

Abstract
Amphiphilic polymers can be used to form micelles to deliver water-insoluble drugs. A biodegradable poly(ethylene glycol) (PEG)-poly(beta-amino ester) (PBAE)-PEG triblock copolymer was developed that is useful for drug delivery. It was shown to successfully encapsulate and pH-dependently release a water-insoluble, small molecule anti-cancer drug, verteporfin. PEG-PBAE-PEG micelle morphology was also controlled through variations to the hydrophobicity of the central PBAE block of the copolymer in order to evade macrophage uptake. Spherical micelles were 50 nm in diameter, while filamentous micelles were 31 nm in width with an average aspect ratio of 20. When delivered to RAW 264.7 mouse macrophages, filamentous micelles exhibited a 89% drop in cellular uptake percentage and a 5.6-fold drop in normalized geometric mean cellular uptake compared to spherical micelles. This demonstrates the potential of high aspect ratio, anisotropic shaped PEG-PBAE-PEG micelles to evade macrophage-mediated clearance. Both spherical and filamentous micelles also showed therapeutic efficacy in human triple-negative breast cancer and small cell lung cancer cells without requiring photodynamic therapy to achieve an anti-cancer effect. Both spherical and filamentous micelles were more effective in killing lung cancer cells than breast cancer cells at equivalent verteporfin concentrations, while spherical micelles were shown to be more effective than filamentous micelles against both cancer cells. Spherical and filamentous micelles at 5 and 10 μM respective verteporfin concentration resulted in 100% cell killing of lung cancer cells, but both micelles required a higher verteporfin concentration of 20 μM to kill breast cancer cells at the levels of 80% and 50% respectively. This work demonstrates the potential of PEG-PBAE-PEG as a biodegradable, anisotropic drug delivery system as well as the in vitro use of verteporfin-loaded micelles for cancer therapy.
Keywords: anisotropic micelles, poly(beta-amino ester), verteporfin, cancer therapy
Graphical Abstract
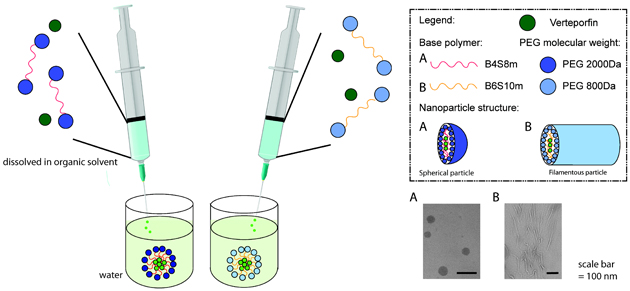
Introduction
Polymeric nano-vehicles have been investigated as gene and drug delivery systems due to their small size and high loading capacity.1,2 Specifically, amphiphilic block copolymers self-assemble into nano-sized structures due to the hydrophobic effect when exposed to an aqueous environment. This process produces micelles with a hydrophobic core and hydrophilic shell. As the majority of small molecule drugs have low solubility in aqueous medium, there is a significant need to engineer delivery vehicles capable of encapsulating poorly water-soluble drugs and enabling administration of these drugs at physiologically relevant dosages.3 In addition, when designing a nanomedicine, it has been demonstrated that a hydrophilic corona, often consisting of poly(ethylene glycol) (PEG), can promote colloidal stability and neutralize surface charge, and consequently reduce clearance by the reticuloendothelial (RES) system, increase passive accumulation at neovasculature around a tumor, and improve diffusion through the interstitial space.4,5
Poly(beta-amino ester)s (PBAE) compose a class of cationic biodegradable polymers, that due to their positive charge, have been used by researchers to form polyplexes for the delivery of hydrophilic anionic nucleic acid cargo, but have not been well-investigated for the potential to carry non-genetic cargo.6,7 The capacity to synthesize libraries of PBAEs with distinct chemical properties through the use of monomers with differential structures has been an asset to explore structure/function relationships for gene delivery.6,8 PBAE copolymers can be further developed through the synthesis of hydrophobic PBAE blocks combined with hydrophilic blocks to create an amphiphilic copolymer capable of forming micellar structures.9 Tuning hydrophobicity of the PBAE segment of PBAE-based amphiphilic copolymers can affect the packing parameter thermodynamic equilibrium of the polymers in solution, and change the shape of the resulting micelles, such as spheres, filaments, and multilamellar vesicles.10 Different shapes of particles have been previously shown to exhibit varying pharmacokinetics and biodistribution.11 Also, one of the critical properties of gene delivery cationic polymers is their tertiary amines along the backbone that provide the pH buffering capacity to facilitate endosomal escape.12 For PBAE-based micelles, the many tertiary amines can enable similar endosomal escape. In addition, the pH-sensitive ester linkages of PBAEs can allow environmentally-triggered release as pH-sensitive cargo release has been demonstrated in the more acidic tumor microenvironment with this class of materials.5
