

Abstract
Aging of the vasculature plays a central role in morbidity and mortality of older people. In order to develop novel treatments for amelioration of unsuccessful vascular aging and prevention of age-related vascular pathologies it is essential to understand the cellular and functional changes that occur in the vasculature during aging. In this review, the pathophysiological roles of fundamental cellular and molecular mechanisms of aging, including oxidative stress, mitochondrial dysfunction, impaired resistance to molecular stressors, chronic low-grade inflammation, genomic instability, cellular senescence, epigenetic alterations, loss of protein homeostasis, deregulated nutrient sensing and stem cell dysfunction in the vascular system are considered in terms of their contribution to the pathogenesis of both micro- and macrovascular diseases associated with old age. The importance of pro-geronic and anti-geronic circulating factors in relation to development of vascular aging phenotypes are discussed. Finally, future directions and opportunities to develop novel interventions to prevent/delay age-related vascular pathologies by targeting fundamental cellular and molecular aging processes are presented.
Keywords: geroscience, vascular inflammation, vascular senescence, endothelial dysfunction, atherosclerosis
Introduction
Cardiovascular and cerebrovascular diseases are the most common cause of death among older people in the United States 1 accounting for approximately 1/3 of all deaths in the US at the age of 65 and nearly 2/3 at an age of 85. With a projected increase in the number of adults over 65 years old increasing from 12 % to 22% in the next 30 years, addressing age related vascular diseases is of critical importance as the annual cost to care for these older people are projected to more than double over that time2. As many age-related cardiovascular and cerebrovascular diseases are due to alterations in arterial function or are exacerbated by arterial functional and phenotypic changes, it is important to better elucidate mechanisms underlying arterial aging3. Furthermore, as the microcirculation is pervasive, being present in every tissue in the body, it has a unique ability to influence the local environment of the majority of tissues and organs. As such, aging-induced functional and structural alterations of the microcirculation contribute to the pathogenesis of range of age-related diseases including vascular cognitive impairment, Alzheimer’s disease, sarcopenia, kidney and eye disease. Therefore, it is also critical to explore the spectrum of age-related microvascular functional and phenotypic changes from subclinical dysfunction to manifested disease as to better predict and prevent microvascular contributions to the pathogenesis of multiple diseases associated with old age. A better mechanistic understanding of macro- and microvascular aging processes is critical to find and evaluate both lifestyle and pharmacological countermeasures to treat this growing health issue.
Rapid advances in geroscience in the last 25 years, including studies on invertebrate models of aging, long-lived mammals, transgenic mouse strains and interventional studies, have led to the identification of evolutionarily conserved pathways involved in lifespan regulation, as well as common denominators of aging in different organisms4. In this review, the pathophysiological roles of these aging mechanisms, including oxidative stress, mitochondrial dysfunction, impaired resistance to molecular stressors, chronic low-grade inflammation, genomic instability, telomere attrition and cellular senescence, epigenetic alterations, loss of protein homeostasis (“proteostasis”), deregulated nutrient sensing, stem cell exhaustion, and altered intercellular communication in the vascular system are considered in terms of their contribution to the pathogenesis of both micro- and macrovascular diseases (Figure 1). The interconnectedness between the potential mechanisms of vascular aging and the interaction between the cellular and molecular aging processes and disease-specific pathways are discussed. Likewise, the implications of molecular, cellular and system theories of aging for vascular aging phenotypes are considered. Finally, based on our current mechanistic understanding of vascular aging, potential novel targets for intervention to improve cardiovascular and cerebrovascular health are identified. As such, it is critical to move these potential new therapies forward to reduce the morbidity and mortality associated with cardiovascular and cerebrovascular dysfunction/disease and thereby improving healthspan in an increasingly aged population. We will not discuss in detail data on the effects of preventive measures already available and in clinical use (including physical exercise, smoking cessation, dietary regimens, inhibitors that disrupt the renin – angiotensin system, statins) on vascular aging phenotypes, as these have been the subject of a recent comprehensive reviews5–8. The great deal of phenomenological work has been performed on the effects of aging on specific cell types within the vascular wall and on structural alterations and the hemodynamic consequences that result from arterial stiffening. We refer the interested reader to excellent reviews on these topics6, 9–11.
Figure 1: Conceptual model for the role of cell-autonomous and non-cell-autonomous mechanisms in vascular aging.
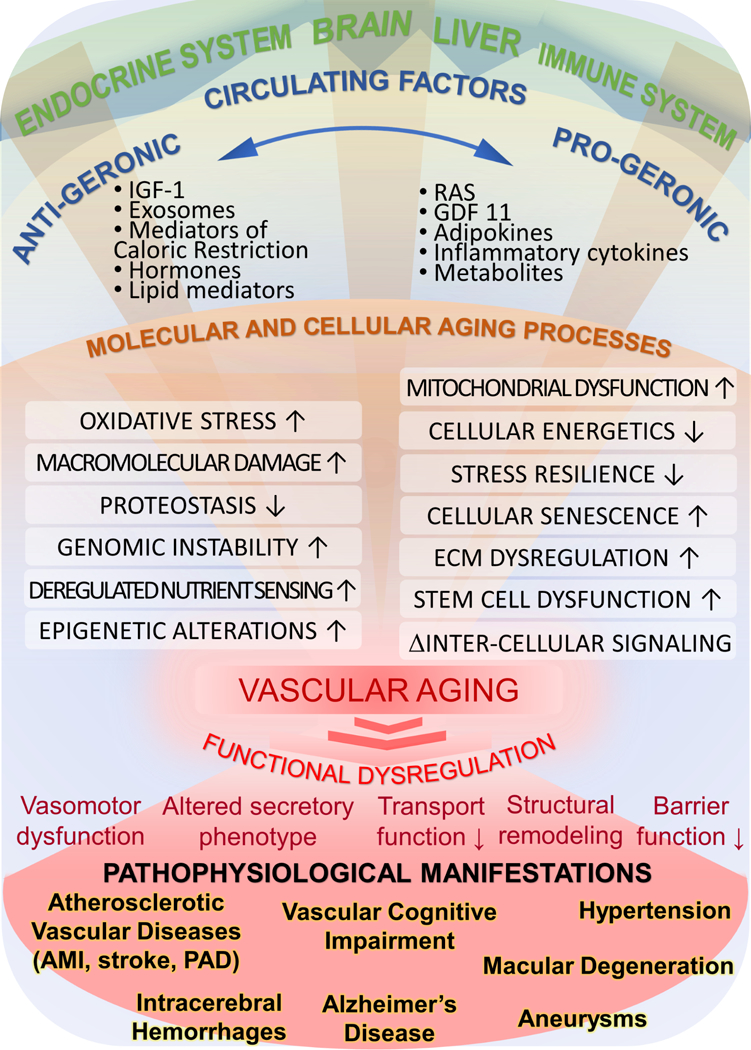
The model predicts that circulating pro-geronic (e.g. inflammatory cytokines, RAS/renin-angiotensin system, aldosterone) and anti-geronic factors (e.g. IGF-1, mediators of caloric restriction, estrogen) derived from the brain, the endocrine system, cells of the immune system and/or the adipose tissue orchestrate aging processes simultaneously in the endothelial and smooth muscle cells within the large vessels and microcirculation. The hierarchical regulatory cascade for vascular aging involves modulation of cell-autonomous cellular and molecular aging processes. The resulting functional dysregulation of vascular cells (i.e. impaired vasomotor, barrier, secretory and transport functions of the vasculature as well as adverse structural remodeling) promote the development of a wide range of age-related vascular pathologies.
Molecular and cellular mechanisms of vascular aging
Role of oxidative and nitrative stress
Since Denham Harman first proposed the free radical theory of aging in the 1950s12 a large amount of data has been published implicating oxidative stress in vascular aging processes (Figure 2). In particular, there is strong evidence that increased production of reactive oxygen species (ROS) by NAD(P)H oxidases13–17 and mitochondria18, 19 contributes to endothelial dysfunction and large elastic artery stiffening with advancing age both in laboratory animals13, 14, 20–24 and humans 16, 25. Whilst oxidative stress may influence many facets of vascular function with advancing age via oxidation of critical proteins or induction of redox sensitive transcription factors, one of its most potent effects is inactivation of endothelium-derived nitric oxide (NO). Impaired bioavailability of NO is responsible for age-related reduction in endothelium dependent dilation, enhanced vasoconstriction and dysregulation of tissue perfusion13, 14, 20, 26–30. There is strong evidence that endothelial dysfunction caused by increased oxidative stress contributes significantly to both impaired dilation of coronary arteries13, promoting myocardial ischemia and neurovascular uncoupling, impairing the moment-to-moment adjustment of cerebral blood flow to increased oxygen and nutrient demand that occurs with neuronal activation31, 32. In addition to inactivation of NO by ROS, alterations in eNOS activation status, substrate (L-arginine) and cofactor (BH4) availability, increased endothelin-126 and/or reduced expression of eNOS 33–35 may also contribute to age-related reduction in NO bioavailability. NO exerts potent anti-inflammatory, anti-thrombotic and anti-leukocyte adhesion effects, thus reduction in NO likely promotes a pro-atherogenic vascular phenotype in aging36–38.
Figure 2: Proposed scheme for mechanisms and pathological consequences of age-related oxidative stress in vascular endothelial cells.
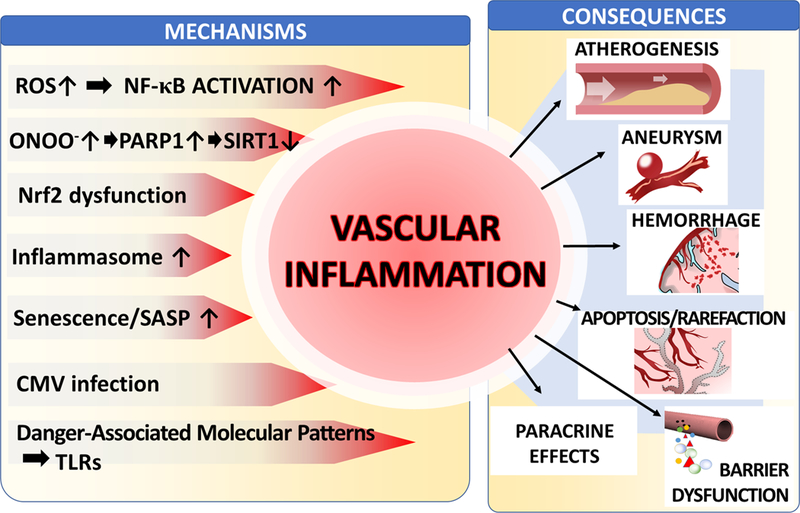
The model predicts that in aged endothelial cells dysfunctional mitochondria and NAD(P)H oxidases are critical sources of increased ROS production. Increased levels of O2.- generated by the electron transport chain are dismutated to H2O2, which can penetrate the mitochondrial membrane increasing cytoplasmic H2O2 levels. Increased oxidative stress is exacerbated by age-related impairment of Nrf2-dependent homeostatic antioxidant defense mechanisms. H2O2 plays important signaling roles, including activation of NF-κB, which contribute to age-associated low grade chronic vascular inflammation. Increased levels of O2.- generated by NAD(P)H oxidases (stimulated by elevated TNFα levels and/or by the activated local renin-angiotensin system [RAS] in the vascular wall) decrease the bioavailability of NO by forming ONOO-. Increased nitrative stress lead to PARP-1 activation, which promotes vascular inflammation and contributes to cellular energetic dysfunction by consuming NAD+ , compromising sirtuin-mediated anti-aging pathways. Impaired bioavailability of NO promotes vasodilator dysfunction and compromises endothelial viability. In addition, increased vascular oxidative stress in aging also induces MMP activation, promoting the pathogenesis of intracerebral hemorrhages, aneurysm formation and blood brain barrier disruption.
Many of the results of vascular oxidative stress are mediated via production of the highly reactive oxidant peroxynitrite (ONOO-), the reaction product of NO and superoxide39, 40 which has been well documented in older endothelial cells and arteries 13, 15, 20, 22. The mechanisms by which peroxynitrite contributes to vascular aging are multifaceted and include direct cytotoxic effects, adverse effects on mitochondrial function, and activation of inflammatory pathways (Figure 2). In particular, oxidative stress and the consequent activation of redox-sensitive cellular signaling pathways, including NF-kB, are thought to be implicated in the inflammatory process in the aged vasculature. This inflammatory process is characterized by increased endothelial activation41 and pro-inflammatory changes in the cytokine expression profile of aged vascular cells18, 19, 42. Increased vascular oxidative stress has also been linked to activation of matrix metalloproteinases (MMPs) and consequential disruption of the structural integrity of aged arteries, potentially contributing large artery stiffening 43 and the pathogenesis of aortic aneuryms44. Furthermore, in the cerebral circulation aging- and hypertension-dependent activation of the ROS-MMP axis promotes the development of cerebral microhemorrhages45, which contribute to cognitive decline, geriatric psychiatric syndromes and gait disorders46. Importantly, in preclinical models similar antioxidative treatments were reported to prevent large artery stiffening 47, cerebral microhemorrhages 45 and aortic aneurysms44, providing further evidence for shared pathomechanisms.
Role of mitochondrial dysfunction
Mitochondria play central role in regulation of aging processes4, 48, including regulation of lifespan49. As mammals age, the efficacy of the respiratory chain diminishes, promoting electron leakage and increased ROS production and reducing cellular ATP generation.
Recent studies suggest that mitochondrial ROS production (mtROS) has an important role in age-related vascular dysfunction18, 50–52. In the aged vasculature increased mtROS has been attributed to a dysfunctional electron transport chain53 and is likely exacerbated by peroxynitrite-mediated nitration and inhibition of MnSOD14, decline in cellular glutathione content54, down-regulation of p66Shc55, and/or impaired Nrf2-mediated antioxidant defense responses18, 56, 57. There is also evidence that aging is associated with impaired mitochondrial biogenesis in endothelial and smooth muscle cells both in conduit arteries53 and the capillaries58, 59, which is likely to negatively impact cellular energetics and also may increase mitochondrial ROS production by increasing electron flow through the deficient electron transport chain. Importantly, mtROS can be pharmacologically targeted for vasoprotection. For example, treatment with the mitochondrial antioxidant MitoQ60, resveratrol61(which substantially attenuates mtROS production in endothelial and smooth muscle cells19, 62) and the potent mitochondria-targeted antioxidative tetrapeptide SS-3151 has been shown to improve endothelial function in arteries from rodent models of aging. Treatment with SS-31 was also shown to restore endothelium-dependent regulation of cerebral blood flow via neurovascular coupling, improving cognitive function in aged mice51. There is evidence that mitochondria-derived H2O2 promotes low grade vascular inflammation in aging by inducing NF-κB, activation in endothelial and smooth muscle cells19, 24 (Figure 2). Recent studies also link increased hypertension-induced mtROS production in aged vascular smooth muscle cells50 to increased MMP activation in the vascular wall and consequential exacerbation of cerebral microhemorrhages46. Another potentially important link between mtROS production and vascular aging is the induction of apoptosis via a Bcl-2 dependent pathway63.
It is increasingly realized that the functional integrity of the vasculature, including regulation of membrane transport and barrier functions, depends of normal cellular energy metabolism. Thus, dysfunctional mitochondria and impaired mitochondrial energy metabolism can potentially contribute to vascular aging in addition to the effects of increased mitochondrial release of ROS. The mitochondrial DNA (mtDNA) has a very high mutation rate, due to the proximity of mtDNA to sites of ROS production in the mitochondria, the lack of protective histone coverage in the mtDNA, and the limited efficiency of mitochondrial DNA repair mechanisms. There is growing evidence that aging increases mutations and deletions in mtDNA, eroding mitochondrial energy production and contributing to aging processes48. mtDNA damage is likely a result of increased mitochondrial oxidative stress64. Although aging is associated with significant mitochondrial oxidative stress in the vasculature18, 19, 24, 50, the role of mtDNA damage is development of vascular aging phenotypes is not well understood. To date, only few studies have focused on the link between mitochondrial heteroplasmy and atherogenesis65. Mitochondrial DNA deletions have been detected in human atherosclerotic lesions65, 66, which may contribute to impaired cellular metabolism in the vascular wall. Recent studies provide direct evidence that mitochondrial mutations play a causal role in atherogenesis in mouse models of the disease67. Importantly, transgenic mice ApoE−/− harboring a version of the mitochondrial DNA polymerase (polG) deficient in proof-reading activity accumulate mutations in their mtDNA and exhibit accelerated atherosclerosis, associated with impaired proliferation and apoptosis of VSMCs67.
