

Summary
Deregulated HER2 is a target of many approved cancer drugs. We analyzed 111,176 patient tumors and identified recurrent HER2 transmembrane domain (TMD) and juxtamembrane domain (JMD) mutations, including G660D, R678Q, E693K and Q709L. Using a saturation mutagenesis screen and testing of patient-derived mutations we found several activating TMD and JMD mutations. Structural modeling and analysis showed that the TMD/JMD mutations function by improving the active dimer interface or stabilizing an activating conformation. Further, we found that HER2 G660D employed asymmetric kinase dimerization for activation and signaling. Importantly, anti-HER2 antibodies and small molecule kinase inhibitors blocked the activity of TMD/JMD mutants. Consistent with this, a G660D germline mutant lung cancer patient showed remarkable clinical response to HER2 blockade.
Keywords: ERBB2/HER2, ERBB2 activation, transmembrane domain (TMD) mutation, juxtamembrane (JMD) domain mutation, HER2 germline mutation, HER2 somatic mutation, anti-HER2 antibodies, HER2 kinase inhibitors, ERBB2 structure
Summary
Pahuja et al. show that recurrent HER2 transmembrane domain and juxtamembrane domain mutations enhance HER2 activity by improving the active dimer interface or stabilizing an activating conformation. Importantly, HER2 inhibiting antibodies and small molecules can block the activity of these mutants.
Graphical Abstract
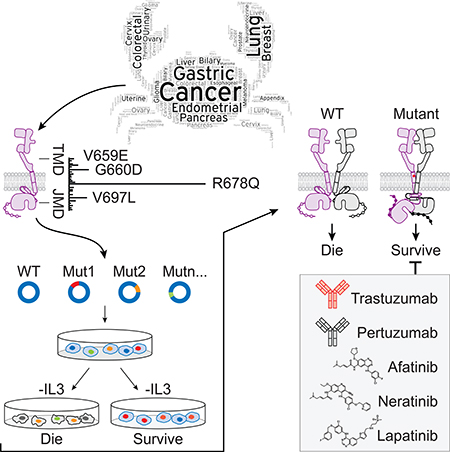
Introduction
The human epidermal growth factor receptor (HER) tyrosine kinase family consists of ERBB1/EGFR/HER1, ERBB2/HER2, ERBB3/HER3 and ERBB4/HER4. These receptors play an important role in cellular processes including growth, proliferation, differentiation and survival (Baselga and Swain, 2009; Hynes and Lane, 2005). ERBB receptors contain an extracellular domain (ECD), a transmembrane domain (TMD), an intracellular region that consists of a juxtamembrane domain (JMD), a kinase domain (KD) and a carboxy terminal tail domain (CTD) (Kovacs et al., 2015). The ECD is comprised of four subdomains (I-IV). In the absence of ligand, the ECD adopts an auto-inhibited tethered (closed) conformation that involves domain II and IV. Upon ligand binding between domains I and III, the dimerization arm in domain II is untethered, leading to receptor homo or heterodimerization, allosteric kinase activation, CTD phosphorylation and downstream signaling (Kovacs et al., 2015).
HER2 is an atypical member of the ERBB family, as its ECD adopts an untethered conformation constitutively (Roskoski, 2014). Unlike the other ERBB family members, HER2 does not have a ligand. HER2 preferentially heterodimerizes with ligand bound untethered (open) HER3 or EGFR to initiate cellular signaling, although HER2 homodimers capable of signaling have been reported in HER2 overexpressing cells (Brennan et al., 2000; Roskoski, 2014).
The transforming ability of HER2 was originally discovered in a nitrosoethylurea (neu)-induced glioblastoma rat model (Schubert et al., 1974; Schechter et al., 1985). Cloning and analysis of the HER2 sequence in this model revealed a Val to Glu mutation at codon 664 (V664E; V659E in humans) in the TMD of HER2 (Bargmann et al., 1986). Subsequent studies showed amplification and overexpression of the HER2 gene (ERBB2) as the oncogenic driver in ~20% of human breast and gastric cancers (Roskoski, 2014). Its established role as a potent oncogene has made HER2 a major target for therapy (Stern, 2012). Three HER2 antibody drugs trastuzumab, adotrastuzumab emtansine (T-DM1) and pertuzumab, and two small molecule HER2 kinase inhibitors lapatinib and neratinib have been approved by the FDA for use in the clinic for treating HER2 driven tumors (Gianni, 2018).
While overexpression remains a major mechanism of HER2-driven tumorigenesis, recent large-scale sequencing efforts have identified oncogenic mutations in the ECD and KD (Greulich et al., 2012; Zabransky et al., 2015; Ross et al., 2016). Mutations in the TMD and JMD have also been reported, albeit at a low frequency, and their relevance in oncogenesis is not fully understood (Bose et al., 2013; Yamamoto et al., 2014; Kavuri et al., 2015; Ou et al., 2017; Chang et al., 2018).
We hypothesized that HER2 TMD and JMD play significant role in oncogenesis and recurrent mutations in these domains are candidates of anti-HER2 therapy.
Results
Recurrent somatic HER2 TMD and JMD mutations in human cancers
To understand the spectrum of HER2 somatic alterations in sporadic cancers we analyzed targeted exome data from 111,176 tumors representing 54 disease groups and about 400 cancer types (Frampton et al., 2013). We detected HER2 mutations in 3,851 tumors (3.5%). While the ECD and KD accounted for the majority of these mutations (~40% each), the TMD and JMD accounted for ~2.8% and ~7.7% of the mutations, respectively (Figure 1A, B and Table S1). Since the KD and ECD mutants have been studied extensively, we focused on the TMD and JMD mutations. In the TMD we found mutations in 24 of the 27 amino acids. The most frequent mutations occurred at codons 652 (Thr), 653 (Ser), 659 (Val) and 660 (Gly) (Figure 1B and Table S1). The recurrent HER2 TMD mutations included P650L/S (2 cases), T652M/R (7 cases), S653C/P (12 cases), I654L/M/T (5 cases), I655M (5 cases), A657V (2 cases), V659E (19 cases), V659D/ins (7 cases), G660D (16 cases), G660R (1 case), L662V (3 cases), V664F/I (4 cases), V665M/del (5 cases), G668E/R (2 cases), V669A/L (3 cases), G672R (2 cases), I673F/M/V (3 cases) and L674I/V (3 cases) (Table S1). Additional mutation data sources such as cBioPortal (http://www.cbioportal.org; August 28, 2017), confirmed the presence of recurrent mutations in the HER2 TMD (7/27 residues) (Table S2), including G660D/R in 5 cases and V659E/D in 3 cases (Cerami et al., 2012). Recently, the HER2 G660D mutation was reported in lung adenocarcinoma cases from a family of Japanese descent (Yamamoto et al., 2014). The adjacent residue, Val at codon 659, was previously found mutated in a rat model of chemically induced carcinoma (Bargmann and Weinberg, 1988).
Figure 1. HER2 mutations in patient tumors.

(A) Mutations observed in HER2 shown across a diagram of the HER2 protein. (B) Mutational hotspots observed in HER2 TMD/JMD regions across different cancers. Only mutations found in at least 4 samples are shown. (C) Amino acid composition of the TMD in WT HER2 (PDB ID: 2JWA) and in V659E, G660D, or G660R mutants, highlighting the relative arrangement of side chain atoms of polar (oxygen (red) and nitrogen (blue) atoms shown as spheres) and apolar (carbon atoms (green) shown as sticks) residues. See also Tables S1 and S2.
Further, we found mutations in 30 of the 39 amino acids that comprise the JMD. (Figure 1B and Table S1). Among the most recurrent JMD mutations were R678Q/P/W (192 cases), V697L/M/del (24 cases) and P702L/S (9 cases), R677L/Q (8 cases), R683Q/W (6 cases), E693G/K (5 cases), Q709L/K (5 cases), I675L/M/T (4 cases), T686A/M/R (4 cases), R688L/Q/W (4 cases), R713L/Q/W (4 cases) and Q711H (3 cases). Additional JMD mutations found in at least 2 cases included Q679E/H, I682M/T, Y685H, R689I/K, E695K, P699S/del, G704E/R, A705V, A710V and M712L. The R678Q mutation in the JMD was the third most common mutation among all the non-synonymous mutations observed in HER2.
To better understand recurrent TMD mutations within the context of HER2 structure, we assessed the local environment of the recurring TMD mutations in an NMR structure of the HER2 TMD (PDB ID: 2JWA; Figure 1C). We observed that the TMD possesses an amphiphilic quality resulting from a stretch of polar residues along one face of the N-terminus of the TMD helix (S649, T652, S653, S656; Figure 1C). The most frequent mutations identified in patient data set (V659E and G660D/R) extend this polar strip. This observation implies the polar character of these mutations amplifies intrinsic properties of the native TMD which may be an essential feature leading to the activating effect of these mutations on HER2.
Saturation mutagenesis identifies activating mutations in HER2 TMD/JMD
Since we found recurrent mutations in the HER2 TMD and JMD in cancer patients we hypothesized that perturbing residues in the TMD and JMD could lead to HER2 activation. To test this, we mutagenized the entire TMD (27 amino acids, Ala 648 to Leu 674) and adjoining flanking regions corresponding to the C-terminal portion of domain IV (residues 641 – 647) and JMD region (residues 675 – 684) (Figure 2A, B). Using a pool of oligonucleotides, each replacing one amino acid at every position along the region Gly 641 to Lys 684 with one of the other possible 19 amino acids, we generated a library of 836 HER2 mutants. We assessed our pool for the representation of the mutant alleles by deep sequencing and detected 466 of the possible 836 mutants in the library. The retroviral pool of mutants was then used to generate stable BaF3 cells either with or without WT HER2 (Figure 2B) as these were heterozygous mutations in patient tumors. The stable BaF3 cells expressing the mutants were placed on Interleukin-3 (IL-3) free media for four days. We used DNA extracted from the surviving cells to amplify the HER2 TMD region and then deep sequenced the amplified region to assess the allele frequency of HER2 mutants.
