

Abstract
Interleukin (IL)- 18 is a cytokine previously demonstrated to participate in neuroinflammatory processes. Since the components of the IL-18 receptor complex are expressed in neurons throughout the brain, IL-18 is also believed to directly influence neuronal function. Here we tested this hypothesis on mouse hippocampal neurons by measuring the effects of IL-18 on three pathways previously shown to be regulated by this cytokine in non-neuronal cells: the MAPK pathways, p38 and ERK1/2 MAPKs, STAT3 and NF-κB. Experiments were carried out in vitro using the immortalized hippocampal neuronal line HT-22 or in vivo following i.c.v. injection with recombinant mouse IL-18. We showed that IL-18 did not activate NF-κB in HT-22 cells whereas it induced a rapid (within 15 minutes) activation of the MAPK pathways. Moreover, we demonstrated that IL-18 treatment enhanced P-STAT3 (Tyr705)/ STAT3 ratio in the nucleus of HT-22 cells after 30–60 minutes of exposure. A similar increase in P-STAT3 (Tyr705)/ STAT3 ratio was observed in the whole hippocampus one hour after i.c.v. injection. These data demonstrate that IL-18 can act directly on neuronal cells affecting the STAT3 pathway; therefore, possibly regulating the expression of specific genes within the hippocampus. This effect may help to explain some of the IL-18- induced effects on synaptic plasticity and functionality within the hippocampal system.
Keywords: IL-18, hippocampus, brain, HT-22, STAT-3, signaling, IL-18R alpha, IL-18R beta, isoforms, NF-κB
1. INTRODUCTION
Interleukin (IL)- 18 is a pleiotropic cytokine belonging to the IL-1 family that has several established roles both in innate and adaptive immune responses (Nakanishi et al., 2001). IL-18 mediates a variety of effects in virtually every organ system of the body by interacting with its heterodimer receptor (IL-18R) that consists of a subunit α (type I IL-18Rα, required for binding), and a subunit β (full-length IL-18Rβ, responsible for signal transduction) (Sergi and Pentilla, 2004). Besides the periphery, and the cells of the immune system, the IL-18/ IL-18R system is expressed and active also in the brain in both glia cell populations (including microglia, astrocyte and ependymal cells) and neurons. Interestingly, the central action of IL-18 appears to be tightly regulated. Indeed, aside the first characterized negative regulator of IL-18- mediated actions, the IL-18 binding protein (BP) (Novick et al., 1999), a short transcript IL-18Rα that lacks the TIR domain (type II IL-18Rα) and a soluble form IL-18Rβ (small IL-18Rβ) exists (Alboni et al., 2009; Alboni et al., 2011).
In the last few years it has been demonstrated that the IL-18/ IL-18R system may have broad-ranging actions in the healthy as well as the pathological brain (Alboni et al., 2010). On the “physiological” side, it has been reported that this cytokine affects neuronal differentiation, survival and synaptic plasticity. Liu and colleagues reported that IL-18 may reduce survival and attenuate neuronal cell fate in cultured embryonic rat-derived neuronal progenitor cells (Liu et al., 2005). However, very recently it has been demonstrated that hippocampal levels of IL-18 correlate positively with hippocampal neurogenesis following physical exercise in aging rats (Speisman et al., 2013). Moreover, the role of IL-18 in synaptic plasticity in the dentate gyrus (DG) of the hippocampus system has been demonstrated (Curran and O’Connor, 2001; Cumiskey et al., 2007; del Rey et al., 2013). Beside its role in long-term potentiation (LTP), this pro-inflammatory mediator is involved in certain types of learning. A role for IL-18 in favoring spatial memory was described by Yaguchi and co-workers (2010) while very recently it has been demonstrated that during learning the expression levels of IL-18 increase in the dorsal hippocampus, a possible key event promoting spatial memory (del Rey et al., 2013). On the “pathological” side, a lot of literature reported the involvement of IL-18 in neuropathological conditions. These include neuroinflammatory (Abdul-Careem et al., 2006; von Giesen et al., 2004) and autoimmune diseases (Huang et al., 2004; Losy and Niezgoda, 2001) as well as neurodegenerative (Bossu et al, 2008; Iannello et al, 2009; Sugama et al., 2004; Sutinen et al., 2012) and neuropsychiatric disorders (Haastrup et al., 2012; Kroes et al., 2006; Lu et al., 2004; Kokai et al, 2002; Shelton et al., 2011). Indeed, increased levels of IL-18 were found in the presence of cognitive dysfunction (Oztürk et al., 2007; Kumar et al., 2007) and for Alzheimer’s disease the IL-18 levels correlate with cognitive decline (Bossu et al 2008), providing evidence linking IL-18 also to cognitive impairment. Moreover, IL-18 actions shift from promoting neuroinflammation and/or neurodegeneration to promoting neuroprotection; for example, after kainic acid administration or following status epilepticus (Andoh et al., 2008, Jung et al., 2012, Ryu el al., 2010, Yaguchi et al., 2010, Zhang et al., 2007), demonstrating that IL-18/ IL-18R system act within the brain in a more complex way than simple disease-promoting or inhibiting and that its outcomes depend on the concentration, cell targets, stimulus duration as well as other disease-associated factors.
Nevertheless, despite the suggested key role for IL-18 in participating in the brain function and dysfunction, the IL-18 system has been poorly studied in brain and the molecular mechanisms through which IL-18 may regulate CNS functions remain largely unknown. The initiating signaling pathways that ultimately lead to IL-18-mediated effects in the brain and in particular in neurons are poorly explored. Indeed, while IL-18 signaling has been extensively studied in immune cells, signaling events activated through IL-18/ IL-18R in neuronal cells may differ from those activated in cells of the immune system, thus leading to transcriptional modification characteristic of the brain. Within the brain, the hippocampus is particularly vulnerable to inflammatory challenge and plays a key role in regulating many functions impaired in central nervous system diseases (including neurodegenerative and psychiatric diseases). Therefore, the present study was designed to investigate the signaling triggered by IL-18 particularly in the hippocampus given the role played by this cytokine in this brain area. Experiments were performed on the hippocampal neuronal cell line HT-22, or in vivo after intracerebroventricular (i.c.v.) injection of IL-18. HT-22 cells are an immortalized hippocampal cell line (Davis and Maher, 1994) often used as an hippocampal cell model to investigate molecular mechanisms underlying neurotoxic, or even neuroprotectiv effects induced by different stimuli (including cytokines) in this area (Hu et al., 2009; Pace et al 2011; Schmidt et al 2005). These cells could provide a simple and useful tool to explore the molecular events triggered by IL-18 in hippocampal neuronal cells and their physiological and pathological significance. Moreover, in vivo, i.c.v., studies were performed to explore the signaling pathway triggered by IL-18 in a non-homogeneous population. Indeed, the hippocampal system is formed by phenotypically different cells that include neurons but also glia cells. Treatments were followed by evaluation of the state of signaling pathways that has been demonstrated to be activated by IL-18 in peripheral (i.e. immune) cells, in particular the nuclear factor κB (NFκB), but also Mitogen Activated Protein Kinase (MAPKs) p38 and Extracellular Regulated Kinases (ERK1/2) and Signal Transducer and Activator of Transcription (STAT)-3 (Fortin et al., 2009; Lee et al., 2004; Matsumoto et al., 1997; Morel et al., 2001; Kalina et al., 2000; Kroeger et al., 2009; Olee et al., 1999; Reddy et al., 2010; Sahar et al., 2005; ; Tsuji-Takayama et al., 1999; Yu et al., 2012).
2. METHODS
2.1. Cell culture and treatment
The mouse clonal hippocampal neuronal cell line HT-22 was a generous gift from Dr Pamela Maher (The Salk Institute for Biological Studies, La Jolla, San Diego, CA). Cells were maintained at 37°C and 5% CO2 in complete medium containing DMEM high glucose supplemented with 10% heat-inactivated (56°C, 30 min) fetal bovine serum (FBSEuroclone® Italy, Milan), 50 U/ml penicillin, 50mg/ml streptomycin and passed by tripsinization. Cells (2×106) were seeded into Petri dishes and grown for 20–24 h until 70–80% confluent for protein analysis. For gene expression analysis cells were plated at a density of 1×106 cells per well on 6-well plates in 1 mL of complete medium and treated the day after plating. Treatments for protein analyses were performed using 100 ng/ml of IL-18 mouse recombinant (Immunological Sciences®, Rome, Italy) after 5, 15, 30 and 60 minutes, while the control group (0 min) received phosphate-buffered saline (PBS). Three independent experiments were performed. The dose was decided after preliminary titration tests and according to published works (Kalina et al., 2000; Nakahira et al., 2002; Sugimoto et al., 2003).
