in 10 s trains delivered every 11 s) in the CA3 region of the ventral hippocampus for 60–90 min to induce status epilepticus according to a well-established protocol (De Simoni et al., 2000; Noé et al., 2008). EEG was recorded in each rat every 10 min epoch for 1 min in the absence of electrical stimulation, i.e. the ‘stimulus-off’ period. All rats used for subsequent analysis showed an EEG pattern of uninterrupted bilateral spikes in the hippocampi during the ‘stimulus-off’ period, starting between the first and the fourth epoch of stimulation onwards. These criteria selected rats developing status epilepticus that remitted spontaneously within 24 h from the initial stimulation then leading to subsequent epilepsy development (De Simoni et al., 2000; Noé et al., 2008). Status epilepticus was defined by the appearance of continuous spike activity with a frequency >1.0 Hz intermixed with high amplitude and frequency discharges lasting for at least 5 s, with a frequency of ≥8 Hz. Spikes were defined as sharp waves with amplitude at least 2.5-fold higher than baseline and duration lower than 100 ms, or as a spike-and-wave with duration lower than 200 ms (Pitkanen et al., 2005). The end of status epilepticus was defined by the occurrence of interspike intervals longer than 1 s. No pharmacological intervention was done to stop status epilepticus since no mortality is observed in this model. Status epilepticus was evaluated by EEG analysis and its total duration and the number of spikes were quantified during the first 24 h using Clampfit 9.0 program (Axon Instruments). Power spectral density (PSD) distribution of four frequency bands (delta: 1–4 Hz; theta: 4–8 Hz; alpha: 8–13 Hz; beta-gamma: 13–40 Hz) was calculated during 9 h segmented in temporal windows of 1 h each. Fast Fourier transforms (FFTs) were computed by 50% overlapping sliding windows (1024 data-point each) with Hanning windowing function. EEG data were normalized by dividing the EEG power density at each frequency with the EEG power density averaged across all frequencies (Pitkanen et al., 2005).
Spontaneous seizures detection and quantification
Rats exposed to status epilepticus were continuously video-EEG recorded (24 h/day) from status epilepticus induction until the first two spontaneous seizures occurred, at least 48 h apart from status epilepticus induction (epilepsy onset). All EEG seizures were associated with generalized motor seizures (forelimb clonus with or without rearing and/or falling) (Gorter et al., 2001; Noé et al., 2008). After epilepsy onset in each rat, video-EEG monitoring was discontinued and resumed at 2 and 5 months post-status epilepticus to determine spontaneous recurrent seizure (SRS) frequency by continuous EEG monitoring for 2 weeks (24 h/7 days). Spontaneous seizures were discrete EEG ictal episodes lasting on average 60 s, characterized by high-frequency and high-voltage synchronous spike activity and/or multi-spike complexes (Noé et al., 2008). EEG was recorded using the TWin EEG Recording System connected with a Comet AS-40 32/8 Amplifier (sampling rate 400 Hz, high-pass filter 0.3 Hz, low-pass filter 70 Hz; Grass-Telefactor). Digitized EEG data were processed using the TWin record and review software. EEG was visually inspected for seizure detection and quantification was performed by two independent expert operators blinded to the treatments. If there was lack of concordance then a third expert operator was consulted.
In vivo study design and drug treatment schedule
We investigated whether oxidative stress generated in the hippocampus during epileptogenesis in the electrical status epilepticus model was reduced by the antioxidant drugs NAC and SFN. Electrode-implanted rats not exposed to status epilepticus were used as controls (Sham); these rats were sacrificed at the corresponding time points of the respective status epilepticus-exposed rats after 10-day recovery period following surgery. Experimental rats were exposed to status epilepticus and sacrificed after 4 days (n = 9) or 14 days (n = 5) for HPLC analysis of reduced (GSH), oxidized (GSSG) glutathione and glutathionylated proteins (GS-Pro) and compared to sham rats (n = 15) (Figs 1A, 2A and 5A) (Pastore et al., 1998; Liang and Patel, 2016; Supplementary material). A different group of rats was sacrificed 4 days post-status epilepticus (n = 5; sham = 4) for immunohistochemical analysis of markers of oxidative stress, such as inducible nitric oxide (iNOS), the cysteine transporter (Xct), the transcriptional nuclear factor (erythroid-derived 2)-like 2 (Nrf2) (Fig. 1B) and HMGB1 (Fig. 5B) (Supplementary material).
Figure 1.
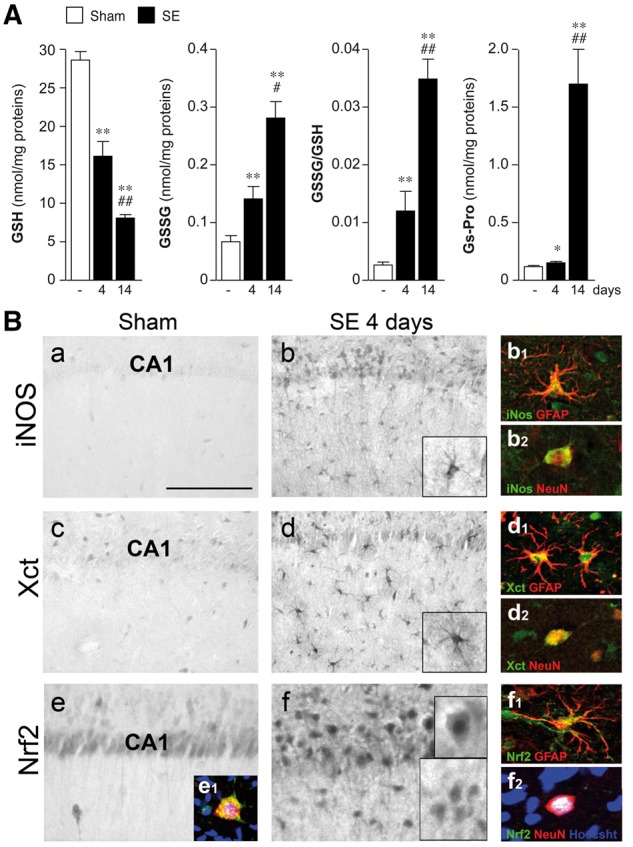
Generation of oxidative stress in hippocampal tissue of rats exposed to electrical status epilepticus. (A) HPLC analysis of reduced (GSH) and oxidized (GSSG) glutathione levels, and their ratio, and the level of glutathionylated proteins (GS-Pro) in the rat hippocampus at Days 4 (n = 9) and 14 (n = 5) after status epilepticus (SE) onset compared to corresponding baseline levels in sham rats (electrode-implanted but not stimulated, n = 15). Sham values are pooled (since they did not differ) from rats sacrificed at 4 days (n = 10) or 14 days (n = 5) after 10-day recovery period following surgery to match the same time points of the corresponding status epilepticus-exposed rats. Data are mean ± SEM. *P < 0.05; **P < 0.01 versus sham by Kruskal-Wallis followed by Dunn’s post hoc test; #P < 0.05; ##P < 0.01 versus Day 4 by Mann-Whitney test. (B) Representative immunohistochemical micrographs of the CA1 region depicting the expression of iNOS, the Xct and Nrf2 in sham (a, c and e) and 4 days post-status epilepticus (b, d and f) (n = 4–5). (b, d and f) Show the increase in the respective marker expression in GFAP-positive activated astrocytes (b1, d1 and f1) and in neurons (b2, d2 and f2). Activated astrocytes are defined by hypertrophic cell body with thick processes. Nrf2 expression is increased in neuronal nuclei (f2 versus e1) indicating increased transcriptional activation of detoxifying enzymes. Scale bars: B(a–f) = 25 µm; inset in B(b, d and f) = 15 µm; immunofluorescence inset = 10 µm.
Figure 2.