Figure 2. Saturation mutagenesis of HER2 TMD/JMD.

(A) Schematic of the mutagenesis screen. (B) Bar plot representing the allele frequency of HER2 mutations identified in the screen 4 days following IL-3 removal. Screen was done without or with co-expressed WT HER2. HER2 protein sequence where mutations were observed in patient tumors is shown within shaded boxes below the bar plot. See also Table S1.
Among the enriched mutant alleles were G660D and V659E (Figure 2B) both in the presence and absence of WT HER2. Additionally, we identified G641S, A644F, E645K, A648L, S649T, L651W, L663H, V664F, V665D, F671N, L674H, I675M, R677T, Q680F and I682G mutants to be enriched in parental BaF3 cells expressing the mutant library (Figure 2B). Interestingly, certain mutants such as R678Q and R647T were highly enriched in the presence of WT HER2 (Figure 2B). In addition, we found E645F, V659E, G660D and K676T to be enriched in the presence of WT HER2. Several of the recurrent mutations identified in patient tumors were enriched in saturation mutagenesis screen. These include G641S, A644F, E645K, R647T, V659E, G660D, R678Q, K676T and R677T indicating that these are likely drivers (Figure 2B and Table S1).
Clinical HER2 TMD/JMD mutations are constitutively active
Saturation mutagenesis studies identified several activating mutations in the TMD/JMD (Figure 2B) suggesting that these alterations might serve as a mechanism for HER2 activation in cancer. We therefore generated 72 (43 unique mutants) stable BaF3 cell lines each expressing a HER2 TMD or JMD mutant identified in cancer patients (Table S2), either in the presence or the absence of WT HER2 and tested their survival signaling following IL-3 withdrawal.
We found that the TMD mutants S653C, S656C, V659E, G660D and G660R, and JMD mutants R677L, R678W, T686A and E693K promoted significant cell survival compared to WT HER2 (Figure 3A). In the presence of WT HER2, S649T, P650S, L651V, V659G, G660D, G660R, L663P and L674V TMD mutants showed significant survival compared to WT HER2. Several mutants detected in patient tumors in the JMD including R677L, R678Q, R683Q, E693K, Q709L and A710V were found to support cell survival in the presence of WT HER2 (Figure 3A). Concordant with the survival signaling, we found elevated levels of pHER2 in cells expressing active HER2 mutants (Figure 3A-C). Given that WT HER2 amplification and overexpression is linked to oncogenesis (Arteaga et al., 2011; Muthuswamy et al., 2001), we assessed the expression levels of WT HER2 in the parental and engineered BaF3 cells using flow cytometry and western blot (Figure S1A-C) (Onsum et al., 2013; Schoeberl et al., 2009). We did not detect endogenous HER2 in parental BaF3 cells, consistent with a previous report (Riese et al., 1995). Even though the stable expression of WT HER2 in BaF3 cells was comparable to that observed in HER2-amplified cells it did not support IL-3 independent survival (Figure S1D) as previously reported (Jaiswal et al., 2013; Riese et al., 1995).
Figure 3. HER2 TMD/JMD mutants promote cell survival.

(A) Relative survival of BaF3 cells expressing indicated HER2 mutants in the absence or presence of WT HER2. Residues mutated in patient tumors in the TMD and JMD are shown in shaded boxes below the bar plot. Solid green bars indicate mutants that showed statistically significant (two-tailed Student t-test; Bonferroni adjusted p value < 0.05) survival when compared to WT HER2 alone expressing BaF3 cells. Grey bars indicate mutants that did not show significant survival compared to WT HER2 alone. Data are presented as mean ± Standard Deviation (SD) of relative cell survival of twelve replicates and representative of three independent experiments. Circles inset with the single letter amino acid code representing the mutants below the bar plot shows the amount of pHER2 observed in BaF3 cells expressing the mutants. (B,C) Western blots showing the expression of total HER2, pHER2 and β-actin in the indicated stable BaF3 cells in the absence (B) or presence (C) of WT HER2. (D) MCF10A cell lines expressing the indicated HER2 mutants tested in the absence or presence of WT HER2. Scale bar shown at the bottom of each image represent 400 μm. Assay was performed in the absence of exogenous EGF or other growth factors. EV- empty vector. See also Figure S1 and Table S2.
MCF10A breast epithelial cells form acinar-cell spheroids when cultured in the presence of EGF on a reconstituted three-dimensional (3D) basement membrane such as matrigel (Debnath et al., 2003). However, the expression of oncogenes in MCF10A can render them EGF-independent and lead to the formation of multi-acinar structures (Debnath et al., 2003; Wang et al., 2006). To further confirm the effect of the mutations characterized using the BaF3 system, we stably expressed the indicated HER2 mutants in MCF10A cells (Figure S1E) and plated them on matrigel in the absence of any EGF. In agreement with the survival signaling activity observed in BaF3 cells, the HER2 TMD/JMD mutants G660D, L663P, R678Q and Q709L led to the formation of large multi-acinar bodies when compared to cells expressing WT HER2 or empty vector, confirming the oncogenic potential of these mutants (Figure 3D). Previously, WT HER2 overexpression in MCF10A in the presence of EGF and serum was reported to disrupt acinar formation (Muthuswamy et al., 2001). However, we do not observe this effect in our assays as we performed the assay in the absence of EGF, as described previously (Jaiswal et al., 2013). The acinar disruption by the TMD/JMD mutants was observed in both the absence and presence of exogenous WT HER2 expression in MCF10A and was more pronounced in the case of R678Q when it was expressed in the presence of exogenous WT HER2. These findings further confirmed the oncogenic potential of the HER2 TMD/JMD mutants.
Conformational analysis of TMD/JMD mutations
Having defined the most prevalent and activating HER2 TMD/JMD mutations in cancer (Figure 2, 3, 4A), we sought to understand the mechanisms by which they promote HER2 signaling. Both the TMD and JMD have been shown to play structural roles in the formation of active ERBB receptor complexes through interactions that stabilize the asymmetric kinase domain dimer, where one kinase domain (activator kinase) allosterically activates the other (receiver kinase) (Jura et al., 2009; Red Brewer et al., 2009; Zhang et al., 2006). Specifically, the TMD has been suggested to regulate receptor dimerization through an N-terminal Sm-xxx-Sm motif (“Sm” for small amino acid) (Bocharov et al., 2008; Burke et al., 1997; Fleishman et al., 2002; Sternberg and Gullick, 1989). This motif is thought to dictate a specific orientation between TM helices, which in turn can affect the conformations of juxtamembrane and catalytic domains and thereby influence their activation state (Bell et al., 2000). Investigations of EGFR have shown that the JMD is essential for ligand-dependent activation through stabilization of the asymmetric kinase dimer, which involves an intermolecular interaction between a segment within the JMD, called the juxtamembrane latch, and the C-lobe of another kinase (Jura et al., 2009; Littlefield et al., 2014; Red Brewer et al., 2009). Because the juxtamembrane latch sequence of HER2 is almost identical to EGFR (Figure 4B), the role of the latch in HER2 activation is predicted to be the same.
Figure 4. Conformational analysis of HER2 TMD/JMD mutants.

(A) Representative TMD/JMD germline and somatic mutations mapped onto a structural model of full-length HER2 composed using the crystal structures of the HER2 extracellular domain (PDB ID: 1N8Z), HER2 transmembrane domain (PDB ID: 2JWA) and HER2 kinase domain (PDB ID: 3PP0). (B) Sequence alignment of the HER family receptors. (C) MD simulations were performed using the coordinates of the WT HER2 TMD dimer (PDB ID 2JWA, residues 649 – 679) as a starting model. (D) Overview of the final state obtained at the end of a 100 ns simulation of the WT HER2 TMD dimer (left), a heterodimer between a WT TMD and an activating C-terminal R678Q TMD mutant (WT/R678Q) (middle) and a homodimer of the activating N-terminal G660D TMD mutant (G660D/G660D) (right). (E) Overlay of the final states observed for five independent MD simulations of the G660D/G660D HER2 TMD dimer (sim1-sim5). (F) Surface representation of the C-lobe of the HER2 kinase domain bound to a juxtamembrane latch binding region to depict interactions mediated by Q709 and L709. The interface was modeled by aligning structures of the HER2 kinase domain (PDB ID: 3PP0) on both activator and receiver kinases in the structure of the HER3/EGFR asymmetric dimer (PDB ID: 4RIW). See also Figure S2.
We used molecular dynamics (MD) simulations to evaluate the possible effects of the most activating mutations (G660D, G660R, R678Q) on the dimeric TMD conformation in the activated receptor predicted by NMR studies (Bocharov et al., 2008). In the putative activated dimer, the TMD helices of HER2 interact through an S656-xxx-G660 motif, a variation of the higher-affinity G-xxx-G dimerization motif (Lemmon et al., 1994; Senes et al., 2000). Thus, any mutation that might increase the stability of the HER2 S656-xxx-G660 motif should stabilize the activated state. In the S653C mutant, mutation of a polar serine to a relatively hydrophobic cysteine converts the HER2 S656-xxx-G660 to a strong glycophorin A-like Sm-xxx-Sm motif (Lu et al., 2010) leading to activation, presumably by stabilizing the receptor dimer.