2.2. Animals
Adult male C57BL/6J (JAX Laboratories) mice of 8 weeks of age were used in this study. Animals were housed in polycarbonate cages (28 × 17 × 12 cm) with ad libitum access to food and water throughout the study, and maintained under a 12:12 light-dark cycle in an ambient temperature of 21 ± 3 °C with relative humidity controlled. Animals were checked for signs of discomfort as indicated by animal care and use guidelines [National Academy of Sciences. Guide for the care and use of laboratory animals, 1998, “Guidelines for the Care and Use of Mammals in Neuroscience and Behavioral Research” (National Research Council 2003)]. EC guidelines (EEC Council Directive 86/609 1987), Italian legislation on animal experimentation (Decreto Legislativo 116/92) were followed throughout the whole experiment. Procedures adhered to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by The Scripps Research Institute Animal Care and Use Committee.
2.3. Surgery and i.c.v. injection
Intracerebroventricular (i.c.v.) administration of IL-18 was performed through a lateral ventricular guide cannula stereotaxically secured at anterior/posterior −0.34 from Bregma and ventral −1.7 from dura (Franklin and Paxinos, 2001). Recombinant endotoxinfree mouse IL-18 was obtained commercially (BioSource International, Camarillo, CA, USA). Twenty-five µg of IL-18 was dissolved in 12.5 µL normal artificial cerebrospinal fluid (aCSF: NaCl 126 mM, KCl 3.5 mM, CaCl2 2 mM, MgSO4 1 mM, NaH2PO4 1.25 mM, NaHCO3 26 mM, and glucose 10 mM (pH 7.4)). Bilaterally, one µL of this mrIL-18 solution (N= 3) or vehicle (aCSF alone) (N= 3) was injected (1.0 µL/min) (2.48 nmol/mouse) four days after surgery. One hour after injection, animals were sacrificed and the brain regions rapidly dissected as previously described (Blom et al. 2006). All tissues were stored at −80°C.
2.4. RNA extraction and RT-PCR
Total RNA extraction and DNAse treatment were performed as previously described (Alboni et al., 2013): 1 µg of total RNA was reverse transcribed with High Capacity cDNA Reverse Transcription Kit (Life Technologies Corporation, Carlsbad, CA, USA) in 20 µL of reaction mix.
2.5. Real Time RT-PCR
Real Time PCR was performed in ABI PRISM 7900 HT (Applied Biosystems) using Power SYBR Green mix (Applied Biosystems) as previously described (Benatti et al., 2011). The following primer pairs were used for the IL-18\ IL-18R system: IL-18 forward: TGA AGA AAA TGG AGA CCT GGA, IL-18 reverse: GGC TGT CTT TTG TCA ACG AAG (NCBI GenBank accession number: NM_008360); IL-18BP forward: TGC CAC TGA ATG GAA CTC TG; IL-18BP reverse: CTG GGA GGT GCT CAA TGA AG (NCBI GenBank accession number: NM_010531); type I IL-18Rα forward: GAG TAA CTG TGC TTG TTC TCG CCT CTG T, type I IL-18Rα reverse: GGG TAA CGT CTC CAC ACG AAA AGT AT (NCBI GenBank accession number: NM_008365), type II IL-18Rα forward: GGC ACC CTA GCT CAT GTT TT; type II IL-18Rα reverse: AAC GAG GCT CAG AGA TCA TTA GT (NCBI GenBank accession number: BC023240); full-length (canonical) IL-18Rβ forward: CCT ATC TGA TGT CCA GTG GT; full-length (canonical) IL-18Rβ reverse: GGG GGC TCC TAA TTC TGG G; short IL-18Rβ forward: CCT ATC TGA TGT CCA GTG GT; small IL-18Rβ reverse: GTC CTG TGA GCA CTT GTC TA (NCBI GenBank accession number: NM_010553). The cycling parameters were: 95°C 10 min and 95 °C 15 s, 60° 1 min for 40 cycles. Single PCR products were subjected to a heat dissociation protocol (gradual increase of temperature from 60°C to 95 °C) and to agarose gel separation to verify the absence of artifacts, such as primer-dimers or non-specific products. Direct detection of PCR products was monitored by measuring an increase in fluorescence intensity caused by binding of SYBR GREEN I dye to neo-formed double strand DNA during the amplification phase. Ct (cycle threshold) values, obtained by an interpolate study, were determined by the SDS software 2.2.2 (Life Technologies Corporation, Carlsbad, CA, USA). The relative quantification of IL-18 and IL-1β mRNAs was analyzed by the ∆∆Ct method using as calibrator average of the IL-18 levels. The mRNA levels of the target genes were normalized on the intensity of the housekeeping gene, cyclophilin A (NCBI GenBank accession number: NM_008907.1). For an appropriate application of comparative ∆∆Ct method, it was demonstrated that the amplification efficiency of the target gene and endogenous control gene was approximately equal. Each cDNA sample was run in triplicate and the mean values were used to calculate the gene expression levels.
2.6. Protein extraction
Hippocampus was homogenized by potter (12 stroke at 600 rpm) in lysis buffer as previously described (Alboni et al., 2011b). For protein extraction HT-22 cell monolayers were rinsed 2 times with 1X PBS and then harvested and pelleted by centrifugation 5 min at 3,000 × g 4°C. After removing PBS, cells were resuspended in 150 µL of omogenization buffer (OB) containing Hepes 20 mM, Sucrose 250 mM, KCl 10 mM, MgCl2 1.5 mM, EDTA 1 mM, EGTA 1 mM, Dithiothreitol 1 mM, Sodium Pyrophosphate 5 mM, NaF 20 mM, Na3VO4 1 mM, and Complete 1X Protease Inhibitor Cocktail Tablets (Roche, Germany) and chilled on ice for at least 30min. Cells were then homogenized with 40 strokes in a Dounce homogenizer (loose pestle) (Incofar, Italy). The homogenates were centrifuged at 900 × g 5min 4°C and the supernatants saved as cytosolic extracts. The nuclear pellets were washed in OB buffer and then resuspended in 100 µL of OB buffer. Nuclei were left for 30 minutes on ice and then homogenized with 40 strokes in a Dounce homogenizer (tight pestle). The samples were then subjected to centrifugation at 14,000 rpm at 4°C for 10min. These supernatants contained nuclear proteins. Protein concentration of the extracts was determined using standard protocol Coomassie® reagent (Pierce). Cytosolic and nuclear fractions were stored at −20°C.
2.7. Western Blot analysis
For hippocampus, western blots were carried out on 20 µg total extract whereas and protein analysis included STAT-3, Phospho (P)-STAT3 (phosphorylated at) Ser727, PSTAT3 Tyr705, P-p38, p38, P-ERK 1/2, ERK 1/2, P-NF-κB Ser536, NF-κB and β-Tubulin. For HT-22 cells western blots were carried out on 15 µg of cytosolic extract, for STAT3, STAT3 Tyr705, P-STAT3 Ser727, P-p38, p38, P-ERK 1/2, ERK1/2 MAPK, P- P-NF-κB Ser536 and NF-κB detection. P-STAT3, STAT3, P-NF-κB Ser536 and NF-κB protein levels were also measured on 15 µg of nuclear enriched extracts. Cytosolic or nuclear extracts were mixed with sample buffer just before the SDS-PAGE. Electrophoresis was performed using Bio-Rad Protean III mini-gel apparatus with 10% polyacrylamide gel run at 200 V for 45 min. Proteins were transferred to PVDF membranes (Amersham®) using Bio-Rad trans-blot apparatus at 100 V for 75 min. The membranes were blocked for 1 h with 5% non-fat dry milk in Tris- buffered saline (TBS)- 0.1% Tween-20 followed by incubation, overnight at 4 °C, with primary antibodies: anti-STAT3 (STAT3 rabbit polyclonal antibody, Cell Signaling®, #9132) 1:1000 in PAD; anti-P-STAT3 Ser727 (Phospho-STAT3 Ser727 rabbit polyclonal antibody, Cell Signaling®, #9134) 1:1000 in PAD; anti-P-p38 (Phospho-p38 MAPK-thr180 tyr182 rabbit polyclonal antibody Tyr180/Tyr182, Cell Signaling®, #9211) 1:1000 in Primary Antibody Dilution (PAD) buffer (1X TBS, 0.1% Tween-20, with 5% BSA); anti p38 (p38 MAPK rabbit polyclonal, Cell Signaling®, #9212) 1:1000 in PAD; anti-P-ERK1/2 MAPK (Phospho-ERK1/2 MAPK-thr202 tyr204 mouse monoclonal antibody, Cell Signaling®, #9106) 1:1000 in PAD; anti-ERK1/2 MAPK (p44/42 MAPK- rabbit polyclonal antibody, Cell Signaling®, #9102) 1:1000 in PAD; anti-P-NF-κB Ser536 (Phospho-NF-κB p65 Ser536 rabbit monoclonal antibody, Cell Signaling®, #3033) 1:500 in PAD; anti-NF-κB (NF-κB p65 rabbit polyclonal antibody, Abcam®, ab7970) 1:500 in Blocking-Buffer; anti-β-tubulin (β-tubulin rabbit polyclonal antibody, Santa Cruz D-10 TEBU-Bio, sc-5274) 1:2000 in Blocking-Buffer. To clean PSTAT3 Tyr705 signal, the blocking section was carried out overnight and the anti-phospo-STAT3 Tyr705 1:1000 in PAD (anti-phospho-STAT3 Tyr705 rabbit polyclonal antibody, Cell Signaling®, #9131) incubation time for 2 h at room temperature. After multiple washings membranes were incubated 1 h at RT with secondary antibodies in BlockingBuffer: anti-rabbit IgG-HRP-linked (Cell Signaling®, #7071) 1:10 000 for STAT3, P-STAT3 Tyr705, P-STAT3 Ser727, p38, ERK 1/2, P-NF-κB Ser536, NF-κB and β-tubulin, 1:5000 for P-p38; anti-mouse IgG-HRP-linked (Santa Cruz®, sc-2005) 1:5000 for P-ERK 1/2. After 3 washing steps, bands were detected using Immobilon Western Chemiluminescent HRP (Millipore®). The protein levels were calculated by measuring the peak densitometric area of the autoradiography analyzed with an image analyzer (GS-690 BIORAD). Each western blot analysis were repeated at least twice. Phosphorylated and un-phosphorylated proteins were run and detected on the same blot after stripping. The optical densities (OD) of P-p38 MAPK, p38 MAPK, P-ERK1/2, ERK1/2 MAPK, P-STAT3s, STAT3 and NF-κB signals were normalized according to the OD of β-tubulin or the OD of phospho-protein were normalized also with the respective OD of un-phosphorylated protein. Ratios were expressed as percentage of control ± S.E.M.
