

CONSPECTUS.
Implementation of any chemical reaction in a structurally complex setting (King, S. M., J. Org. Chem., 2014, 79, 8937) confronts structurally-defined barriers: steric environment, functional group reactivity, product instability, and through-bond electronics. But there are also practical barriers. Late stage reactions conducted on small quantities of material are run inevitably at lower than optimal concentrations. Access to late stage material limits extensive optimization. Impurities from past reactions can interfere, especially with catalytic reactions. Therefore, chemical reactions that can be relied upon at the front lines of a complex synthesis campaign emerge from the crucible of total synthesis as robust, dependable, and widely applied. Trost conceptualized ‘chemoselectivity’ as a reagent’s selective reaction of one functional group or reactive site in preference to others (Trost, B. M., Science, 1983, 219, 245). Chemoselectivity and functional group tolerance can be evaluated quickly using robustness screens (Collins, K. D., Nat. Chem., 2013, 5, 597). A reaction may also be characterized by its ‘chemofidelity’, its reliable reaction with a functional group in any molecular context. For example, ketone reduction by an electride (dissolving metal conditions) exhibits high chemofidelity, but low chemoselectivity: it usually works, but many other functional groups are reduced at similar rates. Conversely, alkene coordination chemistry effected by π Lewis acids can exhibit high chemoselectivity (Trost, B. M., Science, 1983, 219, 245), but low chemofidelity: it can be highly selective for alkenes, but sensitive to substitution patterns (Larionov, E., Chem Comm., 2014, 50, 9816). In contrast, alkenes undergo reliable, robust, and diverse hydrogen atom transfer reactions from metal hydrides to generate carbon-centered radicals. Although there are many potential applications of this chemistry, its functional group tolerance, high rates, and ease of execution have led to its rapid deployment in complex synthesis campaigns. Its success derives from high chemofidelity; its dependable reactivity in many molecular environments and with many alkene substitution patterns. Metal hydride H-atom transfer (MHAT) reactions convert diverse, simple building blocks to more stereochemically and functionally dense products (Crossley, S. W. M., Chem. Rev., 2016, 116, 8912). When hydrogen is returned to the metal, MHAT can be considered the radical equivalent of Brønsted acid catalysis—itself a broad reactivity paradigm. This Account summarizes our group’s contributions to method development, reagent discovery, and mechanistic interrogation. Our earliest contribution to this area—a stepwise hydrogenation with high chemoselectivity and high chemofidelity—has found application to many problems. More recently, we reported the first examples of a dual-catalytic cross-couplings that rely on the merger of MHAT cycles and nickel catalysis. With time, we anticipate MHAT will become a staple of chemical synthesis.
Graphical Abstract
Introduction.
The Drago–Mukaiyama hydration (Figure 1) has emerged as a reliable tool with both high chemoselectivity and chemofidelity.1 It has been used in more than 38 total syntheses.1 Variation of its reactant components has led to methods for C–O, C–N, C–X, C–S, and C–C bond formation. When our group reported a hydrogenation variant in 2014, no broad consensus had coalesced around these reaction mechanisms. Norton2 and Boger,3 however, alluded to H• transfer as a possibility. Our group and Herzon’s group proposed4,5 that the compendium of Mukaiyama-type reactions display reactivity consistent with hydrogen atom transfer from a metal hydride complex, a proposal rapidly taken up by literature highlights and mini-reviews.6 Since that time, our hydrogenation protocol has seen extensive use in the literature due to its stereoselectivity, chemoselectivity, and chemofidelity. Some unusual features that differentiate it from standard alkene hydrogenations are briefly listed below.
Figure 1.

History, attributes and proposed mechanism of Drago-Mukaiyama reactions.
no hydrogen gas is required; liquid silanes (PhSiH3, Ph(iPrO)SiH2) serve as reductants
inexpensive ligands and abundant first row transition metals (Fe, Mn, Co) can be used
benzyl groups and halogens are reliably spared
stereoselectivity is often reversed compared to other common methods
reaction rates are high (e.g. a trisubstituted alkene has been reduced with a second order rate constant of ca. 60 M−1 s−1)7
reactions are unusually tolerant of: reaction concentration; plasticizers or stabilizers; amine or thiol impurities. Water or O2 impurities have minor effects.
In the first part of this Account, we document others’ use of our chemistry to call attention to its broad utility and to encourage further uptake. For so simple a reaction, it has seen extensive use in a short time. Next, we describe how this chemistry can be merged with traditional catalytic cross-coupling cycles. And we also seek to unravel why and how these purported metal hydrides can generate unstabilized carbon radicals through MHAT, instead of self-destruct through hydrogen evolution.
Thermodynamic, Chemoselective Hydrogenation (First Generation).
Hydrogenation aids complex molecule synthesis through retrosynthetic access to alkenes, which can be disconnected many ways. However, when steric constraints dictate approach of a reagent from one face of the alkene, alteration of stereochemistry from this ‘kinetic’ product can prove challenging. Dissolving metal reduction often allows the user to access an alternative ‘thermodynamic’ product but this method is restricted generally to the reduction of conjugated or electron-poor alkenes (Figure 2).8,9 The more negative reduction potential of electron-neutral alkenes necessitates elevated temperatures and other functional groups are reduced preferentially.
Figure 2.

Dissolving metal reduction versus catalytic hydrogenation: stereochemical divergence.
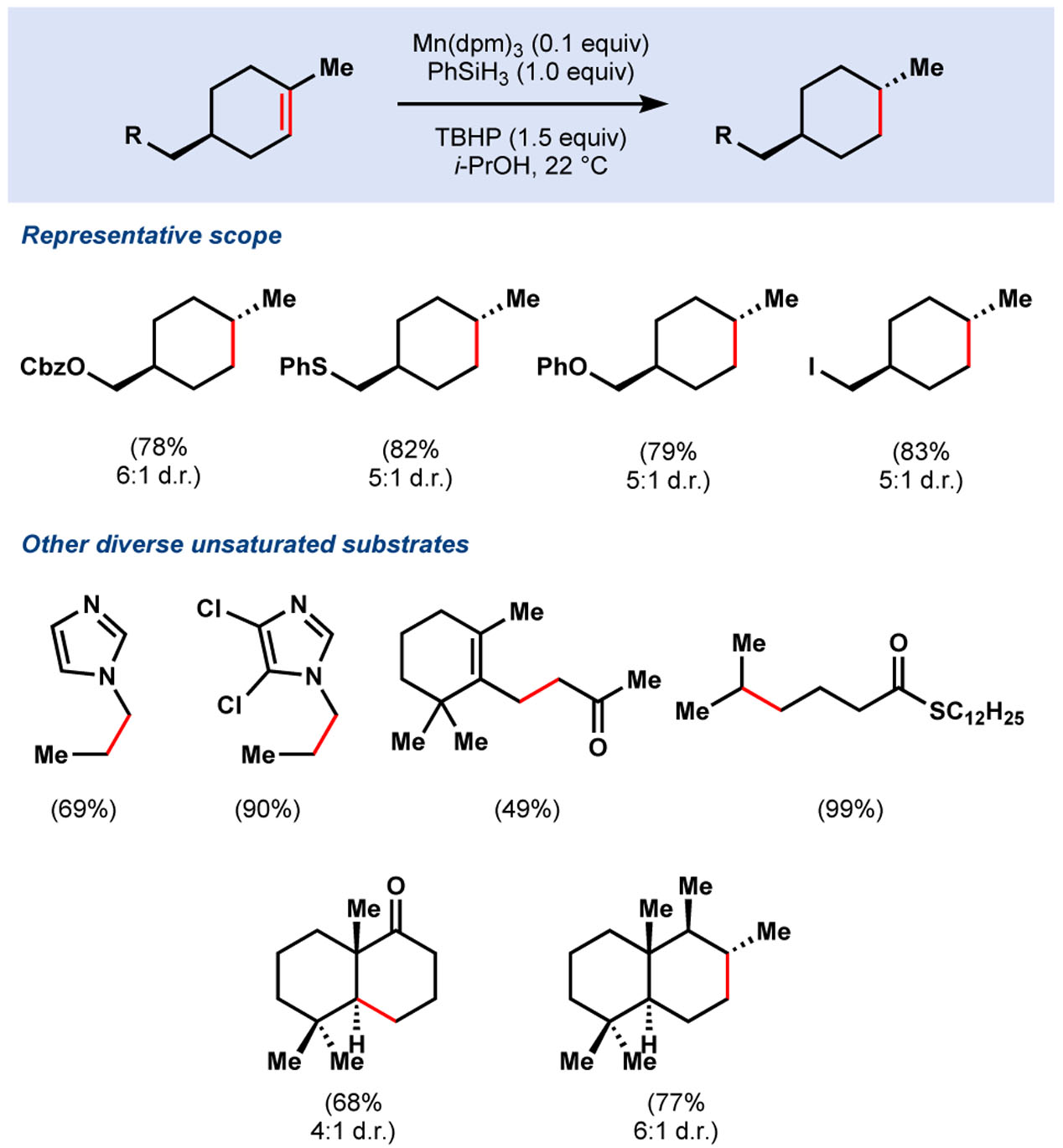
In 2014, our group envisioned that MHAT hydrogenation could maintain the same stereochemical outcome as dissolving metal reduction, but increase functional group tolerance. Our work, based on the redox hydration conditions of Mukaiyama10 and studies by Carreira11 and Magnus,12 identified Mn(dpm)3 and Co(dpm)2 (dpm = dipivaloyl methane), PhSiH3, and superstoichiometric tert-butyl hydroperoxide (TBHP) in isopropanol as an efficient system to reduce electron-neutral alkenes to thermodynamically-preferred stereoisomers. These conditions display improved chemoselectivity over dissolving metal reductions, with tolerance for functional groups such as carboxybenzyl, (thio)phenyl ethers, aryl/alkyl iodides, aryl chlorides, aryl fluorides, electron-rich arenes, and Lewis basic heteroarenes such as imidazole, carbonyls, and thioesters (Table 1). This hydrogenation proved effective for the synthesis of thermodynamically-configured polycyclic systems such as trans-decalone frameworks and petroleum biomarker drimane. In contrast, heterogeneous, hydrogenation catalyst systems (e.g. Pd/C) favor the kinetic products of each.4
Table 1.
First generation hydrogenation conditions
|
The broad functional group compatibility, experimental ease, and orthogonal selectivity of these conditions relative to other hydrogenation methods have aided several total synthesis endeavors (Figure 3).13
Figure 3.