The NAD+-dependent pro-survival enzyme SIRT1 modulates mitochondrial function in the vasculature, controlling mitochondrial biogenesis, mtROS production and cellular energy metabolism42, 62, 68 as well as removal of damaged mitochondria by autophagy69. The mitochondrial sirtuin SIRT3 also regulates many key enzymes involved in mitochondrial energy metabolism. NAD+ is a rate-limiting co-substrate for sirtuin enzymes and there is growing evidence that cellular NAD+ availability decreases in aging70, 71, at least in part, due to overactivation of NAD+ utilizing PARP-141. There is strong evidence that in aged mice enhancing NAD+ biosynthesis (e.g by treatment with nicotinamide mononucleotide, a key NAD+ intermediate70) rescues age-related functional alterations in the aorta72 and cerebral vasculature (Tarantini, Csiszar and Ungvari, unpublished findings, 2017), likely by activating sirtuin mediated pathways and reversing age-related decline in mitochondrial function73. Other potential mechanisms contributing to impaired bioenergetics in aged cells include oxidation/nitration of mitochondrial proteins, destabilization of the macromolecular organization of electron transport chain complexes and impaired mitophagy (a mitochondrion-specific form of autophagy). The combination of increased mitochondrial damage and decreased turnover of mitochondria, due to impaired biogenesis and deficient mitophagy, likely contribute to the accumulation of dysfunctional mitochondria in the vascular cells, exacerbating vascular aging processes.
Vascular inflammation in aging
There is strong experimental and clinical evidence that chronic, sterile, low-grade inflammation is a hallmark of the aging process (“inflammaging”74). Age-related activation of inflammatory processes play a key role in a wide range of macro- and microvascular pathologies, ranging from atherogenesis and aneurysm formation to microvascular dysfunction, blood-brain barrier disruption and Alzheimer’s pathologies41 (Figure 3). Previous studies demonstrate that both in aged laboratory rodents and primates there is a pro-inflammatory shift in the gene expression profile of vascular endothelial and smooth muscle cells, including an induction of inflammatory cytokines (e.g. IL-6, IL-1β, TNFα), chemokines, adhesion molecules, iNOS and other pro-inflammatory mediators13, 24, 33, 61, 75–78. The resulting pro-inflammatory microenvironment in the vascular wall promotes vascular dysfunction79, 80, impairs cellular metabolism, increases apoptosis76, 79 and contributes to the pathogenesis of vascular diseases.
Figure 3: Mechanisms and consequences of age-related vascular inflammation.
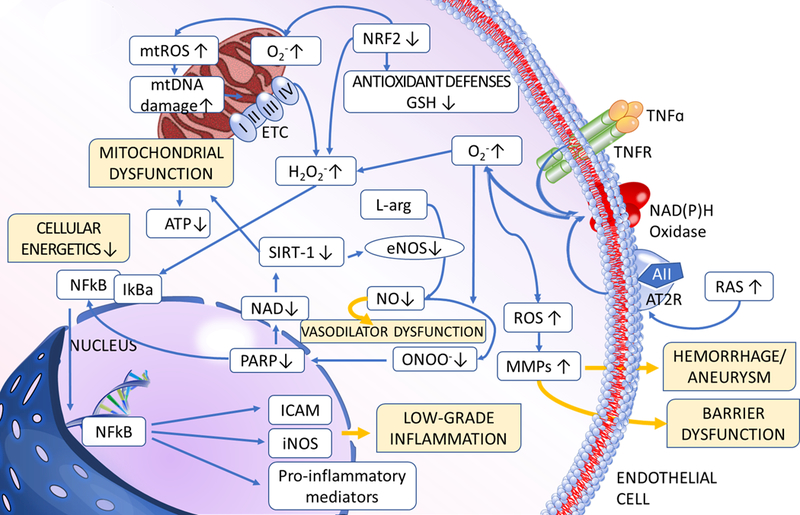
The model predicts that multiple pathways converge on activation of inflammatory processes in the vascular tissue. During aging increased ROS production, exacerbated by Nrf2 dysfunction, enhances NF-κB activation, which promotes inflammatory cytokine and chemokine expression, microvascular endothelial activation, leukocyte adhesion and extravasation. Increased nitrative stress promotes PARP1 activation, which contributes to impaired activity of anti-inflammatory sirtuins. Sterile inflammation in the vascular wall is also exacerbated by increased secretion of inflammatory mediators from senescent cells and danger-associated molecular patterns (DAMPs), which activate innate immune system effectors, including toll-like receptors (TLRs) and the NLRP3 inflammasome complex. The aging vasculature in humans is also affected by the high prevalence of endothelium-trophic persistent cytomegalovirus (CMV) infection. Inflammatory processes contribute to a wide range of macro- and micro-vascular pathologies affecting older people.
The mechanisms contributing to vascular inflammation in aging are likely multifaceted. There is an important cross-talk between increased oxidative stress and activation of inflammatory processes in the aged vascular wall41. First, ROS act as signaling molecules activating pro-inflammatory signaling pathways, including NF-κB24, which regulate endothelial activation and expression of pro-inflammatory paracrine mediators and promote atherogenesis. Importantly, aged endothelial and smooth muscle cells exhibit significant NF-κB activation24, 56 and selective inhibition of NF-κB in the vasculature was shown to improve blood flow regulation, decrease systemic inflammation, exert beneficial metabolic effects and extend health span81. Second, inflammatory mediators are potent inducers of cellular oxidative stress (e.g. TNFα activates NAD(P)H oxidases79).
SIRT1 exerts potent anti-inflammatory effects and decreased SIRT1 activity likely contributes to vascular inflammation in aging42, 82, 83. Importantly, pharmacological activators of SIRT1 were shown to attenuate vascular inflammation in aged mice61, 83.
Endothelial senescence (see below) is associated by a significant increase in the production/release of a wide range of inflammatory cytokines and chemokines84, termed the “senescence-associated secretory phenotype”, or SASP. Induction of SASP is likely mediated by activation of NF-κB, p38MAPK, the DNA damage response pathway and GATA485, 86. There is growing evidence that presence of senescent cells contributes significantly to the heightened inflammatory status of the aged vasculature87.
Sterile inflammation in the vascular wall is also exacerbated by danger-associated molecular patterns (DAMPs), which activate innate immune system effectors, including toll-like receptors (TLRs) and the NLRP3 inflammasome complex. TLRs are activated by a wide range of DAMP ligands (e.g. molecules released to the extracellular matrix from necrotic cells, breakdown products of the extracellular, matrix and/or bacterial breakdown products leaking through a damaged gut barrier) and control the secretion of a number of pro-inflammatory paracrine mediators (e.g. IL-1, IL-6, IL-8, TNFα). There is evidence that aging induces a proinflammatory phenotype in vascular smooth muscle cells, at least in part, due to activation of TLR4-mediated, MyD88-dependent signaling pathways78. Strong data suggest that the canonical Nlrp3 inflammasome contributes to systemic low-grade age-related sterile inflammation in mice88, but further studies are needed to establish the exact role of this mechanism in age-related vascular pathologies.
There is growing evidence that impaired oxidative stress resilience in aging also exacerbates vascular inflammation induced by cardiovascular risk factors, including obesity, metabolic disease and hypertension89–91. The interaction between aging and the effect of environmental inflammatory factors (e.g. particulate exposure92) to exacerbate vascular inflammation warrants further studies.
Further studies are needed to differentiate between adaptive and maladaptive inflammatory responses in the aged vasculature and to define the role of chronic low-grade microvascular inflammation in a wide range of functional impairments in multiple organs. The interaction between cell-autonomous mechanism of age-related vascular inflammation and inflammatory processes induced by bacterial breakdown products getting to the circulation through a leaky intestinal barrier or by chronic infection with viruses that exhibit endothelial tropism in vascular inflammation also has to be further clarified. For example, over 90% of adults 80 years of age or older have persistent human cytomegalovirus (CMV) infection93. CMV replicates in the vascular endothelial cells during the entire life of the host following initial infection and severity of CMV infection (antibody titers) was shown to predict increased incidence of frailty and risk of mortality94.
Maladaptation to molecular stresses: role of Nrf2 dysfunction
Recent progress in geroscience research has identified a critical hallmark of the aging process: impaired ability of aged cells to respond to molecular stresses and return to homeostasis. In young organisms in response to increased generation of ROS in the vascular endothelial and smooth muscle cells adaptive homeostatic mechanisms are invoked that involve activation of Nrf2-driven antioxidant defense pathways56, 57, 95. Nrf2 is an evolutionarily conserved redox sensitive transcription factor, which coordinates the antioxidant response, including up-regulation of enzymes that detoxify ROS and repair ROS-induced macromolecular damage96. In the young vasculature this adaptive homeostatic response serves to reduce oxidative stress and attenuate cellular and macromolecular damage caused by increased ROS levels. Induction of Nrf2 was also shown to exert potent anti-inflammatory97 and pro-angiogenic98 effects. There is strong evidence that aging promotes Nrf2 dysfunction in the vasculature, exacerbating oxidative stress and increasing sensitivity of aged vascular cells to ROS-mediated cellular and molecular damage56, 57. This loss of oxidative stress resilience may be a major determinant for the development of age related vascular pathologies99. Importantly, anti-aging effects of caloric restriction is associated with induction of Nrf2-mediated pathways18, 100. Further studies are warranted to determine how pharmacological activation of Nrf2 exerts anti-aging vasoprotective effects.
Loss of proteostasis in vascular aging
Impaired maintenance of proteostasis is thought to contribute to organismal aging101. There is evidence that activity of proteostasis networks and proteome stability determine healthy cardiac aging102, 103. It is suspected that disequilibrium between protein synthesis, maintenance, and degradation also compromises vascular health. Indeed, increased presence of misfolded protein aggregates is associated with cardiovascular diseases104. Aging impacts multiple components of the proteostasis systems in heart and the vasculature, including chaperones105, the ubiquitin-proteasome and the lysosome-autophagy system101. Chaperones assist the folding, assembly, disassembly and transport of other proteins and play a major role in preventing protein misfolding and aggregation. Many age-related molecular alterations can impact chaperoning activities. For example, aging results in down-regulation of HSP70 in the vascular tissue105. Further, mitochondrial dysfunction and the resulting decline in cellular ATP content likely also impairs the function of ATP-dependent chaperones. The processes of autophagy (macroautophagy, microautophagy, and chaperone-mediated autophagy) allow the degradation and recycling of cellular constituents. Recently hypotheses were put forward that dysregulation of autophagic processes, including mitophagy, may be a common pathway promoting vascular aging and development of age-related vascular diseases106. Experimental findings showing increased oxidative stress, impaired bioavailability of NO and up-regulation of inflammatory mediators in autophagy-deficient endothelial cells support this view107. Further, pharmacological interventions that stimulate autophagy (e.g. trehalose or spermidine treatment) were reported to reverse aspects of arterial aging108, 109. Proteasomes degrade unneeded or damaged proteins by proteolysis. There is evidence that proteasome activity declines in advanced aging101 and is diminished in atherosclerotic plaques of elderly patients110 and in hearts of ageing rats111. In addition to direct effects on protein degradation the ubiquitin-proteasome system (UPS) is critical for the activation of key regulators of atherogenesis and vascular inflammation112. Future studies should identify the role of the UPS and other proteostatic pathways that are impaired in specific vascular disease states associated with aging and elucidate age-related factors, including neuroendocrine factors, that regulate proteostasis machineries in the vascular wall.
Role of genomic instability
Since the original formulation of Somatic Mutation Theory of aging 113, a large amount of data has been published both in support and against a causal role of DNA damage and mutation accumulation in aging114. There is evidence that diverse genetic lesions may accumulate within aged cells, including somatic mutations, chromosomal aneuploidies and copy number variations and telomere shortening. Despite these advances the role of genomic instability in vascular aging is not well understood. Hypotheses that predict that genomic instability plays a role in vascular aging, usually focus on the primary role of oxidative stress-induced DNA damage, illustrating how these hypotheses and the oxidative stress hypothesis of aging are interconnected115. Importantly, endothelial cells appear to have less efficient DNA repair pathways than many other cell types84. In that regard it is interesting that known interventions that cause extensive DNA damage (e.g. whole brain irradiation) result in significant phenotypic and functional alterations in endothelial cells, promoting microvascular rarefaction, impaired vasodilation and pro-inflammatory changes, mimicking several aspects of the aging phenotype84, 116, 117. In vascular endothelial cells DNA damage readily triggers replicative senescence (see below) 84, to prevent propagation of damaged DNA. In light of recent developments it seems to be likely that induction of senescence is a major mechanism by which DNA damage contributes to vascular aging118.
To elucidate the causal role of DNA damage in vascular aging an interesting recent study reported that mouse models with genomic instability resulting from the defective nucleotide excision repair genes (ERCC1, XPD) exhibit aging-like vascular phenotypes, including endothelial dysfunction, increased vascular stiffness, increased presence of senescence cells and hypertension119. However, these mouse models also exhibit severe liver, kidney, bone marrow, neurological and/or bone phenotypes, which associate with reduced lifespan119 and it is unclear how closely these phenotypes indeed mimic aging. Mice with genetic deficiency in the spindle assembly checkpoint protein BubR1, which promotes progressive aneuploidy, were also shown to exhibit aging-like vascular phenotypes, including endothelial dysfunction, increased vascular stiffness, media atrophy and fibrosis120. However, this mouse model also exhibits short life span associated with severe functional deficits in multiple organs, including cachectic dwarfism and lordokyphosis. Thus, the relevance of this model to normal aging remains unresolved. It was also proposed that an association exists between single-nucleotide polymorphisms in human DNA repair genes and vascular stiffness119, yet the mechanistic role of DNA repair pathways in the genesis of age-related vascular diseases in humans remains to be determined. There are studies reporting that increased oxidative DNA damage121 and increased expression of multiple biomarkers of DNA double strand breaks122 are present in atherosclerotic plaques. While accelerating or retarding repair of double strand breaks in transgenic mouse models have lesser effect on atherogenesis, they significantly alter plaque stability122.
Children with Hutchinson-Gilford progeria syndrome and other laminopathies exhibit accelerated vascular pathologies leading to fatal myocardial infarction or stroke at a very young age123. There is evidence implicating DNA damage response induced by genetic nuclear lamina dysfunction in aging-like phenotypic changes in vascular smooth muscle cells124, 125, however, it remains to be demonstrated that these pathways also contribute to “normal” vascular aging.
Impairment of mechanisms responsible for maintaining the appropriate length and functionality of telomeres is thought to play a role in vascular aging and hypertension by inducing cellular senescence (see below)87, 126.
Role of cellular senescence
Cellular senescence is a fundamental aging process in which cells, including vascular endothelial and smooth muscle cells, permanently withdraw from the cell cycle in response to a range of endogenous and exogenous stressors (e.g. ROS, dysfunctional telomeres, DNA damage, paracrine signals) and undergo distinctive phenotypic alterations, including profound pro-inflammatory secretome changes127 (Figure 4). Recent studies demonstrate that elimination of senescent cells expressing p16INK4A extends lifespan and healthspan in mice128, 129, suggesting that cellular senescence plays a fundamental role in physiological decline associated with aging. The available evidence indicate that endothelial senescence also contributes to endothelial dysfunction in aging and pathophysiological conditions associated with accelerated vascular aging117, 130–132. Endothelial senescence has also been implicated in the pathogenesis of heart failure133. Replicative senescence may also be potentially important for impairment of regenerative and angiogenic capacity of the vascular endothelium. Studies using mouse models of irradiation-induced, DNA damage mediated senescence84 demonstrate that induction of cellular senescence in the neurovascular unit associates with significant cerebromicrovascular dysfunction117 and microvascular rarefaction, mimicking the aging phenotype. Advanced atherosclerotic lesions contain senescent cells and recent studies using genetic and pharmacological approaches to eliminate senescent cells in Ldlr−/−mice suggest that senescent cells promote atherogenesis, contributing to plaque instability by up-regulating matrix metalloproteases134 and/or by exacerbating vascular inflammation135. The hypothesis was put forward that pharmacological treatment with senolytic agents to clear senescence cells may exert atheroprotective effects134. Importantly, long-term senolytic treatment was also shown to improve endothelial function in mouse models of aging130. However, the exact role of different senescence mechanisms in age-related vascular pathophysiology is not well understood. Future studies should address the relationship among acquisition of a senescence-associated secretory phenotype (SASP) in the endothelium and the vascular smooth muscle cells and specific disease processes. There is evidence that the SASP can also induce paracrine senescence and/or alter the function of neighboring cells and the role of this mechanism in vascular aging should be further evaluated. The possibility of paracrine transmission of senescence from microvascular endothelial cells to parenchymal cells also requires further investigations. It should be noted that many studies assess only senescence associated β-galactosidase activity as a marker of senescent cells. Future studies assessing should use novel molecular markers of senescence and senescence reporter mouse models and analyze senescence-related gene expression in individual cells.
Figure 4: Conceptual model for the pathogenic role of cellular senescence in vascular aging.