2.8. Statistical analysis
Statistical analyses were performed using Student’s t test for ex vivo i.c.v. experiments and one-way analysis of variance (ANOVA) for in vitro experiments. These analyses were followed by planned pairwise post-hoc comparisons (Dunnett) against the control group for multiple comparisons between different time points. Values were expressed as percentage with respect to controls ± SEM (Standard Error of the Mean). All differences were considered statistically significant if p < 0.05.
3. RESULTS
3.1. HT-22 cells expressed the IL-18/ IL-18R system
We first characterized the clonal murine hippocampal cell line HT-22 for the expression of the entire IL-18\ IL-18R system by real-time PCR. We found that this line constitutively expressed IL-18, type I IL-18Rα, type II IL-18Rα, full-length IL-18Rβ, the small IL-18Rβ and IL-18BP transcripts in HT-22 cells (Tab. 1). IL-18 and its natural inhibitor IL-18BP were expressed at similar level in HT-22 cells at basal conditions when comparing each other. Measurement of IL-18Rα isoforms showed that both were low expressed but the truncated isoform was expressed at higher levels than the canonical isoform (ct mean ± SEM 36.52 ± 0.29 for type I IL-18Rα and 32.70 ± 0.50 for type II IL-18Rα). Basal levels of full-length IL-18Rβ were higher if compared to those of its truncated isoform, small IL-18Rβ, in HT-22 cells. In particular, our data showed th e highest expression levels for the full-length IL-18Rβ among the components of IL-18 system tested (ct mean 23.91 ± 0.12), thus comparing full-length IL-18Rβ transcript levels to those of type I IL-18Rα (the two canonic chains of the IL-18R) we observed that the full-length IL-18Rβ/type I IL-18Rα ratio was very high (data not shown). Finally in HT-22 cells grown in standard conditions the expression levels of IL-18 mRNA were significantly higher (t = −28,73476; p < 0.00001) than those of IL-1β mRNA (data not shown).
Table 1. Summary of expression levels of IL-18 system components in HT-22 cells.
The table shows for IL-18, IL-18BP, type I IL-18Rα, type II IL-18Rα, full length IL-18Rβ and small IL-18Rβ the respective Ct (cycle threshold ± S.E.M.) value obtained from Real time RT-PCR analysis (starting from cDNA equivalent to 100 ng total RNA) and thus utilized to calculate mRNA fold changes using the delta delta ct (∆∆Ct) method (see Methods for further details). The putative relative function described in different published works for each IL-18 member and the respective NCBI accession number are also indicated.
| Gene(transcript) | Putative Function | Ct(mean*)±S.E.M. | Genebank (accession number) |
|---|---|---|---|
| IL-18 | Pro-inflammatory cytokine | 26.35±0.11 | NM_008360 |
| IL-18BP | Inhibitor of IL-18-mediated activity | 26.88±0.13 | NM_010531 |
| IL-18Rα Type I | IL-18R binding chain | 36.52±0.29 | NM_008365 |
| IL-18Rα Type II | Inhibitor of IL-18-mediated activity | 32.70±0.50 | BC023240 |
| Full-length IL-18Rβ | Signaling receptor activity | 23.91±0.12 | NM_010553 |
| Small IL-18Rβ | Inhibitor of IL-18-mediated activity | 31.71±0.15 | NA |
NA= not available
mean of 10–12 samples from 4 independent experiments.
Transcripts with an average Ct value of ≥ 35 were considered as low expression genes (Italic), genes with Ct values between 29 and 34 were of medium expression and those with Ct values of ≤ 28 were highly expressed (Bold). NA: not available.
3.2. Mouse recombinant (mr) IL-18 treatment did not activate NF-κB in HT-22 cells
Nuclear factor-κB (NF-κB) is a protein complex that controls the transcription of genes codifying for proteins that regulate inflammation and immune responses but also cell proliferation and apoptosis. NF-κB is retained in a latent state in the cytoplasm by specific inhibitory proteins of the IκB family. In response to a variety of stimuli including pro-inflammatory cytokines and bacterial lipopolysaccaride, IκB proteins are degraded and the free NF-κB translocates to the nucleus where it initiates gene transcription (Lappas et al., 2002; Li and Verma, 2002; Natoli et al., 2005). Different evidence demonstrated that IL-18 activates NF-κB in several types of peripheral cells like immune cells (Matsumoto et al., 1997; Tsuji-Takayama et al., 1999; Yu et al., 2012; Fortin et al., 2009), chondrocytes (Olee et al., 1999) and fibroblasts (Morel et al., 2002). Considering these data, we wanted to examine first whether NF-κB could be activated in the clonal murine hippocampal HT-22 cells 5, 15, 30 and 60 minutes after mrIL-18 (100 ng/mL) treatment. Western blot analysis showed no difference in the level of NF-κB protein in the cytoplasmic or in the nuclear fractions at any of the time-points investigated (Fig. 1). We also found that IL-18 had no effects on levels of NF-κB phosphorylation at ser536 (P-NF-κB Ser536), a modification able to modulate the transactivation of this transcription factor (Buss et al., 2004; Viatour et al., 2005) (Fig. 1).
Figure 1. Treatment with mouse recombinant IL-18 does not affect either NF-κB translocation into the nucleus or NF-κB phosphorylation at serine 536 of in HT-22 cells.

(A and D) Following treatments with mrIL-18 (100 ng/mL) we did not observed NFκB translocation in to the nucleus or any changes in NF-κB levels in nuclear enriched or cytosolic extracts from HT-22 cells at the time points evaluated (5, 15, 30 or 60 minutes) compared to the control (0 min) group (open bar). (B and E) The levels of P-NF-κB Ser536 were not significantly affected by exposure to mrIL-18 (100 ng/mL) at the indicated time (5, 15, 30 and 60 minutes) either in nuclear enriched or in cytosolic extracts from mrIL-18 stimulated HT-22 cells compared to control (0 min) cells. Phosphorylated and unphosphorylated proteins were detected on the same blot. HT-22 cells were treated with mrIL-18 (100ng/ml) for 5, 15, 30 and 60 minutes after which proteins were measured in the cytoplasmic extracts by western blot. Optical density was expressed as % of control (vehicle treated) group. (C and F) Representative western blots showing NF-κB and P-NFκB Ser536 in nuclear enriched or cytosolic extracts of HT-22 cells expose to mrIL-18 (100 ng/mL) at different time points (5, 15, 30 and 60 minutes). Time 0 correspond to untreated HT-22. The relative bands for β-tubulin are also represented.