Select examples of our MHAT-mediated hydrogenation in others’ syntheses
Krische and coworkers reported the total synthesis of orimadicyn A, triptoquinones B and C, and isoiresin in 2016.14 The group achieved selective reduction of the less-substituted olefin to favor the thermodynamic stereochemistry of isoiresin. Luo and coworkers found that use of Fe(acac)3 instead of [Co] or [Mn] complexes with our conditions afforded (+)-ibogamine in 26% yield from the olefin.15
MHAT hydrogenation has also found application in the pharmaceutical industry. In 2017, Novartis reported the synthesis of a series of Raf kinase inhibitors for the treatment of cell proliferation disorders. They found that MHAT reduction proceeded without reduction of the aryl bromide or inhibition by the Lewis basic pyridine moiety.16
A recent synthesis of (+)-pleuromutilin from the Reisman group features MHAT hydrogenation conditions.17 The authors found that use of other conditions preferentially reduced the more sterically accessible terminal olefin, while use of our MHAT-initiated hydrogenation conditions proceeded via a transannular [1,5]-hydrogen transfer. The Herzon group also employed our MHAT-hydrogenation conditions during their studies on the late-stage oxidation of (+)-pleurumutilin.18
During the total synthesis of (–)-xestosaprol N and O by Gao and coworkers,19 use of our hydrogenation conditions overcame carbonyl reduction problems encountered with heterogeneous catalysts (e.g. Pd/C).
In 2018, Dethe and coworkers reported the enantioselective total synthesis of (+)-taondiol, which required installation of trans stereochemistry at the decalin ring junction.20 The authors found that our hydrogenation conditions selectively reduced the olefin without removal of the benzyl protecting group. During the synthesis of the proposed structure of navirine C, Ma and coworkers21 employed our hydrogenation conditions to yield the desired imine. For the first time, a cyano group was shown to work efficiently as a radical acceptor under MHAT conditions.
Synthesis of 8-arylmenthol derivatives.
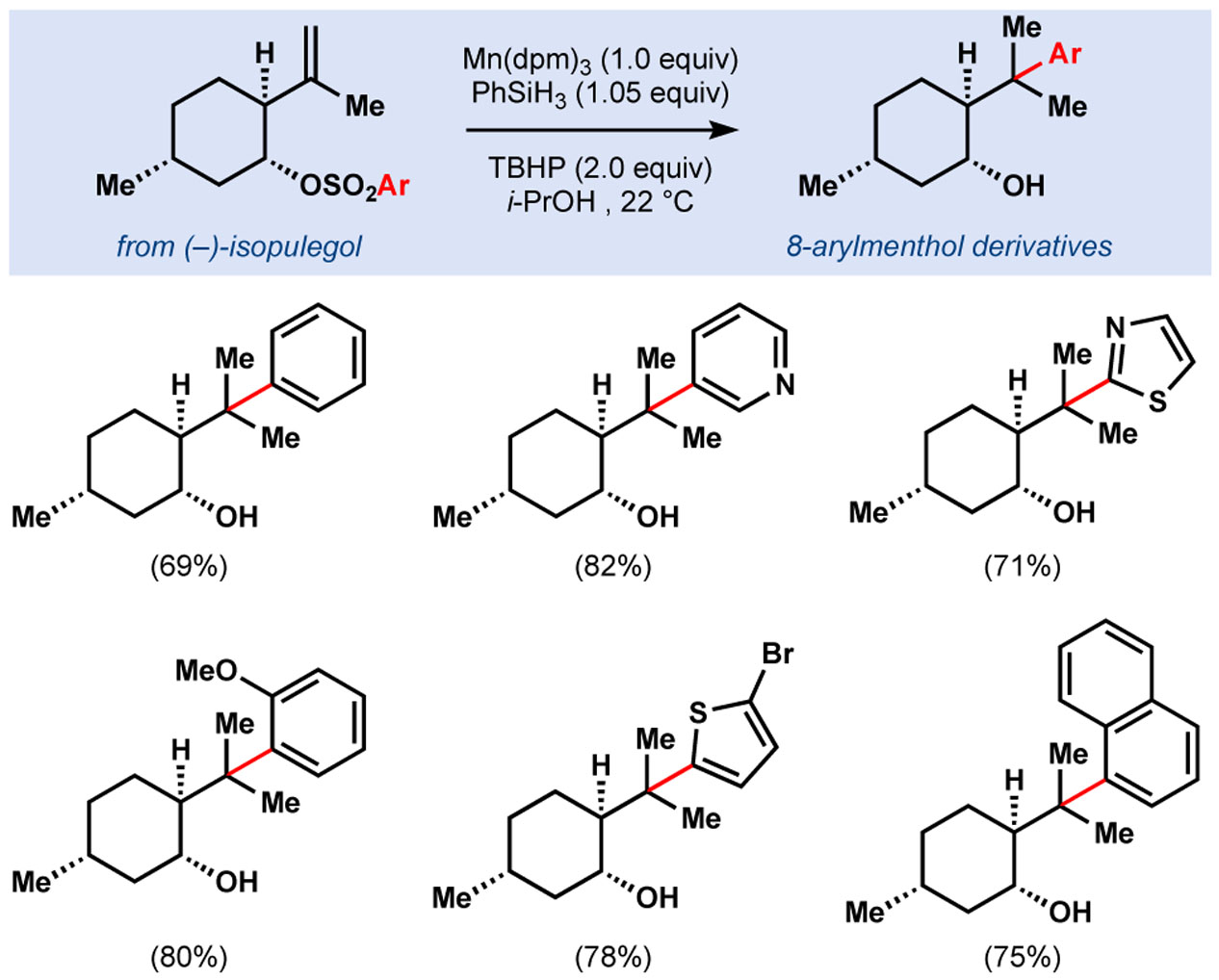
When faced with material supply problems of 8-phenylmenthol in the synthesis of diisocyanoadociane, we hypothesized that MHAT-mediated addition of radicals into pendant arenes could provide a more expedient approach to the synthesis of 8-phenylmenthol. Gratifyingly, our first generation hydrogenation conditions of Mn(dpm)3 and PhSiH3 induced a Smiles–Truce rearrangement of homoallyl arylsulfonates (Table 2).22 Whereas previous syntheses required 5 to 6 steps, this method can be used for a 2-step synthesis of 8-arylmenthol derivatives, privileged scaffolds for asymmetric induction.
Table 2.
Synthesis of 8-arylmenthol derivatives.
|
Second generation MHAT-mediated hydrogenation conditions.
During analysis of the hydrogenation reagents in the absence of olefin, we noticed that phenylsilane was slowly converted to its di-solvolysis product, diisopropoxyphenyl silane. Curiously, the mono-solvolysis product, monoisopropoxyphenyl silane (Ph(iPrO)SiH2), was not observed and led us to speculate that this species was the kinetically-preferred reductant. Independently synthesized Ph(iPrO)SiH2 was found to be exceptionally active and allowed for the use of more solvents and a wider variability in temperature.7 Ph(iPrO)SiH2 can be easily prepared on multigram scale (Figure 4a) and is stable for months when stored under inert atmosphere.
Figure 4.