Proper positioning of the S656-xxx-G660 motif for productive TMD dimer formation is highly dependent on the orientation and geometry of the monomeric TMD helices, defined by basic residues near the interfacial regions between the cytoplasm and head group region of the bilayer (Gleason et al., 2013; Hristova and Wimley, 2011; Kim et al., 2011). We expected that activating HER2 mutations such as R678Q might have a significant effect on the TMD geometry and dimerization. To test this, we performed allatom 100 ns MD simulations for wild-type (WT) and the WT/R678Q HER2 TMD dimers in a phospholipid bilayer as the R678Q mutant was active in our functional assays in the presence of WT HER2 (Figure 2B and 3A). The coordinates of the HER2 TM dimer in the putative activated conformation determined by NMR (PDB ID: 2JWA) were used as the starting positions in the simulations (Figure 4C). As expected, the conformation of the WT HER2 TMD homodimer (WT/WT) remains stable over the course of the simulation as evidenced by the lack of fluctuation in the Cα RMSD (Figure S2A). In the WT/R678Q TMD heterodimer, the S656-xxx-G660 motif remained engaged for the duration of the simulation, albeit through different interactions. However, the R678Qcontaining region of the C-termini separated by several angstroms compared to the WT homodimer (Figure 4D; S2A). Despite these differences, in both WT/WT and WT/R678Q dimers, the conformations observed in the final state are consistent with a geometry proposed to support an activated, asymmetric configuration of the cytoplasmic kinase domains, and suggests that the enhanced activity of the mutant may be the result of its stabilizing effect on the specific heterodimeric configuration required for signaling.
A second possibility is that oncogenic mutations are able to stabilize an alternate activated dimeric TMD conformation. It is well known that polar interactions strongly support helix association both in conformations that cooperate with small Sm-xxx-Sm motif dimerization or in entirely unique geometries (Brooks et al., 2014; Goldberg et al., 2010; Gordeliy et al., 2002). To see the effect of a polar mutation on HER2 TMD dimers, we simulated the G660D mutant. MD simulations demonstrated that the introduction of the protonated aspartate disrupts the native dimeric configuration (Figure 4D). In five independent 100 ns simulations, the TMD dimer configuration gradually drifted away from the starting configuration sampled for WT/WT HER2 and without achieving a common final state (Figure 4E, Figure S2A-C). A simulation of the HER2 G660R mutant (Figure 3A) revealed similar structural consequences as the G660D mutation (Figure S2D, E). On a 100 ns time scale it was not possible, however, to predict with certainty the final geometry of a HER2 TMD dimer in the presence of the G660D/R mutations, but these results suggest that polar mutations at position 660 alter the WT HER2 TMD geometry.
To understand the activating effect of the juxtamembrane latch mutant, Q709L, we analyzed the interface between the juxtamembrane latch and the C-lobe of the activator kinase in a model of the HER2 homodimer constructed using the crystal structure of the EGFR/HER3 heterodimer (PDB ID: 4RIW) in which HER3 in the activator position was replaced with the HER2 kinase domain (PDB ID: 3PP0), and the juxtamembrane latch sequence of EGFR in the receiver position was replaced with that of HER2 (Figure 4F). From this model we observed that Q709 does not optimally participate in the dimer interface. Calculation of the HER2 C-lobe electrostatic surface reveals a hydrophobic pocket in the vicinity of the large polar side chain of Q709 likely suboptimal for hydrophobic packing between the C-lobe and the juxtamembrane latch. In contrast, the presence of a hydrophobic side chain such as leucine appears to be more optimal for favorable interactions with such a hydrophobic pocket, and is predicted to stabilize the dimerization interface essential for the allosteric activation of HER receptor kinase activity.
Dimerization and allosteric activation of HER2 TMD mutant promotes survival signaling
We sought to understand if signaling by the HER2 TMD mutants required its kinase activity. To test this we stably expressed a kinase-impaired K753M/G660D double mutant (G660D.KI) HER2 in BaF3 cells and assessed it for survival signaling in the absence of IL-3 (Figure 5A-C). Compared to HER2 G660D, the G660D.KI mutant did not support IL-3 independent survival of BaF3 cells (Figure 5B, C and S3). Although the expression of G660D.KI was low (Figure S3) we found HER2 kinase small molecule inhibitors to be effective in blocking the G660D mutant (see Figure 6 below). Taken together these results confirm that the kinase activity of G660D is essential for its oncogenic activity. Structure-guided point mutations in the receiver and/or activator interface of the kinase domains (Figure 5A) have been used to confirm the role of the asymmetric dimers in the allosteric activation of EGFR (Zhang et al., 2006) and chimeric EGFR/HER2 receptors (Ward and Leahy, 2015). We stably expressed HER2 G660D carrying an N-terminal I714Q mutation in the receiver interface (RM) or the C-terminal V956R mutation in the activator interface (AM) alone, together, or in combination with WT HER2 in BaF3 cells and assayed for cell survival activity. Expression of receiverimpaired mutant HER2 G660D-I714Q (RM) or activator-impaired HER2 mutant G660DV956R (AM) on its own did not promote BaF3 cell survival following IL-3 withdrawal (Figure 5B, C). However, combined expression of HER2 G660D-I714Q (RM) and HER2 G660D-V956R (AM) in BaF3 cells restored the cell survival signaling activity of HER2 G660D (Figure 5B, C; lane 9), confirming the allosteric activation of the kinase domain following HER2 G660D dimerization promotes cell survival signaling. Consistent with survival signaling, we observed elevated levels of pHER2 in cells coexpressing HER2 G660D-I714Q (RM) and HER2 G660D-V956R (AM) mutants (Figure S3). Since the HER2 G660D mutant can promote survival signaling in the presence of WT HER2 in BaF3 cells (Figure 3A and 5B) we tested if it preferentially functioned as a receiver or activator in the presence of WT HER2. Expression of HER2 G660D-I714Q (RM) in BaF3 cells in the presence of WT HER2 did not promote cell survival (Figure 5B, C), revealing that it was not able to function as a WT HER2 activator. However, HER2 G660D-V956R (AM) promoted cell survival in the presence of WT HER2 (Figure 5B,C), indicating that the HER2 G660D is predisposed to adopt a receiver conformation.
Figure 5. G660D HER2 activation involves asymmetric dimerization of the KD.

(A) Active HER2 asymmetric dimer model. Residues involved in the asymmetric kinase domain interactions in HER2 deduced from EGFR structural studies are shown (inset). (B) IL-3 independent cell survival of BaF3 cells stably expressing the indicated kinase, activator or receiver mutants. Data are presented as mean ± Standard Deviation (SD) of relative cell survival of twelve replicates and representative of three independent experiments. (C) Cartoon representation of the proposed mechanism of activation of HER2 G660D. The number shown below corresponds to the lane number in (B). See also Figure S3.
Figure 6. HER2 inhibitory drugs are effective in blocking HER2 TMD/JMD mutants.

(A) Cartoon of HER2 depicting binding of trastuzumab and pertuzumab to ECD domain IV and domain II, respectively. (B) Effect of indicated anti-HER antibodies and the Fab portion of pertuzumab on survival of BaF3 cells expressing HER2 G660D mutant. (C-F) Effect of trastuzumab (C), pertuzumab (D), neratinib (E), and afatinib (F) on IL-3 independent survival of BaF3 cell expressing the indicated HER2 mutants. Data shown in (B-F) are mean ± SEM, where SEM is standard error of mean of four technical replicates and representative of experiment repeated three independent times. RLU = Relative luciferase units; EV = empty vector; WT = wild-type HER2. (G) Kaplan-Meier survival curves for cohorts of mice (n = 10) implanted with HER2 G660D expressing BaF3 cells that were either untreated or treated with anti-Ragweed (control) or trastuzumab. WT HER2 expressing BaF3 cells served as controls. See also Figures S4, S5 and Tables S3, S4.