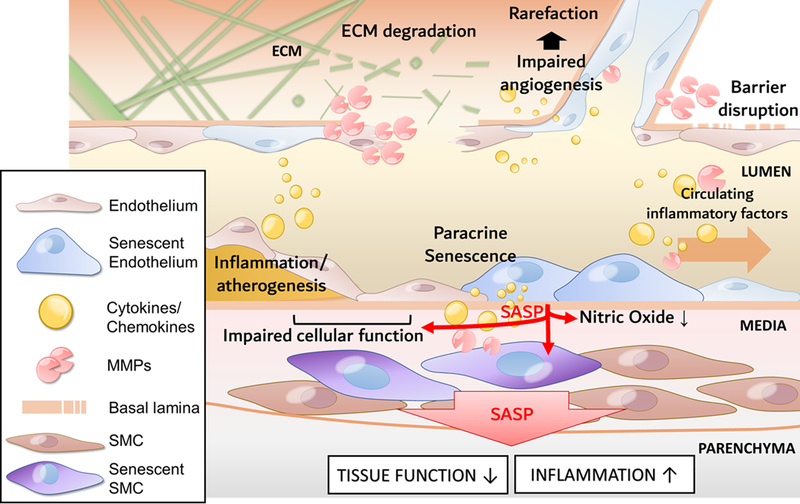
The model predicts that increased presence of senescent endothelial and/or smooth muscle cell (SMC) in the aged vasculature and their proinflammatory secretome (SASP: senescence-associated secretory phenotype) contributes to impaired angiogenesis and microvascular rarefaction, pathological remodeling of the ECM, barrier disruption, chronic inflammation and atherogenesis.
Role of increased apoptosis and necroptosis
Apoptosis is an evolutionarily conserved cell death program that is tightly regulated and executed through the interaction of extrinsic and intrinsic signaling pathways136. There is strong evidence that alterations in apoptotic potential contribute to a number of aging phenotypes across species, including the genesis of age-related cardiovascular pathologies. In the vasculature there is an increased presence of apoptotic endothelial cells, which has been attributed to impaired bioavailability of NO, up-regulation of TNFα and/or mitochondrial oxidative stress61, 76, 79, 137. Increased apoptotic cell death likely contributes to aging-induced microvascular rarefaction and the pathogenesis of atherosclerotic vascular diseases and aneurysm formation11.
Necroptosis is a newly identified form of programmed cell death that does not involve caspase activation but critically depends on receptor-interacting serine-threonine kinase 3 and mixed lineage kinase domain-like (MLKL) and is characterized by morphological features of necrosis138. Necroptosis plays a role in inflammaging by promoting pro-inflammatory phenotypic changes in tissues due to the release of cell debris (damage‐associated molecular patterns, DAMPs)139. DAMPs are a major activator of NLRP3 inflammasome, which is an important mechanism involved in low‐grade chronic inflammation in aging (see above). Inhibition of necroptosis either genetically, pharmacologically or by dietary means was shown to reduce inflammation in mouse models138, 139. Importantly, biomarkers of necroptosis are evident in the atherosclerotic plaques in apolipoprotein E (ApoE)-knockout mice and inhibition of necroptosis pathways reduce atherosclerosis burden and increases the lifespan in these models138. Biomarkers of necroptosis are also increased in human aorta aneurysmal tissues and up-regulation of necroptosis pathways promote aorta aneurysm progression in mouse models140.
Taken together, pathways involved in programmed cell death are promising targets for interventions in multiple aging-related diseases, including cardiovascular protection as well as prevention of cell loss leading to muscle atrophy and neurodegeneration.
Role of epigenetic alterations
A wide range of epigenetic alterations affect the cells during the lifespan, which may modulate vascular aging phenotypes141. Epigenetic changes that may contribute to vascular aging processes involve alterations in DNA methylation patterns, posttranslational modification of histones, microRNAs, long noncoding RNAs and chromatin remodeling.
DNA methylation is thought to be a central regulator of genome function. Aging is associated with complex, likely sexually dimorphic alterations in methylation patterns in many organs, which can be partially reversed by anti-aging interventions (e.g. caloric restriction) 142. Also, in animal models of aging altered methylation of genes important for vascular function has been observed143, 144. There is preliminary evidence that in vascular diseases DNA methylation patterns of cells within the vascular wall are altered145, however, understanding the pathogenic role of these changes in vascular aging requires further studies.
Understanding how post-translational histone modifications (lysine methylation, acetylation), which were demonstrated to regulate lifespan in lower organisms and to modulate aging phenotypes in mammals, modulate vascular aging processes is an area of intense current research4. Histone acetylation is dynamically regulated by histone acetyltransferases and histone deacetylases (HDACs). There is particularly strong data that decreased activity/expression of class III HDACs (the NAD+ utilizing sirtuin family) contributes to vascular aging42, 72, 82, 83, 146.
DNA- and histone-modifying enzymes act in concert with key epigenetic factors that determine changes in chromatin architecture, regulating lifespan and healthspan in evolutionarily diverse organisms4. Such chromatin remodeling factors include the Polycomb group proteins, which can remodel chromatin such that epigenetic silencing of genes takes place. The role of the Polycomb group proteins in regulation of endothelial progenitor cell function has been extensively studied147. There is evidence that expression/activity of many of these chromatin remodeling factors are altered in aging4, yet, their mechanistic role in vascular aging has yet to be elucidated.
The expression of 60% of human protein coding genes is controlled through post-transcriptional repression by microRNAs [miRNAs], a class of small non-coding RNAs. There is a complex interplay between miRNAs and other epigenetic factors, and dysregulation of miRNA expression is an emerging field in age-related epigenetics. In the vasculature miRNAs contribute to the regulation of important biological processes, including angiogenesis148, atherogenesis149 and restenosis150. There is growing evidence that aging is associated with dysregulation of miRNA expression in vascular endothelial and smooth muscle cells, which likely contributes to age-related impairment of angiogenic processes151, decreased cellular stress resilience18 and plaque formation, destabilization and rupture152. Importantly, there is also evidence that IGF-1 deficiency during a critical period during early in life results in persistent changes in post-transcriptional miRNA-mediated control of critical target genes for vascular health, which likely contribute to the deleterious late-life cardiovascular effects known to occur with developmental IGF-1 deficiency153.
Long noncoding RNAs (lncRNAs) are transcripts longer than 200 nucleotides, which regulate multiple aspects of RNA transcription and translation. There is growing evidence that lncRNAs interact with pro-inflammatory signaling pathways and regulate senescence, however, their role on regulation of vascular aging processes is virtually unknown154. Interestingly, there is initial evidence linking the expression of the lncRNA Meg3 to age-related impairment of angiogenic capacity of endothelial cells155.
Further studies are definitely needed to understand the contribution of alteration of epigenetic patterns to the development of various age-related vascular pathologies and to elucidate age-related changes in cellular mechanisms assuring the generation and maintenance of epigenetic patterns (e.g. DNA methyltransferases, histone acetylases and deacetylases, methylases, and demethylases, protein complexes implicated in chromatin remodeling). Epigenetic alterations are reversible, thus efforts should persist to develop epigenome-influencing interventions for prevention/treatment of age-related vascular diseases.
Role of dysregulated nutrient sensing pathways in vascular aging: mTOR, sirtuins, AMPK
Evolutionarily highly conserved cellular energy sensing pathways were reported to regulate fundamental aging processes by controlling cellular responses to nutrient availability and growth signals, including mTOR (mechanistic/mammalian target of rapamycin) signaling156, adenosine monophosphate protein kinase (AMPK), and sirtuins.
It is now recognized that mTOR acts as a critical molecular regulator of key anabolic processes, controlling biosynthesis of proteins, lipids and nucleic acids. In low nutrient conditions, mTOR activity is reduced, which relieves mTOR-dependent inhibition of autophagy, a major catabolic cellular process. Reduced activity of the mTOR pathway has been reproducibly shown to regulate aging and extend lifespan in invertebrates and in mice157. Consistent with the concept that mTOR regulates fundamental molecular processes of aging, there is growing evidence that attenuating mTOR activity inhibits or delays the pathogenesis of age-associated diseases, including Alzheimer’s disease158, 159. There is growing evidence that mTOR inhibition also confers protective, anti-aging vascular effects, delaying endothelial cell senescence160, 161 and promoting endothelium-mediated, NO-dependent vasodilation159, 162–165. mTOR inhibition also regulates phenotypic switch of vascular smooth muscle cells166. Recently it was reported that chronic treatment with the mTOR inhibitor rapamycin reverses age-associated arterial dysfunction, decreasing vascular stiffness and oxidative stress167. The potential significance of these findings for human aging stems from the availability of inhibitors for mTOR signaling pathways that are approved for clinical use. There is considerable excitement about the potential of mTOR inhibitors to treat cancer and neurological diseases of aging, and potentially to improve health span in elderly patients. Preliminary preclinical studies suggest that mTOR inhibition may also exert certain beneficial effects on cardiovascular pathologies associated with old age, including stroke168–171. More importantly, recent studies159, 172 identified mTOR as a major regulator of brain vascular damage and dysfunction in Alzheimer’s disease models159, 173–175 and in models of atherosclerosis176. In mouse models of Alzheimer’s disease mTOR inhibition with rapamycin was shown to reduce Aß vascular pathology and improve cerebral blood flow via an endothelial NO-dependent mechanism159, 172, significantly improving cognitive outcomes. Thus, while further studies are needed to define the role of mTOR in vascular aging, the available evidence indicates that mTOR has an important role in cerebrovascular dysfunction associated with neurodegenerative diseases.
Sirtuins and AMPK are critical cellular energy sensors that are activated by cellular metabolic factors, such as NAD+ in the case of sirtuins and the AMP:ATP ratio for AMPK. These pathways are thought to be activated in times of low nutrient status in order to provide the cell with enhanced stress resistance to prevent cellular loss or derangement. Both pathways been shown to be enhanced in response to longevity promoting interventions (eg calorie restriction) and are known to interact with one another. Activators of both the sirtuins, specifically Sirt-1 and −6, and AMPK have been effective in improving endothelial function, enhancing NO bioavailability, and reducing oxidative stress and inflammation. Sirtuins are evolutionarily conserved NAD+-dependent protein deacetylases and ADP-ribosyltransferases, which regulate multiple pathways involved in the aging process. There is strong evidence that in the vasculature, activation of members of the sirtuin family, particularly Sirt-1, confers multifaceted anti-aging effects42, 61, 62, 68, 72, 83, 146, 177–180. Sirt-1 specifically contributes to the vasoprotective effects of calorie restriction42, 181, augments eNOS activation 146, reduces oxidative stress, exerts anti-inflammatory effects180, is anti-apoptotic, reduces DNA damage, and promotes telomere stability. Sirt-6 also exerts endothelial protective and anti-atherogenic effects in mice182, 183. In addition to regulating redox homeostasis, mitochondrial function, endothelial vasodilation and protecting against apoptosis and senescence, Sirt-1 and −6 have been shown to regulate the DNA damage response in VSMCs184. Studies also suggest that Sirt-1 is reduced in human atherosclerosis and that Sirt-1 exerts anti-atherogenic effects in mouse models by protecting against DNA damage184. Importantly, the activity of many sirtuins, including Sirt-1 and −6, is dependent on the cellular NAD+ supply, which is known to decline with age.
Due to the multitude of beneficial effects, many sirtuin activators have been utilized to determine if sirtuin activation can reverse the vascular aging phenotype. Resveratrol, a naturally occurring polyphenol, can activate sirtuins and numerous other pathways and has been used extensively to reverse arterial aging 185, 186. Additionally, more specific and potent Sirt-1 activators, such as SRT1720, have recapitulated the beneficial effects afforded by resveratrol treatment, demonstrating anti-oxidant and anti-inflammatory effects. However, although SRT1720 rescued endothelial function in aged mice, that was the result of enhanced cyclooxegenase vasodilators instead of increased NO bioavailability. Supplementation with nicotinamide mononucleotide (NMN), a key NAD+ intermediate, has also been shown increase sirtuin activation187, resulting in reversal of mitochondrial dysfunction in advanced age70. In the aged vasculature, 8 weeks of NMN supplementation increases SIRT-1 activity and reverses age-related endothelial dysfunction and oxidative stress72. In summary, Sirt-1 and other sirtuins are considered promising drug targets180 and future studies should evaluate the efficacy other sirtuin activating compounds for cardiovascular protection in older persons.
AMPK signaling is an important energy sensing pathway that is involved in regulation of aging processes, integrating energy balance, metabolism and cellular stress resistance 188, 189. In lower organisms increased AMPK activity was shown to extend lifespan. Treatment of aged mice with pharmacological activators of AMPK, including metformin or aminoimidazole carboxamide ribonucleotide (AICAR), was also shown to confer significant health benefits. There is currently an aging clinical trial, TAME (targeting aging with metformin), that aims to determine if metformin can delay the onset of age-related diseases in humans190. In the context of vascular aging, AMPK was shown to confer vasoprotective effects, augmenting eNOS activation191–194. Furthermore, inhibition of NF-κB signaling by AMPK suppresses inflammatory processes. Importantly, AMPK activity is reduced in the aorta195 and cerebral arteries 196 of old rodents. Pharmacological activation of AMPK by AICAR was shown to restore endothelium-dependent vasodilation in old mice 195, suggesting that inactivation of arterial AMPK contributes to age-associated endothelial dysfunction. Still, little is known regarding the effects of AMPK activators on other vascular functions and much more research is needed to better understand the similarities and differences between the effects of various AMPK activators.
Role of the renin-angiotensin system in vascular aging
In model organisms (C. elegans) reducing the activity of acn-1, a homologue of the angiotensin converting enzyme (ACE), results in significant extension of lifespan suggesting that peptide hormones produced by these enzymes regulate fundamental aging processes197. There is initial evidence that the anti-aging effects of pharmacological ACE inhibitors are mediated by pathways that partially overlap with other evolutionarily conserved mechanisms involved in regulation of lifespan (e.g. FOXO signaling197). Studies on laboratory rodents extend these findings showing that inhibition of the renin-angiotensin system (RAS) pharmacologically or by genetic means exerts significant anti-aging effects, extending lifespan and reversing age-related phenotypic and functional changes in the aged vasculature198–200. There is growing evidence demonstrating that up-regulation of tissue RAS plays a role in vascular aging promoting intimal thickening and remodeling in large conduit arteries of aged animals and elderly human subjects77, 201–203. Proof-of-concept is derived from studies demonstrating that infusion of angiotensin II into young rats promotes aging-like changes in the vascular phenotype, promoting carotid media thickening and intima infiltration by vascular smooth muscle cells203. Further, pharmacological inhibition of RAS activity can reduce arterial stiffness in aged animals and elderly humans independently of changes in blood pressure204, 205. Up-regulation of RAS in the vascular wall likely also promotes chronic low-grade vascular inflammation and oxidative stress, enhancing the vascular response to injury and rendering the aged vascular wall susceptible to atherogenesis77, 202. Activation of RAS/increased angiotensin II levels in aging have also been linked to induction of mitochondrial oxidative stress in the vasculature206, development of cerebral microhemorrhages45 and disruption of the blood brain barrier207.
More recently the view has emerged that an extended renin-angiotensin-aldosterone system, including local expression of mineralocorticoids and their receptors in the vasculature, plays a tissue-specific role in regulation of aging processes. It has been well established that circulating aldosterone regulates water and electrolyte homeostasis and thereby controls blood pressure. In addition, aldosterone also promotes structural and functional alterations in the vasculature, including inflammation and pathological remodeling208. Recent studies demonstrate that age-associated changes of aldosterone and mineralocorticoid receptor signaling dysregulation occur in vascular smooth muscle cells, which may contribute to age-associated arterial remodeling208. These findings encourage further experimentation aiming to better characterize the pathophysiological relevance of aging-induced alterations in the extended renin-angiotensin-aldosterone system and to the efficiency of treatments targeting the extended renin-angiotensin-aldosterone system for cardiovascular protection in older individuals.
Role of ECM remodeling in vascular aging
The ECM is an important contributor to health and longevity. This non-cellular compartment, ubiquitous to all tissues and organs does not only provide essential mechanical scaffolding but mediates highly dynamic bio-mechanical and bio-chemical signals required for tissue homeostasis, morphogenesis and cell differentiation. Studies on model organisms suggest that evolutionarily conserved pathways regulate ECM remodeling during aging and that promotion of ECM youthfulness by anti-aging interventions is an essential signature of longevity assurance209. Aging in mammals also result in significant changes in ECM biosynthesis, postsynthetic modifications of ECM components and alterations of cell-matrix interactions, which contribute to the development of a spectrum of age-related pathologies210.