Second generation MHAT-initiated hydrogenation conditions
Our first generation MHAT hydrogenation conditions reduced terpineol (Figure 4b) using a catalyst loading of 10 mol% in isopropanol, the sole competent solvent for manganese. In contrast, Ph(iPrO)SiH2 caused full consumption of terpineol with only 0.1 mol% catalyst loading (or lower) in hexanes. In contrast to the first generation conditions, the second-generation reduction could now be run at higher concentration (1M) or lower temperature, which may suit larger scale applications.7
Due to the increased solvent versatility permitted by Ph(iPrO)SiH2, a number of new radical-polar crossover methods23 and applications in total synthesis have been developed (Figure 5).24,25 In 2015, the Pronin group reported the synthesis of emindole SB.24 A MHAT-driven polycyclization delivered the tricyclic core of the indole diterpene; however, low efficiency and competitive formation of the wrong diastereomer plagued initial efforts. Pronin and coworkers found that combination of Fe(III) catalysis with isopropoxy(phenyl)silane significantly improved the yield and purity of the cyclized core.
Figure 5.

Application of Ph(iPrO)SiH2 in total synthesis
Zhang and coworkers utilized Ph(iPrO)SiH2 in their total synthesis of aplydactone, a biscyclobutane-containing natural product.25 Comparison between phenylsilane and isopropoxy(phenyl)silane showed superior performance of the latter with respect to yield and diastereoselectivity in the reduction of the vinyl bromide, though both could provide the desired product without cleavage of the strained cyclobutane rings or reductive dehalogenation.
Li and coworkers devised an efficient route to aplysiasecosterol by merger of its two halves through Reformatsky aldol and MHAT-initiated intramolecular Giese reaction.26 Diastereoselectivity was not, however, straightforward, since three new stereocenters emerged in the process. Use of Ph(iPrO)SiH2, however, improved diastereoselectivity. The authors also noted that Fe(dpm)3 enhanced diastereoselectivity relative to Fe(acac)3, which may indicate a possible secondary role of iron in Lewis acid activation of the enone.
In brief, Ph(iPrO)SiH2 can be useful when alcoholic solvents are problematic, lower temperatures are desirable, functional groups undergo competitive reaction, or hydrosilylation becomes competitive. Its lower catalyst loading requirements and compatibility with less expensive catalysts [e.g. Mn(acac)3] commend Ph(i PrO)SiH2 for large scale application.
Isomerization, cycloisomerization, and retrocycloisomerization.
The supposition of an MHAT mechanism in our hydrogenation led us to imagine that the corresponding isomerization may be possible through catalyst modification, as demonstrated by Norton27,28 and others29 (Figure 6a). A mild, radical olefin isomerization would be valuable as canonical transition metal-catalyzed methods30 require coordination of the metal to the olefin and can be inhibited by strong Lewis bases like amines.31 In contrast, metal hydrides that engage in MHAT do not coordinate to olefins and can display high functional group tolerance.32,1 As demonstrated by Norton,28 the fate of a radical can undergo numerous pathways in the absence of a traditional radical trap (e.g. O2): back transfer (kbackH[M⋅]) resulting in the starting olefin, hydrogenation (khyd[M—H], isomerization (kiso[M⋅ ]), and cyclization (kcycl). Theoretically, isomerization should be favored by low concentrations of metal hydride and slow M–C bond formation (Figure 6b).
Figure 6.

MHAT for the (cyclo)isomerization of olefins
We discovered that catalytic Co(SaltBu,tBu)Cl and PhSiH3 under anaerobic conditions effected the isomerization of terminal olefins by one position, with minimal reduction of the double bond.33 Co(SaltBu,tBu)Cl reacted preferentially with 1,1-disubstituted olefins in the presence of trisubstituted olefins due to the unfavorable steric interaction between the bulky ligand and the more hindered olefin. As a result, thermodynamically unfavorable products, such as 1,3-skipped dienes and β,γ-unsaturated ketones could be obtained (Figure 7a). As expected for carbon-centered radicals, pendant olefins were engaged to provide the equivalents of a pericyclic ene reaction, or an intramolecular Friedel–Crafts alkylation under mild conditions (Figure 7b), resulting in a mechanistically unique polyene cyclization. Adjacent strained rings were also cleaved, an observation that led to an expedient synthesis of humulene oxide from caryophyllene oxide (Figure 7c).
Figure 7.

Co(salen) mediated (cyclo)isomerization of olefins
Although 1,1-disubstituted olefins isomerized efficiently at room temperature, mono-substituted olefins required elevated temperatures. Interestingly, a competition experiment between a mono- and a 1,1-disubstituted olefin completely suppressed isomerization, which led us to suspect formation of an off-cycle organometallic intermediate (Figure 8). Heating the reaction mixture yielded product, presumably through homolysis of the Co–C bond and concomitant hydrogen atom abstraction to regenerate a cobalt hydride and isomeric olefin.
Figure 8.

Plausible mechanistic scheme for terminal olefins.
Like MHAT hydrogenation, the utility of MHAT isomerization is best appreciated by its rapid uptake and application to natural product synthesis (Figure 9). In our initial report, β-funebrene was isomerized to α-funebrene without decomposition of the strained core, whereas Brønsted acids caused decomposition.33 Sun, Li, and co-workers modified the Co(SalMeO,tBu)Cl-catalyzed cycloisomerization to forge the cyclohexyl D ring of the notoamides I, R and F.34 The authors indicated that attempts to form this bond using alternative radical methods or Friedel-Crafts alkylation were unsuccessful. Metz and coworkers found use of our method for the synthesis of 3β-hydroxy-7β-kemp-8(9)-en-6-one.35 In contrast, rhodium-catalyzed isomerization leads to the conjugated olefin.36
Figure 9.

MHAT isomerization in total synthesis.
The classic homoallyl radical rearrangement has also been encountered using MHAT catalysis. For example, a side chain migration was observed during the synthesis of the asmarines,37 a pathway that was unfortunate for us but could be beneficial for others. In a beautiful application of MHAT chemistry, the Newhouse and Maimone groups effected a homoallyl radical rearrangement to synthesize andrastatin D and terretonin L (Figure 10).38
Figure 10.

Homoallyl rearrangement via MHAT.
The rate of these cycloisomerization reactions is of special interest: ongoing studies in our laboratory have demonstrated that some substrates reach >90% conversion in under 1 minute. Our current work pertaining to the cycloisomerization33 has yielded an overall bimolecular rate constant on the order of 101-102 M−1s−1. The high reactivity, chemoselectivity, and tolerance for numerous solvents, including water, invites use of these reactions for bioconjugation.39 These high rates are not limited to cobalt, since both iron- and manganese-catalyzed MHAT reactions have been reported to be completed on the order of minutes to hours.1,7,40
MHAT and Ni Dual Catalysis.
Prior to 2016, MHAT-based methods utilized well-established stoichiometric radical traps to install heteroatomic functional groups.1 We thought a step-change in the field might be enabled by a transition metal co-catalyst to engage traditional cross-coupling partners. Nickel offered a good starting point41,42 due to its precedence in radical cross-coupling reactions with polar electrophiles, such as aryl and alkyl halides43 (Figure 11). Barriers to this schema included: (1) kinetically competent intersection of two catalytic cycles; (2) redox compatibility of two metal complexes; and (3) suppression of undesired reactivity accessible by each complex in isolation, such as Ni-reductive Heck reactivity, isomerization, and hydrogenation.
Figure 11.