HER2 TMD/JMD mutants respond to anti-HER2 antibodies and kinase inhibitors
Several targeted therapeutics have been approved for treating breast cancers overexpressing HER2. Trastuzumab, an anti-HER2 ECD domain IV binding antibody, and pertuzumab, an antibody that binds to HER2 ECD domain II, have been approved for treatment of HER2 positive breast cancers (Gianni, 2018). Trastuzumab has also been approved for treating HER2 positive metastatic gastric cancers (Van Cutsem et al., 2016). While trastuzumab blocks HER2 signaling through diverse mechanisms, pertuzumab blocks ligand-driven HER2 heterodimerization and signaling (Arteaga et al., 2011). Unlike antibody drugs, small molecule inhibitors such as lapatinib and neratinib approved for treatment of HER2+ breast cancers bind to the kinase domain and block its activity (Gianni, 2018). We tested trastuzumab, pertuzumab, and cetuximab (an antiEGFR antibody) for their ability to block the TMD/JMD HER2 mutant activity (Figure 6 and S4). We also tested lapatinib (an FDA approved HER2 ATP-competitive kinase inhibitor), neratinib (an FDA approved HER2 non-ATP competitive inhibitor), afatinib (an EGFR inhibitor that is also active against HER2), erlotinib and gefitinib (both are FDA approved EGFR kinase small molecule inhibitors for treating lung cancer) for their activity against HER2 JMD/TMD mutants (Ellis et al., 2015; Gianni, 2018). Among the anti-HER antibodies (Figure 6A), we found trastuzumab to be effective in blocking cell proliferation and survival of HER2 V659E, G660D, G660R, L663P, R678Q and Q709L (IC50 0.5–7ng/ml; Figure 6B-C and Table S3). Surprisingly, though pertuzumab was effective in blocking L663P, R678Q and Q709L (IC50 1.316.7ng/ml; Table S3), it stimulated the growth of V659E and G660D expressing BaF3 cells in a dose dependent manner (Figure 6B, D). Also, pertuzumab was not effective in blocking G660R (Figure 6D). Pertuzumab binds to domain II of HER2 ECD and is a bivalent antibody capable of binding two HER2 receptor molecules simultaneously. This may facilitate the formation of productive homodimers and/or stabilization of the active mutant homodimers, contributing to the observed survival stimulating effect in the context of some mutants. To test this, we generated a monovalent Fab fragment of pertuzumab and assessed its effect on BaF3 cells expressing HER2 G660D. We found that the Fab was not stimulatory and was able to partially inhibit cell survival and proliferation at higher concentrations (Figure 6B). Further, we found that neither cetuximab nor anti-HER3 antibodies blocked the viability of BaF3 cells expressing G660D HER2 (Figure 6B), confirming that the observed cell survival effect was due to HER2 mutant homodimers. Consistent with the observed effect of the anti-HER2 antibodies on cell survival, trastuzumab blocked colony formation by HER2 G660D expressing BaF3 cells, while pertuzumab was stimulatory (Figure S4A). Further, cetuximab was not effective in blocking colony formation or cell survival signaling by TMD/JMD mutants (Figure S4A and S4B). Among the small molecule kinase inhibitors tested, we found neratinib (IC50 0.12–0.58 nM), afatinib (IC50 0.26–18 nM) and lapatinib (IC50 0.5–18 nM) to be effective in blocking the survival of BaF3 cells expressing HER2 mutants (Figure 6E, 6F, S4C and Table S4), though neratinib was the most effective inhibitor against all the mutants tested (Hyman et al., 2018). Erlotinib and gefitinib, both EGFR kinase inhibitors, were not effective against the HER2 mutants tested (Figure S4D, S4E and Table S4), confirming that the survival effect observed in BaF3 cells is primarily mediated by HER2.
Previous studies have shown that BaF3 cells expressing oncogenes when implanted in nude mice promote a leukemia-like disease leading to their reduced overall survival (Jaiswal et al., 2013). We used this in vivo system and found that the HER2 G660D mutant BaF3 cells led to a reduced median overall survival of 22–23 days compared to mice receiving WT HER2 expressing BaF3 (Figure 6G). As expected, the histological analysis of spleen from mice implanted with HER2-G660D cells showed sheets of large mononuclear cells with irregular nuclear contours, nucleoli and scattered mitotic figures suggesting tumor cell infiltration (Figure S5). Liver sections from these mice showed congestion of sinusoids and involvement of lobules by monomorphic cells with enlarged nuclei and nucleoli (Figure S5). The spleen and liver from mice receiving WT HER2 cells showed normal architecture devoid of infiltrating tumor cells.
Using this in vivo model we also tested the efficacy of trastuzumab and found that it prolonged the survival of mice implanted with BaF3 HER2 G660D cells, while the control anti-Ragweed antibody was not effective, as expected (Figure 6G). Histological analysis revealed that mice implanted with HER2-G660D cells and then treated with trastuzumab did not show significant spleen and liver infiltration when compared to the control antibody treated mice (Figure S5).
Germline HER2 G660D lung cancer patient responds to therapy
Familial lung cancer cases are rare. Two siblings and a first cousin in an AsianIndian family were diagnosed with stage IV lung cancer at the Tata Memorial hospital in India (Figure 7A). The affected patients at diagnosis were 41 (female), 47 (female) and 53 (male) years old. This was much earlier than the typical age of 65 or above at which sporadic lung cancer is generally diagnosed. All the familial lung cancer patients were non-smokers. Though familial form of lung cancer is rare, the disease occurrence within multiple family members, the early age of onset and family history suggested that there was a common genetic risk factor within the family.
Figure 7. Germline HER2 G660D lung cancer patient responds to therapy.

(A) Pedigree of a family in which multiple members were diagnosed with lung cancer. Solid black and grey circles (females) and squares (males) indicate affected individuals. Blood samples were obtained from affected individuals represented by solid black circle or squares. Slash mark indicates deceased individuals. (B) Flowchart depicting the exome analysis. (C) Chest CT scan of the patient before and after 12 weeks of treatment with afatinib. See also Figures S6, S7 and Table S5.
We performed whole exome sequencing using DNA obtained from peripheral blood samples from the three affected patients (88–99X fold coverage; Figure S6A-C). Joint variant calling resulted in 551,896 variants (Figure 7B). After filtering out common variants present at MAF >= 1% frequency in the ExAC database (Lek et al., 2016) or 1000 genomes (1000 Genomes Project Consortium et al., 2015), we obtained 60,688 rare variants. Of these, we found 2,645 variants (~4%) to be protein-altering or potentially protein-altering. We then focused on 282 variants from the set of 2,645 that were shared among all 3 patients. We assessed the distribution of the 282 variants among a curated list of 138 cancer core genes (Vogelstein et al., 2013) and identified G660D, a missense variant in HER2. We also performed exome sequencing on DNA obtained from formalin fixed tumor available from one of the patients (III.3) and confirmed that the G660D mutation was present in the tumor (Table S5). Additionally, we observed that proportions of somatic mutations among the possible six classes of base substitution (C>A, C>G, C>T, T>A, T>C, T>G) were similar between patient III.3 tumor and non-smoker TCGA lung adenocarcinoma samples (Cancer Genome Atlas Research, 2014) (Figure S7A,B).
The efficacy of various drugs against the activity of the oncogenic G660D HER2 mutation in vitro (Figure 6A-G) suggested that patients carrying this mutation might benefit from a HER2 targeted therapy. Patient III.3 (Figure 7A and S6A) prior to the genomics analysis was treated with pemetrexed and carboplatin combination chemotherapy followed by erlotinib. Following the identification of G660D HER2 mutation, this patient was started on fourth line afatinib 40 mg once daily. Within 30 days the patient’s chest pain and her shortness of breath was resolved. Computed tomography (CT) of the chest 12 weeks following treatment showed 21% reduction in the tumor measurement by RECISTv1.1 criteria (Figure 7C). The side effects observed were minimal with complaints of nausea and occasional skin rashes. The treatment was well tolerated by the patient and overall general condition improved with no appearance of any fresh lesions. The patient response was durable and lasted for over 15 months. These results indicate that the HER2 G660D germline mutation was the driver in the patient tumor and such patients can benefit from HER2 targeted therapy. Discussion
Analysis of sequence data from ~111,000 tumors representing ~400 cancer types identified many recurrent somatic mutations in the TMD and JMD of HER2 that included G660D, V659E, R678Q and Q709L. Functional analysis of the mutants observed in patients showed that a majority of the recurrent mutations were activating and are likely drivers. Both V659 and G660 are part of the N-terminal S656-xxx-G660 motif in the HER2 TMD important for receptor dimerization, kinase activation, and signaling (Arkhipov et al., 2013; Endres et al., 2013; Fleishman et al., 2002; Ou et al., 2017). We observed a striking relationship between the chemical nature of the TMD mutations and the potency with which they activate HER2. Substitutions to polar residues (S653C, V659E, G660D/R, E693K) exert a much stronger activating effect on HER2 than apolar mutations (L651V, V659G, L674V). These polar mutations cluster at the N-terminus of the TMD, extending the existing stretch of polar residues on one face of the amphiphilic transmembrane helix. The significance of this pattern is unclear, but our MD data suggest that polar residues support rearrangements of the TMD helices and deviation from the N-terminally mediated dimer, a phenomenon that has previously been observed in other membrane proteins carrying polar TMD mutations (Brooks et al., 2014; Goldberg et al., 2010; Gordeliy et al., 2002).
Among the HER2 mutations reported in this study, the JMD mutant R678Q is the third most recurrent HER2 mutation. Basic residues (such as arginine) play an important role in anchoring TMD helices of single TM membrane proteins to the bilayer (Gleason et al., 2013; Hristova and Wimley, 2011; Kim et al., 2011) with an orientation tilted relative to the plane of the membrane leaflet (Barrett et al., 2012; Monk et al., 2014; Vostrikov et al., 2010). Their mutation could affect receptor activation by releasing constraints on the helical tilt angle and promote new TMD orientations. Our analysis of the R678Q HER2 mutation at the TMD/JMD interface suggests that this mutation indeed stabilize the native TMD dimer in alternative conformation. A similar mutation of a lysine at the periphery of the bilayer in the constitutively dimeric integrin receptor complexes leads to spontaneous receptor activation, presumably due to the destabilization of the inactive TMD dimers in the membrane (Tohyama et al., 2003).
The juxtamembrane region plays a critical role in EGFR activation and is highly conserved in HER2 (Jura et al., 2009; Red Brewer et al., 2009). The constitutively active juxtamembrane latch mutation Q709L is predicted to fit well within a hydrophobic pocket in the activator kinase and leading to activation by enhancing the receiver-activator interaction. A similar set of mutations introducing hydrophobic residues in the juxtamembrane latch to optimize binding to other small hydrophobic pockets along the juxtamembrane latch-binding groove on the C-lobe of the activator kinase have been identified as cancer mutations in EGFR and HER3 (Littlefield et al., 2014; Red Brewer et al., 2009). Hence, the Q709L mutation seems to utilize a similar strategy to drive constitutive HER2 receptor signaling in cancer.