Verteporfin (VP) is a small molecule also known as benzoporphyrin derivative monoacid ring A, which belongs to the porphyrin family of photosensitizers in photodynamic therapy (PDT). Due to VP’s low solubility of 13.6 μg/mL in aqueous medium, liposomal VP formulations were developed to as a PDT agent in neovascular age-related macular degeneration (NVAMD) and tumor.13,14 Although Visudyne® remains the only FDA-approved liposomal VP for PDT against NVAMD, other nanoparticle VP formulations has been tested in PDT against subcutaneous cancer in vivo, such as Meth-A sarcoma and rhabdomyosarcoma.15,16 These PDT-based approaches permit localized tissue necrosis using low energy laser where the agent is administered and/or sequestered. Clinically, such procedures pose issues of photosensitivity and exposure to sunlight during treatment periods, often requiring patients to remain quarantined in a limitedly lighted environment. As such, although some existing therapies exploit the PDT properties of VP in NVAMD and cancer, recent reports have investigated and demonstrated the utility and benefits of this drug in a non-PDT regiment against cancer. These reports reveal the ability of VP to decrease cancer cell proliferation and attenuate tumor growth in vivo without requiring photoactivation, making it a safe and effective method distinct from PDT therapy that can be employed clinically. Specifically, it was recently demonstrated that VP leads to inhibition of growth and proliferation of human retinoblastoma cells and a number of central nervous system-derived cancer cells in the absence of light activation.17,18 VP has been associated with down-regulation of the yes-associated protein-transcriptional enhancer factor domain (YAP-TEAD) complex, which is involved in the Hippo pathway in cancer cells to induce uncontrolled proliferation, but is inactive in healthy tissues.18–20 This mechanism facilitates specific anti-cancer treatment of cancer cells without harming healthy cells.21 YAP signaling is also known to be hyperactivated in epithelial-derived carcinomas. As liposomal VP formulations can readily destabilize in the presence of blood plasma22 and can be quickly cleared by the reticuloendothelial system, there is a need to develop safe and effective alternative delivery vehicles to administer VP locally or systemically for specific treatment against cancer.
In this study, stable polymeric micelles encapsulating VP were formulated with a novel PEG-PBAE-PEG triblock copolymer. Given the above-mentioned benefits of non-PDT utility of VP, we evaluated the properties and efficacy of these VP-loaded micelles as non-photodynamic chemotherapeutic agents in two epithelial cancer cells, human small cell lung cancer (H446) and human triple-negative breast cancer (MDA-MB 231) cells. Furthermore, two different types of the triblock copolymers were synthesized to generate spherical and anisotropic micelles that showed shape-dependent differential uptake by macrophages.
Materials and Methods
Materials
1,4-butanediol diacrylate (B4), octylamine (S8m), decylamine (S10m), pyrene (Sigma-Aldrich), 1,6-hexanediol diacrylate (B6), 1-(3-aminopropyl)-4-methylpiperazine (E7) (Alfa Aesar), dimethyl sulfoxide (DMSO), dimethyl formamide (DMF), hexane, methoxy poly(ethylene glycol) thiol (2 kDa and 800 Da) (Laysan Bio, Inc), and Verteporfin (VP) (U.S. Pharmacopeial Convention, Inc.) were purchased and used as received. CellTiter 96 AQueous One MTS assay (Promega, Fitchburg, WI) was used per manufacturer’s instructions.