Effect of drug combination versus single drug alone on oxidative stress markers. (A) GSH and GSSG levels, and their ratio, and GS-Pro levels in the hippocampus of rats exposed to status epilepticus versus sham rats as assessed by HPLC analysis. Status epilepticus-exposed rats (n = 5 each group) received either vehicle combination, or NAC (500 mg/kg, i.p., twice daily for 7 days) or SFN (5 mg/kg, i.p., daily for 14 days) or their combination (NAC + SFN for 7 days followed by SFN alone for additional 7 days). Controls were sham rats injected with vehicle (n = 5). The drug combination reduced oxidative stress to a greater extent than each drug given alone. Data are mean ± SEM. *P < 0.01 versus Sham; °P < 0.01 versus status epilepticus + NAC + SFN; #P < 0.01 versus status epilepticus + vehicle by Kruskal-Wallis followed by Dunn’s post hoc test. (B) Bargrams depict low Mg2+-induced mitochondrial membrane potential changes of neocortical cell cultures (Supplementary Fig. 3B–E). Preincubation of neurons with SFN (5 µM, 24 h) decreased the rate of depolarization at all time points. Addition of NAC (10 mM, acutely) evoked hyperpolarization in the mitochondrial membrane potential. Pretreatment with SFN (5 µM, 24 h) with addition of NAC (10 mM, acutely) showed similar effect as NAC alone, with significantly higher hyperpolarization after 25 and 30 min. Data are mean ± SEM. *P < 0.01 versus artificial CSF (aCSF); #P < 0.01 versus low Mg2+; °P < 0.01 versus NAC + SFN by Kruskal-Wallis followed by Dunn’s post hoc test. SE = status epilepticus.
Figure 5.

Effect of antioxidant treatment on HMGB1 isoforms in brain and blood. Histogram in A shows the increase in GSSG/GSH ratio during epileptogenesis (4 days post-status epilepticus, n = 9) and its reduction to baseline (Sham + vehicle, n = 10) by 4 day treatment with NAC (5 mg/kg, i.p. twice daily) + SFN (5 mg/kg, i.p., once daily) (n = 11) (treatment was started 1 h post-status epilepticus). A similar antioxidant effect was observed when treatment was started 24 h after status epilepticus (Supplementary Table 1). Data are mean ± SEM. **P < 0.01 versus Sham + vehicle; ##P < 0.01 versus status epilepticus + vehicle by Kruskal-Wallis followed by Dunn’s post hoc test. (B) Representative photomicrographs of CA1-CA3 stratum lacunosum-moleculare of hippocampi from control rats (Sham) or status epilepticus-exposed rats treated with vehicle or NAC + SFN (n = 4–5; treatment protocol as in A). (a) HMGB1 immunoreactivity is localized in cell nuclei in sham rats; (b) HMGB1 immunoreactivity is increased in cytoplasm of glial cells (arrows, b) following status epilepticus; (c) reduced cytoplasmatic staining in status epilepticus-exposed rats treated with NAC + SFN denoting inhibiton of HMGB1 nuclear-to-cytoplasm translocation. Second row: HMGB1 signal (green) in OX-42-positive microglia (red), GFAP-positive astrocytes (red) and EBA-positive endothelial cells (red); co-localization signal is depicted in yellow (merge). White arrows represent cytoplasmic staining. Hoechst-positive nuclei are shown in blue. Scale bars: first row = 25 µm; second row = 12 µm. Histograms in C and D show levels (mean ± SEM, n = 9–11) of HMGB1 (acetylated, disulfide and reduced) isoforms in brain tissue (hippocampus, C) and corresponding blood (D) of rats during epileptogenesis (i.e. 4 days post-status epilepticus), and the effect of treatment. NAC + SFN abolished the increase in acetylated (releasable) and disulfide (pathogenic) HMGB1 isoforms in brain (C) and in blood (D). Total and reduced HMGB1 were also decreased by the treatment. **P < 0.01 versus Sham + vehicle; ##P < 0.01 versus status epilepticus + vehicle by Kruskal-Wallis followed by Dunn’s post hoc test. SE = status epilepticus.
In parallel experiments, status epilepticus-exposed rats were treated with NAC (Sigma-Aldrich; 500 mg/kg dissolved in H2O, pH 7.4) and SFN (LKT Laboratories; 5 mg/kg dissolved in 0.1% DMSO in buffered saline, pH 7.4) intraperitoneally (i.p.) either alone or in combination, or their vehicles. For determining the effect of each drug alone, or their combination, on oxidative stress (Fig. 2A), a cohort of rats (n = 5 rats in each group) was treated with either NAC alone (twice daily 6 h apart) for 7 days (protocol in Supplementary Fig. 1A) or SFN alone (5 mg/kg, i.p., once daily) for 14 days (Supplementary Fig. 1A), or NAC + SFN combination for 7 days (same schedule as each drug given alone) followed by SFN alone for an additional 7 days (Supplementary Fig. 1A). In each treatment schedule, the first drug dose was injected 1 h after status epilepticus onset. In the combination protocol, SFN was injected 1 h after the first NAC administration. Rats were sacrificed at the end of each treatment for HPLC analysis of glutathione forms.
For determining if oxidative stress was associated with the generation of disulfide HMGB1 (Fig. 5 and Supplementary Fig. 1B), a group of status epilepticus-exposed rats was treated with NAC + SFN (same schedule as each drug given alone) or their vehicles for 4 days (n = 9–11 each group), and compared to time-matched sham rats (n = 9). Both glutathione forms and HMGB1 levels were measured in the hippocampus; HMGB1 was also measured in corresponding venous blood in each animal (Antoine et al., 2009, 2012; Palmblad et al., 2015) (Supplementary material). Additionally, to verify whether oxidative stress promotes HMGB1 translocation, a rat cohort (n = 5) was treated with the drug combination (NAC + SFN) for 4 days and their brain analysed by immnohistochemistry (Fig. 5B).
All rats used for immunohistochemical or biochemical analysis in cross-sectional studies were EEG recorded from status epilepticus induction until 4 days post-status epilepticus, and no SRS were observed in the various experimental groups.
To assess NAC + SFN therapeutic effects on spontaneous seizures, cognitive deficits, and cell loss, the drugs (same dose of each drug given alone) were co-administered for 7 days post-status epilepticus followed by SFN administered alone for an additional 7 days (Figs 3, 4 and Supplementary Fig. 1C). Rats were randomly assigned 1 h after status epilepticus onset to either drug (n = 9) or vehicle groups (n = 9), and longitudinally followed by video-EEG recording as described above. At the end of EEG recording, rats underwent the T-maze test, then they were sacrificed and brains were collected for subsequent histological analysis.
Figure 3.

Therapeutic effects of antioxidant drug combination in status epilepticus-exposed rats. (A) A typical EEG recorded spontaneous seizure in a chronically epileptic rat injected with vehicle. LHP and RHP are left and right hippocampus, respectively. (B) Kaplan-Meier survival curve reporting the per cent of rats developing the first spontaneous seizure as a function of days post-status epilepticus: status epilepticus + vehicle, 100% rats developed the first spontaneous seizure by Day 12 versus status epilepticus + NAC + SFN by Day 16 (P < 0.01 by Log-rank test, n = 9 each group). (C) The number of SRS during 2-week EEG recording at 2 and 5 months post-status epilepticus in vehicle- and drug-treated rats. The data show that seizure progression was prevented by the treatment resulting in 70% SRS reduction at 5 months versus vehicle injected status epilepticus-exposed rats (**P < 0.01 versus 2 months; #P < 0.05 versus status epilepticus + vehicle by Mann-Whitney test). (D) Depicts the average cumulative duration of SRS recorded by EEG for 2 weeks at 5 months post-status epilepticus; this parameter was reduced by treatment versus vehicle (*P < 0.05 versus status epilepticus + vehicle by Mann-Whitney test). (E) The rat performance in the T-maze showing the average per cent of correct alternation in the each arm in the various experimental groups. The drug combination rescued the behavioural deficit in the epileptic rats. Data are mean ± SEM. **P < 0.01 versus Sham (n = 9); ##P < 0.01 versus status epilepticus + vehicle by Kruskal-Wallis followed by Dunn’s post hoc test. SE = status epilepticus.
Figure 4.

Histological analysis and quantification of cell loss in the hippocampus of status epilepticus-exposed rats treated with the antioxidant drugs versus vehicles. Panels depict representative microphotographs of Nissl-stained neurons in CA1 pyramidal layer (A) and in the hilus (B) and calretinin-stained hilar interneurons (C) in control (Sham + vehicle) and epileptic rats treated with vehicle (status epilepticus + vehicle) or the antioxidant drugs (status epilepticus + NAC + SFN; same rats of Fig. 3). Bargrams (mean ± SEM, n = 9 each group) report the correspondent quantification of cell loss showing the neuroprotective effect of the treatment. Scale bars: A and B = 100 µm; C = 50 µm. GC = granule cell layer; h = hilus; CA1 pyramidal layer. **P < 0.01 versus Sham + vehicle; #P < 0.05, ##P < 0.01 versus status epilepticus + vehicle by Kruskall-Wallis followed by Dunn’s post hoc test. SE = status epilepticus.