Besides amplification and mutations in the KD and ECD, mutations in the TMD/JMD domains expand the list of actionable HER2 alterations. We found that they occur in multiple cancers. In addition to somatic HER2 mutations, we investigated a heterozygous germline HER2 TMD mutation, G660D that we identified in a family from India, where the individuals carrying the mutation developed lung cancer at an early age. Germline mutations in other oncogenes such as AKT1 E17K (Lindhurst et al., 2011), EGFR T790M (Bell et al., 2005), PIK3CA G118D (Orloff et al., 2013), BRAF G469E and KRAS G60R (Niihori et al., 2006) are known to contribute to familial diseases including cancers. The HER2 G660D mutation is a very rare variant and is not found in the normal population. It was previously reported in a Japanese family where individuals developed lung cancer (Yamamoto et al., 2014). A limited number of genetic variants associated with hereditary forms of lung cancer are known and they include a dominantly inherited germline EGFR T790M variant (Bell et al., 2005; Gazdar et al., 2014). Interestingly, the EGFR and HER2 mutant hereditary cancer patients are predominantly never-smokers, including affected individuals studied here (Gazdar et al., 2014). Individuals carrying the HER2 G660D mutation are normal at birth, suggesting the expression of this mutant allele is not developmentally detrimental despite the potential expression of a constitutively active HER2 during development. Similarly, germline expression of the HER2 TMD V659E (neu) mutant cDNA in the presence of endogenous mouse ERBB2 did not affect the development of mice (Andrechek et al., 2004). Unlike human subjects with this mutation that develop lung cancer, the mice were free of cancer for over 24 months (Andrechek et al., 2004). These observations suggest that further studies are needed to establish the secondary events leading to carcinogenesis in HER2 mutant carriers.
Our data also highlight important clinical considerations for treating the TMD and JMD activating mutations in HER2. Pertuzumab further stimulated signaling by G660D HER2 in BaF3 cells. Pertuzumab is a bivalent antibody and works by blocking liganddependent HER2/HER3 heterodimer signaling. However, in the context of G660D HER2 expressing BaF3 cells, it likely promotes HER2 activation by further clustering homodimers stabilized by TMD interactions leading to elevated sustained signaling. In contrast, the germline G660D HER2 patient reported here responded well to afatinib in a fourth line setting. Similarly, patients having lung cancer with somatic HER2 V659E or V659E/G660R mutations showed a durable response to afatinib (Ou et al., 2017). Taken together, our results suggest that HER2 TMD mutant, and also likely the JMD mutant patients are candidates for targeted anti-HER2 therapy in the clinic.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Somasekar Seshagiri (sekar@gene.com).
Experimental models and subject details Tumor and blood samples
Tumor samples were processed in one of the two broad protocols applicable to solid tumors or hematologic cancers as previously described (Frampton et al., 2013; Hartmaier et al., 2017). Samples were submitted to a Clinical Laboratory Improvement Amendment (CLIA)-certified, New York State-accredited, and CAP-accredited laboratory (Foundation Medicine) for hybrid capture followed by next-generation sequencing (NGS). All samples that advanced to DNA extraction contained a minimum of 20% tumor cells. DNA was extracted from formalin fixed paraffin embedded 10-μm sections. Approval for this study, including a waiver of informed consent and a HIPAA waiver of authorization, was obtained from the Western Institutional Review Board (protocol number 20152817).
In this study we analyzed blood samples from three patients (III.1, III.2 and III.3) and tumor DNA from one patient (III.3). The patients at diagnosis were 41 (female), 47 (female) and 53 (male) years old. The study was conducted with the approval of Tata Memorial hospital institutional ethics committee (IEC) and was monitored by the data safety and monitoring subcommittee (see below).
Human Subject
Two siblings and a first cousin in an Asian-Indian family were diagnosed with stage IV lung cancer at the Tata Memorial hospital in India. The affected patients at diagnosis were 41 (female), 47 (female) and 53 (male) years old. The study was conducted with the approval of Tata Memorial hospital institutional ethics committee (IEC) and was monitored by the data safety and monitoring subcommittee. The trial was conducted according to the principles laid down by the 18th Helsinki World Medical Assembly (1964) and its amendments. This study was approved by IEC with project no - 108 dated June 6, 2013. Written patient informed consent was obtained for genetic analysis.
Mouse studies
Balb/C nude mice (female, 8–12 week old) were used for tail vein injection and followed for survival of the mice. Each arm in the study contained ten mice. All animal studies were conducted under protocols approved by Genentech’s Institutional Animal Care and Use Committee (IACUC) guidelines.
Cell lines
All the cell lines were obtained from ATCC and authenticated by Genentech internal cell line repository, gCell. Cell lines used in this study were mouse pro-B cell line BaF3, Phoenix retrovirus producer cell line, and a mammary epithelial cell line, MCF10A. BaF3 cells were cultured in complete RPMI media and IL-3. MCF10A cells were cultured in normal growth media containing 5% horse serum and EGF. Phoenix retrovirus producer cell lines were cultured in complete DMEM media.
Method Details Plasmids and antibodies
The pLPCX retroviral vector (Clontech, CA) expressing full length HER2 with an N-terminal herpes simplex glycoprotein D (gD) tag was constructed and used for site directed mutagenesis. A retroviral vector, pRetro-IRES-GFP (Jaiswal et al., 2013), was used to stably express N-terminally FLAG-tagged HER2 WT.
Antibodies that recognize pHER2 (Y1221/22), pERK1/2, total ERK, HER2 and βactin were obtained from Cell Signaling Technology, MA. Anti-gD antibody (Genentech Inc., CA), anti-FLAG-M2 (Sigma, MO), and HRP-conjugated secondary antibodies (Pierce Biotechnology, IL) were also used in the study.
Mutagenesis Screen
HER2 saturation scanning mutagenesis library was generated by replacing each residue in the TMD/JMD and flanking region (amino acids G641-K684) with possible 19 amino acids (836 possible single site mutants). Full length HER2 with an N-terminal gD (herpex simplex glycoprotein D) tag cloned into pLPCX retroviral vector (Clontech) was used as a plasmid template to generate mutants. The 836 mutagenic oligonucleotides and amplification primers needed to amplify the mutant primers were purchased from Agilent, CA (custom Quikchange HT protein engineering system™). The mutagenic oligos were amplified using a pair of flanking primers and used to mutagenize gDtagged HER2 cloned into the pLPCX retroviral vector. The mutant HER2 constructs were then transformed into Solopack Gold supercompetent E. coli cells (Agilent) and amplified. The retroviral vector DNA carrying the mutant HER2 pool was sequenced on Hiseq2500 (Illumina) to assess the mutation prevalence. Control mutant constructs carrying mutations were generated by site-directed mutagenesis and used as spike-in controls prior to generation of the retrovirus pool.
The retroviral plasmid library was transfected using Fugene6 (Roche, CA) into four 10 cm tissue culture dishes containing 0.25 × 106 phoenix amphoteric packaging cells. BaF3 parental cells or those expressing Flag tagged WT HER2 were infected with the retroviral pool at a multiplicity of infection (MOI) of ~1 per cell using spinfection at 1,800rpm for 45 min, as previously described (Jaiswal et al., 2013). Pool of BaF3 stables cells was derived by culturing the cells in media supplemented with 1 μg/ml of puromycin for 14 days. Stable cells were washed twice with PBS and plated in quadruplicates in RPMI media devoid of IL-3. Surviving cells were collected at day 4 and genomic DNA was prepared (DNeasy Blood and Tissue Kit, Qiagen) and used to amplify the HER2 TMD/JMD regions. A 243–249bp amplicon was prepared from each replicate using a pool of three staggered primer pairs, F1 (5’–CTTGCCCCATCAACTGCAC 3’), F2 (5’-CCTTGCCCCATCAACTGCAC-3’), F3 (5’AGCCTTGCCCCATCAACTGCAC-3’), R1 (5’-TCGCTCCGCTAGGTGTCAGCGGCT-3’); R2 (5’-ATCGCTCCGCTAGGTGTCAGCGGCT-3’) and R3 (5’-GCATCGCTCCGCTAGGTGTCAGCGGCT-3’) and 50ng of genomic DNA using standard PCR conditions. Amplicons were column purified, quantified on Qubit fluorometer, pooled and used to construct libraries (NuGen Ovation Library System (NuGen) cat# 9092–256) with 75ng of DNA as the input. The libraries were then sequenced on HiSeq 2500 (Illumina) to generate 2 × 75-bp paired-end data. The reads were aligned to the HER2 sequence (Ensembl ID: ENST00000269571) using BWAMEM (version 0.7.10; http://arxiv.org/abs/1303.3997) with default parameters. For each mutant, the reads observed were counted using only high-quality bases (Q score > 30) from high-quality alignments (mapping quality >30). The frequency of each mutant was calculated as the number of observed mutant-reads divided by the total number of reads at that locus. The mutant allele-frequency at day 4 was used to estimate the enrichment of mutants in the pool.
Cell survival assay
BaF3 cells stably expressing HER2 mutants were washed twice with PBS and plated in 96-well plates (10,000 cells/well) in replicates of 12 in complete RPMI medium without IL-3. Cell viability was measured using the Cell Titer Glo Luminescence Cell Viability Kit (Promega), and plates were read on a Synergy 2 (Biotek Instruments) luminescence plate reader. Relative survival reported in cell survival studies is a ratio of relative luciferase activity (RLU) at day 4 over RLU measured at the time (day 0) the experiment was initiated.