Synthesis of PEG-PBAE-PEG triblock copolymer
A PBAE-based triblock amphiphilic copolymer was synthesized by a two-step polymer synthesis. First, 1,4-butanediol diacrylate (B4) was reacted with octylamine (S8m) by Michael addition reaction at molar ratio of 1.15:1 at 90oC for 72 h to yield acrylate-terminated hydrophobic PBAE base polymer (B4S8m) as shown in Figure 1A/B. The B4S8m base polymer was subsequently precipitated in hexane twice and then dried under vacuum with desiccant overnight. The structure and molecular weight of the base polymer was confirmed using Bruker Avance III 500 MHz 1H NMR spectrometer in CDCl3. Following the procedure described by Kim et al., the base polymer reacted with 2 kDa mPEG-thiol in DMSO with trace amounts of 1-(3-aminopropyl)-4-methyl-piperazine (E7) as a primary amine-containing catalyst by thiol-ene Michael addition reaction at molar ratio of 1:6:0.25 (B4S8m : PEG : catalyst) while stirring at 50°C for 24 h.23 The B4S8m triblock copolymer with PEG 2 kDa, named PP1, was purified by first removing DMSO with a rotary evaporator and then precipitating with hexane twice. The structure of the PEG-PBAE-PEG triblock copolymer was confirmed using 1H NMR in CDCl3. The identical procedure was used for synthesis of B6S10m triblock copolymer with PEG 800 Da, named PP2. The partition coefficient of each copolymer was determined using ChemBioDraw software.
Figure 1. Schematic diagram of polymer synthesis.

(A) Two-step Michael addition reaction for PEG-PBAE-PEG synthesis, (B) chemical structures of diacrylate and primary amine monomers used in the synthesis, and (C) nomenclature of final polymers.
Formulation of micelles
Spherical VP-loaded micelles (sVPM) and filamentous VP-loaded micelles (fVPM) were prepared using nanoprecipitation method. First, PEG-PBAE-PEG triblock copolymer was dissolved in DMF at 20 mg/mL. Next, an equivalent volume of 1 mg/mL VP solution in DMSO was added to the polymer solution and vortexed. Each nanoprecipitation reaction was limited to 5 mg of the 10 mg/mL VP-polymer solution. 500 μL (5 mg) of the VP-polymer solution was added slowly using an insulin syringe into 3x volume of ultrapure water, while stirring at 500 rpm. Spherical blank micelle (sBM) and filamentous blank micelles (fBM) batches were identically synthesized excluding the presence of VP. Immediately after adding the solution, the reaction vial was placed in a water bath sonicator for 1 min and then placed back on stir plate for 4 h. The solution was then added to a 10 kDA MWCO filter (EMD Millipore, Burlington, MA) and spun for 15 minutes. Next, the remaining solution was added to a Sephadex column with Sephadex S-500 High Resolution. After spinning for 3 minutes at 800 g, the filtrate was spun at 17,000 g for 15 min. The supernatant was collected and then filtered through a 0.22 μm PTFE syringe filter. The micelle solution was lyophilized with 10% final sucrose solution as a cryoprotectant. Several aliquots were lyophilized without sucrose to measure loading and release of VP.
Characterization of micelles
Critical micelle concentration (CMC) of sVPM was measured by spectrofluorophotometry using pyrene as the indicator of micelle formation. Pyrene emission peaks shift depending on the polarity of the local environment. Briefly, 185.8 ng of pyrene in acetone was left to dry and 1.5 mL of water was added. Organic phase with a range of polymer concentrations in DMSO: DMF (1:1 v/v) was added to the aqueous solution as described above and sonicated for 1 min. Following the 4 h stirring step, excitation spectrum of the samples was recorded with a constant emission value of 390 nm. The intensity ratios of the excitation peaks at 339 and 335 nm were calculated and then plotted as a function of log[PEG-PBAE-PEG]. The inflection point of the fitted sigmoidal graph was determined as the CMC.
Micelle size and morphology was determined by transmission electron microscopy (TEM). 0.5% uranyl acetate was used as a negative stain for TEM. ImageJ was used to determine aspect ratio distribution of fVPM. Dynamic light scattering (DLS) with Malvern Zetasizer Nano ZS (Malvern Instruments, Malvern, U.K.) was used to determine initial size and particle stability at 0, 1, 3, 5, 12 and 31 h time points of micelle formulations. To determine particle stability, sVPM and sBM micelles were reconstituted with deionized water to reach isotonic concentration of 10% w/v sucrose and then further with four different mediums to reach a polymer concentration of 1 mg/mL: 1) 10% sucrose solution, 2) 1x PBS, and 3) human serum plasma. Zeta potential was determined with Malvern Zetasizer by preparing micelle sample in 10 mM NaCl at 1 mg/mL.