3.3. IL-18 treatment affected p38 and Erk1\2 MAPK pathways in HT-22 cells
P38 and Erk1\2 belong to a system of intracellular components known as mitogenactivated protein kinase (MAPK) signaling cascade that governs a diverse array of cellular processes including cell proliferation, differentiation and development. At least two independent groups showed that IL-18 can also activate p38 and Erk1\2 MAPKs in immune and epithelial cells (Lee et al., 2004; Arend et al., 2008). Here we tested whether the same effects could be seen in HT-22 cells treated with rmIL-18 (100 ng/ml) for 5, 15, 30 and 60 minutes. We observed a main effect among the time-point tested for the Pp38/p38 ratio [one-way ANOVA F (4;29) = 6,398, p = 0.001] in the cytoplasmic fraction (Fig. 2A and 2B). In particular, post hoc Dunnett test revealed that P-p38 protein levels increased 5 minutes following the treatment (p = 0.003) (Fig. 2A and 2B). Furthermore, we found a significative effect of IL-18 treatment on P-ERK1/ERK1 ratio [One-way ANOVA: F (4;14) = 4,220, p = 0.029] (Fig. 2C and 2D) as well as for P-ERK2/ERK2 ratio [F (4;14) = 19,746, p = 0.000] (Fig.2 and 2F) in the cytoplasmic fraction. Interestingly, post hoc Dunnett analysis demonstrated that IL-18 treatment enhanced P-ERK2 protein levels above those of total ERK2 after 5 minutes (p = 0.004) and 15 minutes (p = 0.034) with respect to the control group (Fig. 2E and 2F).
Figure 2. Treatment with mouse recombinant IL-18 positively regulates MAPKs (p38 and ERK1/2) phosphorylation at early time points.
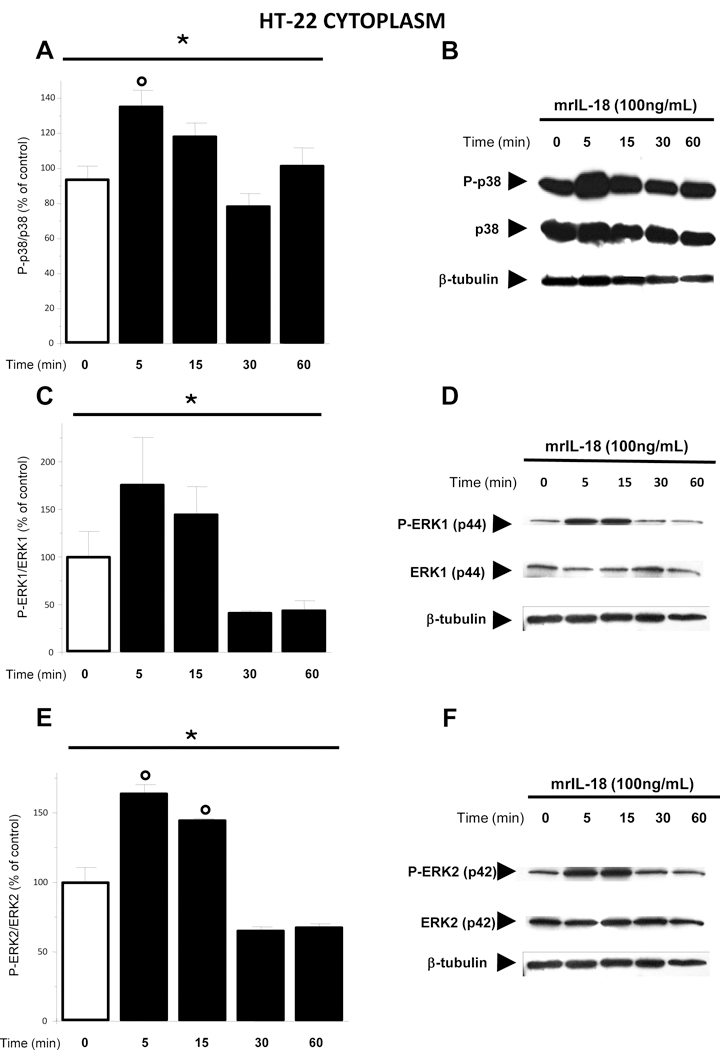
(A) P-p38\ p38 MAPK protein ratio was significantly enhanced 5 minutes after the treatment with rmIL-18 (100 ng/mL) with respect to the control (0 min) group (open bar). (C) Treatment of HT-22 cells with rmIL-18 affected P-ERK1/ERK1 MAPK ratio. (E) P-ERK2ERK2 MAPK ratio increased 5 and 15 minutes when compared to the control (0 min) group (open bar). (B, D and F) Representative western blots showing P-p38, p38, P-ERK1, ERK1, P-ERK2, and ERK2 respectively. The relative bands for β-tubulin are also represented. Phosphorylated and un-phosphorylated proteins were detected on the same blot. HT-22 cells were treated with IL-18 (100ng/mL) for 5, 15, 30 and 60 minutes after which proteins were measured in the cytoplasmic extracts by western blot. Optical density was expressed as % of control (vehicle treated) group. Each column represents mean ± S.E.M.; *p < 0.05 among the groups (one-way ANOVA). °p < 0.05 with respect to the control group (post hoc Dunnett test).
3.4. IL-18 treatment enhanced P-STAT3 (Tyr705)/ STAT3 ratio in the nucleus of HT-22 cells
STAT3 is a transcription factor of the Janus kinase/signal transducer and activator of transcription (JAK/STAT) activated by several cytokines and growth factors. Following tyrosine-residue phosphorylation by the activated JAKs, STAT3 homo- or heterodimerizes, translocates to the nucleus and binds to specific consensus sequences of target-gene promoters (Cattaneo et al., 1999). STAT3 can be phosphorylated at two positions: Ser727 and at Tyr705. While phosphorylation of Tyr705 activates STAT3, the significance of the phosphorylation at Ser727 is less clear (Decker and Kovarik, 2000) as it was proposed to negatively regulate STAT3 activity (Chung et al., 1997). We investigated the effects of IL-18 (100 ng/mL) in HT-22 cells at 5, 15, 30 and 60 minutes of treatment. One-way ANOVA revealed a main effect for P-STAT3 (Tyr705)/ STAT3 ratio specifically in the nucleus [F (4;22) = 5.825 p = 0.002] (Fig. 3A and 3B), whereas no significant effect was found in the cytoplasm (data not shown). Moreover, post hoc analysis showed that IL-18 treatment enhanced P-STAT3 (Tyr705) protein levels above those of total STAT3 after 30 (p = 0.028) and 60 (p = 0.034) minutes with respect to the control group (Fig. 3A and 3B). For P-STAT3 (Ser727) we found a statistically significant effect of time on P-STAT3 (Ser727)/ STAT3 ratio in the nucleus of HT-22 cells exposed to mrIL-18 compared to control cells (One-way ANOVA F (4;14) = 5,1968; p = 0.01897). At the later time point (60 minutes) the P-STAT3 (Ser727)/ STAT3 ratio was significantly lower in the nucleus of the HT-22 mrIL-18 exposed with respect to vehicle exposed cells as revealed by post hoc analysis (p = 0.00196) (Fig. 3C and 3D). In the cytosol IL-18 increased the P-STAT3 (Ser727)/ STAT3 ratio (one-way ANOVA F (4;14) = 3.54; p = 0.048) after 15 (p = 0.031) and 30 (p = 0.011) minutes (data not shown).
Figure 3. Treatment of HT-22 cells with mouse recombinant IL-18 increases P-STAT3 (Tyr705)/STAT3 ratio at 30 minutes and reduces P-STAT3 (Ser727)/STAT3 ratio after 60 minutes when compared to the control (0 min) group in the nucleus.

(A and C) Histograms showing P-STAT3 (Tyr705)/ STAT3 (upper left panel) and P-STAT3 (Ser727)/ STAT3 (lower left panel) protein levels, respectively in HT-22 cell nucleus after different time points from IL-18 treatment. HT-22 cells were treated with rmIL-18 (100 ng/mL) (filled bars) or with vehicle (open bars) then proteins were measured by western blotting in the nuclear enriched fractions. Optical density was expressed as % of control group. β-tubulin protein levels were not affected by the treatment with IL-18 in every time point considered. Phosphorylated and un-phosphorylated protein were detected on the same blot (one-way ANOVA). °p < 0.05 with respect to the control (0 min) group (post hoc Dunnett test). (B and D) Representative blots of P-STAT3 (Tyr705) (upper right panel) and P-STAT3 (Ser727) (lower right panel), respectively and their relative STAT3 and β-tubulin bands, detected in the nucleus after the indicated IL-18 treatment.
Finally, considering that the cytokine interferon-alpha has been described to activate STAT5 in the nucleus of mouse HT-22 cells (Hu et al., 2009), we also analyzed IL-18 effects on STAT5 and P-STAT5, but we did not find any significant effects (data not shown).