Expression analysis using flow cytometry
HER2 expression levels were quantified by incubating cells with saturating amounts of an anti-HER2-PE antibody (BioLegend), washed, and read by flow cytometry (FACS Calibur) to determine the mean fluorescence intensities (MFI).
Phospho-HER2 quantitation
BaF3 cells stably expressing HER2 mutants were starved of IL-3 for 24 hours. Cells bearing HER2 mutants without and with WT HER2 were lysed at 1 × 106 and 0.25 × 106 cells per 30 μl Tris lysis buffer, respectively, supplemented with phosphatase inhibitors II and III (Sigma-Aldrich), and protease inhibitor (Roche). Lysates were centrifuged 5 min at 14,000 rpm to remove cell debris and 25 μl per well added to plates coated with antibodies to HER2 p-Tyr1248 and total HER2. Bound HER2 was detected with a Sulfo-Tag-labeled secondary antibody. Percent p-HER2 was calculated as follows: ((2 x Phospho-signal) / (Phospho-signal + Total signal)) x 100.
Western blotting
Western blots were performed as previously described (Jaiswal et al., 2013). For Western blot studies, BaF3 cells were grown in absence of IL-3 for 18 h. MCF10A cells were cultured in normal growth media containing 5% horse serum and EGF.
Molecular Dynamics simulation experiments
The input models were built using a truncated form (residues 649 – 679) of the median model reported in the NMR ensemble of a structure determined for wild-type HER2 dimer (PDBID: 2JWA) as our template. The orientation of the TMD dimer in the membrane was predicted using the OPM ppm server (Lomize et al., 2012). The proteinbilayer system was built by combining the oriented protein coordinates with a 60 × 60 membrane slab of POPC lipids and solvated with 15 Å of water containing 0.15 M NaCl on each side of the bilayer, removing overlapping lipids. Each system contained approximately 20,500 atoms. Langevin dynamics simulations were run under NPT conditions with a Berendsen barostat and Langevin thermostat (Berendsen et al., 1984) using the CHARMM36 force field (Huang and MacKerell, 2013) and ACEMD (Harvey et al., 2009) with a 4 fs time step using the hydrogen mass repartitioning scheme (Feenstra et al., 1999). Model equilibration for simulations of WT/WT, G660D/G660D, G660R/G660R, and WT/R678Q HER2 TMD dimers was conducted for 10 ns with simple harmonic positional constraints of backbone Cα atoms with a Langevin damping constant of 1.0 ps−1. Constraints were then lifted for 90 ns production simulations, with Langevin damping constant of 0.1 ps−1. A 400 fs Berendsen barostat relaxation time was used throughout. The stability of the final model was evaluated by calculating the backbone Cα root mean square deviation (RMSD) between the simulation at each time step and either the input model or the final model obtained at the end of the simulation. To check the robustness of the observed conformation in the G660D HER2 TMD homodimer, we repeated the simulation an additional 4 times and calculated pairwise RMSD values for each final state observed to assess the level of convergence between simulations. In all G660D mutant simulations, the side chain of the aspartate residues was protonated.
HER2 inhibitor testing
BaF3 cells stably expressing HER2 mutants either with or without WT HER2 were plated in 96 well plates in 100 μl of RPMI lacking IL-3. The cells were then treated with indicated concentrations of trastuzumab, pertuzumab, cetuximab, anti-ERBB3 antibodies (ERBB3.1(YW55.87.5) and ERBB3.2 (YW57.88.5 )) or Fab fractions of pertuzumab antibody or small molecule inhibitors. The Fab fraction of the pertuzumab antibody was generated and purified using papain, a non-specific thiol-enodpeptidase, immobilized on agarose resin (Pierce Fab preparation kit, Thermo Scientific).
Viable cell number was assessed 4 days after treatment using Cell Titer-Glo Luminescent cell viability assay kit (Promega Corporation) as described earlier (Jaiswal et al., 2013). Non-linear regression plot of antibodies and their fractions or of inhibitors were generated. Calculation of IC50 was performed using GraphPad Prism 5.00 software. Data are presented as mean ± SEM of at least 3 replicates of a representative experiment that was repeated at least three times.
Three-dimensional morphogenesis assay
MCF10A cells stably expressing WT HER2 or mutant HER2 either alone or in combination with WT HER2 were seeded on growth factor reduced Matrigel (BD Biosciences) in the absence of any exogenous EGF or growth factor in a 8-well chamber slide as previously described (Jaiswal et al., 2013). Acini morphology was photographed on day 12 using EVOS microscope (Thermo Fischer) with a 10x objective.
Colony formation assay
About 20,000 BaF3 cells stably expressing either WT HER2 or G660D HER2 were plated on IL-3 free methylcellulose (Stemcell Technologies) in the absence or presence of various HER2 inhibitors and assessed for colony formation as previously described (Jaiswal et al., 2013). The plates were incubated at 37°C for 2 weeks and colonies were photographed using Gel count Imager (Oxford Optronix Ltd, UK).
In vivo survival studies
BaF3 in vivo survival studies using cells expressing HER2 mutants were performed as previously described (Jaiswal et al., 2013). Briefly, 2 × 106 BaF3 cells expressing either empty vector (EV), WT HER2 or mutant HER2 were implanted into 812 week old Balb/C nude mice by tail vein injection and followed for survival of the mice. For in vivo antibody efficacy study, 10 mg/kg anti-Ragweed antibody (control) or 10 mg/kg trastuzumab was administered intra-peritoneal once a week starting at 4 days post cell implantation. Each arm in the study contained ten mice. All animal studies were conducted under protocols approved by Genentech’s Institutional Animal Care and Use Committee (IACUC) guidelines.
Samples, DNA and RNA preps
In this study we analyzed blood samples from three patients (III.1, III.2 and III.3) and tumor DNA from one patient (III.3; Figure 1A)). The study was conducted with IRB approval and written patient informed consent.
Exome capture and sequencing
Exome capture was performed using the Agilent SureSelect Human All Exome kit (50 Mb). Exome capture libraries were sequenced on HiSeq 2500 (Illumina) to generate 2 × 75 bp paired-end data. A targeted mean coverage of 88x with 94% bases covered at ≥10x was achieved for exome libraries.
Sequence data processing
All sequencing reads were evaluated for quality using the Bioconductor ShortRead package (Morgan et al., 2009). An all-against-all sample comparison was done on germline variants to confirm the patient matched tumor-normal pairing prior to additional data analysis.
Variant calling
Whole exome sequencing data were processed using the Genome Analysis Toolkit (GATK) (version v3.5–0-g36282e4) following best practices recommendations (DePristo et al., 2011; Van der Auwera et al., 2013). Reads were mapped to the human reference genome GRCh37 using BWA-MEM (version 0.7.10; http://arxiv.org/abs/1303.3997). Duplicate alignments were marked and removed using Picard tool (version 1.126) (http://broadinstitute.github.io/picard/) followed by indel realignment and base quality score recalibration. Haplotype Caller was used to generate gVCFs for all samples. Joint variant calling was performed for the 3 samples using GATK Genotype GVCFs. Variant quality score recalibration was carried out to estimate the confidence of variants called in the discovery cohort. Variant annotation was carried out using SnpEff (version 4.2) (Cingolani et al., 2012). Additional variant filtering and interpretation was done using Qiagen’s Ingenuity Variant Analysis software (www.qiagen.com/ingenuity). A set of final variants were manually reviewed using Integrative Genomics Viewer (IGV) (Robinson et al., 2011).
Targeted sequencing of tumor samples
Samples were processed in one of two broad protocols applicable to solid tumors or hematologic cancers as previously described (Frampton et al., 2013). Samples were submitted to a Clinical Laboratory Improvement Amendment (CLIA)-certified, New York State-accredited, and CAP-accredited laboratory (Foundation Medicine) for hybrid capture followed by next-generation sequencing (NGS). All samples that advanced to DNA extraction contained a minimum of 20% tumor cells. DNA was extracted from formalin fixed paraffin embedded 10-μm sections. Adaptor-ligated DNA underwent hybrid capture for all coding exons of 287 or 395 cancer-related genes plus select introns from 19 or 31 genes frequently rearranged in cancer. Captured libraries were sequenced to a median exon coverage depth of >500x using Illumina HiSeq sequencing technology.
Quantification and Statistical Analysis
Quantification and statistical analysis were done using R software. Comparison of relative cell survival between HER2 mutant and WT was performed using two-tailed Student t-test. Resulting p values were corrected using Bonferroni method. Corrected p value of 0.05 was used as significance threshold. Error bars (as shown in Figure 3) represent standard deviation of relative cell survival.
Supplementary Material
Highlights.
Recurrent HER2 transmembrane/juxtamembrane domain (TMD/JMD) mutations identified
TMD/JMD activating mutations identified in multiple cancers
Transmembrane HER2 mutant homodimerizes leading to allosteric activation of kinase
TMD/JMD mutants respond to targeted HER2-therapy
Significance.
We have identified several recurrent actionable activating HER2 TMD and JMD mutants in patient tumors from multiple cancers. Our data indicate that patients with HER2 TMD/JMD mutant are likely candidates for approved HER2-targeted therapies. Based on the mutation frequency, we estimate >6000 TMD/JMD mutant cancer patients are likely to benefit annually from targeted HER2 therapy. Our findings will aid in the implementation of precision medicine in cancer by matching patient mutations with targeted therapy.