Combination of MHAT and Ni: hypothetical catalytic cycle.
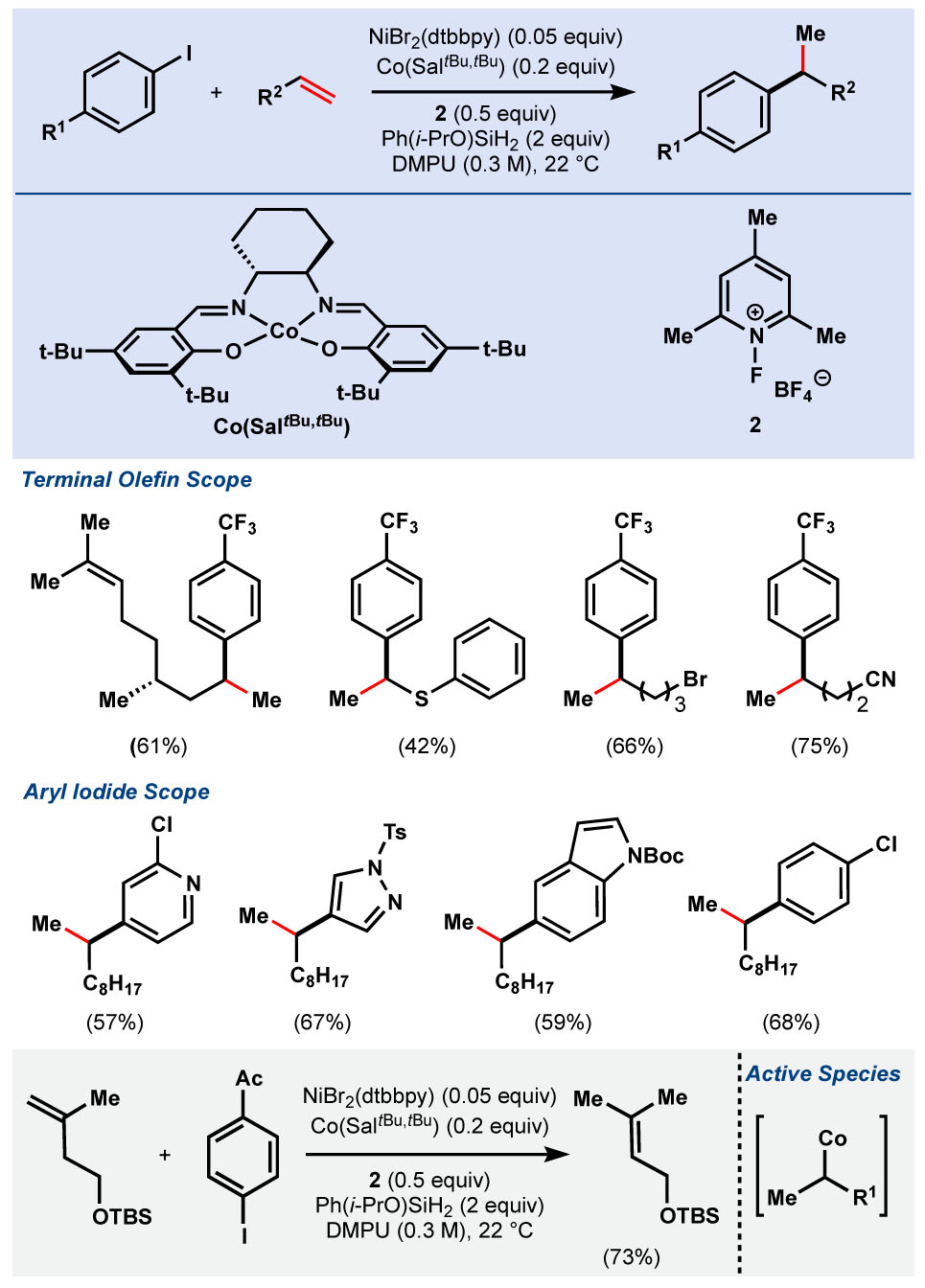
Our first investigation in this area entailed a method for the hydroarylation of unactivated terminal olefins,44 utilizing Ni-catalyzed electrophile-electrophile conditions pioneered by Weix.41 Co(SaltBu,tBu) was the only effective MHAT catalyst under the reaction conditions and required fluoropyridinium 2 as an oxidant.45 Ph(iPrO)SiH2 was most effective in the reaction,7 whereas PhSiH3 produced a significant amount of competitive hydrosilylation. A variety of terminal olefins containing numerous functional groups were tolerated, including thioethers, alkyl bromides, nitriles, and unprotected alcohols (Table 3). Although electron-deficient arenes were superior substrates for the arylation, both electron-rich and -neutral arenes gave moderate yields. Additionally, pharmaceutically-relevant heterocycles such as pyridine and pyrazoles proved compatible.
Table 3.
Co/Ni-Dual catalysis hydroarylation of unactivated olefins.
|
Mechanistic interrogation of Co-/Ni-hydroarylation indicated that the reaction proceeded via direct cobalt-alkyl to nickel transmetalation,46 a step that has few precedents in the literature.47 Kinetic analysis (RPKA)48 indicated an apparent rate law that was first order in both cobalt and nickel, ruling out a mechanism whereby rate-limiting cobalt–carbon bond homolysis led to freely-diffusing alkyl radical (Figure 12a, i). Furthermore, hydroarylation of a “radical clock” with varying concentrations of nickel pre-catalyst indicated minimal direct relationship between loading and unrearranged:rearranged product ratios (Figure 12b).49 These results ruled out a radical cage escape mechanism (Figure 12a, ii) but were consistent with a cage rebound mechanism (Figure 12a, iii).
Figure 12a-c.

Mechanistic investigation of Co/Ni system.
Attempted replication of the transmetalation in a stoichiometric experiment with a pre-formed nickel oxidative addition intermediate with a cobalt alkyl complex proved informative. The reaction of Co(III)(SaltBu,tBu)(i-Pr) (4) with an isolated dtbbpyNi(II)ArI complex (3) led to cross-coupled product (5) in only 10% yield, far from the quantitative yield expected if these two species were involved in the rate-determining transmetalation step (Figure 12c). Additionally, use of dtbbyNi(II)ArI as the nickel pre-catalyst in the catalytic reaction exhibited an induction period.
The combination of these results – the low yield of the stoichiometric experiment and the induction period under catalytic conditions – provided evidence against a Ni(0)/Ni(II) catalytic cycle and led us to consider the likelihood of a Ni(I)/Ni(III) reaction pathway (Figure 13). Here, oxidative addition across a Ni(I) complex generates a Ni(III)–aryl intermediate, which can act as a single electron oxidant50 of the Co(III)–alkyl species to generate a Ni(II)–aryl/Co(IV)–alkyl pair. Rapid homolysis of the cobalt alkyl bond generates an alkyl radical that is immediately captured by the Ni(II) aryl species to generate a Ni(III)aryl(alkyl) species, followed by reductive elimination to product. Co(III) and Ni(I) are regenerated after the transmetalation, reflected in our ability to run the hydroarylation reaction without exogenous oxidant when starting with a Co(III) pre-catalyst. The facility of solvent cage collapse of a carbon radical with its Ni(II)aryl partner is supported by the high bimolecular rate constant (1 × 108 M–1 s–1) calculated from Weix’s data.49a
Figure 13.