Fluoro-Jade® analysis of cell loss was done to determine the effect of the first day drug treatment on status epilepticus-induced acute injury (Ravizza et al., 2008) (Supplementary material). An additional group of rats (n = 6) was injected 1 h after status epilepticus onset with NAC (500 mg/kg, i.p.) followed by a second NAC dose 6 h apart. One hour after the first NAC dose, rats were injected with SFN (5 mg/kg; i.p.), then they were killed 24 h post-status epilepticus onset (protocol in Supplementary Fig. 1B). Rats exposed to status epilepticus and injected with corresponding vehicles and sham rats (n = 5 each group) were used as controls.
Finally, we determined whether a delayed treatment schedule also reduced hippocampal oxidative stress and cell loss in rats injected with NAC + SFN until Day 4 and starting treatment 24 h post-status epilepticus (protocol in Supplementary Fig. 1B). For measuring GSH and GSSG, we prepared new cohorts of NAC + SFN treated rats (24 h post-status epilepticus until Day 4; n = 8–10) and time-matched status epilepticus + vehicle and sham rats (n = 9–10) (Supplementary Table 1) and compared them with NAC + SFN rats depicted in Fig. 5A (1 h post-status epilepticus). For measuring hippocampal cell loss, two new rat cohorts were treated with NAC + SFN starting either 1 h or 24 h (n = 4–5 each group) post-status epilepticus and compared with time-matched status epilepticus + vehicle and sham rats (n = 5 each group) (Supplementary Table 2).
Human subjects
The cases included in this study were obtained from the archives of the Departments of Neuropathology of the Academic Medical Center (AMC, Amsterdam, The Netherlands) and the VU University medical center (VUmc, Amsterdam, The Netherlands). A total of five hippocampal specimens (removed from patients undergoing surgery for drug-resistant epilepsy) and 11 hippocampal specimens obtained at autopsy from patients who died after status epilepticus were examined. Control material was obtained at autopsy from age-matched control patients, without a history of seizures or other neurological diseases. All autopsies were performed within 24 h after death. Tissue was obtained and used in accordance with the Declaration of Helsinki and the AMC Research Code provided by the Medical Ethics Committee.
All cases were reviewed independently by two neuropathologists and the classification of hippocampal sclerosis was based on analysis of microscopic examination as described by the International League Against Epilepsy (Blumcke et al., 2013). The clinical features of the cases analysed is reported in Supplementary Table 3.
Statistical analysis
Sample size was a priori determined based on previous experience with the rat epilepsy model as well as following the principles of the three Rs (Replacement, Reduction and Refinement; https://www.nc3rs.org.uk/the-3rs). Endpoints (outcome measures) and statistical tests were prospectively selected. A simple random allocation was applied to assign a subject (rat) to a particular experimental group. All efforts were made to minimize the number of animals used and their suffering. Data acquisition and analysis were done blindly.
Statistical analysis was performed by GraphPad Prism 6 (GraphPad Software, USA) for Windows using absolute values. Data are presented as mean ± standard error of the mean (SEM) (n = number of individual samples). Mann–Whitney test for two independent groups and Kruskal-Wallis followed by Dunn’s post hoc test for more than two independent groups were used for statistical analysis of data. In the longitudinal study, changes in time to seizure onset were analysed by Log-rank (Mantel Cox) test. The temporal distribution of spikes during status epilepticus was analysed by two-way ANOVA followed by Bonferroni’s multiple comparisons test. Differences were considered significant with a P < 0.05.
Results
Assessment of oxidative stress during epileptogenesis
We studied the generation of oxidative stress during epileptogenesis in status epilepticus-exposed rats by measuring the hippocampal levels of GSSG and GSH and their ratio (GSSG/GSH), which is an established indicator of ROS production (Liang and Patel, 2006; Ryan et al., 2014). We choose two time points post-status epilepticus reflecting early epileptogenesis before the onset of epilepsy (4 days) and shortly after disease onset (14 days, the time at which the treatment was stopped). In accordance with other rodent models of epileptogenesis, the levels of both GSH and GSSG significantly changed (P < 0.01) between 4 days (n = 9) and 14 days post-status epilepticus (n = 5) resulting in a progressive 3- to 14-fold increase in GSSG/GSH ratio above control values (in sham rats not exposed to status epilepticus, n = 15) (Fig. 1A). A concomitant increase in glutathionylated proteins (Gs-Pro), another indicator of oxidative stress, was measured in the same hippocampal tissue (Fig. 1A).
To determine which cell types underwent oxidative stress, immunohistochemical analysis was done in the hippocampus in a group of rats killed 4 days post-status epilepticus. Figure 1B depicts the cellular expression of molecular markers of oxidative stress: iNOS [Fig. 1B(a,b)], the cystine transporter [Xct; Fig. 1B(c,d)] and the transcriptional factor Nrf2 [Fig. 1B(e,f)] in the representative CA1 hippocampal region. These molecules were induced in activated GFAP-positive astrocytes [Fig. 1B(b1,d1,f1)] as well as in NeuN-positive neurons [Fig. 1B(b2,d2,f2)] but they were not detected in OX-42-positive microglia (not shown). Nrf2 staining was increased in neuronal nuclei [Fig. 1B (f2 versus e1)] reflecting transcriptional activation of antioxidant genes in response to ROS production (Abbas et al., 2011; Mazzuferi et al., 2013; Steele et al., 2013). These changes were similarly observed in the various CA3/CA4 hippocampal subfields, in the hilus, subiculum/parasubiculum and entorhinal cortex (not shown).
Antioxidant drug combination versus single treatment
To design the optimal treatment protocol for the therapeutic study, we tested whether a combination of NAC and SFN, two antioxidant drugs with complementary mechanism of action, was more effective in reducing oxidative stress than each drug given alone. We designed a treatment schedule lasting 14 days to encompass the epileptogenesis phase between the acute injury (status epilepticus) and the aftermath of disease onset. Based on the available pharmacokinetic and pharmacodynamic information (Holdiness, 1991; Farr et al., 2003; Hu et al., 2004; Arakawa and Ito, 2007; Harvey et al., 2008; Wang et al., 2011), we treated different cohorts of rats with either NAC (500 mg/kg, i.p., twice a day for 7 days) or SFN alone (5 mg/kg, i.p., once daily for 14 days), or their combination (NAC + SFN injected for 7 days followed by SFN injected alone for an additional 7 days). Treatment began 1 h after the onset of status epilepticus (protocol in Supplementary Fig. 1A). NAC, the precursor of GSH, which represents the major non-enzymatic antioxidant pathway of the body, was administered to rats for 1 week to attain a rapid scavenging action of ROS during status epilepticus (Sun, 2010). SFN, an activator of Nrf2-dependent transcription of detoxification enzymes, was administered for one additional week after NAC withdrawal to provide a sustained antioxidant effect (Houghton et al., 2013). Figure 2A shows that GSH and GSSG were modified by status epilepticus resulting in a significant increase in GSSG/GSH ratio (P < 0.01 versus sham, n = 5 each group). Each drug alone increased GSH and reduced GSSG compared to status epilepticus-exposed rats receiving vehicles (P < 0.01, n = 5 each group). However, the combination of NAC and SFN showed a greater effect versus single drugs (P < 0.01) in normalizing both GSH and GSSG levels and their ratio. Similarly, Gs-Pro level returned to sham value after the drug combination whereas it was still significantly elevated in rats treated with each drug alone.
We further tested whether the drug combination was more effective than the individual drugs in preventing mitochondrial dysfunction using primary neuronal cortical cultures where epileptiform activity was induced by removing extracellular Mg2+(Supplementary Fig. 3 and Supplementary material) (Haynes, 1999). We measured the changes in the mitochondrial inner membrane potential evoked by epileptiform activity and the effects of drugs. Figure 2B shows a progressive increase in mitochondrial membrane depolarization, which was positively correlated with the time of exposure to low Mg2+-induced epileptiform activity (from 10 to 30 min; Supplementary Fig. 3B). This effect was significantly reduced by preincubation with SFN or NAC alone; notably, NAC induced membrane hyperpolarization (P < 0.01 versus respective artificial CSF in Fig. 2B and Supplementary Fig. 3D and E). The combination of NAC + SFN was more effective in preventing inner membrane depolarization and increasing membrane hyperpolarization than each drug alone (at 25 and 30 min; P < 0.01 versus each drug alone; Fig. 2B and Supplementary Fig. 3E). These compounds were not found to have an anti-seizure effect in vitro (not shown).