Loading and release kinetics of VP
Dilution series of lyophilized VPM were made by dissolving in DMSO at the highest polymer concentration of 1.0 mg/mL. Fluorescence intensity was measured with excitation wavelength of 420 nm and emission wavelength of 680 nm using Synergy 2 plate reader (Biotek). Each concentration was tested as triplicates, and fluorescence intensity was translated to VP mass by using a standard curve. The DLC (drug loading content) and the DLE (drug loading efficiency) were then calculated according to the following formulas:
Micelles were dissolved in a mixed buffer of citric acid monohydrate and sodium phosphate dibasic at three different pH’s (5.0, 6.5, 7.4) at 0.1 mg/mL and 1 mL of each was transferred into separate scintillation vials. The vials were incubated on a shaker at 37°C. At time points of 1, 3, 5, and 12 h entire volume was spun down at 500k g for 20 min at 4°C to pellet remaining micelles. Released VP in the supernatant was stored in a separate tube for fluorescence measurement. Pelleted micelles were resuspended in 1mL of fresh buffer and transferred to a new scintillation vial for incubation at 37°C oven until the next time point. The release samples (supernatant solutions) were read under plate reader (Biotek) and concentration of VP was calculated based on the VP standard curve.
Cell culture
Human triple-negative breast cancer cells MDA-MB231 and murine macrophages RAW 264.7 (ATCC) were grown in high-glucose Dulbecco’s modified Eagle medium (Invitrogen, Carlsbad, CA) with 10% FBS and 1% penicillin/streptomycin at 37°C and 5% CO2.
Human small cell lung cancer cells H446 (ATCC) were grown with ATCC-modified RPMI 1640 media (Life Technologies, Carlsbad, CA), supplemented with 10% FBS and 1% penicillin/streptomycin, at 37°C and 5% CO2.
Cellular uptake of VPM, BM, and free VP
MDA-MB 231, H446, and RAW 264.7 cells were seeded in 96 well plates at 15,000 cells per well in 100 μL of media, and incubated for 24 h at 37°C with 5% CO2. Cells were then treated with free VP in 0.4% DMSO solution, sVPM and sBM in 10% sucrose solution, and fVPM and fBM in 1X PBS at 2-fold increasing final concentrations from 1.25 μM - 20 μM for 1.5 h. VPM and BM were sonicated for 10 s at 20% amplitude just prior to being added to cells. MDA-MB 231 and H446 cells were then washed twice with heparin-containing PBS (50 μg/mL) to remove VP adhered to cells’ surface, trypsinized, resuspended with 170 μL of FACS buffer (PBS with 2% FBS), transferred to a round-bottom 96-well plate, centrifuged at 800 rpm for 5 min, resuspended in 30 μL FACS buffer, and analyzed by flow cytometry (BD Accuri C6 with HyperCyt adaptor). The same procedure was followed for RAW 264.7 except vigorous trituration was performed to detach cells from the plate rather than trypsinization. The results were analyzed by FlowJo 7.6.5 software using FSC-H vs. SSC-H gating for singlet cells and FL3 vs. FSC-H gating for VP-positive cells. Wells with more than 500 singlet events were counted in analysis. All conditions were tested in quadruplicates.
Cell killing with VPM
MDA-MB 231, H446, and RAW 264.7 cells were seeded in 96-well plates at 15,000 cells per well and incubated for 24 h. Free VP, VPM, and BM were added to each well at 2-fold increasing final VP concentrations from 2.5 μM – 20 μM. VPM and BM were sonicated for 10s at 20% amplitude just prior to being added to cells. Following 2 h incubation, cells were washed with 1X PBS and replenished with fresh media. Cells were observed under bright-field microscope for viability at 2, 6, 24, and 48 h post-treatment. At each time-point, cell killing was measured by CellTiter 96AqueousOne MTS assay. Cells were incubated with 100 μL of media and cell titer reagent solution (6:1 v/v) at 37°C for 2 h, and absorbance at 490 nm was measured using Synergy 2 plate reader. All conditions were tested in quadruplicates.