3.5. Phosphorylated STAT3 (Tyr705)/ STAT3 ratio increased in mouse hippocampus one hour after i.c.v. injection of mrIL-18
We previously demonstrated that IL-18Rs are expressed in the hippocampal neurons of the adult mouse (Alboni et al., 2009; Alboni et al., 2011a). Here, we investigated whether IL-18 could induce STAT3 phosphorylation in Tyr705 and Ser727 amino acid residues. Analysis was performed on protein extract of hippocampi collected one hour after i.c.v. injection of mrIL-18 (2,48 nmol of mrIL-18 /mouse) or vehicle (artificial cerebrospinal fluid- aCSF). IL-18 treatment significantly elevated the level of P-STAT3 Tyr705 over pan-STAT3 (t = 2,34612; p = 0,032) (Fig. 4A). No statistically significant changes were observed for P-STAT3 Ser727\pan-STAT3 (Fig. 4B), P-p38\ p38 ratio (Fig. 4C), P-ERK1\ ERK1 or P-ERK2\ ERK2 ratio (Fig. 4D and 4E respectively) or P-NF-κB Ser536\ NF-κB (Fig. 4F).
Figure 4. Intracerebroventricular (i.c.v.) treatment with recombinant IL-18 increases P-STAT3 (Tyr705) protein levels in hippocampus of i.c.v. injected mice with respect to vehicle (aCSF) i.c.v. injected mice.
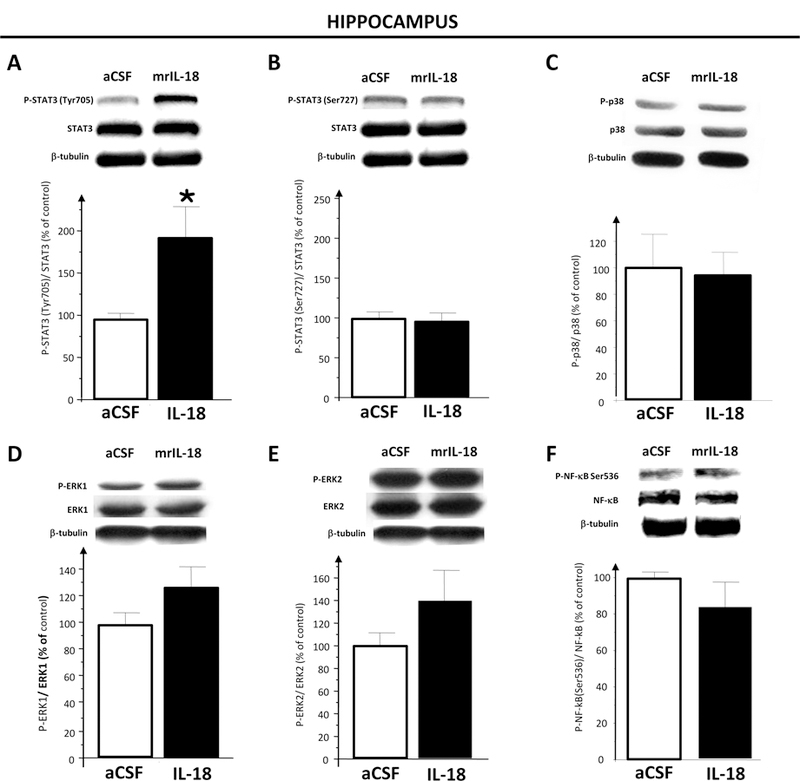
(A, C and D) A representative western blots of PSTAT3 (Tyr705), P-STAT3 (Ser727), STAT3, P-p38, p38, P-Erk1\2 MAPKs and β-tubulin detected in the hippocampal whole protein extract 1 hour after the artificial cerebro-spinal fluid (aCSF; n = 3) and IL-18 (n = 3) treatments is shown. (B) Bar graph demonstrating the increment of P-STAT3 (Tyr705)/ STAT3 protein ratio (left graph) and no significant effect for P-STAT3(Ser727)/ STAT3 protein ratio (right graph) in hippocampus of rmIL-18 injected mice (filled bars) compared to aCFS injected animals (open bars). (D and F) Histograms showing that IL-18 did not affect either P-p38\ P38 nor P-Erk1\ 1Erk and PErk2\ Erk2 ratio in the hippocampus of rmIL-18 injected mice (filled bars) compared to aCFS injected mice (open bars). Optical density was expressed as % of control (aCFStreated) group. Each column represents mean ± S.E.M.; *p < 0.05 among the two groups (t-Test analysis). Optical density was expressed as % of control (aCFS-treated) group. Each column represents mean ± S.E.M.; *p < 0.05 among the two groups (t-Test analysis).
4. DISCUSSION
The current study was designed to explore some intracellular signaling pathways possibly triggered by IL-18 at hippocampal level because this cytokine seems to play an important role in regulating mechanisms involved in function or dysfunction of this brain area. The main findings of our investigation were that mrIL-18 did not activate the nuclear factor κB but rather increased the tyrosine 705 phosphorylation of the signal transducer and activator of transcription 3 in the mouse hippocampal cell line HT-22. We also found that in a non homogenous cellular population, such as the hippocampus system, i.c.v. injection of mrIL-18 still increased STAT-3 phosphorylation at Tyr705 without significantly changing levels of NF-κB or NF-κB phosphorylation at serine 536.
Herein we demonstrated that hippocampal HT-22 neuronal cells expressed the components of the IL-18\ IL-18R system. Interestingly, both transcripts of IL-18 and its IL-18R complex were present in these cells suggesting that they could not only be responsive to IL-18, but could also produce this cytokine under basal conditions. Furthermore, our data showed that HT-22 cells under basal conditions expressed high to medium levels of IL-18BP and small IL-18Rβ mRNAs coding for proteins that were characterized as negative regulators of IL-18 action (Kim et al., 1999; Andre et al., 20003; Fiszer et al., 2007; Alboni et al., 2011). These data may indicate that IL-18 system in HT-22 could be regulated by the expression of its natural inhibitors. The alpha chain of IL-18R complex, that was widely demonstrated to be necessary for the binding of IL-18, was barely expressed in HT-22 cells. Higher levels, but still low, of expression were measured for the mRNA codifying for the type II IL-18Rα. Type II IL-18Rα is a short isoform of the alpha chain of the IL-18R lacking the intracellular TIR domain that has been believed to be a decoy receptor that can bind to IL-18 but fails to initiate the signal transduction (Alboni et al., 2009). However, we can not exclude that the binding to IL-18 to the TIR-less splice variant of the alpha chain may eventually build a signaling complex triggering the activation of transcription factors possibly different from those activated via full IL-18Rα as reported for other TIR-less binding chains (Han et al., 2010). Moreover, given the very low levels of the expression of the canonical binding chain of the IL-18R in HT-22 cells, it is possible to hypothesize that IL-18 might bind to a different receptor thus trigging STAT-3 activation.
IL-18 activates NF-κB signaling in immune cells such as T helper type 1 (Matsumoto et al., 1997; Tsuji-Takayama et al., 1999), macrophage-derived foam cells (Yu et al., 2012) and neutrophils (Fortin et al., 2009), in particular chondrocytes (Olee et al., 1999), in primary cardiomyocytes (Reddy et al., 2010), in EL4/6.1 thymoma cells (Thomassen et al., 1998) and in rheumatoid arthritis synovial fibroblasts (Morel et al., 2002). However it has been found that in natural killer cell line 92 IL-18 strongly enhanced tyrosine phosphorylation of STAT3 (Kalina et al., 2000). Moreover, the failure of IL-18 to induce NF-κB was previously discovered by Lee and co-workers in brain cells of epithelial origin such as those of the hypothalamic thermoregulatory center (Lee et al., 2004). Our results indicate that also in hippocampal (HT-22) cells and in the whole hippocampus IL-18 triggers intracellular signal pathways different from NF-κB such as that of STAT3. We found an increased P-STAT3 (Tyr705)\ STAT3 ratio on nuclear enriched extracts from HT-22 neurons after 30 minutes of exposure to rmIL-18. We also demonstrated that P-STAT3 Tyr705 protein levels, above those of total STAT3, were enhanced in the total hippocampus of mice one hour after i.c.v. injection of recombinant IL-18. Phosphorylation on Tyr705 has been considered as the most important event that activates STAT3 and results in DNA binding and gene expression regulation. However, another phosphorylation site in Ser727 has been described for STAT3 and the most recent finding reported that it regulated negatively the duration of STAT3 activity in HepG2 cell line (Wakahara et al., 2012). We found decreased levels of P-STAT3 (Ser727)\ STAT3 specifically in the nucleus of HT-22 cells treated with mrIL-18 compared to vehicle exposed cells. These results reinforce the hypothesis that IL-18 promotes STAT3 long-lasting activation by increasing the amount of Tyr705 rather than Ser727-phosphorylated protein in the nuclear compartment of HT-22 cells (Wakahara et al., 2012). Although further studies are needed to understand in depth the role of phosphorylation on Ser727 site of STAT3 our data demonstrated that IL-18 treatment specifically affected STAT3 residue phosphorylation possibly regulating the expression amount of specific genes within the hippocampus.