Therapeutic effects of antioxidant drug combination in status epilepticus-exposed rats
The combined treatment protocol was applied starting 1 h after status epilepticus onset for 14 days, then treatment was stopped to determine if therapeutic effects occurred after drug withdrawal (protocol in Supplementary Fig. 1C). The drug combination did not attenuate the overall severity of the acute injury, namely the duration of status epilepticus or the frequency of spikes and their total number, as quantified by continuous 24 h EEG analysis from status epilepticus onset (Supplementary Fig. 4B and C). No difference was detected between the treatment and vehicle groups in the relative power distribution for each frequency band during status epilepticus (Supplementary Fig. 4D). Moreover, the treatment did not affect the acute cell loss induced by status epilepticus assessed by quantification of Fluoro-Jade-positive cells in the stimulated hippocampus 24 h post-status epilepticus (Supplementary Fig. 4E).
Spontaneous seizures onset and their progression
Rats treated for 14 days during epileptogenesis with NAC + SFN showed a significant delay in the time to spontaneous seizure onset (11.7 ± 1.1 days, n = 9, P < 0.01) compared to status epilepticus-exposed rats injected with vehicle (8.6 ± 0.7 days, n = 9). According to the Kaplan-Meier survival curve, 100% rats developed the first spontaneous seizure by Day 12 post-status epilepticus in the status epilepticus + vehicle group versus Day 16 in the drug-treated status epilepticus rats (Fig. 3B). This epilepsy model is characterized by an average 5-fold increase in SRS frequency between 2 months (5.1 ± 1.8 SRS/2 weeks) and 5 months (24.2 ± 7.7 SRS/2 weeks, P < 0.01) post-status epilepticus while the average seizure duration did not change (2 months, 46.8 ± 3.5 s; 5 months, 56.0 ± 4.7 s). Although the number of seizures was not significantly modified by the drugs during the 14 days of treatment (status epilepticus + vehicle, 2.4 ± 0.5; status epilepticus + NAC + SFN, 3.6 ± 0.9, n = 9/each group), or at 2-month follow-up after treatment withdrawal (SRS/2 weeks, status epilepticus + vehicle, 5.1 ± 1.8; status epilepticus + NAC + SFN, 4.9 ± 2.5), the SRS progression between 2 and 5 months was prevented (Fig. 3C, P < 0.01 versus vehicle). Overall, drug-treated rats showed ∼ 70% SRS reduction at 5 months post-status epilepticus compared to vehicle-injected rats (P < 0.05; Fig. 3C) although the proportion of rats with epilepsy did not change. Cumulative SRS duration during 2-week EEG recording at 5 months was significantly reduced by drugs compared to vehicle controls (P < 0.05; Fig. 3D) while the average duration of seizures was not affected by the treatment (status epilepticus + vehicle, 56.0 ± 4.7 s; status epilepticus + NAC + SFN, 49.5 ± 3.3 s, n = 9/each group). Similarly, drugs did not affect average seizure duration at 2-month follow-up (status epilepticus + vehicle, 46.8 ± 3.5 s; status epilepticus + NAC + SFN, 55.1 ± 5.8 s, n = 9/each group).
Cognitive deficits
Rats were tested in the T-maze at the end of EEG recordings (i.e. 5.5 months post-status epilepticus). Status epilepticus-exposed animals treated with vehicles showed an impairment of spatial memory in the T-maze, as shown by failure of correct alternation in the entry arm of the maze (40.5 ± 2.8% correct alternation, n = 9, P < 0.01) compared to sham rats (66.8 ± 3.9% correct alternation, n = 9) (Fig. 3E). The drug combination rescued this behavioural deficit as shown by the correct alternation rate (68.4 ± 5.0%, n = 9) of treated rats, which was similar to sham rats (Fig. 3E). No difference in locomotion was detected among the experimental groups in the open field task (not shown). All rats were confirmed to be epileptic before the T-maze test (Fig. 3C) but they did not show behavioural seizures during the test.
Neurodegeneration in epileptic rats
At the end of behavioural testing, rats were killed for quantitative analysis of cell loss in Nissl-stained forebrain sections (Filibian et al., 2012) of the ventral pole of the stimulated hippocampus (Fig. 4; rats are the same of Fig. 3). Vehicle-injected rats showed significant pyramidal cell loss in CA1 (Fig. 4A) and CA3/CA4 pyramidal cell layers (22 ± 8% decrease in cell number versus Sham, P < 0.05; not shown) and hilar interneurons (Fig. 4B). The antioxidant treatment reduced cell loss by half in CA1 (P < 0.05; Fig. 4A) and virtually prevented the neurodegeneration of hilar interneurons (P < 0.01; Fig. 4B). In particular, hilar calretinin- (but not somatostatin, not shown) positive cells were significantly protected by the drug combination (Fig. 4C). No significant neuroprotection was observed in CA3/CA4, the region of electrical stimulation, or in adjacent entorhinal cortex (not shown).
Effects of the antioxidant drug combination on the redox state of HMGB1
We tested the novel hypothesis that reduction of oxidative stress during epileptogenesis prevents the generation of the pathologic disulfide HMGB1 isoform in the brain. We found that NAC + SFN decreased oxidative stress already 4 days post-status epilepticus (Fig. 5A, n = 9–11; protocol in Supplementary Fig. 1B); this antioxidant effect was similar when treatment was started 1 h (Fig. 5A) or 24 h (Supplementary Table 1) after status epilepticus onset. At 4 days post-status epilepticus, we found immunohistochemical evidence of nucleus-to-cytoplasm translocation of HMGB1 in activated astrocytes, microglia and brain endothelium of status epilepticus-exposed rats [n = 5; Fig. 5B(b versus a)], a phenomenon indicative of HMGB1 extracellular release. Clusters of CA1/CA3 pyramidal neurons with cytoplasmatic HMGB1 staining was observed in two of five status epilepticus-exposed rats (not shown). Such signal was absent in sham control rats (n = 4). Accordingly, LC-MS/MS measurements of HMGB1 isoforms in the hippocampus (Supplementary Fig. 2) showed a 10-fold increase of the acetylated (releasable) HMGB1 isoform 4 days post-status epilepticus compared to sham rats (Fig. 5C; P < 0.01 versus sham; n = 9–11). We also found that the pathologic disulfide HMGB1 isoform, which is absent in control brain tissue, is generated during epileptogenesis (Fig. 5C; P < 0.01 versus sham; n = 9–11). The increase in both acetylated and disulfide HMGB1 (Fig. 5C), as well as the cytoplasmatic translocation of HMGB1 [Fig. 5B(c versus b)], were abolished by NAC + SFN. We also detected a minor but significant increase (P < 0.01) in reduced HMGB1 during epileptogenesis (Fig. 5C). This likely reflects increased HMGB1 biosynthesis since reduced HMGB1 is the constitutive isoform bound to nuclear chromatin. NAC + SFN blocked also the reduced HMGB1 increase (Fig. 5C). Notably, the changes in brain HMGB1, and the effects of treatment, were similarly detected in the blood of the same animals (Fig. 5D).
Based on these findings, we measured the GSSG/GSH ratio as well as total HMGB1 and its isoforms in the blood of status epilepticus-exposed rats undergoing the therapeutic trial (same rats of Fig. 3). Blood was drawn by tail vein at the end of treatment (14 days post-status epilepticus). GSSG/GSH ratio (Fig. 6A; P < 0.05), total HMGB1 and its isoforms (Fig. 6B; P < 0.01) were increased in blood of status epilepticus-exposed rats compared to sham rats. All these effects were prevented by drug treatment (Fig. 6A and B). There was no correlation between the blood levels of total or disulfide HMGB1 and total seizure number, their frequency and total and average seizure duration in rats (Spearman correlation test; not shown).
Figure 6.

Oxidative stress markers and HMGB1 isoforms in blood. Histograms (mean ± SEM, n = 9 each group) report GSSG/GSH ratio (A) and total HMGB1 and its isoforms (B) in blood of status epilepticus-exposed rats injected with either vehicle or antioxidant drug combination (NAC + SFN) (same rats as in Fig. 3) compared to baseline values in sham rats (Sham + vehicle). Blood was drawn by the tail vein at the end of treatment (i.e. 14 days post-status epilepticus) then rats were followed up for monitoring SRS at 2 and 5 months post-status epilepticus. The blood levels of the molecules reflect their brain changes (Figs 1, 2 and 5) and, as in brain, they were normalized by the treatment. **P < 0.01 versus Sham; #P < 0.05, ##P < 0.01 versus status epilepticus + vehicle by Kruskal-Wallis followed by Dunn’s post hoc test. SE = status epilepticus.