Statistics
GraphPad Prism 6 software package was used to perform statistical analysis. One-way ANOVA with Dunnett post-hoc test was performed to compare multiple conditions against the control group, Tukey post-hoc test to compare all pairs, or Student’s t-test to compare two conditions. (* = p < 0.05)
Results
Synthesis and characterization of PBAE and PEG-PBAE-PEG triblock copolymer
Both hydrophobic PBAE backbone polymers, B4S8m and B6S10m, were synthesized with a 1.15:1 ratio of diacrylate to alkylamine monomer. Resulting molecular weights of each PBAE polymer were 5100 and 4300 Da for B4S8m and B6S10m, respectively, as determined by 1H NMR (Figure S1A–C). The partition coefficients determined from the chemical structure of B4S8m and B6S10m matched the predicted hydrophobicity of both polymers (Figure S1C). B4S8m and B6S10m were then endcapped with 2000 Da and 800 Da mPEG-thiol to create PP1 and PP2 triblock copolymers, respectively (Figure 1C). PP2 exhibits greater hydrophobic proportion in the amphiphilic polymer chain in comparison to PP1. The thiol-ene Michael addition of PEG to both ends of PBAE was confirmed using 1H NMR by the disappearance of the peaks from the acrylates (Figure S1A/B).
Formulation and characterization of micelles
To measure the critical micelle concentration (CMC), pyrene was loaded into the micelles at increasing concentrations of PP1 polymer, while keeping the organic solvent to water ratio and the final volume constant. The log of the polymer concentration is plotted against the ratio of the intensity values at two wavelengths. In Figure 2A, the resulting sigmoidal plot of log[PP1] vs. I339/I336, has an inflection point at an exact concentration of 0.056 mg/mL. The increase in the intensity ratio as polymer concentration is increased corresponds to the shift of pyrene excitation peak. This demonstrates micelle formation and pyrene encapsulation. To further corroborate micelle formation and to confirm the morphology, resulting micelle structures pre- and post- lyophilization were visualized under TEM (Figure 2B). Micelles were spherical in shape, and there was no significant difference in shape or size between loaded micelles with VP (sVPM) and unloaded micelles (sBM), and between pre-lyophilized and post-lyophilized samples.
Figure 2. Spherical PP1 micelle characterization.

Stable, spherical micelles of approximately 50 nm in size were formulated using PP1 polymer. (A) Critical micelle concentration (CMC) measured by pyrene sensitivity assay, (B) TEM images of both sBM and sVPM pre- and post-lyophilization (scale bar = 100 nm), (C) DLS mean size and PDI of both (sBM) and (sVPM) (n=3, mean ± SD, Student’s t-test), (D) zeta potential of sBM and sVPM measured with Zetasizer, and (E) stability of sBM and sVPM in 1x PBS and human serum (45% serum, 55% 1X PBS) at room temperature for 31 hours (n=3, mean ± SD, One-way ANOVA with Dunnett post-hoc test comparing size measurement at different timepoints for each sample to its initial size at t = 0 h).
The size, polydispersity index (PDI), and zeta potential of micelles were measured with DLS. In order to be an effective vehicle for VP delivery via passive targeting, the size of our micelles should be below 200 nm to enter into the small pores on tumor tissue.24 Before lyophilization and after filtration and removal of unloaded VP, spherical micelles sVPM and sBM were both sized at 50 nm in water (Figure 2C), which is similar to the size visualized under TEM. sVPM showed a lower PDI and standard deviation between multiple batch measurements, which can possibly be explained by more stable micellar structure with the presence of VP which increases the hydrophobic force the micelle formation. The surface charge of sVPM and sBM in 10 mM NaCl were approximately neutral at −3 ± 4 mV and 3 ± 5 mV, respectively, demonstrating that the PEG is effective at shielding the particle (Figure 2D).
Particle stability was determined by time-course DLS size measurement of lyophilized micelles that are reconstituted in different medium (Figure 2E). sVPM showed excellent size stability in 55:45 v/v human serum:1x PBS over 31 hr, with no aggregation occurring over time. The control solution of human serum and PBS solution contained particulates with a size of approximately 50 nm, which likely represents the various serum protein aggregates and extracellular vesicles present in the serum. In 1x PBS, sVPM was approximately stable at a moderately larger size of 160 nm. Both blank sBM samples were initially smaller than their respective loaded samples, which is attributed to particle size needing to be larger to accommodate the VP cargo. While particle size is stable with time for sVPM samples in human serum and PBS, size increased over time for the empty sBPM particles, which signifies that the VP helped to improve the stability and minimize the aggregation of sVPM over time, likely due to the interactions between the hydrophobic VP in the hydrophobic core of the sVPMs. The amount of VP molecule encapsulated in VPM was measured by dissolving VPM with DMSO to release the VP and measuring the intrinsic fluorescence of VP molecule. Loading capacity is the amount of VP loaded per mass of particles, while loading efficiency is the amount of VP loaded per starting mass of VP. The loading capacity and efficiency of sVPM were 5.36% and 43.7%, respectively.