Age-related alterations of the ECM, including the subendothelial basement membrane, intima, media, adventitia, and interstitial matrix (which constitute more than half of the mass of the vascular tissue), play a fundamental role in impairment of both structural and regulatory homeostasis of the vasculature211. With age, the expression of growth factors that regulate ECM biosynthesis is altered45 and the synthesis of many ECM components (e.g. elastin) declines, which impairs elasticity and resilience of the vascular wall to mechanical damage and rupture induced by bursts in wall-tension due to pulsatile pressure waves211. Age-related ECM changes also likely alter vascular mechano-transduction, dysregulating cell responses to alterations in the hemodynamic environment. Additionally, aging and cellular senescence alter the secretory phenotype of vascular endothelial and smooth muscle cells, increasing MMP secretion45. This, together with increased MMP activation211 induced by high ROS levels compromises the structural integrity of the vasculature and promotes pathological remodeling (e.g. in hypertension), resulting in increased likelihood of aneurysm formation and vessel rupture, including the development of cerebral microhemorrhages45. The available evidence suggests that many of these age-related ECM alterations are governed by circulating factors and factors produced in the vascular wall, including the extended renin-angiotensin-aldosteron system (see above) and an age-related decline in circulating IGF-1 212.
Collagen synthesis is also dysregulated with age in the vascular wall likely due to the effects of increased paracrine action of TGF-β126, which contributes to vascular fibrosis and arterial stiffening211. Additional features that contribute to increased arterial stiffness include decreased elastin synthesis, elastin degradation and fragmentation, elastin calcification, alterations in cross-linking of extracellular matrix components (e.g. by increased presence of advanced glycation end-products [AGEs])211, 213, 214.
The pathophysiological consequences of age-related ECM remodeling and arterial stiffening have been the subject of a recent comprehensive review by AlGhatrif and Lakatta6. In brief, as the large conduit arteries stiffen in aging, aortic pulse wave velocity, systolic pressure and pulse pressure significantly increase215, whereas diastolic pressure decreases. Decreased diastolic pressure leads to a decline in coronary blood flow. Increased systolic pressure promotes left ventricular remodeling, diastolic dysfunction and exacerbates atherogenesis. Due to the dilation of conduit arteries wall tension significantly increases, contributing to the development of aneurysms. In addition to alterations in the biomechanical properties of large arteries, age-related ECM remodeling likely also affects microvascular transport and barrier functions216. Age-related alteration of the ECM structure and composition are also manifested in the wall of veins, contributing to the pathogenesis of varicosities217.
Role of pro-geronic and anti-geronic circulating factors: lessons learnt from heterochronic parabiosis and caloric restriction
There is growing evidence that non-cell-autonomous mechanisms play a critical role in orchestrating vascular aging processes (Figure 1). Aging-induced alterations in vasoprotective endocrine factors are of particular importance. Such changes include an age-related decline in circulating levels of GH218, IGF-1219 and estrogens, all of which regulate multiple aspects of endothelium-dependent vasodilation220, autoregulation of blood flow221, vascular structural remodeling, atherogenesis222 and angiogenic processes223.
The impact of circulating factors on aging phenotypes was also demonstrated by studies using mice with heterochronic parabiosis, which involves surgically connecting the circulatory system of a young and an aged mouse224. Cerebromicrovascular density typically declines with advanced age225, and there is initial evidence that circulating “anti-geronic” factors (which reverse/ prevent development of aging phenotypes) present in young mice can rejuvenate microvascular network architecture in aged heterochronic parabionts224. The anti-geronic circulating factors present in young mice are currently unknown, and the previously proposed role for GDF11224 remains controversial. Future studies should identify additional anti-geronic factors that might be targeted by interventions to extend vascular health-span. Pro-geronic circulating factors increase with age and impair tissue homeostasis in young animals. There is initial evidence that mediators secreted by senescent cells (e.g. inflammatory cytokines, such as TNFα79) may serve as pro-geronic circulating factors. Further studies are warranted to identify additional pro-geronic proteins and determine their impact on atherogenesis, endothelial function, blood brain barrier integrity and microvascular function in aging.
Additional evidence to support a central role of anti-geronic circulating factors governing vascular aging processes is derived from studies on caloric restriction, a dietary regimen, which improves health and slow the aging process in evolutionarily distant organisms226. Caloric restriction was shown to promote a youthful endothelial phenotype by up-regulating and activating eNOS in aged animals226–228 and perhaps humans229. A critical role of anti-geronic circulating factors in vasculoprotective phenotypic responses induced by caloric restriction was first indicated by the observations that in vitro treatment of cultured aged endothelial cells with sera derived from caloric restricted animals mimics phenotypic effects observed in vivo during caloric restriction, promoting anti-inflammatory and pro-angiogenic effects42, 230. Treatment with sera derived from caloric restricted animals up-regulates SIRT-1231, however, the exact nature of the circulating factor responsible for this effect remains elusive. One potential candidate anti-geronic protein mediating the vasoprotective effects of caloric restriction is adiponectin, whose serum level is known to be increased by caloric restriction232–234.
Human studies are needed to identify novel pro-geronic and anti-geronic circulating factors and their cofactors, activators or inhibitors/antagonists and to seek associations with vascular aging phenotypes. Future studies should also identify cellular origins of circulating pro-geronic and anti-geronic factors that impact vascular aging and characterize pathological conditions that alter their levels in circulation with aging. Further, mechanistic studies describing the cellular effects of pro-geronic and anti-geronic circulating factors in the vascular wall are warranted.
Progenitor cell exhaustion
On the basis of the stem cell theory of aging it is postulated that the inability of various types of vascular progenitor cells to continue to replenish the circulatory system with functional differentiated endothelial and smooth muscle cells compromises the biological functions of the aged vasculature. Importantly, aging impairs neovascularization, which depends on the function of highly proliferative endothelial progenitor cells (EPCs). Previous studies provide evidence that aging compromises the function of circulating EPCs 235–237, likely by altering the production of factors promoting cell proliferation, migration, and survival (e.g. IGF-1), and/or by enhancing inflammation and oxidative stress, activating the renin-angiotensin system and promoting senescence. There are data demonstrating that sera derived from young animals improves the function of cultured EPCs isolated from aged rats 238, suggesting a key role for circulating factors. There are also preclinical data extant suggesting that progressive progenitor cell deficits may contribute to the development of atherosclerosis237. In particular, treatment with bone marrow–derived progenitor cells isolated from young mice prevents atherosclerosis progression in ApoE−/− mice, whereas treatment with progenitor cells from older mice is ineffective237. It should be noted that role of EPCs in the genesis of vascular aging phenotypes in humans is note well understood. For example, patients with peripheral artery disease exhibit low EPC numbers239, while patients with chronic myocardial ischemia from coronary microvascular disease240 or abdominal aorta aneurism have significantly higher levels of circulating progenitor cells than age‐matched controls241. Future studies should investigate the synergistic effects of aging and associated cardiovascular risk factors in humans and determine how declining EPC function contributes to different vascular aging phenotypes.
Future directions
Although significant progress has been achieved in characterizing aging-induced changes in vascular function and phenotype, research efforts should persist in this direction to develop innovative strategies based on recent achievements in the biology of aging to improve vascular health-span. Understanding the interaction of processes of aging and chronic diseases and should be a high priority. Better alignment of preclinical studies on vascular aging and human investigations is needed. Limitations of translating the results of pre-clinical studies should be recognized. An important recent example is caloric restriction242. While caloric restriction confers significant lifespan extension and cardiovascular protection in laboratory rodents5, 18, 42, 100, 226, 243, 244 and in certain cohorts of non-human primates230, 245, its protective effects in non-human primates in other studies246 and in patients with multiple cardiovascular risk factors are less evident247. Additionally, in cross-sectional studies the older groups may represent a selected long-lived subset of the younger population. There are existing longitudinal studies in humans (e.g. InCHIANTI study) and non-human primates, and important information related to mechanisms of vascular aging could be derived from add-on studies to these existing cohorts.
Critical areas of vascular aging research include the role of senescence, epigenetics, stress resilience, inflammation, macromolecular damage, proteostasis, mitochondrial and metabolic dysfunction and impaired stem cell biology. The specific roles for cell-autonomous and non-cell-autonomous mechanisms contributing to vascular aging need to be elucidated further. The role of signal transduction pathways linked to regulation of cellular energetics in the vascular aging process should be better defined. Future studies should also lead to improved understanding of the role circadian clocks to vascular aging. New studies investigating cellular heterogeneity in vascular aging are warranted. Stochastic macromolecular damage leads to regional variability in the presence of senescent cells, cells with altered metabolism, mitochondrial dysfunction and increased ROS production. Such regional variability likely contributes to the focal development of vascular pathologies, ranging from atherosclerotic plaques to microhemorrhages. Single-cell gene expression analysis should facilitate better understanding of the pathophysiological role of functional heterogeneity. Finally, the impact of environmental factors and lifestyle choices impact the vascular aging processes should be better understood.
The disposable soma theory of aging predicts age-related functional decline occurs due to the accumulation of random macromolecular damage and that the proportion of energy investment into cellular maintenance and repair processes will determine longevity of the organism. Numerous studies investigating cellular and molecular stress responses in the vasculature agree with the predictions of this theory. For example, cells of the long-lived muroid rodent species white-footed mouse (Peromyscus leucopus; maximum lifespan: >8 years) exhibits decreased production of ROS, improved antioxidant defense mechanisms, increased resistance against oxidative stressors, superior DNA repair mechanisms and more efficient mitochondria than the shorter-lived Mus musculus248–250. Similar findings were reported in the vasculature of the longest-living rodent species, the naked mole-rat (Heterocephalus glaber; maximum lifespan: >30 years)251. Importantly virtually all of the mechanistic hypotheses related to vascular aging are tested solely in Mus musculus (one of the least successfully aging species), which is potentially a source for significant bias and limit the translatability of the results to the clinical scenario. Therefore, there is a great need for interspecies comparative studies using animals with disparate longevity as well as human studies to understand generalized mechanisms of vascular aging and identify translationally relevant treatments for the promotion of vascular health in older adults.
The same cellular and molecular aging processes that affect arterial vessels and capillaries also affect veins and the lymphatic/glymphatic system, likely contributing to various disease pathologies. Examples include the potential role of cerebral venules in neuroinflammation, Alzheimer’s disease and cerebral microhemorrhages46 and the potential link between age-related glymphatic dysfunction and amyloid pathologies252. These areas are attractive targets for future studies.
Taken together, instead of targeting a single disease, interventions that target fundamental aging processes have the potential to prevent/delay a range of vascular pathologies and other age-related diseases simultaneously. In recent years a variety of candidate drugs/interventions have emerged from basic research and translational studies that may target aging processes. These interventions can be adapted for prevention/treatment of age-related vascular pathologies.
Acknowledgement
This work was supported by grants from the American Heart Association (to ST), the National Institute on Aging (NIA; R01-AG055395, R01-AG047879; R01-AG038747; 1R01AG057964–01, 1R21AG055090–01A1, 2AG013319–21, R44-AG053131, K02-AG045339, R01-AG050238), the National Institute of Neurological Disorders and Stroke (NINDS; R01-NS056218, R01-NS100782), Veterans Administration Merit Award 1 I01 BX002211–01A2, the Oklahoma Center for the Advancement of Science and Technology, the Presbyterian Health Foundation, the EU-funded grant EFOP-3.6.1–16-2016–00008, the Owens Foundation, Kleberg Foundation, San Antonio Medical Foundation and the Robert L. Bailey and daughter Lisa K. Bailey Alzheimer’s Fund in memory of Jo Nell Bailey. The authors acknowledge the support from the NIA-funded Geroscience Training Program in Oklahoma (T32AG052363).
Footnotes
Non-standard abbreviation and non-standard acronym: none
Disclosures: None
References
- 1.Health, United States, 2016: With Chartbook on Long-term Trends in Health Hyattsville (MD); 2017. [PubMed]
- 2.Heidenreich PA, Trogdon JG, Khavjou OA, Butler J, Dracup K, Ezekowitz MD, Finkelstein EA, Hong Y, Johnston SC, Khera A, Lloyd-Jones DM, Nelson SA, Nichol G, Orenstein D, Wilson PW, Woo YJ, American Heart Association Advocacy Coordinating C, Stroke C, Council on Cardiovascular R, Intervention, Council on Clinical C, Council on E, Prevention, Council on A, Thrombosis, Vascular B, Council on C, Critical C, Perioperative, Resuscitation, Council on Cardiovascular N, Council on the Kidney in Cardiovascular D, Council on Cardiovascular S, Anesthesia, Interdisciplinary Council on Quality of C and Outcomes R. Forecasting the future of cardiovascular disease in the United States: a policy statement from the American Heart Association. Circulation. 2011;123:933–44. [DOI] [PubMed] [Google Scholar]
- 3.Lakatta EG and Levy D. Arterial and Cardiac Aging: Major Shareholders in Cardiovascular Disease Enterprises: Part I: Aging Arteries: A “Set Up” for Vascular Disease. Circulation. 2003;107:139–146. [DOI] [PubMed] [Google Scholar]
- 4.Lopez-Otin C, Blasco MA, Partridge L, Serrano M and Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alfaras I, Di Germanio C, Bernier M, Csiszar A, Ungvari Z, Lakatta EG and de Cabo R. Pharmacological Strategies to Retard Cardiovascular Aging. Circ Res. 2016;118:1626–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.AlGhatrif M and Lakatta EG. The conundrum of arterial stiffness, elevated blood pressure, and aging. Curr Hypertens Rep. 2015;17:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang M, Monticone RE and Lakatta EG. Arterial aging: a journey into subclinical arterial disease. Curr Opin Nephrol Hypertens. 2010;19:201–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seals DR, Desouza CA, Donato AJ and Tanaka H. Habitual exercise and arterial aging. J Appl Physiol. 2008;105:1323–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brandes RP, Fleming I and Busse R. Endothelial aging. Cardiovasc Res. 2005;66:286–94. [DOI] [PubMed] [Google Scholar]