Proposed mechanism for Co/Ni hydroarylation.
This understanding of the Co-/Ni-transmetalation provides a template from which to design new dual catalytic reactions. However, the well-precedented instability of tertiary alkylcobalt complexes51 limited this catalytic system to terminal olefins. A complete reconstruction of the reaction conditions was necessary to develop a reaction in which a trisubstituted carbon is captured by a nickel(II) organometallic.44 We determined that a freely-diffusing radical (as opposed to carbon radical/ nickel(II) cage pair) would more closely resemble the compendium of traditional Ni cross-coupling reactions and pursued metal complexes that might allow radical cage escape.
Iron-Based MHAT/Ni chemistry.
Transition to a free radical capture mechanism required the use of β-diketonate ligands on the MHAT catalyst, complexes that have been implicated in nonorganometallic, radical reactivity.1 Achieving a similar reaction rate between the MHAT and Ni cycle was crucial for success. A screen of MHAT catalysts resulted in a “Goldilocks Scenario”: Co was too fast, Mn was too slow, but Fe was just right.52
Internal olefins, substrates that were incompatible with Co(SaltBu,tBu), could be coupled in high yield (Table 4). The reaction proved tolerant to many functional groups such as carbamates, perfluorinated olefins, redox-active esters, amides, vinyl thioethers, vinyl boronates, and nitriles. This method offered a complementary approach to similar dehalogenative41 or decarboxylative approaches;53 however, in the case where the corresponding alkyl–X components were unknown, as in the case of the fused cyclobutene, this method offered a viable way to synthesize otherwise inaccessible motifs. More surprisingly, hydroarylation of 1,1-di- or trisubstituted olefins successfully forged arylated quaternary carbons, a difficult synthetic problem remaining in the field of Ni catalysis.54 Olefins offer the unique benefit of starting from prochiral sp2 carbons, rather than a preformed tetrasubstituted carbon like tertiary alkyl bromides,54a,b which can be laborious to synthesize or unstable. Both cyclic and linear substrates could be derived from Wittig olefinations of commercially available ketones, and coupled with equal efficiency and high regioselectivity. Unsaturated heterocycles – oxetanes, tetrahydrofurans, pyrans, thianes, pyrrolidines, and piperidines, – could all be coupled with a range a (hetero)aryl iodides and bromides.
Table 4.
Fe/Ni hydroarylation of mono-, di-, and trisubstituted olefins.
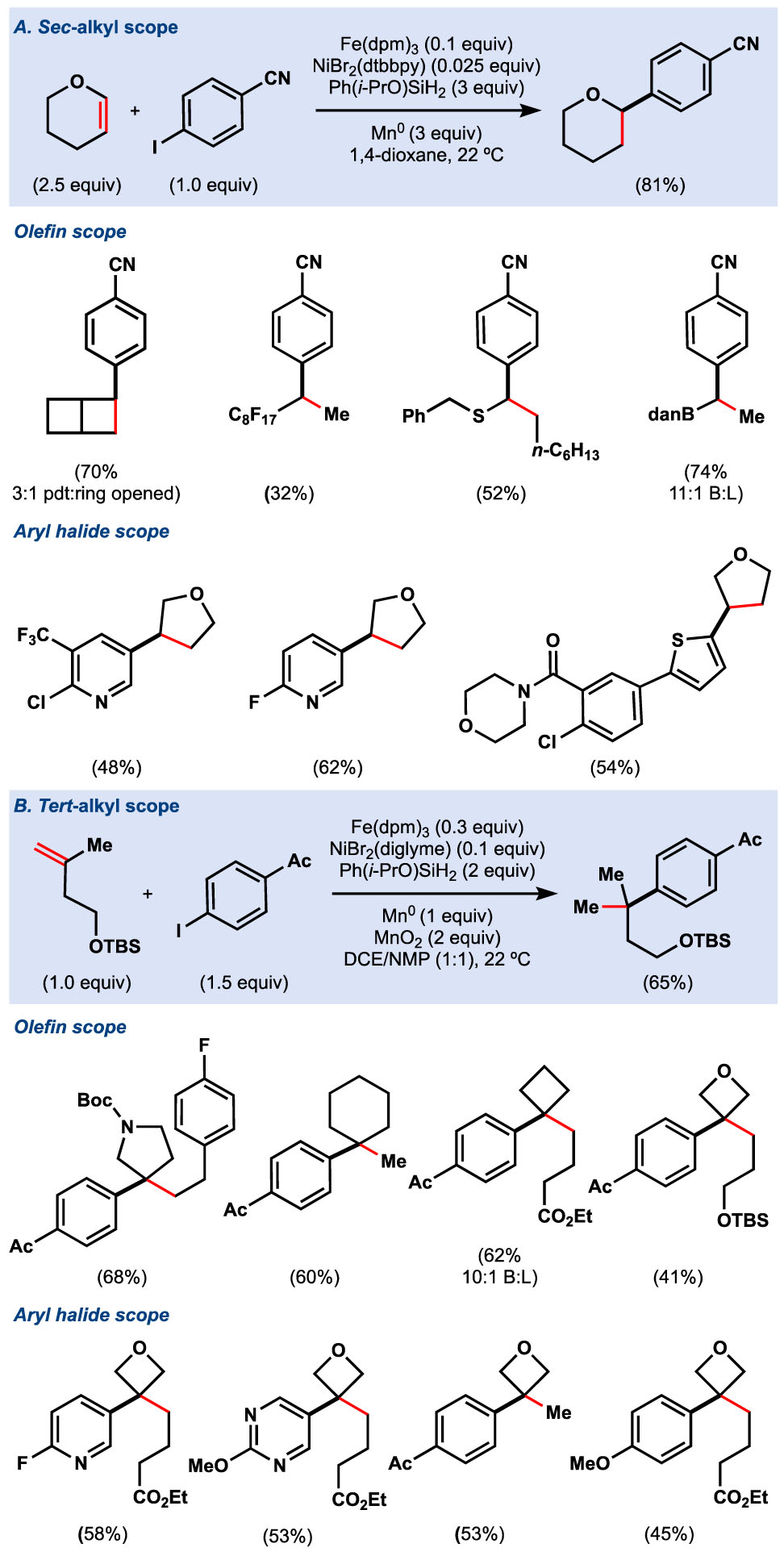
|
The Fe/Ni system accelerated access to motifs found in numerous pharmaceutical patents. Our arylation method constructed oxetane- and piperidine-bearing quaternary centers in fewer steps and at a fraction of the cost of the initial patent reports (Figure 14).55,56,57 We hope to extend our dual catalytic system to engage unactivated alkyl counterparts resulting in a hydroalkylation of unactivated olefins. Such sp3-sp3 coupling when applied to natural product synthesis could allow unusual retrosynthetic disconnections to yield functional group handles that are chemically innocuous and ubiquitous in nature.
Figure 14.

Application of Fe/Ni dual catalysis to biologically active targets.
Thermochemistry Discussion.
Both our group4 and the Herzon group5 have proposed that carbon-centered radical intermediates formed in Drago-Mukaiyama reactions1 arise from a MHAT elementary step rather than coordinative hydrometallation followed by metal-carbon bond homolysis (Figure 15).
Figure 15.

MHAT versus coordinative hydrometallation
Successful MHAT to electron-neutral alkenes implies that the intermediate metal hydrides must have BDFE values (i.e. ΔG°MH) that are weaker than any characterized metal hydride bond58,59 but outcompete bimolecular hydrogen evolution (Figure 16). Weak metal hydrides (<56 kcal/mol) are thermodynamically unstable to hydrogen evolution via bimolecular interaction of two metal hydrides60 and C–H bonds adjacent to carbon-centered radicals have BDFE values on the order of ~40 kcal/mol.61 The weakest well-characterized metal hydride has a BDFE value of 50.3 kcal/mol but does not add across unactivated alkenes at an appreciable rate.58 How can MHAT outcompete hydrogen evolution?
Figure 16.

Thermochemical implications of a MHAT mechanism
Hydrogen evolution is observed in some Drago-Mukaiyama reactions4,40—consistent with weak M–H BDFE values—particularly at higher pre-catalyst loadings, even while the desired hydrofunctionalization reaction proceeds (Figure 17). The predominance of MHAT pathways may be due to 1) a lower kinetic driving force from bimolecular hydrogen evolution based on its second order rate law in [M–H] (low [M–H] and high [alkene]), and/or 2) a lower rate constant for hydrogen evolution than MHAT to alkenes. Hydrogen evolution should impart a high entropic cost (ΔS‡) through an ordered bimolecular reductive elimination transition state.62,63 In contrast, MHAT across an alkene should exhibit a lower ΔS‡ cost due to lower steric and symmetry demands, especially for metal complexes with sterically bulky supporting ligands.62,63 This rationale may explain observations4,33 that bulkier ligands like dpm and salentBu,tBu versus acac and salenH,H have generally allowed lower catalyst loadings for the same hydrofunctionalization reactions (Figure 17).1,4,33 Bulky ligands may also prevent dimerization of the reduced metal,64 which requires reoxidation to the active precatalyst.
Figure 17.