pH-sensitive release of VP from sVPM
PBAE-based polymers consist of tertiary amines along the backbone, enabling pH buffering at acidic conditions.25 The release kinetics of VP from sVPM at pH 7.4, 6.5, and 5.0 at 37 °C in citrate-phosphate buffer was evaluated to simulate the intravenous, tumoral, and lysosomal environments, respectively. The release plot in Figure 3 shows that pH 5.0 has the fastest release rate, 6.5 the slowest, and 7.4 has intermediate release. All three pH conditions followed similar kinetic trends, only varying in the absolute mass of VP released. Interestingly, the micelles showed the least total release of VP at pH 6.5 and an intermediate release at pH 7.4.
Figure 3. pH-sensitive VP release kinetics.

VP is released from sVPM in pH-sensitive manner, with the fastest release at pH 6.5 and slowest at pH 5.0. (A) Release of VP from sVPM at 37oC at 1, 3, 5, and 12 hour timepoints in buffers prepared to pH 7.4, 6.5, and 5.0 (n=3, mean ± SD), and (B) statistical comparisons between pH at each timepoint (Two-way ANOVA with Tukey post-hoc test).
Filamentous micelle characterization and shape dependence of macrophage uptake
We next investigated micelle shape as a parameter by tuning the hydrophobicity of the amphiphilic triblock copolymer to create high-aspect ratio anisotropic micelles. The morphology of VP-loaded micelles formulated with PP2 was determined with TEM (Figure 4A). Analysis using ImageJ confirmed that the filamentous VP-loaded micelles (fVPM) had an average length of 651 nm, width of 31 nm, and resulting average aspect ratio (AR) of 20±10 (Figure 4B). The size of lyophilized fVPM determined using DLS was 69 nm with greater PDI than sVPM (Figure 4C). This is expected because DLS estimates the hydrodynamic diameter assuming that the sample being measured is spherical so filamentous micelles with variation to aspect ratio would be instead detected as spheres of varying sizes. There was no difference in particle size following lyophilization and re-suspension (Figure 4C).
Figure 4. Filamentous PP2 micelle (fVPM) characterization.

Filamentous micelles with aspect ratio of 20 were formulated with PP2 polymer and shown to have lower cellular uptake by macrophages than spherical micelle. (A) TEM image of fVPM (inset: sVPM for comparison, scale bar = 100 nm), (B) aspect ratio (AR) distribution of fVPM post-lyophilization reconstituted in water, (C) DLS mean size and PDI of fVPM following reconstitution with 1x PBS (n=3, mean ± SD), and (D) sVPM and fVPM uptake efficiency in RAW 264.7 cells and geometric mean of uptake normalized to untreated cells measured by flow cytometry after treatment for 1 hr at equivalent VP concentrations (n=4, mean ± SD, One-way ANOVA with Tukey post-hoc test).
After this filamentous morphology was confirmed, the cellular uptake to RAW 264.7 macrophages was compared between sVPM and fVPM to evaluate the potential advantage of anisotropic PEG-PBAE-PEG micelles at reducing non-specific uptake, which could be useful to better evade the immune system. As shown in Figure 4D, fVPM treatment resulted in a significantly lower percentage of macrophages that internalized the fVPM compared with sVPM at equivalent VP concentrations evaluated. Similarly, the normalized geometric mean values for internalized VP using fVPM as the vehicle were 15 – 20% of that for sVPM at all concentrations evaluated, demonstrating that there is significantly less VP uptake per cell in fVPM-treated cells (Figure 4D). Interestingly, as the concentration of VP decreased from 0.08 to 0.04 μM, the uptake percentage for fVPM shows a sharp decrease from 55% to 6% in comparison to little decrease for sVPM. There was no cytotoxicity of macrophages resulting from delivered VP at the concentrations tested by either type of particle (Figure S2).