- 10.Lacolley P, Regnault V and Avolio AP. Smooth muscle cell and arterial aging: basic and clinical aspects. Cardiovasc Res. 2018;114:513–528. [DOI] [PubMed] [Google Scholar]
- 11.Ungvari Z, Kaley G, de Cabo R, Sonntag WE and Csiszar A. Mechanisms of vascular aging: new perspectives. J Gerontol A Biol Sci Med Sci. 2010;65:1028–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harman D Aging: A Theory Based on Free Radical and Radiation Chemistry. J Gerontol. 1956:298–300. [DOI] [PubMed] [Google Scholar]
- 13.Csiszar A, Ungvari Z, Edwards JG, Kaminski PM, Wolin MS, Koller A and Kaley G. Aging-induced phenotypic changes and oxidative stress impair coronary arteriolar function. Circ Res. 2002;90:1159–66. [DOI] [PubMed] [Google Scholar]
- 14.van der Loo B, Labugger R, Skepper JN, Bachschmid M, Kilo J, Powell JM, Palacios-Callender M, Erusalimsky JD, Quaschning T, Malinski T, Gygi D, Ullrich V and Lüscher TF. Enhanced peroxynitrite formation is associated with vascular aging. J Exp Med. 2000;192:1731–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adler A, Messina E, Sherman B, Wang Z, Huang H, Linke A and Hintze TH. NAD(P)H oxidase-generated superoxide anion accounts for reduced control of myocardial O2 consumption by NO in old Fischer 344 rats. Am J Physiol Heart Circ Physiol. 2003;285:H1015–22. [DOI] [PubMed] [Google Scholar]
- 16.Donato AJ, Eskurza I, Silver AE, Levy AS, Pierce GL, Gates PE and Seals DR. Direct evidence of endothelial oxidative stress with aging in humans: relation to impaired endothelium-dependent dilation and upregulation of nuclear factor-kappaB. Circulation research. 2007;100:1659–66. [DOI] [PubMed] [Google Scholar]
- 17.Jacobson A, Yan C, Gao Q, Rincon-Skinner T, Rivera A, Edwards J, Huang A, Kaley G and Sun D. Aging enhances pressure-induced arterial superoxide formation. Am J Physiol Heart Circ Physiol. 2007;293:H1344–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Csiszar A, Gautam T, Sosnowska D, Tarantini S, Banki E, Tucsek Z, Toth P, Losonczy G, Koller A, Reglodi D, Giles CB, Wren JD, Sonntag WE and Ungvari Z. Caloric restriction confers persistent anti-oxidative, pro-angiogenic, and anti-inflammatory effects and promotes anti-aging miRNA expression profile in cerebromicrovascular endothelial cells of aged rats. Am J Physiol Heart Circ Physiol. 2014;307:H292–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Csiszar A, Sosnowska D, Wang M, Lakatta EG, Sonntag WE and Ungvari Z. Age-associated proinflammatory secretory phenotype in vascular smooth muscle cells from the non-human primate Macaca mulatta: reversal by resveratrol treatment. J Gerontol A Biol Sci Med Sci. 2012;67:811–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun D, Huang A, Yan EH, Wu Z, Yan C, Kaminski PM, Oury TD, Wolin MS and Kaley G. Reduced release of nitric oxide to shear stress in mesenteric arteries of aged rats. Am J Physiol Heart Circ Physiol. 2004;286:H2249–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hamilton CA, Brosnan MJ, McIntyre M, Graham D and Dominiczak AF. Superoxide excess in hypertension and aging: a common cause of endothelial dysfunction. Hypertension. 2001;37:529–34. [DOI] [PubMed] [Google Scholar]
- 22.Francia P, delli Gatti C, Bachschmid M, Martin-Padura I, Savoia C, Migliaccio E, Pelicci PG, Schiavoni M, Luscher TF, Volpe M and Cosentino F. Deletion of p66shc gene protects against age-related endothelial dysfunction. Circulation. 2004;110:2889–95. [DOI] [PubMed] [Google Scholar]
- 23.Csiszar A, Labinskyy N, Orosz Z, Xiangmin Z, Buffenstein R and Ungvari Z. Vascular aging in the longest-living rodent, the naked mole-rat. Am J Physiol. 2007;293:H919–27. [DOI] [PubMed] [Google Scholar]
- 24.Ungvari ZI, Orosz Z, Labinskyy N, Rivera A, Xiangmin Z, Smith KE and Csiszar A. Increased mitochondrial H2O2 production promotes endothelial NF-kB activation in aged rat arteries. Am J Physiol Heart Circ Physiol. 2007;293:H37–47. [DOI] [PubMed] [Google Scholar]
- 25.Jablonski KL, Seals DR, Eskurza I, Monahan KD and Donato AJ. High-Dose Ascorbic Acid Infusion Abolishes Chronic Vasoconstriction and Restores Resting Leg Blood Flow in Healthy Older Men. J Appl Physiol. 2007;103:1715–21. [DOI] [PubMed] [Google Scholar]
- 26.Donato AJ, Gano LB, Eskurza I, Silver AE, Gates PE, Jablonski K and Seals DR. Vascular endothelial dysfunction with aging: endothelin-1 and endothelial nitric oxide synthase. Am J Physiol Heart Circ Physiol. 2009;297:H425–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spier SA, Delp MD, Meininger CJ, Donato AJ, Ramsey MW and Muller-Delp JM. Effects of ageing and exercise training on endothelium-dependent vasodilatation and structure of rat skeletal muscle arterioles. J Physiol. 2004;556:947–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Durrant JR, Seals DR, Connell ML, Russell MJ, Lawson BR, Folian BJ, Donato AJ and Lesniewski LA. Voluntary wheel running restores endothelial function in conduit arteries of old mice: direct evidence for reduced oxidative stress, increased superoxide dismutase activity and down-regulation of NADPH oxidase. The Journal of physiology. 2009;587:3271–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Novella S, Dantas AP, Segarra G, Novensa L, Heras M, Hermenegildo C and Medina P. Aging enhances contraction to thromboxane A2 in aorta from female senescence-accelerated mice. Age. 2013;35:117–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cardillo C, Kilcoyne CM, Cannon RO, 3rd and Panza JA. Interactions between nitric oxide and endothelin in the regulation of vascular tone of human resistance vessels in vivo. Hypertension. 2000;35:1237–41. [DOI] [PubMed] [Google Scholar]
- 31.Park L, Anrather J, Girouard H, Zhou P and Iadecola C. Nox2-derived reactive oxygen species mediate neurovascular dysregulation in the aging mouse brain. J Cereb Blood Flow Metab. 2007;27:1908–18. [DOI] [PubMed] [Google Scholar]
- 32.Toth P, Tarantini S, Tucsek Z, Ashpole NM, Sosnowska D, Gautam T, Ballabh P, Koller A, Sonntag WE, Csiszar A and Ungvari ZI. Resveratrol treatment rescues neurovascular coupling in aged mice:role of improved cerebromicrovascular endothelial function and down-regulation of NADPH oxidas. Am J Physiol Heart Circ Physiol. 2014;306:H299–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cernadas MR, Sanchez de Miguel L, Garcia-Duran M, Gonzalez-Fernandez F, Millas I, Monton M, Rodrigo J, Rico L, Fernandez P, de Frutos T, Rodriguez-Feo JA, Guerra J, Caramelo C, Casado S and Lopez F. Expression of constitutive and inducible nitric oxide synthases in the vascular wall of young and aging rats. Circ Res. 1998;83:279–86. [DOI] [PubMed] [Google Scholar]
- 34.Chou TC, Yen MH, Li CY and Ding YA. Alterations of nitric oxide synthase expression with aging and hypertension in rats. Hypertension. 1998;31:643–8. [DOI] [PubMed] [Google Scholar]
- 35.Csiszar A, Labinskyy N, Smith K, Rivera A, Orosz Z and Ungvari Z. Vasculoprotective effects of anti-tumor necrosis factor-alpha treatment in aging. Am J Pathol. 2007;170:388–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ganz P and Vita JA. Testing endothelial vasomotor function: nitric oxide, a multipotent molecule. Circulation. 2003;108:2049–53. [DOI] [PubMed] [Google Scholar]
- 37.Moncada S and Higgs A. The L-arginine-nitric oxide pathway. N Engl J Med. 1993;329:2002–12. [DOI] [PubMed] [Google Scholar]
- 38.Moncada S, Palmer RM and Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev. 1991;43:109–42. [PubMed] [Google Scholar]
- 39.Pacher P, Beckman JS and Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiological reviews. 2007;87:315–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Csiszar A, Podlutsky A, Wolin MS, Losonczy G, Pacher P and Ungvari Z. Oxidative stress and accelerated vascular aging: implications for cigarette smoking. Front Biosci. 2009;14:3128–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Csiszar A, Wang M, Lakatta EG and Ungvari ZI. Inflammation and endothelial dysfunction during aging: role of NF-{kappa}B. J Appl Physiol. 2008;105:1333–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Csiszar A, Labinskyy N, Jimenez R, Pinto JT, Ballabh P, Losonczy G, Pearson KJ, de Cabo R and Ungvari Z. Anti-oxidative and anti-inflammatory vasoprotective effects of caloric restriction in aging: role of circulating factors and SIRT1. Mech Ageing Dev. 2009;130:518–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moreau KL, Gavin KM, Plum AE and Seals DR. Ascorbic Acid Selectively Improves Large Elastic Artery Compliance in Postmenopausal Women. Hypertension %R 101161/01HYP0000165678633738c. 2005;45:1107–1112. [DOI] [PubMed] [Google Scholar]
- 44.Kaneko H, Anzai T, Morisawa M, Kohno T, Nagai T, Anzai A, Takahashi T, Shimoda M, Sasaki A, Maekawa Y, Yoshimura K, Aoki H, Tsubota K, Yoshikawa T, Okada Y, Ogawa S and Fukuda K. Resveratrol prevents the development of abdominal aortic aneurysm through attenuation of inflammation, oxidative stress, and neovascularization. Atherosclerosis. 2011;217:350–7. [DOI] [PubMed] [Google Scholar]
- 45.Toth P, Tarantini S, Springo Z, Tucsek Z, Gautam T, Giles CB, Wren JD, Koller A, Sonntag WE, Csiszar A and Ungvari Z. Aging exacerbates hypertension-induced cerebral microhemorrhages in mice: role of resveratrol treatment in vasoprotection. Aging Cell. 2015;14:400–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ungvari Z, Tarantini S, Kirkpatrick AC, Csiszar A and Prodan CI. Cerebral microhemorrhages: mechanisms, consequences, and prevention. Am J Physiol Heart Circ Physiol. 2017;312:H1128–H1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fleenor BS, Eng JS, Sindler AL, Pham BT, Kloor JD and Seals DR. Superoxide signaling in perivascular adipose tissue promotes age-related artery stiffness. Aging Cell. 2014;13:576–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Coskun PE, Ladiges W, Wolf N, Van Remmen H, Wallace DC and Rabinovitch PS. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308:1909–11. [DOI] [PubMed] [Google Scholar]
- 50.Springo Z, Tarantini S, Toth P, Tucsek Z, Koller A, Sonntag WE, Csiszar A and Ungvari Z. Aging Exacerbates Pressure-Induced Mitochondrial Oxidative Stress in Mouse Cerebral Arteries. J Gerontol A Biol Sci Med Sci. 2015;70:1355–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tarantini S, Valcarcel-Ares NM, Yabluchanskiy A, Fulop GA, Hertelendy P, Gautam T, Farkas E, Perz A, Rabinovitch PS, Sonntag WE, Csiszar A and Ungvari Z. Treatment with the mitochondrial-targeted antioxidant peptide SS-31 rescues neurovascular coupling responses and cerebrovascular endothelial function and improves cognition in aged mice. Aging Cell. 2018;17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ungvari Z, Orosz Z, Labinskyy N, Rivera A, Xiangmin Z, Smith K and Csiszar A. Increased mitochondrial H2O2 production promotes endothelial NF-kappaB activation in aged rat arteries. Am J Physiol Heart Circ Physiol. 2007;293:H37–47. [DOI] [PubMed] [Google Scholar]
- 53.Ungvari ZI, Labinskyy N, Gupte SA, Chander PN, Edwards JG and Csiszar A. Dysregulation of mitochondrial biogenesis in vascular endothelial and smooth muscle cells of aged rats. Am J Physiol Heart Circ Physiol. 2008;294:H2121–8. [DOI] [PubMed] [Google Scholar]
- 54.Csiszar A, Labinskyy N, Jimenez R, Pinto JT, Ballabh P, Losonczy G, Pearson KJ, de Cabo R and Ungvari Z. Anti-oxidative and anti-inflammatory vasoprotective effects of caloric restriction in aging: role of circulating factors and SIRT1. Mech Ageing Dev. 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Napoli C, Martin-Padura I, de Nigris F, Giorgio M, Mansueto G, Somma P, Condorelli M, Sica G, De Rosa G and Pelicci P. Deletion of the p66Shc longevity gene reduces systemic and tissue oxidative stress, vascular cell apoptosis, and early atherogenesis in mice fed a high-fat diet. Proc Natl Acad Sci U S A. 2003;100:2112–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ungvari Z, Bailey-Downs L, Gautam T, Sosnowska D, Wang M, Monticone RE, Telljohann R, Pinto JT, de Cabo R, Sonntag WE, Lakatta E and Csiszar A. Age-associated vascular oxidative stress, Nrf2 dysfunction and NF-kB activation in the non-human primate Macaca mulatta J Gerontol A Biol Sci Med Sci. 2011;66:866–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ungvari Z, Bailey-Downs L, Sosnowska D, Gautam T, Koncz P, Losonczy G, Ballabh P, de Cabo R, Sonntag WE and Csiszar A. Vascular oxidative stress in aging: a homeostatic failure due to dysregulation of Nrf2-mediated antioxidant response Am J Physiol Heart Circ Physiol. 2011;301:H363–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Burns EM, Kruckeberg TW, Comerford LE and Buschmann MT. Thinning of capillary walls and declining numbers of endothelial mitochondria in the cerebral cortex of the aging primate, Macaca nemestrina. J Gerontol. 1979;34:642–50. [DOI] [PubMed] [Google Scholar]
- 59.Burns EM, Kruckeberg TW and Gaetano PK. Changes with age in cerebral capillary morphology. Neurobiol Aging. 1981;2:283–91. [DOI] [PubMed] [Google Scholar]
- 60.Gioscia-Ryan RA, LaRocca TJ, Sindler AL, Zigler MC, Murphy MP and Seals DR. Mitochondria-targeted antioxidant (MitoQ) ameliorates age-related arterial endothelial dysfunction in mice. J Physiol. 2014;592:2549–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pearson KJ, Baur JA, Lewis KN, Peshkin L, Price NL, Labinskyy N, Swindell WR, Kamara D, Minor RK, Perez E, Jamieson HA, Zhang Y, Dunn SR, Sharma K, Pleshko N, Woollett LA, Csiszar A, Ikeno Y, Le Couteur D, Elliott PJ, Becker KG, Navas P, Ingram DK, Wolf NS, Ungvari Z, Sinclair DA and de Cabo R. Resveratrol Delays Age-Related Deterioration and Mimics Transcriptional Aspects of Dietary Restriction without Extending Life Span. Cell Metab. 2008;8:157–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ungvari Z, Labinskyy N, Mukhopadhyay P, Pinto JT, Bagi Z, Ballabh P, Zhang C, Pacher P and Csiszar A. Resveratrol attenuates mitochondrial oxidative stress in coronary arterial endothelial cells. Am J Physiol Heart Circ Physiol. 2009;297:H1876–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rice KM, Preston DL, Walker EM and Blough ER. Aging influences multiple incidices of oxidative stress in the aortic media of the Fischer 344/NNiaxBrown Norway/BiNia rat. Free Radic Res. 2006;40:185–97. [DOI] [PubMed] [Google Scholar]
- 64.Wenzel P, Schuhmacher S, Kienhofer J, Muller J, Hortmann M, Oelze M, Schulz E, Treiber N, Kawamoto T, Scharffetter-Kochanek K, Munzel T, Burkle A, Bachschmid MM and Daiber A. Manganese superoxide dismutase and aldehyde dehydrogenase deficiency increase mitochondrial oxidative stress and aggravate age-dependent vascular dysfunction. Cardiovasc Res. 2008;80:280–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Botto N, Berti S, Manfredi S, Al-Jabri A, Federici C, Clerico A, Ciofini E, Biagini A and Andreassi MG. Detection of mtDNA with 4977 bp deletion in blood cells and atherosclerotic lesions of patients with coronary artery disease. Mutat Res. 2005;570:81–8. [DOI] [PubMed] [Google Scholar]
- 66.Bogliolo M, Izzotti A, De Flora S, Carli C, Abbondandolo A and Degan P. Detection of the ‘4977 bp’ mitochondrial DNA deletion in human atherosclerotic lesions. Mutagenesis. 1999;14:77–82. [DOI] [PubMed] [Google Scholar]
- 67.Yu E, Calvert PA, Mercer JR, Harrison J, Baker L, Figg NL, Kumar S, Wang JC, Hurst LA, Obaid DR, Logan A, West NE, Clarke MC, Vidal-Puig A, Murphy MP and Bennett MR. Mitochondrial DNA damage can promote atherosclerosis independently of reactive oxygen species through effects on smooth muscle cells and monocytes and correlates with higher-risk plaques in humans. Circulation. 2013;128:702–12. [DOI] [PubMed] [Google Scholar]