In HT-22 cells we also found that IL-18 exposure rapidly induced the phosphorylation of the extracellular signal-regulated kinase 1/2 (Erk1/2) and p38 mitogen activated protein kinase (MAPK) as previously reported in non-neuronal cells (Kalina et al., 2000; Fortin et al., 2009). Interestingly, IL-18 activated predominantly Erk2 in HT-22 cells that appear to be more closely involved in regulating cognitive functions such as learning and memory, while ERK1 is believed to have accessory functions to ERK2 (Satoh et al., 2007). Considering that the onset of STAT3 phosphorylation is delayed with respect to those of p38 and ErK1/2 MAPKs, it is possible that IL-18 could act directly on MAPKs pathways and secondarily may affect STAT3 activation in HT-22 cells. Thus, a cross-talk between STAT3 and the MAPKs pathway can not be excluded. This last hypothesis is in line with the report by Ng and colleagues that described that the activation of Erk and p38 MAPK is essential in regulating a delayed STAT3 phosphorylation following IL-1β treatment in rat primary cultures of myocytes (Ng et al., 2001). Recently, STAT3 is identified as a key molecule downstream of the Erk1/2 survival signal in dissociated rat hippocampal culture (Murase et al., 2012). One hour after mrIL-18 i.c.v. injection we did not find any significant change in the phosphorylation levels of the MAPKs investigated in the whole hippocampus supporting the hypothesis that this represent an early cytokineinduced effect rather than an effect typical for cells of neuronal origin.
5. CONCLUSIONS
To our knowledge, these data represent the first finding demonstrating that in brain cells IL-18 can activate the JAK–STAT pathway through STAT3. This finding is particularly relevant also considering the role played by STAT-3 within the hippocampus system. A number of studies have shown that STAT3 is present in the whole brain including hippocampus and synaptic fractions of neurons (De-Fraja et al., 1998; Hosoi et al., 2004; Murata et al., 2000) wherein its activation seems to be involved in synaptic plasticity (Nicolas et al., 2012). Notably, STAT3 activation has been identified as essential in the maintenance of adult neurogenesis in the dentate gyrus of the hippocampus (Müller et al., 2009) and in the differentiation of hippocampal neural stem cells (Cheng et al., 2011). Intriguingly, STAT3 signaling has been suggested to control the vulnerability of neurons (Murase et al., 2012). To this regard, activation of STAT3 in mouse hippocampus and in the hippocampal cell line HT-22 by IL-18 may represent one of the key pathways leading to the effects of IL-18 in modulating hippocampal function (including learning and memory and thus cognition) and plasticity (including neurogenesis).
Because mouse hippocampus-derived cell line HT-22 cells express the entire IL-18\ IL-18R system and are responsive to IL-18, we can conclude that they may represent (provide) a suitable model system to investigate IL-18 action at the hippocampal level, which could be helpful toward understanding the role of IL-18 in the healthy and pathological hippocampus. For example, because HT-22 cells possess functional cholinergic properties (Liu et al, 2009), this cellular model may represent an useful in vitro tool to unravel IL-18 mediated molecular events in some of the cognitive deficits observed in Alzheimer’s disease, also considering the ability of IL-18 to activate STAT-3 in these cells. Moreover, similarly to what happens with other cytokines (Rankin et al., 2013; Zalcman et al., 1994), IL-18 may act as modulator of the activity of neurotransmitters including acetylcholine.
AKNOWLEDGEMENTS
We thank Sheila Silverstein for editing the manuscript. This study has been partially supported by the ECNP Research Grant for Young Scientists and a fellowship for young scientists in foreign countries from the Italian Society of Pharmacology (SA) and by DK094026 grant (BC).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement: all authors declare that are no conflict of interest.
REFERENCES
- Abdul-Careem MF, Hunter BD, Sarson AJ, Mayameei A, Zhou H, Sharif S, 2006. Marek’s disease virus-induced transient paralysis is associated with cytokine gene expression in the nervous system. Viral Immunol 19, 167–76. [DOI] [PubMed] [Google Scholar]
- Alboni S, Cervia D, Ross B, Montanari C, Gonzalez AS, Sanchez-Alavez M, Marcondes MC, De Vries D, Sugama S, Brunello N, Blom J, Tascedda F, Conti B, 2009. Mapping of the full length and the truncated interleukin-18 receptor alpha in the mouse brain. J Neuroimmunol 214, 43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alboni S, Cervia D, Sugama S, Conti B 2010. Interleukin 18 in the CNS. J Neuroinflammation 7:9. doi: 10.1186/1742-2094-7-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alboni S, Montanari C, Benatti C, Blom JM, Simone ML, Brunello N, Caggia F, Guidotti G, Marcondes MC, Sanchez-Alavez M, Conti B, Tascedda F, 2011a. Constitutive and LPS-regulated expression of interleukin-18 receptor beta variants in the mouse brain 25, 483–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alboni S, Tascedda F, Corsini D, Benatti C, Caggia F, Capone G, Barden N, Blom JM, Brunello N, 2011b. Stress induces altered CRE/CREB pathway activity and BDNF expression in the hippocampus of glucocorticoid receptor-impaired mice. Neuropharmacology 60, 1337–46. [DOI] [PubMed] [Google Scholar]
- Alboni S, Gibellini L, Montanari C, Benatti C, Benatti S, Tascedda F, Brunello N, Cossarizza A, Pariante CM, 2013. N-acetyl-cysteine prevents toxic oxidative effects induced by IFN-α in human neurons. Int J Neuropsychopharmacol 16, 1–17. [DOI] [PubMed] [Google Scholar]
- Andoh T, Kishi H, Motoki K, Nakanishi K, Kuraishi Y, Muraguchi A, 2008. Protective effect of IL-18 on kainate- and IL-1 beta-induced cerebellar ataxia in mice. J Immunol 180, 2322–8. [DOI] [PubMed] [Google Scholar]
- Andre R, Wheeler RD, Collins PD, Luheshi GN, Pickering-Brown S, Kimber I, Rothwell NJ, Pinteaux E, 2003. Identification of a truncated IL-18R beta mRNA: a putative regulator of IL-18 expressed in rat brain. J Neuroimmunol 145:40–45. [DOI] [PubMed] [Google Scholar]
- Arend WP, Palmer G, Gabay C, 2008. IL-1, IL-18, and IL-33 families of cytokines. Immunol Rev 223, 20–38. [DOI] [PubMed] [Google Scholar]
- Benatti C, Alboni S, Montanari C, Caggia F, Tascedda F, Brunello N, Blom JM, 2011. Central effects of a local inflammation in three commonly used mouse strains with a different anxious phenotype. Behav Brain Res 224, 23–34. [DOI] [PubMed] [Google Scholar]
- Bossù P, Ciaramella A, Salani F, Bizzoni F, Varsi E, Di Iulio F, Giubilei F, Gianni W, Trequattrini A, Moro ML, Bernardini S, Caltagirone C, Spalletta G, 2008. Interleukin-18 produced by peripheral blood cells is increased in Alzheimer’s disease and correlates with cognitive impairment. Brain Behav Immun 22, 487–92. [DOI] [PubMed] [Google Scholar]
- Buss H, Dörrie A, Schmitz ML, Hoffmann E, Resch K, Kracht M, 2004. Constitutive and interleukin-1-inducible phosphorylation of p65 NF-{kappa}B at serine 536 is mediated by multiple protein kinases including I{kappa}B kinase (IKK)-{alpha}, IKK{beta}, IKK{epsilon}, TRAF family member-associated (TANK)-binding kinase 1 (TBK1), and an unknown kinase and couples p65 to TATA-binding protein-associated factor II31-mediated interleukin-8 transcription. J Biol Chem 279, 55633–43. [DOI] [PubMed] [Google Scholar]
- Cattaneo E, Conti L, De-Fraja C, 1999. Signalling through the JAK-STAT pathway in the developing brain. Trends Neurosci 22, 365–9. [DOI] [PubMed] [Google Scholar]
- Cheng X, Jin G, Zhang X, Tian M, Zou L, 2011. Stage-dependent STAT3 activation is involved in the differentiation of rat hippocampus neural stem cells. Neurosci Lett 493, 18–23. [DOI] [PubMed] [Google Scholar]