Cellular uptake by MDA-MB 231 and H446 cells
In order to investigate the level of internalized VP in MDA-MB 231 triple-negative human breast cancer cells and H446 human small cell lung cancer cells, the cellular uptake was measured by incubating the cells with equivalent concentrations of VP in the forms of free VP drug, sVPM, and fVPM for 1.5 hr. Figure 5A/C shows that there was near 100% cellular uptake for free VP, and sVPM in both cancer cells. At the concentrations tested, fVPM showed a slight decrease in the cellular uptake percentage. However, on a per cell basis, a much greater decrease in cellular uptake of fVPM occurred compared to free VP and sVPM in MDA-MB 231 cells as demonstrated by the normalized geometric mean fluorescence, which measures the level of VP uptake on a per cell basis in relative fluorescence units (RFU), (Figure 5B). This is in concert with the observation with macrophages. In the H446 cells, there is a similar significant decrease for fVPM as compared to sVPM and free VP in cellular uptake measured by geometric mean fluorescence, and also there is higher per cell uptake as compared to MDA-MB 231 for all VP formulations in general. The difference in cellular uptake is in part demonstrated by the difference in the level of total signal between the two cell types, as indicated by the scale on y-axes (Figure 5B/D).
Figure 5. Cancer cell uptake.

VP-loaded micelles are efficiently taken up by two cancer cell types in dose-dependent manner. (A/C) Percentage of cells that internalize VP in human triple-negative breast cancer (MDA-MB 231) and human small cell lung cancer (H446) cells incubated with free VP, sVPM, and fVPM at equivalent VP concentrations from 2.5–20 μM for 1.5 hr and (B/D) the corresponding geometric mean fluorescence of cellular uptake normalized to the untreated condition (RFU) (n=4, mean ± SD, One-way ANOVA with Tukey post-hoc test).
VP-induced death of MDA-MB 231 and H446 cells
To determine cancer cell death induced by VP-loaded micelles, both cancer cells were incubated with free VP, sVPM and fVPM for 2 hr, and cell viability was measured at 2, 6, 24, and 48 hr post treatment. For MDA-MB 231 cells, free VP and sVPM showed similar cell killing of approximately 85% and 40% at 20 μM and 10 μM VP concentrations at 48 hr, respectively, which corresponds to similar cellular uptake level (Figure 6A / S3A). This suggests that once endocytosed, free VP and sVPM have similar efficiencies of VP ultimately reaching the nucleus. For fVPM, as expected from the lowest cellular uptake, cell killing was either equivalent to or was the the lowest of the formulations at all VP concentrations and timepoints tested, with 20 μM VP concentration measured at 48 hr showing the maximum cell killing of fVPM at 44% of MDA-MB 231 cells. All negative controls, including 0.4% DMSO 99.6% PBS solution for free VP vehicle and blank micelles (sBM and fBM), did not show any toxicity, demonstrating the therapeutic effect is from the VP drug payload (Figure S3).
Figure 6. VP-induced cell death.

sVPM and fVPM are able to induce cancer cell killing without light in dose- and time-dependent manner. (A/C) VP delivery dose-response to cell viability in MDA-MB 231 and H446 cells incubated with free VP, sVPM, and fVPM at equivalent VP concentrations from 2.5– 20 μM for 2 hrs and measured over time, and (B/D) cell killing kinetics measured at 2, 6, 24, and 48 hr timepoints at stated VP concentrations (n=4, mean ± SD, One-way ANOVA with Tukey post-hoc test).
For H446 cells, there was an overall greater sensitivity towards VP as compared with MDA-MB 231 cells, which follows the cellular uptake findings (Figure 5D). All three VP treatments fully killed the human small cell lung cancer cells at the two highest concentrations of 10 μM and 20 μM at the 24 and 48 timepoints (Figure 6C / S3A). Cell killing by sVPM was very effective at even lower concentrations, showing 100% and 69% cell death at 5 and 2.5 μM, respectively. These efficiencies are significantly greater than both free VP and fVPM. sVPM may therefore improve the transport of VP intracellularly to the nucleus compared to free VP. For fVPM, its low cellular uptake can also lead to lower cell killing efficiency compared with sVPM. It is important to also note the difference in cell death kinetics between MDA-MB 231 and H446 cells (Figure 6B/D), which can be due to a combination of downstream steps following cellular uptake and perhaps greater drug resistance to VP by the human triple negative breast cancer cells compared to the human small cell lung cancer cells, or different level of YAP activity between the two cell types. This observation highlights that the same drug and delivery vehicle may require different optimization across different tumors to be most effective.