Comparison between early and delayed antioxidant treatment on hippocampal cell loss
Based on evidence that NAC + SFN reduced oxidative stress also after treatment was delayed to 24 h post-status epilepticus (Supplementary Table 1 versus 1 h post-status epilepticus in Fig. 5A), we determined whether this delayed treatment also afforded neuroprotection. Early treatment for 4 days displayed neuroprotection in CA1 and hilus (Supplementary Table 2, n = 4), and this was similar to epileptic rats treated with NAC + SFN for 14 days (Fig. 4). The delayed treatment did not rescue CA1 pyramidal cell loss while it reduced hilar cell loss similarly to early treatment (Supplementary Table 2, n = 5). CA3/CA4 cell loss was not rescued by treatment either given at 1 h or 24 h post-status epilepticus, as in epileptic rats treated for 14 days.
Oxidative stress in brain specimens from patients with status epilepticus and in temporal lobe epilepsy
We used immunohistochemistry to analyse the presence of oxidative stress markers in autoptic hippocampal specimens from patients experiencing status epilepticus and in surgically resected human hippocampal tissue from temporal lobe epilepsy. Figure 7 shows increased expression of iNOS (Fig. 7A and B), Xct (Fig. 7C and D) and Nrf2 (Fig. 7E and F) in NeuN-positive neuronal cells and GFAP-positive astrocytes in a patient who died 49 days after status epilepticus. Nrf2 signal was increased prominently in cell nuclei, an indication of its nuclear translocation. A similar pattern of cellular expression of these markers was observed in status epilepticus patient specimens evaluated between 1 and 49 days post-status epilepticus and in chronic epilepsy hippocampal tissue from patients with temporal lobe epilepsy (Supplementary Fig. 5). Notably, HMGB1 cytoplasmatic staining was increased in both neurons and astrocytes in adjacent slices (Supplementary Fig. 6), in accordance with previous findings in human temporal lobe epilepsy (Maroso et al. 2010).
Figure 7.
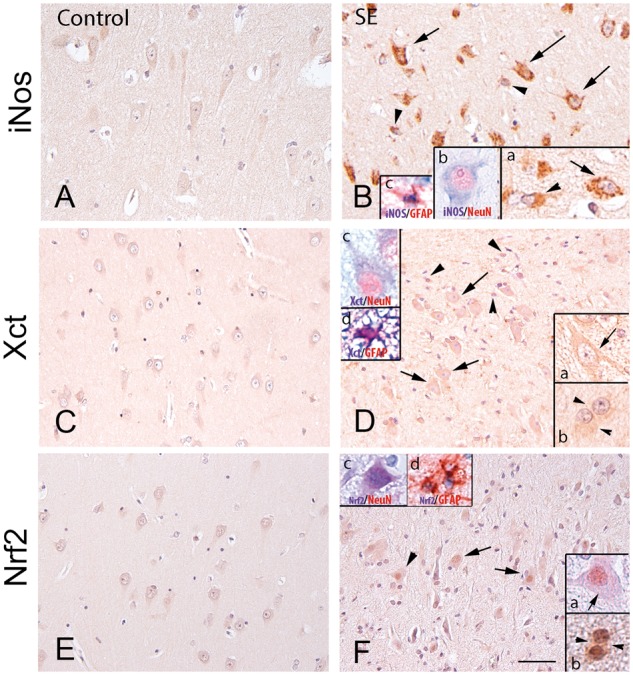
Oxidative stress in the hippocampus of patients with status epilepticus. Representative immunohistochemical micrographs of the CA1 region showing the expression of iNos (A and B), Xct (C and D) and Nrf2 (E and F) in control hippocampus (A, C and E) and in one representative patient who died after status epilepticus (49 days post-status epilepticus; B, D and F). Increased expression of these markers was observed in cells with neuronal (arrows in B, D, F and insets) and glial morphology (arrowheads in B, D, F and insets) compared to controls. Insets in B show iNos-positive neurons (arrow in inset a, and co-localization with NeuN in inset b) and astrocytes (arrowhead in inset a, and co-localization with GFAP in inset c). Insets in D show Xct-positive neurons (arrow in inset a, and co-localization with NeuN in inset c) and astrocytes (arrowhead in inset b, and co-localization with GFAP in inset d). Insets in F show Nrf2-positive neurons (arrow in inset a, and co-localization with NeuN in inset c and astrocytes; arrowhead in inset b and co-localization with GFAP in inset d). Insets in F show nuclear Nrf2 expression, denoting transcriptional activation of detoxifying enzymes. Scale bars in A–F = 80 µm; inset in B = 40 µm; insets in B, D and F = 25 µm. SE = status epilepticus.
Discussion
After brain injury and during seizures, mitochondrial dysfunction and increased NADPH oxidase and xanthine oxidase activities lead to excessive generation of ROS, thereby contributing to neuropathology (Rowley and Patel, 2013; Kovac et al., 2014). Animal models of acquired epilepsy provide evidence of profound changes in mitochondrial function and ROS production as a result of various epileptogenic injuries. These alterations occur rapidly after the inciting event, persist during epileptogenesis and are still observed in the chronic epilepsy phase (Waldbaum et al., 2010; Ryan et al., 2014; Bhuyan et al., 2015). It is likely that the mechanisms leading to oxidative tissue damage vary in the different phases of the epileptic process (Rowley and Patel, 2013). It remains to be determined if, and by which mechanisms, ROS generation contributes to the onset, progression and recurrence of spontaneous seizures. Importantly, whether targeting oxidative stress has any effect on seizure onset or progression is still unresolved.
Our novel findings show that an early and transient intervention with a specific combination of antioxidant drugs, namely NAC and SFN, mediates clinically relevant therapeutic effects in a status epilepticus rat model of acquired epilepsy. This combined treatment was more effective in rescuing mitochondrial dysfunction, and reducing oxidative stress during epileptogenesis than single drugs alone. The drug combination delayed epilepsy onset and blocked seizure progression. Accordingly, spontaneous seizure frequency was not modified at 2-month follow-up while it was drastically reduced at 5-month follow-up. Average SRS duration and incidence of rats developing epilepsy did not change. Notably, the drugs did not affect status epilepticus severity and the associated acute cell loss, nor showed acute antiseizure effects. These data therefore provide evidence for a disease-modification effect mediated by this antioxidant treatment (Varvel et al., 2015) and suggest that oxidative stress may play a role in the mechanisms of disease progression. In support, SFN was reported to suppress the progression of amygdala kindling in mice (Wang et al., 2014) and chronic NAC treatment in neurotrauma-exposed animals normalized seizure threshold, which was reduced by brain injury (Silva et al., 2011). Finally, chronic Nrf2 overexpression attained by gene therapy after epilepsy onset reduced SRS evoked by pilocarpine injection in mice (Mazzuferi et al., 2013). Since Nrf2 coordinates the expression of numerous genes encoding detoxification, antioxidant and anti-inflammatory mediators, these pathways are likely to be relevant for the mechanisms of seizure recurrence (Mazzuferi et al., 2013).
Our findings are apparently at variance with the lack of effect of a catalytic antioxidant porphyrin given post-status epilepticus on SRS, although both neuroprotection and rescue of behavioural deficit were attained in this study (Pearson et al., 2015). It is possible that our drug combination is particularly effective in antagonizing oxidative stress damage contributing to SRS. One major difference may be due to the mechanism of action of direct antioxidant used in a previous study (Pearson et al., 2015) versus the Nrf2 inducer used in our study. The latter induces multiple genes, many of which encode endogenous antioxidants resulting in longer-lasting effects (Houghton et al., 2013). Another factor possibly explaining the difference in results is the longer video-EEG monitoring of our study until the late phases of disease development, which allowed us to determine the effect of treatment on seizure progression. Similar effects on SRS progression were recently reported using an inducible nitric oxide inhibitor reducing reactive nitrogen species (Puttachary et al. 2016).