Acknowledgments
The authors would like to acknowledge Genentech DNA Sequencing, Biologics Resource, Oligo, FACS and Bioinformatics groups for their help with the project. Our thanks to Craig Cumming and Sophia Maund for facilitating our access to large-scale mutation data. We also want to thank Gerard Manning and Gabriele Schaefer for providing critical input during the course of this work. We wish to thank Allison Bruce for her help with the artwork. This work was supported by grants from the National Institute of General Medical Sciences to N.J. (R01 GM109176), Susan G. Komen Foundation Career Award to N.J. (CCR14299947), National Cancer Institute to T.M.T. (F32 CA216928), HHMI Gilliam Fellowship to M.M and UCSF Discovery Fellowship to M.M.
Footnotes
Data and Software availability
Patient exome sequencing data is deposited in European Genome Phenome Archive (EGA) under accession EGAS00001003213.
Declaration of Interests
Genentech authors hold shares in Roche.
URL
Bioconductor, http://www.bioconductor.org; cBioPortal, http://cbioportal.org;
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, and Abecasis GR (2015). A global reference for human genetic variation. Nature 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrechek ER, Hardy WR, Laing MA, and Muller WJ (2004). Germ-line expression of an oncogenic erbB2 allele confers resistance to erbB2-induced mammary tumorigenesis. Proc Natl Acad Sci U S A 101, 4984–4989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkhipov A, Shan Y, Das R, Endres NF, Eastwood MP, Wemmer DE, Kuriyan J, and Shaw DE (2013). Architecture and membrane interactions of the EGF receptor. Cell 152, 557–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arteaga CL, Sliwkowski MX, Osborne CK, Perez EA, Puglisi F, and Gianni L (2011). Treatment of HER2-positive breast cancer: current status and future perspectives. Nat Rev Clin Oncol 9, 16–32. [DOI] [PubMed] [Google Scholar]
- Bargmann CI, Hung MC, and Weinberg RA (1986). Multiple independent activations of the neu oncogene by a point mutation altering the transmembrane domain of p185. Cell 45, 649657. [DOI] [PubMed] [Google Scholar]
- Bargmann CI, and Weinberg RA (1988). Oncogenic activation of the neu-encoded receptor protein by point mutation and deletion. EMBO J 7, 2043–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett PJ, Song Y, Van Horn WD, Hustedt EJ, Schafer JM, Hadziselimovic A, Beel AJ, and Sanders CR (2012). The amyloid precursor protein has a flexible transmembrane domain and binds cholesterol. Science 336, 1168–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baselga J, and Swain SM (2009). Novel anticancer targets: revisiting ERBB2 and discovering ERBB3. Nature Reviews Cancer 9, 463–475. [DOI] [PubMed] [Google Scholar]
- Bell CA, Tynan JA, Hart KC, Meyer AN, Robertson SC, and Donoghue DJ (2000). Rotational coupling of the transmembrane and kinase domains of the Neu receptor tyrosine kinase. Mol Biol Cell 11, 3589–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell DW, Gore I, Okimoto RA, Godin-Heymann N, Sordella R, Mulloy R, Sharma SV, Brannigan BW, Mohapatra G, Settleman J, and Haber DA (2005). Inherited susceptibility to lung cancer may be associated with the T790M drug resistance mutation in EGFR. Nature genetics 37, 1315–1316. [DOI] [PubMed] [Google Scholar]
- Berendsen HJ, Postma J. v., van Gunsteren WF, DiNola A, and Haak J (1984). Molecular dynamics with coupling to an external bath. The Journal of chemical physics 81, 3684–3690. [Google Scholar]
- Bocharov EV, Mineev KS, Volynsky PE, Ermolyuk YS, Tkach EN, Sobol AG, Chupin VV, Kirpichnikov MP, Efremov RG, and Arseniev AS (2008). Spatial structure of the dimeric transmembrane domain of the growth factor receptor ErbB2 presumably corresponding to the receptor active state. J Biol Chem 283, 6950–6956. [DOI] [PubMed] [Google Scholar]
- Bose R, Kavuri SM, Searleman AC, Shen W, Shen D, Koboldt DC, Monsey J, Goel N, Aronson AB, Li S, et al. (2013). Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer discovery 3, 224–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan PJ, Kumagai T, Berezov A, Murali R, and Greene MI (2000). HER2/neu: mechanisms of dimerization/oligomerization. Oncogene 19, 6093–6101. [DOI] [PubMed] [Google Scholar]
- Brooks AJ, Dai W, O’Mara ML, Abankwa D, Chhabra Y, Pelekanos RA, Gardon O, Tunny KA, Blucher KM, Morton CJ, et al. (2014). Mechanism of activation of protein kinase JAK2 by the growth hormone receptor. Science 344, 1249783. [DOI] [PubMed] [Google Scholar]
- Burke CL, Lemmon MA, Coren BA, Engelman DM, and Stern DF (1997). Dimerization of the p185neu transmembrane domain is necessary but not sufficient for transformation. Oncogene 14, 687–696. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N. (2014). Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang MT, Bhattarai TS, Schram AM, Bielski CM, Donoghue MTA, Jonsson P, Chakravarty D, Phillips S, Kandoth C, Penson A, et al. (2018). Accelerating Discovery of Functional Mutant Alleles in Cancer. Cancer discovery 8, 174–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, and Ruden DM (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debnath J, Muthuswamy SK, and Brugge JS (2003). Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods 30, 256–268. [DOI] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature genetics 43, 491498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis PM, Coakley N, Feld R, Kuruvilla S, and Ung YC (2015). Use of the epidermal growth factor receptor inhibitors gefitinib, erlotinib, afatinib, dacomitinib, and icotinib in the treatment of non-small-cell lung cancer: a systematic review. Curr Oncol 22, e183–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endres NF, Das R, Smith AW, Arkhipov A, Kovacs E, Huang Y, Pelton JG, Shan Y, Shaw DE, Wemmer DE, et al. (2013). Conformational coupling across the plasma membrane in activation of the EGF receptor. Cell 152, 543–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feenstra KA, Hess B, and Berendsen HJ (1999). Improving e_ciency of large timescale molecular dynamics simulations of hydrogen-rich systems. [DOI] [PubMed]
- Fleishman SJ, Schlessinger J, and Ben-Tal N (2002). A putative molecular-activation switch in the transmembrane domain of erbB2. Proc Natl Acad Sci U S A 99, 15937–15940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, Schnall-Levin M, White J, Sanford EM, An P, et al. (2013). Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol 31, 1023–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazdar A, Robinson L, Oliver D, Xing C, Travis WD, Soh J, Toyooka S, Watumull L, Xie Y, Kernstine K, and Schiller JH (2014). Hereditary lung cancer syndrome targets never smokers with germline EGFR gene T790M mutations. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer 9, 456–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianni L (2018). Is there room for another HER2-targeting drug? Lancet Oncol 19, 847–849. [DOI] [PubMed] [Google Scholar]
- Gleason NJ, Vostrikov VV, Greathouse DV, and Koeppe RE 2nd (2013). Buried lysine, but not arginine, titrates and alters transmembrane helix tilt. Proc Natl Acad Sci U S A 110, 1692–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg SD, Clinthorne GD, Goulian M, and DeGrado WF (2010). Transmembrane polar interactions are required for signaling in the Escherichia coli sensor kinase PhoQ. Proc Natl Acad Sci U S A 107, 8141–8146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordeliy VI, Labahn J, Moukhametzianov R, Efremov R, Granzin J, Schlesinger R, Buldt G, Savopol T, Scheidig AJ, Klare JP, and Engelhard M (2002). Molecular basis of transmembrane signalling by sensory rhodopsin II-transducer complex. Nature 419, 484–487. [DOI] [PubMed] [Google Scholar]
- Greulich H, Kaplan B, Mertins P, Chen TH, Tanaka KE, Yun CH, Zhang X, Lee SH, Cho J, Ambrogio L, et al. (2012). Functional analysis of receptor tyrosine kinase mutations in lung cancer identifies oncogenic extracellular domain mutations of ERBB2. Proc Natl Acad Sci U S A 109, 14476–14481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmaier RJ, Albacker LA, Chmielecki J, Bailey M, He J, Goldberg ME, Ramkissoon S, Suh J, Elvin JA, Chiacchia S, et al. (2017). High-Throughput Genomic Profiling of Adult Solid Tumors Reveals Novel Insights into Cancer Pathogenesis. Cancer Res 77, 2464–2475. [DOI] [PubMed] [Google Scholar]
- Harvey MJ, Giupponi G, and Fabritiis GD (2009). ACEMD: Accelerating Biomolecular Dynamics in the Microsecond Time Scale. J Chem Theory Comput 5, 1632–1639. [DOI] [PubMed] [Google Scholar]
- Hristova K, and Wimley WC (2011). A look at arginine in membranes. J Membr Biol 239, 49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, and MacKerell AD Jr. (2013). CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J Comput Chem 34, 2135–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman DM, Piha-Paul SA, Won H, Rodon J, Saura C, Shapiro GI, Juric D, Quinn DI, Moreno V, Doger B, et al. (2018). HER kinase inhibition in patients with HER2- and HER3-mutant cancers. Nature 554, 189–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes NE, and Lane HA (2005). ERBB receptors and cancer: the complexity of targeted inhibitors. Nature Reviews Cancer 5, 341–354. [DOI] [PubMed] [Google Scholar]