- 68.Csiszar A, Labinskyy N, Pinto JT, Ballabh P, Zhang H, Losonczy G, Pearson KJ, de Cabo R, Pacher P, Zhang C and Ungvari ZI. Resveratrol induces mitochondrial biogenesis in endothelial cells. Am J Physiol Heart Circ Physiol. 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE, Tsokos M, Alt FW and Finkel T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci U S A. 2008;105:3374–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gomes AP, Price NL, Ling AJ, Moslehi JJ, Montgomery MK, Rajman L, White JP, Teodoro JS, Wrann CD, Hubbard BP, Mercken EM, Palmeira CM, de Cabo R, Rolo AP, Turner N, Bell EL and Sinclair DA. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. 2013;155:1624–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Massudi H, Grant R, Braidy N, Guest J, Farnsworth B and Guillemin GJ. Age-associated changes in oxidative stress and NAD+ metabolism in human tissue. PLoS One. 2012;7:e42357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.de Picciotto NE, Gano LB, Johnson LC, Martens CR, Sindler AL, Mills KF, Imai S and Seals DR. Nicotinamide mononucleotide supplementation reverses vascular dysfunction and oxidative stress with aging in mice. Aging Cell. 2016;15:522–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mills KF, Yoshida S, Stein LR, Grozio A, Kubota S, Sasaki Y, Redpath P, Migaud ME, Apte RS, Uchida K, Yoshino J and Imai SI. Long-Term Administration of Nicotinamide Mononucleotide Mitigates Age-Associated Physiological Decline in Mice. Cell Metab. 2016;24:795–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Franceschi C, Bonafe M, Valensin S, Olivieri F, De Luca M, Ottaviani E and De Benedictis G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci. 2000;908:244–54. [DOI] [PubMed] [Google Scholar]
- 75.Csiszar A, Ungvari Z, Koller A, Edwards JG and Kaley G. Aging-induced proinflammatory shift in cytokine expression profile in rat coronary arteries. Faseb J. 2003;17:1183–5. [DOI] [PubMed] [Google Scholar]
- 76.Csiszar A, Ungvari Z, Koller A, Edwards JG and Kaley G. Proinflammatory phenotype of coronary arteries promotes endothelial apoptosis in aging. Physiol Genomics. 2004;17:21–30. [DOI] [PubMed] [Google Scholar]
- 77.Wang M, Zhang J, Jiang LQ, Spinetti G, Pintus G, Monticone R, Kolodgie FD, Virmani R and Lakatta EG. Proinflammatory profile within the grossly normal aged human aortic wall. Hypertension. 2007;50:219–27. [DOI] [PubMed] [Google Scholar]
- 78.Song Y, Shen H, Schenten D, Shan P, Lee PJ and Goldstein DR. Aging enhances the basal production of IL-6 and CCL2 in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2012;32:103–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Csiszar A, Labinskyy N, Smith K, Rivera A, Orosz Z and Ungvari Z. Vasculoprotective effects of anti-TNFalfa treatment in aging. The American journal of pathology. 2007;170:388–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Arenas IA, Xu Y and Davidge ST. Age-associated impairment in vasorelaxation to fluid shear stress in the female vasculature is improved by TNF-{alpha} antagonism. Am J Physiol Heart Circ Physiol. 2006;290:H1259–63. [DOI] [PubMed] [Google Scholar]
- 81.Hasegawa Y, Saito T, Ogihara T, Ishigaki Y, Yamada T, Imai J, Uno K, Gao J, Kaneko K, Shimosawa T, Asano T, Fujita T, Oka Y and Katagiri H. Blockade of the nuclear factor-kappaB pathway in the endothelium prevents insulin resistance and prolongs life spans. Circulation. 2012;125:1122–33. [DOI] [PubMed] [Google Scholar]
- 82.Chen HZ, Wang F, Gao P, Pei JF, Liu Y, Xu TT, Tang X, Fu WY, Lu J, Yan YF, Wang XM, Han L, Zhang ZQ, Zhang R, Zou MH and Liu DP. Age-Associated Sirtuin 1 Reduction in Vascular Smooth Muscle Links Vascular Senescence and Inflammation to Abdominal Aortic Aneurysm. Circ Res. 2016;119:1076–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gano LB, Donato AJ, Pasha HM, Hearon CM, Jr., Sindler AL and Seals DR. The SIRT1 activator SRT1720 reverses vascular endothelial dysfunction, excessive superoxide production, and inflammation with aging in mice. Am J Physiol Heart Circ Physiol. 2014;307:H1754–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ungvari Z, Podlutsky A, Sosnowska D, Tucsek Z, Toth P, Deak F, Gautam T, Csiszar A and Sonntag WE. Ionizing radiation promotes the acquisition of a senescence-associated secretory phenotype and impairs angiogenic capacity in cerebromicrovascular endothelial cells: role of increased DNA damage and decreased DNA repair capacity in microvascular radiosensitivity. J Gerontol A Biol Sci Med Sci. 2013;68:1443–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Freund A, Patil CK and Campisi J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011;30:1536–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kang C, Xu Q, Martin TD, Li MZ, Demaria M, Aron L, Lu T, Yankner BA, Campisi J and Elledge SJ. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science. 2015;349:aaa5612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Morgan RG, Ives SJ, Lesniewski LA, Cawthon RM, Andtbacka RH, Noyes RD, Richardson RS and Donato AJ. Age-related telomere uncapping is associated with cellular senescence and inflammation independent of telomere shortening in human arteries. Am J Physiol Heart Circ Physiol. 2013;305:H251–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Youm YH, Grant RW, McCabe LR, Albarado DC, Nguyen KY, Ravussin A, Pistell P, Newman S, Carter R, Laque A, Munzberg H, Rosen CJ, Ingram DK, Salbaum JM and Dixit VD. Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metab. 2013;18:519–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bailey-Downs LC, Tucsek Z, Toth P, Sosnowska D, Gautam T, Sonntag WE, Csiszar A and Ungvari Z. Aging exacerbates obesity-induced oxidative stress and inflammation in perivascular adipose tissue in mice: a paracrine mechanism contributing to vascular redox dysregulation and inflammation. J Gerontol A Biol Sci Med Sci. 2013;68:780–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tucsek Z, Toth P, Sosnowska D, Gautam T, Mitschelen M, Koller A, Szalai G, Sonntag WE, Ungvari Z and Csiszar A. Obesity in aging exacerbates blood-brain barrier disruption, neuroinflammation, and oxidative stress in the mouse hippocampus: effects on expression of genes involved in beta-amyloid generation and Alzheimer’s disease. J Gerontol A Biol Sci Med Sci. 2014;69:1212–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tucsek Z, Toth P, Tarantini S, Sosnowska D, Gautam T, Warrington JP, Giles CB, Wren JD, Koller A, Ballabh P, Sonntag WE, Ungvari Z and Csiszar A. Aging exacerbates obesity-induced cerebromicrovascular rarefaction, neurovascular uncoupling, and cognitive decline in mice. J Gerontol A Biol Sci Med Sci. 2014;69:1339–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Orosz Z, Csiszar A, Labinskyy N, Smith K, Kaminski PM, Ferdinandy P, Wolin MS, Rivera A and Ungvari Z. Cigarette smoke-induced proinflammatory alterations in the endothelial phenotype: role of NAD(P)H oxidase activation. American journal of physiology. 2007;292:H130–9. [DOI] [PubMed] [Google Scholar]
- 93.Staras SA, Dollard SC, Radford KW, Flanders WD, Pass RF and Cannon MJ. Seroprevalence of cytomegalovirus infection in the United States,1988–1994. Clin Infect Dis. 2006;43:1143–51. [DOI] [PubMed] [Google Scholar]
- 94.Wang GC, Kao WH, Murakami P, Xue QL, Chiou RB, Detrick B, McDyer JF, Semba RD, Casolaro V, Walston JD and Fried LP. Cytomegalovirus infection and the risk of mortality and frailty in older women: a prospective observational cohort study. Am J Epidemiol. 2010;171:1144–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ungvari ZI, Bailey-Downs L, Gautam T, Jimenez R, Losonczy G, Zhang C, Ballabh P, Recchia FA, Wilkerson DC, Sonntag WE, Pearson KJ, de Cabo R and Csiszar A. Adaptive induction of NF-E2-Related Factor-2-driven antioxidant genes in endothelial cells in response to hyperglycemia. Am J Physiol Heart Circ Physiol. 2011;300:H1133–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Suh JH, Shenvi SV, Dixon BM, Liu H, Jaiswal AK, Liu RM and Hagen TM. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc Natl Acad Sci U S A. 2004;101:3381–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, Liu P, Mostoslavsky G, Franco S, Murphy MM, Mills KD, Patel P, Hsu JT, Hong AL, Ford E, Cheng HL, Kennedy C, Nunez N, Bronson R, Frendewey D, Auerbach W, Valenzuela D, Karow M, Hottiger MO, Hursting S, Barrett JC, Guarente L, Mulligan R, Demple B, Yancopoulos GD and Alt FW. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. 2006;124:315–29. [DOI] [PubMed] [Google Scholar]
- 98.Valcarcel-Ares MN, Gautam T, Warrington JP, Bailey-Downs L, Sosnowska D, de Cabo R, Losonczy G, Sonntag WE, Ungvari Z and Csiszar A. Disruption of Nrf2 signaling impairs angiogenic capacity of endothelial cells: implications for microvascular aging J Gerontol A Biol Sci Med Sci. 2012;67:821–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tarantini S, Valcarcel-Ares MN, Yabluchanskiy A, Tucsek Z, Hertelendy P, Kiss T, Gautam T, Zhang XA, Sonntag WE, de Cabo R, Farkas E, Elliott ME, Kinter MT, Deak F, Ungvari Z and Csiszar A. Nrf2 deficiency exacerbates obesity-induced oxidative stress, neurovascular dysfunction, blood brain barrier disruption, neuroinflammation, amyloidogenic gene expression and cognitive decline in mice, mimicking the aging phenotype. J Gerontol A Biol Sci Med Sci. 2018:in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pearson KJ, Lewis KN, Price NL, Chang JW, Perez E, Cascajo MV, Tamashiro KL, Poosala S, Csiszar A, Ungvari Z, Kensler TW, Yamamoto M, Egan JM, Longo DL, Ingram DK, Navas P and de Cabo R. Nrf2 mediates cancer protection but not prolongevity induced by caloric restriction. Proc Natl Acad Sci U S A. 2008;105:2325–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kaushik S and Cuervo AM. Proteostasis and aging. Nat Med. 2015;21:1406–15. [DOI] [PubMed] [Google Scholar]
- 102.Blice-Baum AC, Zambon AC, Kaushik G, Viswanathan MC, Engler AJ, Bodmer R and Cammarato A. Modest overexpression of FOXO maintains cardiac proteostasis and ameliorates age-associated functional decline. Aging Cell. 2017;16:93–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Dai DF, Karunadharma PP, Chiao YA, Basisty N, Crispin D, Hsieh EJ, Chen T, Gu H, Djukovic D, Raftery D, Beyer RP, MacCoss MJ and Rabinovitch PS. Altered proteome turnover and remodeling by short-term caloric restriction or rapamycin rejuvenate the aging heart. Aging Cell. 2014;13:529–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gouveia M, Xia K, Colon W, Vieira SI and Ribeiro F. Protein aggregation, cardiovascular diseases, and exercise training: Where do we stand? Ageing Res Rev 2017;40:1–10. [DOI] [PubMed] [Google Scholar]
- 105.Chin JH, Okazaki M, Hu ZW, Miller JW and Hoffman BB. Activation of heat shock protein (hsp)70 and proto-oncogene expression by alpha1 adrenergic agonist in rat aorta with age. J Clin Invest. 1996;97:2316–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nussenzweig SC, Verma S and Finkel T. The role of autophagy in vascular biology. Circ Res. 2015;116:480–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bharath LP, Mueller R, Li Y, Ruan T, Kunz D, Goodrich R, Mills T, Deeter L, Sargsyan A, Anandh Babu PV, Graham TE and Symons JD. Impairment of autophagy in endothelial cells prevents shear-stress-induced increases in nitric oxide bioavailability. Can J Physiol Pharmacol. 2014;92:605–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.LaRocca TJ, Henson GD, Thorburn A, Sindler AL, Pierce GL and Seals DR. Translational evidence that impaired autophagy contributes to arterial ageing. J Physiol. 2012;590:3305–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.LaRocca TJ, Gioscia-Ryan RA, Hearon CM, Jr. and Seals DR. The autophagy enhancer spermidine reverses arterial aging. Mech Ageing Dev. 2013;134:314–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Marfella R, Di Filippo C, Laieta MT, Vestini R, Barbieri M, Sangiulo P, Crescenzi B, Ferraraccio F, Rossi F, D’Amico M and Paolisso G. Effects of ubiquitin-proteasome system deregulation on the vascular senescence and atherosclerosis process in elderly patients. J Gerontol A Biol Sci Med Sci. 2008;63:200–3. [DOI] [PubMed] [Google Scholar]
- 111.Bulteau AL, Szweda LI and Friguet B. Age-dependent declines in proteasome activity in the heart. Arch Biochem Biophys. 2002;397:298–304. [DOI] [PubMed] [Google Scholar]
- 112.Wilck N and Ludwig A. Targeting the ubiquitin-proteasome system in atherosclerosis: status quo, challenges, and perspectives. Antioxid Redox Signal. 2014;21:2344–63. [DOI] [PubMed] [Google Scholar]
- 113.Szilard L On the Nature of the Aging Process. Proc Natl Acad Sci U S A. 1959;45:30 –45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Moskalev AA, Shaposhnikov MV, Plyusnina EN, Zhavoronkov A, Budovsky A, Yanai H and Fraifeld VE. The role of DNA damage and repair in aging through the prism of Koch-like criteria. Ageing Res Rev. 2013;12:661–84. [DOI] [PubMed] [Google Scholar]
- 115.Shah AV and Bennett MR. DNA damage-dependent mechanisms of ageing and disease in the macro- and microvasculature. Eur J Pharmacol. 2017;816:116–128. [DOI] [PubMed] [Google Scholar]
- 116.Ashpole NM, Warrington JP, Mitschelen MC, Yan H, Sosnowska D, Gautam T, Farley JA, Csiszar A, Ungvari Z and Sonntag WE. Systemic influences contribute to prolonged microvascular rarefaction after brain irradiation: a role for endothelial progenitor cells. Am J Physiol Heart Circ Physiol. 2014;307:H858–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ungvari Z, Tarantini S, Hertelendy P, Valcarcel-Ares MN, Fülöp GÁ, Logan S, Kiss T, Farkas E, Csiszar A and Yabluchanskiy A. Cerebromicrovascular dysfunction predicts cognitive decline and gait abnormalities in a mouse model of whole brain irradiation-induced accelerated brain senescence. GeroScience. 2017;39:33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Uryga AK and Bennett MR. Ageing induced vascular smooth muscle cell senescence in atherosclerosis. J Physiol. 2016;594:2115–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Durik M, Kavousi M, van der Pluijm I, Isaacs A, Cheng C, Verdonk K, Loot AE, Oeseburg H, Bhaggoe UM, Leijten F, van Veghel R, de Vries R, Rudez G, Brandt R, Ridwan YR, van Deel ED, de Boer M, Tempel D, Fleming I, Mitchell GF, Verwoert GC, Tarasov KV, Uitterlinden AG, Hofman A, Duckers HJ, van Duijn CM, Oostra BA, Witteman JC, Duncker DJ, Danser AH, Hoeijmakers JH and Roks AJ. Nucleotide excision DNA repair is associated with age-related vascular dysfunction. Circulation. 2012;126:468–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Matsumoto T, Baker DJ, d’Uscio LV, Mozammel G, Katusic ZS and van Deursen JM. Aging-associated vascular phenotype in mutant mice with low levels of BubR1. Stroke. 2007;38:1050–6. [DOI] [PubMed] [Google Scholar]
- 121.Martinet W, Knaapen MW, De Meyer GR, Herman AG and Kockx MM. Elevated levels of oxidative DNA damage and DNA repair enzymes in human atherosclerotic plaques. Circulation. 2002;106:927–32. [DOI] [PubMed] [Google Scholar]
- 122.Gray K, Kumar S, Figg N, Harrison J, Baker L, Mercer J, Littlewood T and Bennett M. Effects of DNA damage in smooth muscle cells in atherosclerosis. Circ Res. 2015;116:816 –26. [DOI] [PubMed] [Google Scholar]
- 123.Olive M, Harten I, Mitchell R, Beers JK, Djabali K, Cao K, Erdos MR, Blair C, Funke B, Smoot L, Gerhard-Herman M, Machan JT, Kutys R, Virmani R, Collins FS, Wight TN, Nabel EG and Gordon LB. Cardiovascular pathology in Hutchinson-Gilford progeria: correlation with the vascular pathology of aging. Arterioscler Thromb Vasc Biol. 2010;30:2301 –9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Varga R, Eriksson M, Erdos MR, Olive M, Harten I, Kolodgie F, Capell BC, Cheng J, Faddah D, Perkins S, Avallone H, San H, Qu X, Ganesh S, Gordon LB, Virmani R, Wight TN, Nabel EG and Collins FS. Progressive vascular smooth muscle cell defects in a mouse model of Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2006;103:3250 –5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ragnauth CD, Warren DT, Liu Y, McNair R, Tajsic T, Figg N, Shroff R, Skepper J and Shanahan CM. Prelamin A acts to accelerate smooth muscle cell senescence and is a novel biomarker of human vascular aging. Circulation. 2010;121:2200 –10. [DOI] [PubMed] [Google Scholar]