Oxidative stress has been implicated in cell loss and cognitive dysfunctions developing during epileptogenesis in different animal models (Rong et al., 1999; Liang et al., 2000; Barros et al., 2007; Wang et al., 2014; Pearson et al., 2015). Both NAC and SFN were shown to provide neuroprotection in brain injury models (Knuckey et al., 1995; Hong et al., 2010; Mazzuferi et al., 2013; Wang et al., 2014), which is compatible with the role of ROS in glutamate excitotoxicity and in apoptotic cell death (Lafon-Cazal et al., 1993; Reynolds and Hastings, 1995; Henshall and Murphy, 2008). Our antioxidant drug combination afforded neuroprotection and rescued cognitive deficits in a reference/working memory test by preventing the persistence of oxidative stress during epileptogenesis. We show that calretinin-positive cells in dentate hilus are rescued by our drug combination. These hilar interneurons form a subpopulation of GABAergic cells with frequent axo-dendritic and dendro-dendritric contacts with other inhibitory interneurons. This unique connectivity may enable them to play a crucial role in the generation of synchronous, rhythmic hippocampal activity by controlling other interneurons terminating on dendritic and somatic compartments of principal cells (Gulyas et al., 1999), therefore they are suggested to play a key role in the hippocampal inhibitory network. Notably, the density of calretinin-immunopositive cells is decreased significantly in the sclerotic hippocampus from human temporal lobe epilepsy, a phenomenon that may contribute to seizure generation and recurrence (Toth and Magloczky, 2014).
Early initiation of treatment (1 h post-status epilepticus) was more effective in reducing cell loss in CA1 than a delayed treatment schedule (24 h post-status epilepticus) while loss of hilar interneurons and oxidative stress were similarly prevented as assessed at 4 days post-status epilepticus. Thus, the data suggest that CA1 neurons are especially sensitive to oxidative damage occurring during the first 24 h post-injury. The resolution of oxidative stress at Day 4 post-status epilepticus by the delayed treatment warrents further investigations on its impact on disease onset and SRS progression.
Whether neuroprotection plays a role in the rescue of cognitive deficit in the T-maze remains speculative. Similar positive effects on cognitive dysfunctions were recently reported using SFN in a model of okadaic acid-induced memory impairment (Dwivedi et al., 2015) or using a metalloporphyrin catalytic antioxidant in a rat model of pilocarpine-induced epileptogenesis (Pearson et al., 2015).
Disturbances in the normal redox state of the cells may contribute to the pathologic effects developing during epileptogenesis in various ways. There is evidence of at least two potential links between mitochondrial oxidative stress and increased neuronal excitability, namely bioenergetic failure due to increased demand for neuronal mitochondria to produce cellular energy during hyperexcitability phenomena, and metabolic fuel utilization (Rowley and Patel, 2013). Moreover, neuronal excitability is controlled by glutamate and GABA, the biosynthesis of which depends on mitochondria (Kann et al., 2005). ROS have the potential to influence epileptogenesis also via oxidative damage to macromolecules including proteins, lipids, and DNA. We tested the novel hypothesis that a pathological switch in the redox state of brain tissue during epileptogenesis leads to the generation of disulfide HMGB1, a pro-inflammatory molecules with ictogenic properties (Maroso et al., 2010; Vezzani et al., 2011b; Iori et al., 2013; Balosso et al., 2014). We previously showed that disulfide HMGB1 contributes to seizure generation and excitotoxic cell loss by activation of TLR4 and receptor for advanced glycation end-products (RAGE) (Maroso et al., 2010; Iori et al., 2013), and mice lacking either one of these receptors develop a milder form of epilepsy following status epilepticus (Iori et al., 2013). Moreover, HMGB1 by activating TLR4 and RAGE, mediates cognitive dysfunctions in mice (Mazarati et al., 2011). Overall, this evidence supports the novel concept that disulfide HMGB1 may be a key mediator of the pathological effects of oxidative stress during epileptogenesis. In accordance, we found that the antioxidant effects of our drug combination were associated with prevention of disulfide HMGB1 generation and its translocation/extracellular release in brain tissue. Differently from astrocytes and neurons, we found that the HMGB1 translocation in microglia after status epilepticus was not associated with increased markers of oxidative stress in these cells. There are mechanisms underlying HMGB1 nucleus-to-cytoplasm translocation which may not depend on oxidative stress (Yu et al., 2015). In particular, JAK/STAT1 activation is pivotal for HMGB1 hyperacetylation at nuclear localization sites and subsequent translocation in macrophages (Lu et al., 2014). We cannot exclude, however, that the histological markers we have studied do not detect oxidative stress generated in microglia.
The brain changes measured during epileptogenesis in total HMGB1 and its isoforms, as well as in oxidative stress indicators, were mirrored by similar changes in blood, and the blood levels of these molecules were modified by the antioxidant intervention similarly to the brain. Thus, these molecules may be potential biomarkers for determining the efficacy of the antioxidant drugs on their targets and possibly predicting their therapeutic effects.
The translation of our findings to the clinical setting is supported by our novel evidence that oxidative stress occurs in brain of patients experiencing status epilepticus, as well as in patients with drug-resistant temporal lobe epilepsy, and this phenomenon is associated with cytoplasmatic translocation of HMGB1 in neurons and glia. Moreover, both NAC and SFN have been used in human clinical trials at doses comparable with the effective doses in our study. In particular, after extrapolating the human equivalent dose (Reagan-Shaw et al., 2008), we found that NAC and SFN doses in rats correspond to 5 g twice daily and to 48 mg daily for a 60 kg person, respectively. Interestingly, an intravenous infusion of 150 mg/kg NAC (corresponding to 9 g in a 60 kg person) in healthy individuals or Parkinson’s and Gaucher’s disease patients was well tolerated and resulted in increased brain GSH as assessed by magnetic resonance spectroscopy (Holmay et al., 2013). Moreover, NAC doses up to 3.6 g/day for several weeks have been used in neurological and psychiatric disorders (Deepmala et al., 2015). In epilepsy clinical studies, NAC was used up to 6 g daily for several months in progressive myoclonus epilepsy, in particular in Unverricht–Lundborg disease with evidence of seizure improvement (Ben-Menachem et al., 2000; Deepmala et al., 2015). NAC seemed to be fairly well tolerated with no significant between group differences in most of the controlled trials. As far as SFN is concerned, clinical studies in cancer used daily doses of 60 mg (Cipolla et al., 2015), and up to 27 mg were administered daily in autism spectrum disorders (Singh and Zimmerman, 2015) with signs of improvement and a safety profile.
In summary, our findings have high translational value since we report novel evidence that: (i) oxidative stress markers are induced in the hippocampus of humans who died following status epilepticus or with chronic pharmacoresistant epilepsy; and (ii) the drug doses we used in animals are compatible with human doses in the therapeutic range given for protracted treatment periods. Noteworthy, symptomatic (structural/lesional) epilepsies are often associated with a worse prognosis, therefore providing an ideal patients population for testing antioxidant drugs with potential disease-modifying properties (Pitkanen and Sutula, 2002; Schmidt and Sillanpaa, 2005).
Funding
This work was supported by the European Union’s Seventh Framework Programme (FP7/2007-2013) under grant agreement n°602102 (EPITARGET to A.V., M.C.W., E.A.V., E.A.), Citizen United for Research in Epilepsy (CURE) (A.V.), Epilepsy Research UK (M.C.W.), and in part by NIH grants RO1 NS086423 and RO1NS039587 (M.P.).
Supplementary material
Supplementary material is available at Brain online.