- Jaiswal BS, Kljavin NM, Stawiski EW, Chan E, Parikh C, Durinck S, Chaudhuri S, Pujara K, Guillory J, Edgar KA, et al. (2013). Oncogenic ERBB3 mutations in human cancers. Cancer Cell 23, 603–617. [DOI] [PubMed] [Google Scholar]
- Jura N, Endres NF, Engel K, Deindl S, Das R, Lamers MH, Wemmer DE, Zhang X, and Kuriyan J (2009). Mechanism for activation of the EGF receptor catalytic domain by the juxtamembrane segment. Cell 137, 1293–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavuri SM, Jain N, Galimi F, Cottino F, Leto SM, Migliardi G, Searleman AC, Shen W, Monsey J, Trusolino L, et al. (2015). HER2 activating mutations are targets for colorectal cancer treatment. Cancer discovery 5, 832–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C, Schmidt T, Cho EG, Ye F, Ulmer TS, and Ginsberg MH (2011). Basic amino-acid side chains regulate transmembrane integrin signalling. Nature 481, 209–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs E, Zorn JA, Huang Y, Barros T, and Kuriyan J (2015). A structural perspective on the regulation of the epidermal growth factor receptor. Annu Rev Biochem 84, 739–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’DonnellLuria AH, Ware JS, Hill AJ, Cummings BB, et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon MA, Treutlein HR, Adams PD, Brunger AT, and Engelman DM (1994). A dimerization motif for transmembrane alpha-helices. Nat Struct Biol 1, 157–163. [DOI] [PubMed] [Google Scholar]
- Lindhurst MJ, Sapp JC, Teer JK, Johnston JJ, Finn EM, Peters K, Turner J, Cannons JL, Bick D, Blakemore L, et al. (2011). A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N Engl J Med 365, 611–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littlefield P, Liu L, Mysore V, Shan Y, Shaw DE, and Jura N (2014). Structural analysis of the EGFR/HER3 heterodimer reveals the molecular basis for activating HER3 mutations. Sci Signal 7, ra114. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- Lomize MA, Pogozheva ID, Joo H, Mosberg HI, and Lomize AL (2012). OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res 40, D370–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Mi LZ, Grey MJ, Zhu J, Graef E, Yokoyama S, and Springer TA (2010). Structural evidence for loose linkage between ligand binding and kinase activation in the epidermal growth factor receptor. Mol Cell Biol 30, 5432–5443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monk BC, Tomasiak TM, Keniya MV, Huschmann FU, Tyndall JD, O’Connell JD 3rd, Cannon RD, McDonald JG, Rodriguez A, Finer-Moore JS, and Stroud RM (2014). Architecture of a single membrane spanning cytochrome P450 suggests constraints that orient the catalytic domain relative to a bilayer. Proc Natl Acad Sci U S A 111, 3865–3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan M, Anders S, Lawrence M, Aboyoun P, Pages H, and Gentleman R (2009). ShortRead: a bioconductor package for input, quality assessment and exploration of highthroughput sequence data. Bioinformatics 25, 2607–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthuswamy SK, Li D, Lelievre S, Bissell MJ, and Brugge JS (2001). ErbB2, but not ErbB1, reinitiates proliferation and induces luminal repopulation in epithelial acini. Nat Cell Biol 3, 785–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niihori T, Aoki Y, Narumi Y, Neri G, Cave H, Verloes A, Okamoto N, Hennekam RC, Gillessen-Kaesbach G, Wieczorek D, et al. (2006). Germline KRAS and BRAF mutations in cardio-facio-cutaneous syndrome. Nature genetics 38, 294–296. [DOI] [PubMed] [Google Scholar]
- Onsum MD, Geretti E, Paragas V, Kudla AJ, Moulis SP, Luus L, Wickham TJ, McDonagh CF, Macbeath G, and Hendriks BS (2013). Single-cell quantitative HER2 measurement identifies heterogeneity and distinct subgroups within traditionally defined HER2positive patients. Am J Pathol 183, 1446–1460. [DOI] [PubMed] [Google Scholar]
- Orloff MS, He X, Peterson C, Chen F, Chen JL, Mester JL, and Eng C (2013). Germline PIK3CA and AKT1 mutations in Cowden and Cowden-like syndromes. Am J Hum Genet 92, 76–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou SI, Schrock AB, Bocharov EV, Klempner SJ, Haddad CK, Steinecker G, Johnson M, Gitlitz BJ, Chung J, Campregher PV, et al. (2017). HER2 Transmembrane Domain (TMD) Mutations (V659/G660) That Stabilize Homo- and Heterodimerization Are Rare Oncogenic Drivers in Lung Adenocarcinoma That Respond to Afatinib. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer 12, 446–457. [DOI] [PubMed] [Google Scholar]
- Red Brewer M, Choi SH, Alvarado D, Moravcevic K, Pozzi A, Lemmon MA, and Carpenter G (2009). The juxtamembrane region of the EGF receptor functions as an activation domain. Mol Cell 34, 641–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riese DJ 2nd, van Raaij TM, Plowman GD, Andrews GC, and Stern DF (1995). The cellular response to neuregulins is governed by complex interactions of the erbB receptor family. Mol Cell Biol 15, 5770–5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat Biotechnol 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roskoski R Jr. (2014). The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol Res 79, 34–74. [DOI] [PubMed] [Google Scholar]
- Ross JS, Gay LM, Wang K, Ali SM, Chumsri S, Elvin JA, Bose R, Vergilio JA, Suh J, Yelensky R, et al. (2016). Nonamplification ERBB2 genomic alterations in 5605 cases of recurrent and metastatic breast cancer: An emerging opportunity for anti-HER2 targeted therapies. Cancer 122, 2654–2662. [DOI] [PubMed] [Google Scholar]
- Schechter AL, Hung MC, Vaidyanathan L, Weinberg RA, Yang-Feng TL, Francke U, Ullrich A, and Coussens L (1985). The neu gene: an erbB-homologous gene distinct from and unlinked to the gene encoding the EGF receptor. Science 229, 976–978. [DOI] [PubMed] [Google Scholar]
- Schoeberl B, Pace EA, Fitzgerald JB, Harms BD, Xu L, Nie L, Linggi B, Kalra A, Paragas V, Bukhalid R, et al. (2009). Therapeutically targeting ErbB3: a key node in ligandinduced activation of the ErbB receptor-PI3K axis. Sci Signal 2, ra31. [DOI] [PubMed] [Google Scholar]
- Schubert D, Heinemann S, Carlisle W, Tarikas H, Kimes B, Patrick J, Steinbach JH, Culp W, and Brandt BL (1974). Clonal cell lines from the rat central nervous system. Nature 249, 224–227. [DOI] [PubMed] [Google Scholar]
- Senes A, Gerstein M, and Engelman DM (2000). Statistical analysis of amino acid patterns in transmembrane helices: the GxxxG motif occurs frequently and in association with betabranched residues at neighboring positions. J Mol Biol 296, 921–936. [DOI] [PubMed] [Google Scholar]
- Stern HM (2012). Improving treatment of HER2-positive cancers: opportunities and challenges. Sci Transl Med 4, 127rv122. [DOI] [PubMed] [Google Scholar]
- Sternberg MJ, and Gullick WJ (1989). Neu receptor dimerization. Nature 339, 587. [DOI] [PubMed] [Google Scholar]
- Tohyama Y, Katagiri K, Pardi R, Lu C, Springer TA, and Kinashi T (2003). The critical cytoplasmic regions of the alphaL/beta2 integrin in Rap1-induced adhesion and migration. Mol Biol Cell 14, 2570–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Cutsem E, Sagaert X, Topal B, Haustermans K, and Prenen H (2016). Gastric cancer. The Lancet 388, 2654–2664. [DOI] [PubMed] [Google Scholar]
- Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, et al. (2013). From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Current protocols in bioinformatics 43, 11 10 11–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr., and Kinzler KW (2013). Cancer genome landscapes. Science 339, 1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vostrikov VV, Daily AE, Greathouse DV, and Koeppe RE 2nd (2010). Charged or aromatic anchor residue dependence of transmembrane peptide tilt. J Biol Chem 285, 3172331730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SE, Narasanna A, Perez-Torres M, Xiang B, Wu FY, Yang S, Carpenter G, Gazdar AF, Muthuswamy SK, and Arteaga CL (2006). HER2 kinase domain mutation results in constitutive phosphorylation and activation of HER2 and EGFR and resistance to EGFR tyrosine kinase inhibitors. Cancer Cell 10, 25–38. [DOI] [PubMed] [Google Scholar]
- Ward MD, and Leahy DJ (2015). Kinase activator-receiver preference in ErbB heterodimers is determined by intracellular regions and is not coupled to extracellular asymmetry. J Biol Chem 290, 1570–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H, Higasa K, Sakaguchi M, Shien K, Soh J, Ichimura K, Furukawa M, Hashida S, Tsukuda K, Takigawa N, et al. (2014). Novel germline mutation in the transmembrane domain of HER2 in familial lung adenocarcinomas. Journal of the National Cancer Institute 106, djt338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabransky DJ, Yankaskas CL, Cochran RL, Wong HY, Croessmann S, Chu D, Kavuri SM, Red Brewer M, Rosen DM, Dalton WB, et al. (2015). HER2 missense mutations have distinct effects on oncogenic signaling and migration. Proc Natl Acad Sci U S A 112, E6205–6214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Gureasko J, Shen K, Cole PA, and Kuriyan J (2006). An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 125, 1137–1149. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.