- Chung J, Uchida E, Grammer TC, Blenis J, 1997. STAT3 serine phosphorylation by ERK-dependent and -independent pathways negatively modulates its tyrosine phosphorylation. Mol Cell Biol 17, 6508–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cumiskey D, Curran BP, Herron CE, O’Connor JJ, 2007. A role for inflammatory mediators in the IL-18 mediated attenuation of LTP in the rat dentate gyrus. Neuropharmacology 52, 1616–23. [DOI] [PubMed] [Google Scholar]
- Curran B, O’Connor JJ, 2001. The pro-inflammatory cytokine interleukin-18 impairs long-term potentiation and NMDA receptor-mediated transmission in the rat hippocampus in vitro. Neuroscience 108, 83–90. [DOI] [PubMed] [Google Scholar]
- De-Fraja C, Conti L, Magrassi L, Govoni S, Cattaneo E, 1998. Members of the JAK/STAT proteins are expressed and regulated during development in the mammalian forebrain. J Neurosci Res 54, 320–30. [DOI] [PubMed] [Google Scholar]
- Decker T and Kovarik P, 2000. Serine phosphorylation of STATs. Oncogene 19, 262837. [DOI] [PubMed] [Google Scholar]
- del Rey A, Balschun D, Wetzel W, Randolf A, Besedovsky HO, 2013. A cytokine network involving brain-borne IL-1β, IL-1ra, IL-18, IL-6, and TNFα operates during longterm potentiation and learning. Brain Behav Immun 2013. 33, 15–23. [DOI] [PubMed] [Google Scholar]
- Fiszer D, Rozwadowska N, Rychlewski L, Kosicki W, Kurpisz M, 2007. Identification of IL-18RAP mRNA truncated splice variants in human testis and the other human tissues. Cytokine 39, 178–83. [DOI] [PubMed] [Google Scholar]
- Fortin CF, Ear T, McDonald PP, 2009. Autocrine role of endogenous interleukin-18 on inflammatory cytokine generation by human neutrophils. FASEB J 23, 194–203. [DOI] [PubMed] [Google Scholar]
- Franklin KBJ And Paxinos G, 2001. The Mouse Brain in Stereotaxic Coordinates. 2 San Diego, CA: Academic. [Google Scholar]
- Haastrup E, Bukh JD, Bock C, Vinberg M, Thørner LW, Hansen T, Werge T, Kessing LV, Ullum H, 2012. Promoter variants in IL-18 are associated with onset of depression in patients previously exposed to stressful-life events. J Affect Disord 136, 134–8. [DOI] [PubMed] [Google Scholar]
- Han KJ, Yang Y, Xu LG, Shu HB, 2010. Analysis of a TIR-less splice variant of TRIF reveals an unexpected mechanism of TLR3-mediated signaling. J Biol Chem 285, 12543–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang WX, Huang P, Hillert J, 2004. Increased expression of caspase-1 and interleukin-18 in peripheral blood mononuclear cells in patients with multiple sclerosis. Mult Scler 10, 482–7. [DOI] [PubMed] [Google Scholar]
- Hosoi T, Okuma Y, Kawagishi T, Qi X, Matsuda T, Nomura Y, 2004. Bacterial endotoxin induces STAT3 activation in the mouse brain. Brain Res 1023, 48–53. [DOI] [PubMed] [Google Scholar]
- Hu F, Pace TW, Miller AH, 2009. Interferon-alpha inhibits glucocorticoid receptormediated gene transcription via STAT5 activation in mouse HT-22 cells. Brain Behav Immun 23, 455–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iannello A, Samarani S, Debbeche O, Tremblay C, Toma E, Boulassel MR, Routy JP, Ahmad A, 2009. Role of interleukin-18 in the development and pathogenesis of AIDS. AIDS Rev 11, 115–125. [PubMed] [Google Scholar]
- Jung HK, Ryu HJ, Kim MJ, Kim WI, Choi HK, Choi HC, Song HK, Jo SM, Kang TC, 2012. Interleukin-18 attenuates disruption of brain-blood barrier induced by status epilepticus within the rat piriform cortex in interferon-γ independent pathway. Brain Res 1447, 126–34. [DOI] [PubMed] [Google Scholar]
- Kalina U, Kauschat D, Koyama N, Nuernberger H, Ballas K, Koschmieder S, Bug G, Hofmann WK, Hoelzer D, Ottmann OG, 2000. IL-18 activates STAT3 in the natural killer cell line 92, augments cytotoxic activity, and mediates IFNgamma production by the stress kinase p38 and by the extracellular regulated kinases p44erk-1 and p42erk21. J Immunol 165, 1307–1313. [DOI] [PubMed] [Google Scholar]
- Kim YM, Kang HS, Paik SG, Pyun KH, Anderson KL, Torbett BE, Choi I, 1999. Roles of IFN consensus sequence binding protein and PU.1 in regulating IL-18 gene expression. J. Immunol 163, 2000–2007. [PubMed] [Google Scholar]
- Kokai M, Kashiwamura S, Okamura H,Ohara K, Morita Y, 2002. Plasma interleukin18 levels in patients with psychiatric disorders. J Immunother 25 Suppl 1, S68–71. [DOI] [PubMed] [Google Scholar]
- Kroeger KM, Sullivan BM, Locksley RM, 2009. IL-18 and IL-33 elicit Th2 cytokines from basophils via a MyD88- and p38alpha-dependent pathway. J Leukoc Biol 86, 76978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroes RA, Panksepp J, Burgdorf J, Otto NJ, Moskal JR, 2006. Modeling depression: social dominance-submission gene expression patterns in rat neocortex. Neuroscience 137, 37–49. [DOI] [PubMed] [Google Scholar]
- Kumar RA, Cann C, Hall JE, Sudheer PS, Wilkes AR, 2007. Predictive value of IL-18 and SC5b-9 for neurocognitive dysfunction after cardiopulmonary bypass. Br J Anaesth 98, 317–22. [DOI] [PubMed] [Google Scholar]
- Lappas M, Permezel M, Georgiou HM, Rice GE, 2002. Nuclear factor kappa B regulation of proinflammatory cytokines in human gestational tissues in vitro. Biol Reprod 67, 668–73. [DOI] [PubMed] [Google Scholar]
- Lee JK, Kim SH, Lewis EC, Azam T, Reznikov LL, Dinarello CA, 2004. Differences in signaling pathways by IL-1beta and IL-18. Proc Natl Acad Sci U S A 101, 8815–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q and Verma IM, 2002. NF-kappaB regulation in the immune system. Nat Rev Immunol 2, 725–34. [DOI] [PubMed] [Google Scholar]
- Liu YP, Lin HI, Tzeng SF, 2005. Tumor necrosis factor-alpha and interleukin18 modulate neuronal cell fate in embryonic neuralprogenitor culture. Brain Res 1054, 152–8. [DOI] [PubMed] [Google Scholar]
- Losy J, Niezgoda A, 2001. IL-18 in patients with multiple sclerosis. Acta Neurol Scand 104, 171–3. [DOI] [PubMed] [Google Scholar]
- Lu LX, Guo SQ, Chen W, Li Q, Cheng J, Guo JH, 2004. Effect of clozapine and risperidone on serum cytokine levels in patients with first-episode paranoid schizophrenia. Di Yi Jun Yi Da Xue Xue Bao 24, 1251–1254. [PubMed] [Google Scholar]
- Matsumoto S, Tsuji-Takayama K, Aizawa Y, Koide K, Takeuchi M, Ohta T, Kurimoto M, 1997. Interleukin-18 activates NF-kappaB in murine T helper type 1 cells. Biochem. Biophys. Res. Commun 234, 454–457. [DOI] [PubMed] [Google Scholar]
- Morel JC, Park CC, Woods JM, Koch AE, 2001. A novel role for interleukin18 in adhesion molecule induction through NF kappa B and phosphatidylinositol (PI) 3kinase-dependent signal transduction pathways. J Biol Chem 276, 37069–75. [DOI] [PubMed] [Google Scholar]
- Morel JC, Park CC, Zhu K, Kumar P, Ruth JH, Koch AE, 2002. Signal transduction pathways involved in rheumatoid arthritis synovial fibroblast interleukin-18induced vascular cell adhesion molecule-1 expression. J Biol Chem 277, 34679–91. [DOI] [PubMed] [Google Scholar]
- Müller S, Chakrapani BP, Schwegler H, Hofmann HD, Kirsch M, 2009. Neurogenesis in the dentate gyrus depends on ciliary neurotrophic factor and signal transducer and activator of transcription 3 signaling. Stem Cells 27, 431–41. [DOI] [PubMed] [Google Scholar]
- Murase S, Kim E, Lin L, Hoffman DA, McKay RD, 2012. Loss of Signal Transducer and Activator of Transcription 3 (STAT3) Signaling during Elevated Activity Causes Vulnerability in Hippocampal Neurons. J Neurosci 32,15511–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata S, Usuda N, Okano A, Kobayashi S, Suzuki T, 2000. Occurrence of a transcription factor, signal transducer and activators of transcription 3 (Stat3), in the postsynaptic density of the rat brain. Brain Res Mol Brain Res 78, 80–90. [DOI] [PubMed] [Google Scholar]