Supplementary Material
Glossary
Abbreviations
- GSH
glutathione
- NAC
N-acetylcysteine
- ROS
reactive oxygen species
- SFN
sulforaphane
- SRS
spontaneous recurrent seizure
References
- Abbas K, Breton J, Planson AG, Bouton C, Bignon J, Seguin C. et al. Nitric oxide activates an Nrf2/sulfiredoxin antioxidant pathway in macrophages. Free Radic Biol Med 2011; 51: 107–14. [DOI] [PubMed] [Google Scholar]
- Antoine DJ, Jenkins RE, Dear JW, Williams DP, McGill MR, Sharpe MR. et al. Molecular forms of HMGB1 and keratin-18 as mechanistic biomarkers for mode of cell death and prognosis during clinical acetaminophen hepatotoxicity. J Hepatol 2012; 56: 1070–9. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Antoine DJ, Williams DP, Kipar A, Jenkins RE, Regan SL, Sathish JG. et al. High-mobility group box-1 protein and keratin-18, circulating serum proteins informative of acetaminophen-induced necrosis and apoptosis in vivo. Toxicol Sci 2009; 112: 521–31. [DOI] [PubMed] [Google Scholar]
- Arakawa M, Ito Y. N-acetylcysteine and neurodegenerative diseases: basic and clinical pharmacology. Cerebellum 2007; 6: 308–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronica E, Crino PB. Inflammation in epilepsy: clinical observations. Epilepsia 2011; 52 (Suppl 3): 26–32. [DOI] [PubMed] [Google Scholar]
- Balosso S, Liu J, Bianchi ME, Vezzani A. Disulfide-containing high mobility group box-1 promotes N-methyl-d-aspartate receptor function and excitotoxicity by activating Toll-like receptor 4-dependent signaling in hippocampal neurons. Antioxid Redox Signal 2014; 21: 1726–40. [DOI] [PubMed] [Google Scholar]
- Barros DO, Xavier SM, Barbosa CO, Silva RF, Freitas RL, Maia FD. et al. Effects of the vitamin E in catalase activities in hippocampus after status epilepticus induced by pilocarpine in Wistar rats. Neurosci Lett 2007; 416: 227–30. [DOI] [PubMed] [Google Scholar]
- Ben-Menachem E, Kyllerman M, Marklund S. Superoxide dismutase and glutathione peroxidase function in progressive myoclonus epilepsies. Epilepsy Res 2000; 40: 33–9. [DOI] [PubMed] [Google Scholar]
- Bhuyan P, Patel DC, Wilcox KS, Patel M. Oxidative stress in murine Theiler’s virus-induced temporal lobe epilepsy. Exp Neurol 2015; 271: 329–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumcke I, Thom M, Aronica E, Armstrong DD, Bartolomei F, Bernasconi A. et al. International consensus classification of hippocampal sclerosis in temporal lobe epilepsy: a task force report from the ILAE commission on diagnostic methods. Epilepsia 2013; 54: 1315–29. [DOI] [PubMed] [Google Scholar]
- Cardenas-Rodriguez N, Coballase-Urrutia E, Perez-Cruz C, Montesinos-Correa H, Rivera-Espinosa L, Sampieri A III. et al. Relevance of the glutathione system in temporal lobe epilepsy: evidence in human and experimental models. Oxid Med Cell Longev 2014; 2014: 759293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipolla BG, Mandron E, Lefort JM, Coadou Y, Della Negra E, Corbel L. et al. Effect of Sulforaphane in men with biochemical recurrence after radical prostatectomy. Cancer Prev Res 2015; 8: 712–19. [DOI] [PubMed] [Google Scholar]
- De Simoni MG, Perego C, Ravizza T, Moneta D, Conti M, Marchesi F. et al. Inflammatory cytokines and related genes are induced in the rat hippocampus by limbic status epilepticus. Eur J Neurosci 2000; 12: 2623–33. [DOI] [PubMed] [Google Scholar]
- Deepmala, Slattery J, Kumar N, Delhey L, Berk M, Dean O. et al. Clinical trials of N-acetylcysteine in psychiatry and neurology: a systematic review. Neurosci Biobehav Rev 2015; 55: 294–321. [DOI] [PubMed] [Google Scholar]
- Dwivedi S, Rajasekar N, Hanif K, Nath C, Shukla R. Sulforaphane ameliorates okadaic acid-induced memory impairment in rats by activating the Nrf2/HO‐1 antioxidant pathway. Mol Neurobiol 2015; 53: 5310–23. doi:10.1007/s12035‐015‐9451‐4. [DOI] [PubMed] [Google Scholar]
- Farr SA, Poon HF, Dogrukol-Ak D, Drake J, Banks WA, Eyerman E. et al. The antioxidant alpha-lipoic acid and N-acetylcysteine reverse memory impairment and brain oxidative stress in aged SAMP8 mice. J Neurochem 2003; 84: 1173–83. [DOI] [PubMed] [Google Scholar]
- Filibian M, Frasca A, Maggioni D, Micotti E, Vezzani A, Ravizza T. In vivo imaging of glia activation using 1 H-magnetic resonance spectroscopy to detect putative biomarkers of tissue epileptogenicity. Epilepsia 2012; 53: 1907–16. [DOI] [PubMed] [Google Scholar]
- Gorter JA, van Vliet EA, Aronica E, Lopes da Silva FH. Progression of spontaneous seizures after status epilepticus is associated with mossy fibre sprouting and extensive bilateral loss of hilar parvalbumin and somatostatin-immunoreactive neurons. Eur J Neurosci 2001; 13: 657–69. [DOI] [PubMed] [Google Scholar]
- Gulyas AI, Megias M, Emri Z, Freund TF. Total number and ratio of excitatory and inhibitory synapses converging onto single interneurons of different types in the CA1 area of the rat hippocampus. J Neurosci 1999; 19: 10082–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey BH, Joubert C, du Preez JL, Berk M. Effect of chronic N-acetyl cysteine administration on oxidative status in the presence and absence of induced oxidative stress in rat striatum. Neurochem Res 2008; 33: 508–17. [DOI] [PubMed] [Google Scholar]
- Haynes LW. The neuron in tissue culture. New York, NY: Wiley; 1999. [Google Scholar]
- Henshall DC, Murphy BM. Modulators of neuronal cell death in epilepsy. Curr Opin Pharmacol 2008; 8: 75–81. [DOI] [PubMed] [Google Scholar]
- Holdiness MR. Clinical pharmacokinetics of N-acetylcysteine. Clin Pharmacokinet 1991; 20: 123–34. [DOI] [PubMed] [Google Scholar]
- Holmay MJ, Terpstra M, Coles LD, Mishra U, Ahlskog M, Oz G. et al. N-Acetylcysteine boosts brain and blood glutathione in Gaucher and Parkinson diseases. Clin Neuropharmacol 2013; 36: 103–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong Y, Yan W, Chen S, Sun CR, Zhang JM. The role of Nrf2 signaling in the regulation of antioxidants and detoxifying enzymes after traumatic brain injury in rats and mice. Acta Pharmacol Sin 2010; 31: 1421–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houghton CA, Fassett RG, Coombes JS. Sulforaphane: translational research from laboratory bench to clinic. Nutr Rev 2013; 71: 709–26. [DOI] [PubMed] [Google Scholar]
- Hsieh HL, Yang CM. Role of redox signaling in neuroinflammation and neurodegenerative diseases. Biomed Res Int 2013; 2013: 484613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu R, Hebbar V, Kim BR, Chen C, Winnik B, Buckley B. et al. In vivo pharmacokinetics and regulation of gene expression profiles by isothiocyanate sulforaphane in the rat. J Pharmacol Exp Ther 2004; 310: 263–71. [DOI] [PubMed] [Google Scholar]
- Iori V, Maroso M, Rizzi M, Iyer AM, Vertemara R, Carli M. et al. Receptor for advanced glycation endproducts is upregulated in temporal lobe epilepsy and contributes to experimental seizures. Neurobiol Dis 2013; 58: 102–14. [DOI] [PubMed] [Google Scholar]
- Kann O, Kovacs R, Njunting M, Behrens CJ, Otahal J, Lehmann TN. et al. Metabolic dysfunction during neuronal activation in the ex vivo hippocampus from chronic epileptic rats and humans. Brain 2005; 128: 2396–407. [DOI] [PubMed] [Google Scholar]
- Knuckey NW, Palm D, Primiano M, Epstein MH, Johanson CE. N-acetylcysteine enhances hippocampal neuronal survival after transient forebrain ischemia in rats. Stroke 1995; 26: 305–10; discussion 311. [DOI] [PubMed] [Google Scholar]
- Kovac S, Domijan AM, Walker MC, Abramov AY. Seizure activity results in calcium- and mitochondria-independent ROS production via NADPH and xanthine oxidase activation. Cell Death Dis 2014; 5: e1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan P, Arzimanoglou A, Berg AT, Brodie MJ, Allen Hauser W, Mathern G. et al. Definition of drug resistant epilepsy: consensus proposal by the ad hoc task force of the ILAE commission on therapeutic strategies. Epilepsia 2010; 51: 1069–77. [DOI] [PubMed] [Google Scholar]
- Lafon-Cazal M, Culcasi M, Gaven F, Pietri S, Bockaert J. Nitric oxide, superoxide and peroxynitrite: putative mediators of NMDA-induced cell death in cerebellar granule cells. Neuropharmacology 1993; 32: 1259–66. [DOI] [PubMed] [Google Scholar]
- Liang LP, Ho YS, Patel M. Mitochondrial superoxide production in kainate-induced hippocampal damage. Neuroscience 2000; 101: 563–70. [DOI] [PubMed] [Google Scholar]
- Liang LP, Patel M. Seizure-induced changes in mitochondrial redox status. Free Radic Biol Med 2006; 40: 316–322. [DOI] [PubMed] [Google Scholar]