- 126.Morgan RG, Ives SJ, Walker AE, Cawthon RM, Andtbacka RH, Noyes D, Lesniewski LA, Richardson RS and Donato AJ. Role of arterial telomere dysfunction in hypertension: relative contributions of telomere shortening and telomere uncapping. J Hypertens. 2014;32:1293 –9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Erusalimsky JD. Vascular endothelial senescence: from mechanisms to pathophysiology. J Appl Physiol. 2009;106:326 –32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness RA, Jeganathan KB, Verzosa GC, Pezeshki A, Khazaie K, Miller JD and van Deursen JM. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature. 2016;530:184 –9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Jeon OH, Kim C, Laberge RM, Demaria M, Rathod S, Vasserot AP, Chung JW, Kim DH, Poon Y, David N, Baker DJ, van Deursen JM, Campisi J and Elisseeff JH. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat Med. 2017;23:775 –781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Roos CM, Zhang B, Palmer AK, Ogrodnik MB, Pirtskhalava T, Thalji NM, Hagler M, Jurk D, Smith LA, Casaclang-Verzosa G, Zhu Y, Schafer MJ, Tchkonia T, Kirkland JL and Miller JD. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell. 2016;15:973 –7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bhayadia R, Schmidt BM, Melk A and Homme M. Senescence-Induced Oxidative Stress Causes Endothelial Dysfunction. J Gerontol A Biol Sci Med Sci. 2016;71:161 –9. [DOI] [PubMed] [Google Scholar]
- 132.Rossman MJ, Kaplon RE, Hill SD, McNamara MN, Santos-Parker JR, Pierce GL, Seals DR and Donato AJ. Endothelial cell senescence with aging in healthy humans: prevention by habitual exercise and relation to vascular endothelial function. Am J Physiol Heart Circ Physiol. 2017;313:H890 –H895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Gevaert AB, Shakeri H, Leloup AJ, Van Hove CE, De Meyer GRY, Vrints CJ, Lemmens K and Van Craenenbroeck EM. Endothelial Senescence Contributes to Heart Failure With Preserved Ejection Fraction in an Aging Mouse Model. Circ Heart Fail. 2017;10. [DOI] [PubMed] [Google Scholar]
- 134.Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J and van Deursen JM. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science. 2016;354:472 –477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gardner SE, Humphry M, Bennett MR and Clarke MC. Senescent Vascular Smooth Muscle Cells Drive Inflammation Through an Interleukin-1alpha-Dependent Senescence-Associated Secretory Phenotype. Arterioscler Thromb Vasc Biol. 2015;35:1963 –74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Joaquin AM and Gollapudi S. Functional decline in aging and disease: a role for apoptosis. J Am Geriatr Soc. 2001;49:1234 –40. [DOI] [PubMed] [Google Scholar]
- 137.Asai K, Kudej RK, Shen YT, Yang GP, Takagi G, Kudej AB, Geng YJ, Sato N, Nazareno JB, Vatner DE, Natividad F, Bishop SP and Vatner SF. Peripheral vascular endothelial dysfunction and apoptosis in old monkeys. Arterioscler Thromb Vasc Biol. 2000;20:1493–9. [DOI] [PubMed] [Google Scholar]
- 138.Meng L, Jin W and Wang X. RIP3-mediated necrotic cell death accelerates systematic inflammation and mortality. Proc Natl Acad Sci U S A. 2015;112:11007–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Deepa SS, Unnikrishnan A, Matyi S, Hadad N and Richardson A. Necroptosis increases with age and is reduced by dietary restriction. Aging Cell. 2018:e12770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wang Q, Liu Z, Ren J, Morgan S, Assa C and Liu B. Receptor-interacting protein kinase 3 contributes to abdominal aortic aneurysms via smooth muscle cell necrosis and inflammation. Circ Res. 2015;116:600 –11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Costantino S, Camici GG, Mohammed SA, Volpe M, Luscher TF and Paneni F. Epigenetics and cardiovascular regenerative medicine in the elderly. Int J Cardiol. 2018;250:207–214. [DOI] [PubMed] [Google Scholar]
- 142.Unnikrishnan A, Jackson J, Matyi SA, Hadad N, Wronowski B, Georgescu C, Garrett KP, Wren JD, Freeman WM and Richardson A. Role of DNA methylation in the dietary restriction mediated cellular memory. Geroscience. 2017;39:331–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Nguyen A, Leblond F, Mamarbachi M, Geoffroy S and Thorin E. Age-Dependent Demethylation of Sod2 Promoter in the Mouse Femoral Artery. Oxid Med Cell Longev. 2016;2016:8627384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Connelly JJ, Cherepanova OA, Doss JF, Karaoli T, Lillard TS, Markunas CA, Nelson S, Wang T, Ellis PD, Langford CF, Haynes C, Seo DM, Goldschmidt-Clermont PJ, Shah SH, Kraus WE, Hauser ER and Gregory SG. Epigenetic regulation of COL15A1 in smooth muscle cell replicative aging and atherosclerosis. Hum Mol Genet. 2013;22:5107–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Toghill BJ, Saratzis A, Harrison SC, Verissimo AR, Mallon EB and Bown MJ. The potential role of DNA methylation in the pathogenesis of abdominal aortic aneurysm. Atherosclerosis. 2015;241:121–9. [DOI] [PubMed] [Google Scholar]
- 146.Donato AJ, Magerko KA, Lawson BR, Durrant JR, Lesniewski LA and Seals DR. SIRT-1 and vascular endothelial dysfunction with ageing in mice and humans. J Physiol. 2011;589:4545–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Maeng YS, Kwon JY, Kim EK and Kwon YG. Heterochromatin Protein 1 Alpha (HP1alpha: CBX5) is a Key Regulator in Differentiation of Endothelial Progenitor Cells to Endothelial Cells. Stem Cells. 2015;33:1512–22. [DOI] [PubMed] [Google Scholar]
- 148.Suarez Y and Sessa WC. MicroRNAs as novel regulators of angiogenesis. Circ Res. 2009;104:442–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Feinberg MW and Moore KJ. MicroRNA Regulation of Atherosclerosis. Circ Res. 2016;118:703–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Gareri C, De Rosa S and Indolfi C. MicroRNAs for Restenosis and Thrombosis After Vascular Injury. Circ Res. 2016;118:1170–84. [DOI] [PubMed] [Google Scholar]
- 151.Ungvari Z, Tucsek Z, Sosnowska D, Toth P, Gautam T, Podlutsky A, Csiszar A, Losonczy G, Valcarcel-Ares MN and Sonntag WE. Aging-Induced Dysregulation of Dicer1-Dependent MicroRNA Expression Impairs Angiogenic Capacity of Rat Cerebromicrovascular Endothelial Cells. J Gerontol A Biol Sci Med Sci. 2013;68:877–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Menghini R, Stohr R and Federici M. MicroRNAs in vascular aging and atherosclerosis. Ageing Res Rev. 2014;17:68–78. [DOI] [PubMed] [Google Scholar]
- 153.Tarantini S, Giles CB, Wren JD, Ashpole NM, Valcarcel-Ares MN, Wei JY, Sonntag WE, Ungvari Z and Csiszar A. IGF-1 deficiency in a critical period early in life influences the vascular aging phenotype in mice by altering miRNA-mediated post-transcriptional gene regulation: implications for the developmental origins of health and disease hypothesis. Age (Dordr). 2016;38:239–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Gupta SK, Piccoli MT and Thum T. Non-coding RNAs in cardiovascular ageing. Ageing Res Rev. 2014;17:79–85. [DOI] [PubMed] [Google Scholar]
- 155.Boon RA, Hofmann P, Michalik KM, Lozano-Vidal N, Berghauser D, Fischer A, Knau A, Jae N, Schurmann C and Dimmeler S. Long Noncoding RNA Meg3 Controls Endothelial Cell Aging and Function: Implications for Regenerative Angiogenesis. J Am Coll Cardiol. 2016;68:2589–2591. [DOI] [PubMed] [Google Scholar]
- 156.Sabatini DM. Twenty-five years of mTOR: Uncovering the link from nutrients to growth. Proc Natl Acad Sci U S A. 2017;114:11818–11825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Johnson SC, Sangesland M, Kaeberlein M and Rabinovitch PS. Modulating mTOR in aging and health. Interdiscip Top Gerontol. 2015;40:107–27. [DOI] [PubMed] [Google Scholar]
- 158.Caccamo A, Majumder S, Richardson A, Strong R and Oddo S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J Biol Chem. 2010;285:13107–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lin AL, Zheng W, Halloran JJ, Burbank RR, Hussong SA, Hart MJ, Javors M, Shih YY, Muir E, Solano Fonseca R, Strong R, Richardson AG, Lechleiter JD, Fox PT and Galvan V. Chronic rapamycin restores brain vascular integrity and function through NO synthase activation and improves memory in symptomatic mice modeling Alzheimer’s disease. J Cereb Blood Flow Metab. 2013;33:1412–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Wang CY, Kim HH, Hiroi Y, Sawada N, Salomone S, Benjamin LE, Walsh K, Moskowitz MA and Liao JK. Obesity increases vascular senescence and susceptibility to ischemic injury through chronic activation of Akt and mTOR. Sci Signal. 2009;2:ra11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Yepuri G, Velagapudi S, Xiong Y, Rajapakse AG, Montani JP, Ming XF and Yang Z. Positive crosstalk between arginase-II and S6K1 in vascular endothelial inflammation and aging. Aging Cell. 2012;11:1005–16. [DOI] [PubMed] [Google Scholar]
- 162.Parlar A, Can C, Erol A and Ulker S. Posttransplantation therapeutic rapamycin concentration protects nitric oxide-related vascular endothelial function: comparative effects in rat thoracic aorta and coronary endothelial cell culture. Transplant Proc. 2010;42:1923–30. [DOI] [PubMed] [Google Scholar]
- 163.Cheng C, Tempel D, Oostlander A, Helderman F, Gijsen F, Wentzel J, van Haperen R, Haitsma DB, Serruys PW, van der Steen AF, de Crom R and Krams R. Rapamycin modulates the eNOS vs. shear stress relationship. Cardiovasc Res. 2008;78:123–9. [DOI] [PubMed] [Google Scholar]
- 164.Corbin F, Blaise GA, Parent M, Chen H and Daloze PM. Effect of rapamycin on rat aortic ring vasomotion. Journal of cardiovascular pharmacology. 1994;24:813–7. [DOI] [PubMed] [Google Scholar]
- 165.Milliard S, Silva A, Blaise G, Chen H, Xu D, Qi S and Daloze P. Rapamycin’s effect on vasomotion in the rat. Transplant Proc. 1998;30:1036–8. [DOI] [PubMed] [Google Scholar]
- 166.Ha JM, Yun SJ, Kim YW, Jin SY, Lee HS, Song SH, Shin HK and Bae SS. Platelet-derived growth factor regulates vascular smooth muscle phenotype via mammalian target of rapamycin complex 1. Biochemical and biophysical research communications. 2015;464:57–62. [DOI] [PubMed] [Google Scholar]
- 167.Lesniewski LA, Seals DR, Walker AE, Henson GD, Blimline MW, Trott DW, Bosshardt GC, LaRocca TJ, Lawson BR, Zigler MC and Donato AJ. Dietary rapamycin supplementation reverses age-related vascular dysfunction and oxidative stress, while modulating nutrient-sensing, cell cycle, and senescence pathways. Aging Cell. 2017;16:17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Chauhan A, Sharma U, Jagannathan NR, Reeta KH and Gupta YK. Rapamycin protects against middle cerebral artery occlusion induced focal cerebral ischemia in rats. Behav Brain Res. 2011;225:603–9. [DOI] [PubMed] [Google Scholar]
- 169.Zhang W, Khatibi NH, Yamaguchi-Okada M, Yan J, Chen C, Hu Q, Meng H, Han H, Liu S and Zhou C. Mammalian target of rapamycin (mTOR) inhibition reduces cerebral vasospasm following a subarachnoid hemorrhage injury in canines. Experimental neurology. 2012;233:799–806. [DOI] [PubMed] [Google Scholar]
- 170.Fletcher L, Evans TM, Watts LT, Jimenez DF and Digicaylioglu M. Rapamycin treatment improves neuron viability in an in vitro model of stroke. PLoS One. 2013;8:e68281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Yin L, Ye S, Chen Z and Zeng Y. Rapamycin preconditioning attenuates transient focal cerebral ischemia/reperfusion injury in mice. Int J Neurosci. 2012;122:748–56. [DOI] [PubMed] [Google Scholar]
- 172.Spilman P, Podlutskaya N, Hart MJ, Debnath J, Gorostiza O, Bredesen D, Richardson A, Strong R and Galvan V. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. PLoS One. 2010;5:e9979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Lin AL, Jahrling JB, Zhang W, DeRosa N, Bakshi V, Romero P, Galvan V and Richardson A. Rapamycin rescues vascular, metabolic and learning deficits in apolipoprotein E4 transgenic mice with pre-symptomatic Alzheimer’s disease. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Van Skike CE, Jahrling JB, Olson AB, Sayre NL, Hussong SA, Ungvari ZI, Lechleiter JD and Galvan V. Inhibition of mTOR protects the blood-brain barrier in models of Alzheimer’s disease and vascular cognitive impairment. Am J Physiol Heart Circ Physiol. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Van Skike CE and Galvan V. A Perfect sTORm: The Role of the Mammalian Target of Rapamycin (mTOR) in Cerebrovascular Dysfunction of Alzheimer’s Disease: A Mini-Review. Gerontology. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Jahrling JB, Lin AL, DeRosa N, Hussong SA, Van Skike CE, Girotti M, Javors M, Zhao Q, Maslin LA, Asmis R and Galvan V. mTOR drives cerebral blood flow and memory deficits in LDLR(−/−) mice modeling atherosclerosis and vascular cognitive impairment. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2018;38:58–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Fry JL, Al Sayah L, Weisbrod RM, Van Roy I, Weng X, Cohen RA, Bachschmid MM and Seta F. Vascular Smooth Muscle Sirtuin-1 Protects Against Diet-Induced Aortic Stiffness. Hypertension. 2016;68:775–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Chen YX, Zhang M, Cai Y, Zhao Q and Dai W. The Sirt1 activator SRT1720 attenuates angiotensin II-induced atherosclerosis in apoE(−)/(−) mice through inhibiting vascular inflammatory response. Biochem Biophys Res Commun. 2015;465:732–8. [DOI] [PubMed] [Google Scholar]
- 179.Mattison JA, Wang M, Bernier M, Zhang J, Park SS, Maudsley S, An SS, Santhanam L, Martin B, Faulkner S, Morrell C, Baur JA, Peshkin L, Sosnowska D, Csiszar A, Herbert RL, Tilmont EM, Ungvari Z, Pearson KJ, Lakatta EG and de Cabo R. Resveratrol prevents high fat/sucrose diet-induced central arterial wall inflammation and stiffening in nonhuman primates. Cell Metab. 2014;20:183–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Baur JA, Ungvari Z, Minor RK, Le Couteur DG and de Cabo R. Are sirtuins viable targets for improving healthspan and lifespan? Nat Rev Drug Discov. 2012;11:443–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Liu Y, Wang TT, Zhang R, Fu WY, Wang X, Wang F, Gao P, Ding YN, Xie Y, Hao DL, Chen HZ and Liu DP. Calorie restriction protects against experimental abdominal aortic aneurysms in mice. J Exp Med. 2016;213:2473–2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Cardus A, Uryga AK, Walters G and Erusalimsky JD. SIRT6 protects human endothelial cells from DNA damage, telomere dysfunction, and senescence. Cardiovasc Res. 2013;97:571–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Xu S, Yin M, Koroleva M, Mastrangelo MA, Zhang W, Bai P, Little PJ and Jin ZG. SIRT6 protects against endothelial dysfunction and atherosclerosis in mice. Aging (Albany NY). 2016;8:1064–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Gorenne I, Kumar S, Gray K, Figg N, Yu H, Mercer J and Bennett M. Vascular smooth muscle cell sirtuin 1 protects against DNA damage and inhibits atherosclerosis. Circulation. 2013;127:386–96. [DOI] [PubMed] [Google Scholar]
- 185.Das DK and Maulik N. Resveratrol in cardioprotection: a therapeutic promise of alternative medicine. Mol Interv. 2006;6:36–47. [DOI] [PubMed] [Google Scholar]
- 186.Stivala LA, Savio M, Carafoli F, Perucca P, Bianchi L, Maga G, Forti L, Pagnoni UM, Albini A, Prosperi E and Vannini V. Specific structural determinants are responsible for the antioxidant activity and the cell cycle effects of resveratrol. J Biol Chem. 2001;276:22586–94. [DOI] [PubMed] [Google Scholar]
- 187.Yoshino J, Mills KF, Yoon MJ and Imai S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011;14:528–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Hattori Y, Suzuki K, Hattori S and Kasai K. Metformin inhibits cytokine-induced nuclear factor kappaB activation via AMP-activated protein kinase activation in vascular endothelial cells. Hypertension. 2006;47:1183–8. [DOI] [PubMed] [Google Scholar]
- 189.Hardie DG, Hawley SA and Scott JW. AMP-activated protein kinase--development of the energy sensor concept. J Physiol. 2006;574:7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Barzilai N, Crandall JP, Kritchevsky SB and Espeland MA. Metformin as a Tool to Target Aging. Cell Metab. 2016;23:1060–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Chen ZP, Mitchelhill KI, Michell BJ, Stapleton D, Rodriguez-Crespo I, Witters LA, Power DA, Ortiz de Montellano PR and Kemp BE. AMP-activated protein kinase phosphorylation of endothelial NO synthase. FEBS Lett. 1999;443:285–9. [DOI] [PubMed] [Google Scholar]
- 192.Chen Z, Peng IC, Sun W, Su MI, Hsu PH, Fu Y, Zhu Y, DeFea K, Pan S, Tsai MD and Shyy JY. AMP-activated protein kinase functionally phosphorylates endothelial nitric oxide synthase Ser633. Circ Res. 2009;104:496–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Levine YC, Li GK and Michel T. Agonist-modulated regulation of AMP-activated protein kinase (AMPK) in endothelial cells. Evidence for an AMPK -> Rac1 -> Akt -> endothelial nitric-oxide synthase pathway. J Biol Chem. 2007;282:20351–64. [DOI] [PubMed] [Google Scholar]