- Nakahira M, Ahn HJ, Park WR, Gao P, Tomura M, Park CS, Hamaoka T, Ohta T, Kurimoto M, Fujiwara H, 2002. Synergy of IL-12 and IL-18 for IFN-gamma gene expression: IL-12-induced STAT4 contributes to IFN-gamma promoter activation by upregulating the binding activity of IL-18-induced activator protein 1. J Immunol 168, 114653. [DOI] [PubMed] [Google Scholar]
- Nakanishi K, Yoshimoto T, Tsutsui H,, Okamura H, 2001. Interleukin-18 is a unique cytokine that stimulates both Th1 and Th2 responses depending on its cytokine milieu. Cytokine Growth Factor Rev 12, 53–72. [DOI] [PubMed] [Google Scholar]
- Natoli G, Saccani S, Bosisio D, Marazzi I, 2005. Interactions of NF-kappaB with chromatin: the art of being at the right place at the right time. Nat Immunol 6, 439–45. [DOI] [PubMed] [Google Scholar]
- Ng DC, Long CS, Bogoyevitch MA, 2001. A role for the extracellular signal-regulated kinase and p38 mitogen-activated protein kinases in interleukin-1 beta-stimulated delayed signal tranducer and activator of transcription 3 activation, atrial natriuretic factor expression, and cardiac myocyte morphology. J Biol Chem 276, 29490–8. [DOI] [PubMed] [Google Scholar]
- Nicolas CS, Amici M, Bortolotto ZA, Doherty A, Csaba Z, Fafouri A, Dournaud P, Gressens P, Collingridge GL, Peineau S, 2013. The role of JAK-STAT signaling within the CNS. JAKSTAT 2, e22925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick D, Kim SH, Fantuzzi G, Reznikov LL, Dinarello CA, Rubinstein M, 1999. Interleukin-18 binding protein: a novel modulator of the Th1 cytokine response. Immunity 10, 127–36. [DOI] [PubMed] [Google Scholar]
- Olee T, Hashimoto S, Quach J, Lotz M, 1999. IL-18 is produced by articular chondrocytes and induces proinflammatory and catabolic responses. J Immunol 162, 1096–100. [PubMed] [Google Scholar]
- Oztürk C, Ozge A, Yalin OO, Yilmaz IA, Delialioglu N, Yildiz C, Tesdelen B, Kudiaki C, 2007. The diagnostic role of serum inflammatory and soluble proteins on dementia subtypes: correlation with cognitive and functional decline. Behav Neurol. 2007 18, 207–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace TW, Hu F, Miller AH, 2011. Activation of cAMP-protein kinase A abrogates STAT5-mediated inhibition of glucocorticoid receptor signaling by interferon-alpha. Brain Behav Immun 25,1716–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankin JS, Zalcman SS, Zhu Y, Siegel A, 2013. Short- and long-term effects of interleukin-2 treatment on the sensitivity of periadolescent female mice to interleukin-2 and dopamine uptake inhibitor. PLoS One 8:e64473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy VS, Prabhu SD, Mummidi S, Valente AJ, Venkatesan B, Shanmugam P, Delafontaine P, Chandrasekar B, 2010. Interleukin-18 induces EMMPRIN expression in primary cardiomyocytes via JNK/Sp1 signaling and MMP-9 in part via EMMPRIN and through AP-1 and NF-kappaB activation. Am J Physiol Heart Circ Physiol 299, H1242–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu HJ, Kim JE, Kim MJ, Kwon HJ, Suh SW, Song HK, Kang TC, 2010. The protective effects of interleukin-18 and interferon-γ on neuronal damages in the rat hippocampus following status epilepticus. Neuroscience 170, 711–21. [DOI] [PubMed] [Google Scholar]
- Sahar S, Dwarakanath RS, Reddy MA, Lanting L, Todorov I, Natarajan R, 2005. Angiotensin II enhances interleukin-18 mediated inflammatory gene expression in vascular smooth muscle cells: a novel cross-talk in the pathogenesis of atherosclerosis. Circ Res 96, 1064–71. [DOI] [PubMed] [Google Scholar]
- Satoh Y, Endo S, Ikeda T, Yamada K, Ito M, Kuroki M, Hiramoto T, Imamura O, Kobayashi Y, Watanabe Y, Itohara S, Takishima K, 2007. Extracellular signalregulated kinase 2 (ERK2) knockdown mice show deficits in long-term memory; ERK2 has a specific function in learning and memory. J Neurosci 27, 10765–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt AJ, Krieg JC, Vedder H, 2005. Interleukin-6 induces glutathione in hippocampal cells. Prog Neuropsychopharmacol Biol Psychiatry 29, 321–6. [DOI] [PubMed] [Google Scholar]
- Sergi B, Penttila I, 2004. Interleukin 18 receptor. J Biol Regul Homeost Agents 18, 5561. [PubMed] [Google Scholar]
- Shelton RC, Claiborne J, Sidoryk-Wegrzynowicz M, Reddy R, Aschner M, Lewis DA, Mirnics K, 2011. Altered expression of genes involved in inflammation and apoptosis in frontal cortex in major depression. Mol Psychiatry 16, 751–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speisman RB, Kumar A, Rani A, Foster TC, Ormerod BK, 2012. Daily exercise improves memory, stimulates hippocampal neurogenesis and modulates immune and neuroimmune cytokines in aging rats. Brain Behav Immun 28, 25–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugama S, Wirz SA, Barr AM, Conti B, Bartfai T, Shibasaki T, 2004. Interleukin18 null mice show diminished microglial activation and reduced dopaminergic neuron loss following acute 1-methyl-4-phenyl-1,2,3,6- tetrahydropyridine treatment. Neuroscience 128, 451–458. [DOI] [PubMed] [Google Scholar]
- Sugimoto N, Nakahira M, Ahn HJ, Micallef M, Hamaoka T, Kurimoto M, Fujiwara H. 2003. Differential requirements for JAK2 and TYK2 in T cell proliferation and IFN-gamma production induced by IL-12 alone or together with IL-18. Eur J Immunol 33, 243–51. [DOI] [PubMed] [Google Scholar]
- Sutinen EM, Pirttilä T, Anderson G, Salminen A, Ojala JO, 2012. Pro-inflammatory interleukin-18 increases Alzheimer’s disease-associated amyloid-β production in human neuron-like cells. J Neuroinflammation 16, 9:199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomassen E, Bird TA, Renshaw BR, Kennedy MK, Sims JE, 1998. Binding of interleukin-18 to the interleukin-1 receptor homologous receptor IL-1Rrp1 leads to activation of signaling pathways similar to those used by interleukin-1. J Interferon Cytokine Res 18, 1077–88. [DOI] [PubMed] [Google Scholar]
- Tsuji-Takayama K, Aizawa Y, Okamoto I, Kojima H, Koide K, Takeuchi M, Ikegami H, Ohta T, 1999. Kurimoto M. Interleukin-18 induces interferon-gamma production through NF-kappaB and NFAT activation in murine T helper type 1 cells. Cell Immunol 196, 41–50. [DOI] [PubMed] [Google Scholar]
- Yaguchi T, Nagata T, Yang D, Nishizaki T, 2010. Interleukin-18 regulates motor activity, anxiety and spatial learning without affecting synaptic plasticity. Behav Brain Res 20, 47–51. [DOI] [PubMed] [Google Scholar]
- Yu XH, Jiang HL, Chen WJ, Yin K, Zhao GJ, Mo ZC, Ouyang XP, Lv YC, Jiang ZS, Zhang DW, Tang CK, 2012. Interleukin-18 and interleukin-12 together downregulate ATP-binding cassette transporter A1 expression through the interleukin18R/nuclear factor-κB signaling pathway in THP-1 macrophage-derived foam cells. Circ J 76, 1780–91. [DOI] [PubMed] [Google Scholar]
- Viatour P, Merville MP, Bours V, Chariot A, 2005. Phosphorylation of NF-kappaB and IkappaB proteins: implications in cancer and inflammation. Trends Biochem Sci 30, 43–52. [DOI] [PubMed] [Google Scholar]
- von Giesen HJ, Jander S, Köller H, Arendt G, 2004. Serum and cerebrospinal fluid levels of interleukin-18 in human immunodeficiency virus type 1-associated central nervous system disease. J Neurovirol 10, 383–6. [DOI] [PubMed] [Google Scholar]
- Zalcman S, Green-Johnson JM, Murray L, Nance DM, Dyck D, Anisman H, Greenberg AH, 1994. Cytokine-specific central monoamine alterations induced by interleukin-1, -2 and -6. Brain Res 643, 40–9. [DOI] [PubMed] [Google Scholar]
- Zhang XM, Duan RS, Chen Z, Quezada HC, Mix E, Winblad B, Zhu J, 2007. IL-18 deficiency aggravates kainic acid-induced hippocampal neurodegeneration in C57BL/6 mice due to an overcompensation by IL-12. Exp Neurol 205, 64–73. [DOI] [PubMed] [Google Scholar]
- Wakahara R, Kunimoto H, Tanino K, Kojima H, Inoue A, Shintaku H, Nakajima K, 2012. Phospho-Ser727 of STAT3 regulates STAT3 activity by enhancing dephosphorylation of phospho-Tyr705 largely through TC45. Genes Cells 17, 132–45. [DOI] [PubMed] [Google Scholar]