- Liang LP, Patel M. Plasma cysteine/cystine redox couple disruption in animal models of temporal lobe epilepsy. Redox Biol 2016; 9: 45–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B, Antoine DJ, Kwan K, Lundback P, Wahamaa H, Schierbeck H. et al. JAK/STAT1 signaling promotes HMGB1 hyperacetylation and nuclear translocation. Proc Natl Acad Sci USA 2014; 111: 3068–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B, Wang H, Andersson U, Tracey KJ. Regulation of HMGB1 release by inflammasomes. Protein Cell 2013; 4: 163–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroso M, Balosso S, Ravizza T, Liu J, Aronica E, Iyer AM. et al. Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat Med 2010; 16: 413–19. [DOI] [PubMed] [Google Scholar]
- Mazarati A, Maroso M, Iori V, Vezzani A, Carli M. High-mobility group box-1 impairs memory in mice through both toll-like receptor 4 and receptor for advanced glycation end products. Exp Neurol 2011; 232: 143–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzuferi M, Kumar G, van Eyll J, Danis B, Foerch P, Kaminski RM. Nrf2 defense pathway: experimental evidence for its protective role in epilepsy. Ann Neurol 2013; 74: 560–8. [DOI] [PubMed] [Google Scholar]
- Noé FM, Polascheck N, Frigerio F, Bankstahl M, Ravizza T, Marchini S. et al. Pharmacological blockade of IL-1beta/IL-1 receptor type 1 axis during epileptogenesis provides neuroprotection in two rat models of temporal lobe epilepsy. Neurobiol Dis 2013; 59: 183–93. [DOI] [PubMed] [Google Scholar]
- Noé F, Pool AH, Nissinen J, Gobbi M, Bland RJ, Rizzi M. et al. Neuropeptide Y gene therapy decreases chronic spontaneous seizures in a rat model of temporal lobe epilepsy. Brain 2008; 131: 1506–15. [DOI] [PubMed] [Google Scholar]
- Palmblad K, Schierbeck H, Sundberg E, Horne AC, Harris HE, Henter JI. et al. High systemic levels of the cytokine-inducing HMGB1 isoform secreted in severe macrophage activation syndrome. Mol Med 2015; 20: 538–47. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Pastore A, Massoud R, Motti C, Lo Russo A, Fucci G, Cortese C. et al. Fully automated assay for total homocysteine, cysteine, cysteinylglycine, glutathione, cysteamine, and 2-mercaptopropionylglycine in plasma and urine. Clin Chem 1998; 44: 825–32. [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. New York, NY: Academic Press; 2005. [Google Scholar]
- Pearson JN, Rowley S, Liang LP, White AM, Day BJ, Patel M. Reactive oxygen species mediate cognitive deficits in experimental temporal lobe epilepsy. Neurobiol Dis 2015; 82: 289–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitkanen A, Engel J Jr. Past and present definitions of epileptogenesis and its biomarkers. Neurotherapeutics 2014; 11: 231–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitkanen A, Kharatishvili I, Narkilahti S, Lukasiuk K, Nissinen J. Administration of diazepam during status epilepticus reduces development and severity of epilepsy in rat. Epilepsy Res 2005; 63: 27–42. [DOI] [PubMed] [Google Scholar]
- Pitkanen A, Sutula TP. Is epilepsy a progressive disorder? Prospects for new therapeutic approaches in temporal-lobe epilepsy. Lancet Neurol 2002; 1: 173–81. [DOI] [PubMed] [Google Scholar]
- Puttachary S, Sharma S, Stark S, Thippeswamy T. Seizure-induced oxidative stress in temporal lobe epilepsy. Biomed Res Int 2015; 2015: 745613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puttachary S, Sharma S, Verma S, Yang Y, Putra M, Thippeswamy A. et al. 1400 W, a highly selective inducible nitric oxide synthase inhibitor is a potential disease modifier in the rat kainate model of temporal lobe epilepsy. Neurobiol Dis 2016; 93: 184–200. [DOI] [PubMed] [Google Scholar]
- Ravizza T, Gagliardi B, Noé F, Boer K, Aronica E, Vezzani A. Innate and adaptive immunity during epileptogenesis and spontaneous seizures: evidence from experimental models and human temporal lobe epilepsy. Neurobiol Dis 2008; 29: 142–60. [DOI] [PubMed] [Google Scholar]
- Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J 2008; 22: 659–61. [DOI] [PubMed] [Google Scholar]
- Reynolds IJ, Hastings TG. Glutamate induces the production of reactive oxygen species in cultured forebrain neurons following NMDA receptor activation. J Neurosci 1995; 15: 3318–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong Y, Doctrow SR, Tocco G, Baudry M. EUK-134, a synthetic superoxide dismutase and catalase mimetic, prevents oxidative stress and attenuates kainate-induced neuropathology. Proc Natl Acad Sci USA 1999; 96: 9897–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowley S, Patel M. Mitochondrial involvement and oxidative stress in temporal lobe epilepsy. Free Radic Biol Med 2013; 62: 121–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan K, Liang LP, Rivard C, Patel M. Temporal and spatial increase of reactive nitrogen species in the kainate model of temporal lobe epilepsy. Neurobiol Dis 2014; 64: 8–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt D, Sillanpaa M. Does surgery prevent worsening of epilepsy? Epilepsia 2005; 54: 391. [DOI] [PubMed] [Google Scholar]
- Silva LF, Hoffmann MS, Rambo LM, Ribeiro LR, Lima FD, Furian AF. et al. The involvement of Na+, K+-ATPase activity and free radical generation in the susceptibility to pentylenetetrazol-induced seizures after experimental traumatic brain injury. J Neurol Sci 2011; 308: 35–40. [DOI] [PubMed] [Google Scholar]
- Singh K, Zimmerman AW. Sleep in autism spectrum disorder and attention deficit hyperactivity disorder. Semin Pediatr Neurol 2015; 22: 113–25. [DOI] [PubMed] [Google Scholar]
- Steele ML, Fuller S, Patel M, Kersaitis C, Ooi L, Munch G. Effect of Nrf2 activators on release of glutathione, cysteinylglycine and homocysteine by human U373 astroglial cells. Redox Biol 2013; 1: 441–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S. N-acetylcysteine, reactive oxygen species and beyond. Cancer Biol Ther 2010; 9: 109–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth K, Magloczky Z. The vulnerability of calretinin-containing hippocampal interneurons to temporal lobe epilepsy. Front Neuroanat 2014; 8: 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varvel NH, Jiang J, Dingledine R. Candidate drug targets for prevention or modification of epilepsy. Annu Rev Pharmacol Toxicol 2015; 55: 229–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venereau E, Casalgrandi M, Schiraldi M, Antoine DJ, Cattaneo A, De Marchis F. et al. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J Exp Med 2012; 209: 1519–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzani A, Aronica E, Mazarati A, Pittman QJ. Epilepsy and brain inflammation. Exp Neurol 2013; 244: 11–21. [DOI] [PubMed] [Google Scholar]
- Vezzani A, Dingledine R, Rossetti AO. Immunity and inflammation in status epilepticus and its sequelae: possibilities for therapeutic application. Expert Rev Neurother 2015; 15: 1081–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzani A, French J, Bartfai T, Baram TZ. The role of inflammation in epilepsy. Nat Rev Neurol 2011a; 7: 31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzani A, Maroso M, Balosso S, Sanchez MA, Bartfai T. IL-1 receptor/Toll-like receptor signaling in infection, inflammation, stress and neurodegeneration couples hyperexcitability and seizures. Brain Behav Immun 2011b; 25: 1281–9. [DOI] [PubMed] [Google Scholar]
- Waldbaum S, Liang LP, Patel M. Persistent impairment of mitochondrial and tissue redox status during lithium-pilocarpine-induced epileptogenesis. J Neurochem 2010; 115: 1172–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Lin W, Shen G, Khor TO, Nomeir AA, Kong AN. Development and validation of an LC-MS-MS method for the simultaneous determination of sulforaphane and its metabolites in rat plasma and its apllication in pharmacokinetics studies. J Chromatogr Sci 2011; 49: 801–6. [DOI] [PubMed] [Google Scholar]
- Wang W, Wu Y, Zhang G, Fang H, Wang H, Zang H. et al. Activation of Nrf2-ARE signal pathway protects the brain from damage induced by epileptic seizure. Brain Res 2014; 1544: 54–61. [DOI] [PubMed] [Google Scholar]
- Yang H, Lundback P, Ottosson L, Erlandsson-Harris H, Venereau E, Bianchi ME. et al. Redox modification of cysteine residues regulates the cytokine activity of high mobility group box-1 (HMGB1). Mol Med 2012; 18: 250–9. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Yu Y, Tang D, Kang R. Oxidative stress-mediated HMGB1 biology. Front Physiol 2015; 6: 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.