Kinetic and Thermodynamic analysis of the feasibility of MHAT to alkenes.
The origin of low BDFE of Drago–Mukaiyama M–H complexes1 may be low bond enthalpy and features that Mayer58,65,66,67 suggests can contribute to favorable entropic change upon MHAT: 1) Mn, Fe, or Co metal centers, 2) MIII to MII reduction upon MHAT, 3) ligands which bind the metal center via hard N, O, and Cl atoms, and a feature which Mayer’s complexes do not possess 4) entropically favorable decrease in coordination number upon loss of H• (Figure 17). Formation of these weak M–H bonds is likely driven by Si/B–X and Si/B–O bond formation and cleavage of O–O bonds (from O2/peroxides) through ligand exchange between silanes/boranes and metal pre-catalysts.
Finally, either ligands or the metal center may stabilize a developing radical in the transition state. At the extreme, bonds to carbon could form,33,44 especially at secondary radicals, which would serve to minimize the effective endergonicity by providing kinetically accessible, yet thermodynamically stable intermediates, and decrease the concentration of metal hydride available for unproductive reactions.
Conclusion.
The proposal that metal hydride atom transfer (MHAT) underlies the Drago-Mukaiyama reaction engine provides a predictive, explanatory and exploratory model for future work: it offers answers and also questions. Just as importantly, MHAT catalysis has become a useful, general method in synthetic chemistry. Many contributions preceded our own work and, hopefully, will also succeed it. In addition to advancing MHAT as an organizing principle, our group has added new methods of hydrogenation, isomerization, Smiles-Truce rearrangement, and hydroarylation via secondary metal involvement. En route to these contributions, we have discovered that isopropoxyphenylsilane significantly expands the efficiency and compatibility of MHAT catalysis and enables new reactivity. Mechanistic contributions have included demonstration that HAT is unlikely to occur from a ligand hydrogen,7 our hydrogenation exhibits a small inverse 1H/2H KIE,7 and our Co/Ni dual-catalyzed hydroarylation occurs via direct transmetalation, not free radical diffusion.46 Among the most important attributes of MHAT catalysis, especially hydrogenation, is its chemofidelity: reliable reactivity, no matter the molecular context or alkene substitution pattern. High chemofidelity will no doubt lead to the continued use of MHAT in chemical synthesis, but especially in the crucible of natural products synthesis—an unsparing proving ground for chemical methods. As synthetic space and natural product space begin to merge in the coming era, MHAT may play a critical role.
Acknowledgements.
Dedicated to Professor E. J. Corey on the occasion of his 90th birthday for his many contributions to science and pedagogy.
Funding Sources
We thank NIGMS for their generous support of this work (R01 GM104180, R35 GM122606). Our lab has been the grateful recipient of additional support by Eli Lilly, Boehringer-Ingelheim, Amgen, Bristol-Myers-Squibb, Novartis and the Baxter Foundation.
Abbreviations
- MHAT
Metal hydride H-atom transfer
- acac
acetylatonate
- dpm
dipivaloylmethane
Biography
Samantha A. Green received her B.S. from Emory University in 2013 conducting research under Professor Huw Davies, after which she completed a postbaccalaureate fellowship at the NIH under Dr. Marta Catalfamo. Currently, she is a graduate student in the Shenvi research group investigating new MHAT methods.
Steven W. M. Crossley completed his B.Sc. at UBC and is currently an NSERC post-graduate scholar in the Shenvi lab. His studies involve the development of MHAT methodology and total synthesis.
Jeishla L. M. Matos obtained a B.S. in Chemistry from the University of Puerto Rico, Río Piedras, where she performed research under the supervision of Professor José A. Prieto. She is currently an NSF graduate fellow at TSRI in the Shenvi laboratory, where she is working to expand the chemistry and our understanding of these proposed MHAT reactions. Her major interests are in understanding nature’s chemical reactivity and in improving social equity through scientific developments.
Suhelen Vásquez-Céspedes obtained her B.Sc. and M.Sc. degrees from the University of Costa Rica working with Professor Jorge Cabezas, and received her PhD degree from the Westfälische Willhems Universität Münster (Germany) under the supervision of Professor Frank Glorius. In 2017 she joined the Shenvi group as a postdoctoral associate, focused on the development of novel MHAT-catalyzed transformations.
Sophia Shevick earned a B.S. in Chemical Biology (2013) at the University of California, Berkeley, and performed undergraduate research under the supervision of Professor F. Dean Toste. After three years as a medicinal chemist at Gilead Sciences in Foster City, CA, she joined the Shenvi lab at The Scripps Research Institute. She is currently investigating the mechanism of dual-catalyzed MHAT cross-coupling reactions, and pursuing her interest in natural product synthesis.
Ryan A. Shenvi trained with Ray Funk (BS), Phil Baran (PhD) and E. J. Corey (PD). His independent career began at The Scripps Research Institute in 2010. He has four kids, wonderful students and no time.
Footnotes
The authors declare no competing financial interest.
References
- 1.Crossley SWM; Obradors CL; Martinez RM; Shenvi RA Mn-, Fe-, and Co-Catalyzed Radical Hydrofunctionalizations of Olefins. Chem. Rev 2016, 116, 8912–9000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Choi J; Tang L; Norton JR Kinetics of Hydrogen Atom Transfer from (η5-C5H5)Cr(CO)3H to Various Olefins: Influence of Olefin Structure. J. Am. Chem. Soc 2007, 129, 234–240. [DOI] [PubMed] [Google Scholar]
- 3.Ishikawa H; Colby DA; Seto S; Va P; Tam A; Kakei H; Rayl TJ; Hwang I; Boger DL Total Synthesis of Vinblastine, Vincristine, Related Natural Products, and Key Structural Analogs. J. Am. Chem. Soc 2009, 131, 4904–4916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iwasaki K; Wan KK; Oppedisano A; Crossley SWM; Shenvi RA Simple, Chemoselective Hydrogenation with Thermodynamic Stereocontrol. J. Am. Chem. Soc 2014, 136, 1300–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.King SM; Ma X; Herzon SB A Method for the Selective Hydrogenation of Alkenyl Halides to Alkyl Halides. J. Am. Chem. Soc 2014, 136, 6884–6887. [DOI] [PubMed] [Google Scholar]
- 6. (a).Hoffmann RW Markovnikov Free Radical Addition Reactions, a Sleeping Beauty Kissed to Life. Chem. Soc. Rev 2016, 45, 577–583.; [DOI] [PubMed] [Google Scholar]; (b) Simonneau A; Oestreich M Fascinating Hydrogen Atom Transfer Chemistry of Alkenes Inspired by Problems in TotalSynthesis. Angew. Chem. Int. Ed 2015, 54, 3556–3558.; [DOI] [PubMed] [Google Scholar]; (c) Streuff J; Gansaüer A Metal-Catalyzed β-Functionalization of Michael Acceptors through Reductive Radical Addition Reactions. Angew. Chem. Int. Ed 2015, 54, 14232–14242. [DOI] [PubMed] [Google Scholar]