- 194.Nagata D, Mogi M and Walsh K. AMP-activated protein kinase (AMPK) signaling in endothelial cells is essential for angiogenesis in response to hypoxic stress. J Biol Chem. 2003;278:31000–6. [DOI] [PubMed] [Google Scholar]
- 195.Lesniewski LA, Zigler MC, Durrant JR, Donato AJ and Seals DR. Sustained activation of AMPK ameliorates age-associated vascular endothelial dysfunction via a nitric oxide-independent mechanism. Mech Ageing Dev. 2012;133:368–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Pu Y, Zhang H, Wang P, Zhao Y, Li Q, Wei X, Cui Y, Sun J, Shang Q, Liu D and Zhu Z. Dietary curcumin ameliorates aging-related cerebrovascular dysfunction through the AMPK/uncoupling protein 2 pathway. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology. 2013;32:1167–77. [DOI] [PubMed] [Google Scholar]
- 197.Kumar S, Dietrich N and Kornfeld K. Angiotensin Converting Enzyme (ACE) Inhibitor Extends Caenorhabditis elegans Life Span. PLoS Genet. 2016;12:e1005866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Basso N, Cini R, Pietrelli A, Ferder L, Terragno NA and Inserra F. Protective effect of long-term angiotensin II inhibition. Am J Physiol Heart Circ Physiol. 2007;293:H1351–8. [DOI] [PubMed] [Google Scholar]
- 199.de Cavanagh EM, Inserra F and Ferder L. Angiotensin II blockade: how its molecular targets may signal to mitochondria and slow aging. Coincidences with calorie restriction and mTOR inhibition. Am J Physiol Heart Circ Physiol. 2015;309:H15–44. [DOI] [PubMed] [Google Scholar]
- 200.Benigni A, Corna D, Zoja C, Sonzogni A, Latini R, Salio M, Conti S, Rottoli D, Longaretti L, Cassis P, Morigi M, Coffman TM and Remuzzi G. Disruption of the Ang II type 1 receptor promotes longevity in mice. J Clin Invest. 2009;119:524–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Spinetti G, Wang M, Monticone R, Zhang J, Zhao D and Lakatta EG. Rat aortic MCP-1 and its receptor CCR2 increase with age and alter vascular smooth muscle cell function. Arterioscler Thromb Vasc Biol. 2004;24:1397–402. [DOI] [PubMed] [Google Scholar]
- 202.Wang M, Takagi G, Asai K, Resuello RG, Natividad FF, Vatner DE, Vatner SF and Lakatta EG. Aging increases aortic MMP-2 activity and angiotensin II in nonhuman primates. Hypertension. 2003;41:1308–16. [DOI] [PubMed] [Google Scholar]
- 203.Wang M, Zhang J, Spinetti G, Jiang LQ, Monticone R, Zhao D, Cheng L, Krawczyk M, Talan M, Pintus G and Lakatta EG. Angiotensin II activates matrix metalloproteinase type II and mimics age-associated carotid arterial remodeling in young rats. Am J Pathol. 2005;167:1429–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Hayashi K, Miyagawa K, Sato K, Ueda R and Dohi Y. Temocapril, an Angiotensin converting enzyme inhibitor, ameliorates age-related increase in carotid arterial stiffness in normotensive subjects. Cardiology. 2006;106:190–4. [DOI] [PubMed] [Google Scholar]
- 205.Michel JB, Heudes D, Michel O, Poitevin P, Philippe M, Scalbert E, Corman B and Levy BI. Effect of chronic ANG I-converting enzyme inhibition on aging processes. II. Large arteries. Am J Physiol. 1994;267:R124–35. [DOI] [PubMed] [Google Scholar]
- 206.Dai DF, Rabinovitch PS and Ungvari Z. Mitochondria and cardiovascular aging. Circ Res. 2012;110:1109–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Toth P, Tucsek Z, Sosnowska D, Gautam T, Mitschelen M, Tarantini S, Deak F, Koller A, Sonntag WE, Csiszar A and Ungvari Z. Age-related autoregulatory dysfunction and cerebromicrovascular injury in mice with angiotensin II-induced hypertension. J Cereb Blood Flow Metab. 2013;33:1732–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Krug AW, Allenhofer L, Monticone R, Spinetti G, Gekle M, Wang M and Lakatta EG. Elevated mineralocorticoid receptor activity in aged rat vascular smooth muscle cells promotes a proinflammatory phenotype via extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase and epidermal growth factor receptor-dependent pathways. Hypertension. 2010;55:1476–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Ewald CY, Landis JN, Porter Abate J, Murphy CT and Blackwell TK. Dauer-independent insulin/IGF-1-signalling implicates collagen remodelling in longevity. Nature. 2015;519:97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Phillip JM, Aifuwa I, Walston J and Wirtz D. The Mechanobiology of Aging. Annu Rev Biomed Eng. 2015;17:113–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Jacob MP. Extracellular matrix remodeling and matrix metalloproteinases in the vascular wall during aging and in pathological conditions. Biomed Pharmacother. 2003;57:195–202. [DOI] [PubMed] [Google Scholar]
- 212.Tarantini S, Valcarcel-Ares NM, Yabluchanskiy A, Springo Z, Fulop GA, Ashpole N, Gautam T, Giles CB, Wren JD, Sonntag WE, Csiszar A and Ungvari Z. Insulin-like growth factor 1 deficiency exacerbates hypertension-induced cerebral microhemorrhages in mice, mimicking the aging phenotype. Aging Cell. 2017;16:469–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Dao HH, Essalihi R, Bouvet C and Moreau P. Evolution and modulation of age-related medial elastocalcinosis: impact on large artery stiffness and isolated systolic hypertension. Cardiovasc Res. 2005;66:307–17. [DOI] [PubMed] [Google Scholar]
- 214.Cantini C, Kieffer P, Corman B, Liminana P, Atkinson J and Lartaud-Idjouadiene I. Aminoguanidine and aortic wall mechanics, structure, and composition in aged rats. Hypertension. 2001;38:943–8. [DOI] [PubMed] [Google Scholar]
- 215.Scuteri A, Morrell CH, Orru M, Strait JB, Tarasov KV, Ferreli LA, Loi F, Pilia MG, Delitala A, Spurgeon H, Najjar SS, AlGhatrif M and Lakatta EG. Longitudinal perspective on the conundrum of central arterial stiffness, blood pressure, and aging. Hypertension. 2014;64:1219–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Rosenberg GA. Extracellular matrix inflammation in vascular cognitive impairment and dementia. Clin Sci (Lond). 2017;131:425–437. [DOI] [PubMed] [Google Scholar]
- 217.Pascual G, Mendieta C, Garcia-Honduvilla N, Corrales C, Bellon JM and Bujan J. TGF-beta1 upregulation in the aging varicose vein. J Vasc Res. 2007;44:192–201. [DOI] [PubMed] [Google Scholar]
- 218.Khan AS, Lynch CD, Sane DC, Willingham MC and Sonntag WE. Growth hormone increases regional coronary blood flow and capillary density in aged rats. J Gerontol A Biol Sci Med Sci. 2001;56:B364–71. [DOI] [PubMed] [Google Scholar]
- 219. Tarantini S, Tucsek Z, Valcarcel-Ares MN, Toth P, Gautam T, Giles CB, Ballabh P, Wei JY, Wren JD, Ashpole NM, Sonntag WE, Ungvari Z and Csiszar A. Circulating IGF-1 deficiency exacerbates hypertension-induced microvascular rarefaction in the mouse hippocampus and retrosplenial cortex: implications for cerebromicrovascular and brain aging. Age (Dordr). 2016;38:273–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Toth P, Tarantini S, Ashpole NM, Tucsek Z, Milne GL, Valcarcel-Ares NM, Menyhart A, Farkas E, Sonntag WE, Csiszar A and Ungvari Z. IGF-1 deficiency impairs neurovascular coupling in mice: implications for cerebromicrovascular aging. Aging Cell. 2015;14:1034–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Toth P, Tucsek Z, Tarantini S, Sosnowska D, Gautam T, Mitschelen M, Koller A, Sonntag WE, Csiszar A and Ungvari Z. IGF-1 deficiency impairs cerebral myogenic autoregulation in hypertensive mice. J Cereb Blood Flow Metab. 2014;34:1887–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Higashi Y, Sukhanov S, Anwar A, Shai SY and Delafontaine P. Aging, atherosclerosis, and IGF-1. J Gerontol A Biol Sci Med Sci. 2012;67:626–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Lahteenvuo J and Rosenzweig A. Effects of aging on angiogenesis. Circ Res. 2012;110:1252–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Katsimpardi L, Litterman NK, Schein PA, Miller CM, Loffredo FS, Wojtkiewicz GR, Chen JW, Lee RT, Wagers AJ and Rubin LL. Vascular and neurogenic rejuvenation of the aging mouse brain by young systemic factors. Science. 2014;344:630–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Ingraham JP, Forbes ME, Riddle DR and Sonntag WE. Aging reduces hypoxia-induced microvascular growth in the rodent hippocampus. J Gerontol A Biol Sci Med Sci. 2008;63:12–20. [DOI] [PubMed] [Google Scholar]
- 226.Ungvari Z, Parrado-Fernandez C, Csiszar A and de Cabo R. Mechanisms underlying caloric restriction and lifespan regulation: implications for vascular aging. Circ Res. 2008;102:519–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Nisoli E, Tonello C, Cardile A, Cozzi V, Bracale R, Tedesco L, Falcone S, Valerio A, Cantoni O, Clementi E, Moncada S and Carruba MO. Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science. 2005;310:314–7. [DOI] [PubMed] [Google Scholar]
- 228.Kondo M, Shibata R, Miura R, Shimano M, Kondo K, Li P, Ohashi T, Kihara S, Maeda N, Walsh K, Ouchi N and Murohara T. Caloric restriction stimulates revascularization in response to ischemia via adiponectin-mediated activation of endothelial nitric-oxide synthase. J Biol Chem. 2009;284:1718–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Pierce GL, Beske SD, Lawson BR, Southall KL, Benay FJ, Donato AJ and Seals DR. Weight loss alone improves conduit and resistance artery endothelial function in young and older overweight/obese adults. Hypertension. 2008;52:72–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Csiszar A, Sosnowska D, Tucsek Z, Gautam T, Toth P, Losonczy G, Colman RJ, Weindruch R, Anderson RM, Sonntag WE and Ungvari Z. Circulating factors induced by caloric restriction in the nonhuman primate Macaca mulatta activate angiogenic processes in endothelial cells. J Gerontol A Biol Sci Med Sci. 2013;68:235–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Allard JS, Heilbronn LK, Smith C, Hunt ND, Ingram DK, Ravussin E and de Cabo R. In vitro cellular adaptations of indicators of longevity in response to treatment with serum collected from humans on calorie restricted diets. PLoS ONE. 2008;3:e3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Shinmura K, Tamaki K and Bolli R. Impact of 6-mo caloric restriction on myocardial ischemic tolerance: possible involvement of nitric oxide-dependent increase in nuclear Sirt1. Am J Physiol Heart Circ Physiol. 2008;295:H2348–H2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Shinmura K, Tamaki K, Saito K, Nakano Y, Tobe T and Bolli R. Cardioprotective effects of short-term caloric restriction are mediated by adiponectin via activation of AMP-activated protein kinase. Circulation. 2007;116:2809–17. [DOI] [PubMed] [Google Scholar]
- 234.Niemann B, Silber RE and Rohrbach S. Age-specific effects of short- and long-term caloric restriction on the expression of adiponectin and adiponectin receptors: influence of intensity of food restriction. Exp Gerontol. 2008;43:706–13. [DOI] [PubMed] [Google Scholar]
- 235.Keymel S, Kalka C, Rassaf T, Yeghiazarians Y, Kelm M and Heiss C. Impaired endothelial progenitor cell function predicts age-dependent carotid intimal thickening. Basic Res Cardiol. 2008;103:582–6. [DOI] [PubMed] [Google Scholar]
- 236.Heiss C, Keymel S, Niesler U, Ziemann J, Kelm M and Kalka C. Impaired progenitor cell activity in age-related endothelial dysfunction. J Am Coll Cardiol. 2005;45:1441–8. [DOI] [PubMed] [Google Scholar]
- 237.Rauscher FM, Goldschmidt-Clermont PJ, Davis BH, Wang T, Gregg D, Ramaswami P, Pippen AM, Annex BH, Dong C and Taylor DA. Aging, progenitor cell exhaustion, and atherosclerosis. Circulation. 2003;108:457–63. [DOI] [PubMed] [Google Scholar]
- 238.Zhu G, Song M, Wang H, Zhao G, Yu Z, Yin Y, Zhao X and Huang L. Young environment reverses the declined activity of aged rat-derived endothelial progenitor cells: involvement of the phosphatidylinositol 3-kinase/Akt signaling pathway. Ann Vasc Surg. 2009;23:519–34. [DOI] [PubMed] [Google Scholar]
- 239.Hayek SS, MacNamara J, Tahhan AS, Awad M, Yadalam A, Ko YA, Healy S, Hesaroieh I, Ahmed H, Gray B, Sher SS, Ghasemzadeh N, Patel R, Kim J, Waller EK and Quyyumi AA. Circulating Progenitor Cells Identify Peripheral Arterial Disease in Patients With Coronary Artery Disease. Circ Res. 2016;119:564–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Mekonnen G, Hayek SS, Mehta PK, Li Q, Mahar E, Mou L, Kenkre TS, Petersen JW, Azarbal B, Samuels B, Anderson RD, Sedlak T, Zaya M, Agarwal M, Haftbaradaran A, Minissian M, Handberg E, Pepine CJ, Cogle CR, Bairey Merz CN, Waller EK and Quyyumi AA. Circulating progenitor cells and coronary microvascular dysfunction: Results from the NHLBI-sponsored Women’s Ischemia Syndrome Evaluation - Coronary Vascular Dysfunction Study (WISE-CVD). Atherosclerosis. 2016;253:111–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Dawson J, Tooze J, Cockerill G, Choke E, Loftus I and Thompson MM. Endothelial progenitor cells and abdominal aortic aneurysms. Ann N Y Acad Sci. 2006;1085:327–30. [DOI] [PubMed] [Google Scholar]
- 242.Vaughan KL, Kaiser T, Peaden R, Anson RM, de Cabo R and Mattison JA. Caloric Restriction Study Design Limitations in Rodent and Nonhuman Primate Studies. J Gerontol A Biol Sci Med Sci. 2017;73:48–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Donato AJ, Walker AE, Magerko KA, Bramwell RC, Black AD, Henson GD, Lawson BR, Lesniewski LA and Seals DR. Life-long caloric restriction reduces oxidative stress and preserves nitric oxide bioavailability and function in arteries of old mice. Aging Cell. 2013;12:772–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Edwards AG, Donato AJ, Lesniewski LA, Gioscia RA, Seals DR and Moore RL. Life-long caloric restriction elicits pronounced protection of the aged myocardium: a role for AMPK. Mech Ageing Dev. 2010;131:739–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Colman RJ, Beasley TM, Kemnitz JW, Johnson SC, Weindruch R and Anderson RM. Caloric restriction reduces age-related and all-cause mortality in rhesus monkeys. Nat Commun. 2014;5:3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Cefalu WT, Wang ZQ, Bell-Farrow AD, Collins J, Morgan T and Wagner JD. Caloric restriction and cardiovascular aging in cynomolgus monkeys (Macaca fascicularis): metabolic, physiologic, and atherosclerotic measures from a 4-year intervention trial. J Gerontol A Biol Sci Med Sci. 2004;59:1007–14. [DOI] [PubMed] [Google Scholar]
- 247.Wycherley TP, Brinkworth GD, Noakes M, Buckley JD and Clifton PM. Effect of caloric restriction with and without exercise training on oxidative stress and endothelial function in obese subjects with type 2 diabetes. Diabetes Obes Metab. 2008;10:1062–73. [DOI] [PubMed] [Google Scholar]
- 248.Csiszar A, Labinskyy N, Zhao X, Hu F, Serpillon S, Huang Z, Ballabh P, Levy RJ, Hintze TH, Wolin MS, Austad SN, Podlutsky A and Ungvari Z. Vascular superoxide and hydrogen peroxide production and oxidative stress resistance in two closely related rodent species with disparate longevity. Aging Cell. 2007;6:783–97. [DOI] [PubMed] [Google Scholar]
- 249.Labinskyy N, Mukhopadhyay P, Toth J, Szalai G, Veres M, Losonczy G, Pinto JT, Pacher P, Ballabh P, Podlutsky A, Austad SN, Csiszar A and Ungvari Z. Longevity is associated with increased vascular resistance to high glucose-induced oxidative stress and inflammatory gene expression in Peromyscus leucopus. Am J Physiol Heart Circ Physiol. 2009;296:H946–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Ungvari Z, Krasnikov BF, Csiszar A, Labinskyy N, Mukhopadhyay P, Pacher P, Cooper AJL, Podlutskaya N, Austad SN and Podlutsky A. Testing hypotheses of aging in long-lived mice of the genus Peromyscus: association between longevity and mitochondrial stress resistance, ROS detoxification pathways and DNA repair efficiency AGE. 2008;30:121–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Csiszar A, Labinskyy N, Orosz Z, Xiangmin Z, Buffenstein R and Ungvari Z. Vascular aging in the longest-living rodent, the naked mole rat. American journal of physiology. 2007;293:H919–27. [DOI] [PubMed] [Google Scholar]
- 252.Kress BT, Iliff JJ, Xia M, Wang M, Wei HS, Zeppenfeld D, Xie L, Kang H, Xu Q, Liew JA, Plog BA, Ding F, Deane R and Nedergaard M. Impairment of paravascular clearance pathways in the aging brain. Ann Neurol. 2014;76:845–61. [DOI] [PMC free article] [PubMed] [Google Scholar]