- 7.Obradors CL; Martinez RM; Shenvi RA Ph(i-PrO)SiH2: An Exceptional Reductant for Metal-Catalyzed Hydrogen Atom Transfers. J. Am. Chem. Soc 2016, 138, 4962–4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rao HSP; Reddy KS Palladium Assisted Hydrogenation of Cyclic α,β-Unsaturated Ketones by Ammonium Formate. Tetrahedron Lett. 1994, 35, 171–174. [Google Scholar]
- 9.Solladié-Cavallo A; Jierry L; Klein A; Schmitt M; Welter R α-Fluoro decalones as chiral epoxidation catalysts: fluorine effect. Tetrahedron: Asymmetry 2004, 15, 3891–3898. [Google Scholar]
- 10.Isayama S; Mukaiayama T A New Method for Preparation of Alcohols from Olefins with Molecular Oxygen and Phenylsilane by the Use of Bis(acetylacetonato)cobalt(II). Chem. Lett, 1989, 18, 1071–1074. [Google Scholar]
- 11.Waser J; Carreira EM Catalytic Hydrohydrazination of a Wide Range of Alkenes with a Simple Mn Complex. Angew. Chem., Int. Ed 2004, 43, 4099–4102. [DOI] [PubMed] [Google Scholar]
- 12.Magnus P; Payne AH; Waring MJ; Scott DA: Lynch V. Conversion of α,β-Unsaturated Ketones into a-Hydroxy Ketones using an MnIII Catalyst, Phenylsilane and Dioxygen: Acceleration of Conjugate Hydride Reduction by Dioxygen. Tetrahedron Lett. 2000, 41, 9725–9730. [Google Scholar]
- 13.Including our own:Lu H-H; Pronin SV; Antonova-Koch Y; Meister S; Winzeler EA; Shenvi RA Synthesis of (+)-7,20-Diisocyanoadociane and Liver Stage Antiplasmodial Activity of the ICT Class. J. Am. Chem. Soc 2016, 138, 7268–7271.
- 14.Feng J; Noack F; Krische MJ Modular Terpenoid Construction via Catalytic Enantioselective Formation of All-Carbon Quaternary Centers: Total Synthesis of Oridamycin A, Triptoquinones B and C, and Isoiresin. J. Am. Chem. Soc 2016, 138, 12364–12367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Y; Xue Y; Li G; Yuan H; Luo T Enantioselective Synthesis of Iboga Alkaloids and Vinblastine via Rearrangements of Quaternary Ammoniums. Chem. Sci 2016, 7, 5530–5536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aversa RJ; Burger M; Dillon MP; Dineen TA; Karki R; Ramurthi S; Rauniyar V; Robinson R; Sarver PJ Tricyclic Compounds and Compositions as Kinase Inhibitors. International patent WO2017103824A1. 2017.
- 17.Farney EP; Feng SS; Schäfers F; Reisman SE Total Synthesis of (+)-Pleuromutilin. J. Am. Chem. Soc 2018, 140, 1267–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma X; Kucera R; Goethe OF; Murphy SK; Herzon SB Directed C–H Bond Oxidation of (+)-Pleuromutilin. J. Org. Chem 2018, 83, 6843–6892.. [DOI] [PubMed] [Google Scholar]
- 19.Shi Y; Ji Y; Xin K; Gao S Total Synthesis of (−)-Xestosaprol N and O. Org. Lett 2018, 20, 732–735. [DOI] [PubMed] [Google Scholar]
- 20.Dethe DH; Mahapatra S; Sau SK Enantioselective Total Synthesis and Assignment of the Absolute Configuration of the Meroterpenoid (+)-Taondiol. Org. Lett 2018, 20, 2766–2769. [DOI] [PubMed] [Google Scholar]
- 21.Liu J; Ma D A Unified Approach for the Assembly of Atisine- and Hetidine-type Diterpenoid Alkaloids: Total Synthesis of Azitine and the Proposed Structure of Navirine C. Angew. Chem Int. Ed 2018, 57, 6676–6680. [DOI] [PubMed] [Google Scholar]
- 22.Crossley SWM; Martinez RM; Guevara-Zuluaga S; Shenvi RA Synthesis of the Privileged 8-Arylmenthol Class by Radical Arylation. Org. Lett 2016, 18, 2620–2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Okumura M; Nakamata Huynh SM; Pospech J; Sarlah D Arenophile-Mediated Dearomative Reduction. Angew. Chem. Int. Ed 2016, 55, 15910–15914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.George DT; Kuenstner EJ; Pronin SV A Concise Approach to Paxilline Indole Diterpenes. J. Am. Chem. Soc 2015, 137, 15410–15413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu C; Chen R; Shen Y; Liang Z; Hua Y; Zhang Y Total Synthesis of Aplydactone by a Conformationally Controlled C−H Functionalization. Angew. Chem. Int. Ed 2017, 56, 8187–8190. [DOI] [PubMed] [Google Scholar]
- 26.Lu Z; Zhang X; Guo Z; Chen Y; Mu T; Li A Total Synthesis of Aplysiasecosterol A. J. Am Chem. Soc, 2018, 140, 9211–9218. [DOI] [PubMed] [Google Scholar]
- 27.Hartung J; Pulling ME; Smith DM; Yang DX; Norton JR Initiating radical cyclizations by H• transfer from transition metals. Tetrahedron, 2008, 64, 11822–11830. [Google Scholar]
- 28.Li G; Kuo JL; Han A; Abuyuan JM; Young LC; Norton JR; Palmer JH Radical isomerization and cycloisomerization initiated by H• transfer. J. Am. Chem. Soc, 2016, 138, 7698–7704. [DOI] [PubMed] [Google Scholar]
- 29.Sweany RL; Halpern J Hydrogenation of α-Methylstyrene by Hydridopentacarbonylmanganese (I). Evidence for a Free-Radical Mechanism. J. Am. Chem. Soc 1977, 99, 8335–8337. [Google Scholar]
- 30.Larionov E; Li H; Mazet C Well-defined transition metal hydrides in catalytic isomerizations. Chem. Comm 2014, 50, 9816–9826. [DOI] [PubMed] [Google Scholar]
- 31.Radetich B; Rajanbabu TV Catalyzed Cyclization of α,ω-Dienes: A Versatile Protocol for the Synthesis of Functionalized Carbocyclic and Heterocyclic Compounds. J. Am. Chem. Soc 1998, 120, 8007–8008. [Google Scholar]
- 32.Trost BM Selectivity: A Key to Synthetic Efficiency. Science 1983, 219, 245–250. [DOI] [PubMed] [Google Scholar]
- 33.Crossley SWM; Barabé F; Shenvi RA Simple, Chemoselective, Catalytic Olefin Isomerization. J. Am. Chem. Soc 2014, 136, 16788–16791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang B; Zheng W; Wang X; Sun D; Li C Total Synthesis of Notoamides F, I, and R and Sclerotiamide. Angew. Chem. Int. Ed 2016, 55, 10435–10438. [DOI] [PubMed] [Google Scholar]
- 35.Wang Y; Jaäger A; Gruner M; Lübken T; Metz P Enantioselective Total Synthesis of 3β-Hydroxy-7β-kemp-8(9)-en-6-one, a Diterpene Isolated from Higher Termites. Angew. Chem. Int. Ed 2017, 56, 15861–15865. [DOI] [PubMed] [Google Scholar]
- 36.See also:Reiher CA; Shenvi RA Stereocontrolled Synthesis of Kalihinol C, J. Am. Chem. Soc 2017, 139, 3647–3650.
- 37.Wan KK; Shenvi RA Conjuring a Supernatural Product – Delmarine. SynLett. 2016, 27, 1145–1164. [Google Scholar]
- 38.Xu G; Elkin M; Tantillo DJ; Newhouse TR; Maimone TJ Traversing Biosynthetic Carbocation Landscape in the Total Synthesis of Andrastin and Terretonin Meroterpenes. Angew. Chem. Int. Ed 2017, 56, 12498–12502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saito F; Noda H; Bode JW Critical Evaluation and Rate Constants of Chemoselective Ligation Reactions for Stoichiometric Conjugations in Water. ACS Chem. Biol 2015, 10, 1026–1033. [DOI] [PubMed] [Google Scholar]
- 40.Lo JC; Gui J; Yabe Y; Pan C-M; Baran PS Functionalized Olefin Cross-Coupling to Construct Carbon-Carbon Bonds. Nature 2014, 516, 343–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weix DJ Methods and Mechanisms for Cross-Electrophile Coupling of Csp2 Halides with Alkyl Electrophiles. Acc. Chem. Res 2015, 48, 1767–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gutierrez O; Tellis JC; Primer DN; Molander GA; Kozlowski MC Nickel-Catalyed Cross-Coupling of Photoredox-Generated Radicals: Uncovering a General Manifold for Stereoconvergence in Nickel-Catalyzed Cross-Couplings. J. Am. Chem. Soc 2015, 137, 4896–4899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tasker SZ; Standley EA; Jamison TF Recent Advances in Homogeneous Nickel Catalysis. Nature 2014, 509, 299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Green SA; Matos JLM; Yagi A; Shenvi RA Branch-Selective Hydroarylation: Iodoarene-Olefin Cross-Coupling. J. Am. Chem. Soc 2016, 138, 12779–12782. [DOI] [PubMed] [Google Scholar]
- 45.Shigehisa H; Aoki T; Yamaguchi S; Shimizu N; Hiroya K Hydroalkoxylation of Unactivated Olefins with Carbon Radicals and Carbocation Species as Key Intermediates. J. Am. Chem. Soc. 2013, 135, 10306–10309. [DOI] [PubMed] [Google Scholar]
- 46.Shevick SL; Obradors C; Shenvi RA A Dual-Catalytic Transmetalation in the Cobalt/Nickel Branch-Selective Hydroarylation of Terminal Olefins. J. Am. Chem. Soc, 2018, 140, 12056–12068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. (a).Ram MS; Riordan CG; Yap GPA; Liable-Sands L; Rheingold AL; Marchaj A; Norton JR Kinetics and Mechanism of Alkyl Transfer from Organocobalt(III) to Nickel(I): Implications for the Synthesis of Acetyl Coenzyme A by CO Dehydrogenase. J. Am. Chem. Soc 1997, 119, 1648–1655. [Google Scholar]; (b) Eckert NA; Dougherty WG; Yap GPA; Riordan CG Methyl Transfer from Methylcobaloxime to (Triphos)Ni(PPh3): Relevance to the Mechanism of Acetyl Coenzyme A Synthase. J. Am. Chem. Soc 2007, 129, 9286–9287. [DOI] [PubMed] [Google Scholar]
- 48.Blackmond DG Reaction Progress Kinetic Analysis: A Powerful Methodology for Mechanistic Studies of Complex Catalytic Reactions. Angew. Chem., Int. Ed 2005, 44, 4302–4320. [DOI] [PubMed] [Google Scholar]
- 49. (a).Biswas S; Weix DJ Mechanism and Selectivity in Nickel-Catalyzed Cross-Electrophile Coupling of Aryl Halides with Alkyl Halides. J. Am. Chem. Soc, 2013, 135, 16192–16197. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Breitenfeld J; Ruiz J; Wodrich MD; Hu X Bimetallic Oxidative Addition Involving Radical Intermediates in Nickel-Catalyzed Alkyl-Alkyl Kumada Coupling Reactions. J. Am. Chem. Soc, 2013, 135, 12004–121012. [DOI] [PubMed] [Google Scholar]
- 50.Brodovitch JC; Haines RI; McAuley A Nickel(III)–tris(di-imine) complexes: a series of strong one-electron oxidants. Can. J. Chem 1981, 59, 1610–1614. [Google Scholar]
- 51.Puxeddu A; Costa G; Marsich N Reaction of Electrogenerated [CoI(salen)]- with t-Butyl Bromide and t-Butyl Chloride. J. C. S. Dalton 1980, 1489–1493. [Google Scholar]
- 52.Green SA; Vásquez-Céspedes S; Shenvi RA Iron-Nickel Dual Catalysis: A New Engine for Olefin Functionalization and the Formation of Quaternary Centers. J. Am. Chem. Soc, 2018, 140, 11317–11324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.For representative examples of decarboxylative arylation seeCornella J; Edwards JT; Qin T; Kawamura S; Wang J; Pan C -M.; Gianatassio, R.; Schmidt, M.; Eastgate, M. D.; Baran, P. S. Practical Ni-Catalyzed Aryl–Alkyl Cross-Coupling of Secondary Redox-Active Esters. J. Am. Chem. Soc 2016, 138, 2174–2177.Zuo Z; Ahneman DT; Chu L; Terrett JA; Doyle AG; MacMillan DWC Dual Catalysis. Merging Photoredox with Nickel Catalysis: Coupling of α-carboxyl sp3-carbons with aryl halides. Science 2014, 345, 437–440.
- 54. (a).Zultanski SL; Fu GC Nickel-Catalyzed Carbon–Carbon Bond-Forming Reactions of Unactivated Tertiary Alkyl Halides: Suzuki Arylations. J. Am. Chem. Soc 2013, 135, 624–627. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang X; Wang S; Xue W; Gong H Nickel-Catalyzed Reductive Coupling of Aryl Bromides with Tertiary Alkyl Halides. J. Am. Chem. Soc 2015, 137, 11562–11565. [DOI] [PubMed] [Google Scholar]; (c) Primer DN; Molander GA Enabling the Cross-Coupling of Tertiary Organoboron Nucleophiles through Radical-Mediated Alkyl Transfer. J. Am. Chem. Soc 2017, 139, 9847–9850. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Mei T-S; Patel HH; Sigman MS Enantioselective Construction of Remote Quaternary Stereocenters. Nature 2014, 508, 340–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bull JA; Croft RA; Davis OA; Doran R; Morgan KF Oxetanes: Recent Advances in Synthesis, Reactivity, and Medicinal Chemistry. Chem. Rev 2016, 116, 12150–12233. [DOI] [PubMed] [Google Scholar]
- 56. (a).Ruah SSH; Miller MT; Bear B; McCartney J; Grootenhuis PDJ Modulators of ATP-Binding Cassette Transporters. WO2007087066A2. 2007. [Google Scholar]; (b) Nguyen PX; Cappiello JR; Heidelbaugh TM; Novel Aromatic Thio Compounds as Receptor Modulators. US2014171393A1. 2014. [Google Scholar]
- 57.Carroll FI; Gichinga MG; Kormos CM; Maitra R; Runyon SP; Thomas JB; Mascarella SW; Decker AM; Navarro HA Design, Synthesis, and Pharmacological Evaluation of JDTic Analogs to Examine the Significance of the 3- and 4-methyl substituents. Bioorg. Med. Chem 2015, 23, 6379–6388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Choi J; Pulling ME; Smith DM; Norton JR Unusually weak metal-hydrogen bonds in HV(CO)4(P–P) and their effectiveness as H• donors. J. Am. Chem. Soc 2008, 130, 4250–4252. [DOI] [PubMed] [Google Scholar]
- 59.Uddin J; Morales CM; Maynard JH; Landis CR Computational studies of metal-ligand bond enthalpies across transition metal series. Organometallics 2006, 25, 5566–5581. [Google Scholar]
- 60.Gonzalez AA; Hoff CD Entropy of binding molecular hydrogen and nitrogen in the complexes of tricarbonylbis(tricyclohexylphosphine)transition metal (transition metal chromium, molybdenum, tungsten). Inorg. Chem 1989, 28, 4295–4297. [Google Scholar]
- 61.Blanksby SJ; Ellison GB Bond dissociation energies of organic molecules. Acc. Chem. Res 2003, 36, 255–263. [DOI] [PubMed] [Google Scholar]
- 62.TΔS‡ = ~12 kcal/mol in:Wayland BB; Ba S; Sherry AE Activation of methane and toluene by rhodium(II) porphyrin complexes. J. Am. Chem. Soc 1991, 113, 5305–5311.
- 63.Gu NX; Oyala PH; Peters JC An S=1/2 iron complex featuring N2, thiolate, and hydride ligands: Reductive elimination of H2 and relevant thermochemical Fe–H parameters. J. Am. Chem. Soc 2018, 140, 6374–6382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bullock RM; Samsel EG Hydrogen Atom Transfer Reactions of Transition-Metal Hydrides. Kinetics and Mechanism of the Hydrogenation of α-Cyclopropylstyrene by Metal Carbonyl Hydrides J. Am. Chem. Soc 1990, 112, 6886–6898. [Google Scholar]
- 65.Warren JJ; Tronic TA; Mayer JM Thermochemistry of proton-coupled electron transfer reagents and its implications. Chem. Rev 2010, 110, 6961–7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mader EA; Davidson ER; Mayer JM Large ground-state entropy changes for hydrogen atom transfer reactions of iron complexes. J. Am. Chem. Soc 2007, 129, 5153–5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mader EA; Manner VW; Markle TF; Wu A; Franz JA; Mayer JM Trends in ground-state entropies for transition metal based hydrogen atom transfer reactions. J. Am. Chem. Soc 2009, 131, 4335–4345. [DOI] [PMC free article] [PubMed] [Google Scholar]