Discussion
While polymeric drug delivery systems can provide clear advantages and opportunities for cancer therapy, there is a need to safely optimize these systems with biodegradable materials to minimize delivery to off-target cells and tissues while maximizing delivery to cancer cells.26 We wished to i) evaluate a new putative anti-cancer drug, verteporfin, for efficacy to human triple negative breast cancer cells and human small cell lung cancer cells; ii) evaluate a new biodegradable micelle system for drug delivery based on poly(ethylene glycol)-poly(beta-amino ester)- poly(ethylene glycol) triblock copolymers; and iii) explore the role of shape of PEG-PBAE-PEG micelles in varying cellular uptake and drug delivery. Our proof-of-concept spherical drug delivery system was further optimized by inducing a high aspect ratio morphological shift through chemical modifications to the triblock copolymer backbone. Our group has shown that hard polymeric particles can be produced via a “top-down” stretching platform after synthesis, however chemical tuning of soft nanoparticles via a “bottom-up” approach does not require post-synthesis steps and can also confer greater scalability and manufacturability for this nanomedicine system.27
Two new PEG-PBAE-PEG triblock copolymers were synthesized, each with a varying number of carbons in the diacrylate monomer and the primary amine monomer, and a different PEG molecular weight. This chemical tuning shifts the packing parameter value, which can be used to predict the shape of the resulting self-assembled micelles.11 This parameter value is defined by the effective hydrocarbon volume of the polymer chain V, divided by the product of the area of the hydrophilic headgroup α0 and the fully extended chain length lc.10 PP2 consists of longer hydrocarbon chain monomers for the PBAE backbone as well as smaller molecular weight PEG at both ends, leading to increased packing parameter and more anisotropic structures. This prediction corroborates the observed morphological shift with the PEG-PBAE-PEG micelles.
The observed VP encapsulation efficiency into the micelles is commonly observed in many nanoparticles fabricated using nanoprecipitation. The loading capacity can be increased with a higher mass of VP initially added to the synthesis reaction, however it has been demonstrated that increasing the drug concentration lowers the encapsulation efficiency due to the specific properties of polymers that limit the amount of cargo that can be encapsulated when being used as molecule-carrying vehicles.28 Nevertheless, other techniques to increase the loading capacity of these micelles can be of interest to enhance the therapeutic effect with their treatment. First, a greater loading capacity of VP will permit a lower concentration of micelles to be used in treatments. In our in vitro studies, the killing of MDA-MB 231 cells was lower than that of H446 cells. Since cancer cells differ in their responses to certain types and doses of treatment as mentioned previously, it is likely IC50 of MDA-MB 231 cells is higher than that of H446 cells. Using micelles with a greater VP loading capacity, we can maintain the same nanoparticle concentration in the treatment, and continue to ensure that the polymer itself is not having a toxic effect on the cells. Additionally, it is important for us to consider increasing the loading capacity of the micelles as an option to increase the amount of VP that reaches the tumor upon systemic circulation in vivo, given the limitations such as the low tumor accumulation rate of nanoparticles and the solubility of nanoparticles.29
Interestingly, the novel PEG-PBAE-PEG micelles show biphasic release trends with varying pH. This is consistent with a previous finding from Zhang et al. that a VP-analog molecule has shown to dimerize at pH 6.5, which could affect the drug release from the micelles.30 Also, it has been shown previously that PBAE nanoparticles can release more slowly at weakly acidic conditions.31 Non-protonated amines in the backbone of PBAEs at pH 7.4 can act as weak bases, sequestering protons from water molecules, leaving free hydroxyl groups to act as nucleophiles and degrade ester bonds, and releasing the cargo faster than at weakly acidic pH. However, tertiary amines along the backbone of PBAE become protonated at pH 5 to allow disintegration of micelles and release of VP. This balance between VP dimerization, base-catalyzed hydrolysis, and micelle disassembly cause the pH-sensitive release kinetics. The slower release of VP at pH 6.5 compared to 7.4 and the fastest release at pH 5.0 have an advantage with intracellular VP delivery to cancer cells. The tumor microenvironment can have a pH range from 6.5–7.2, therefore it can be favorable for an intracellular drug, such as VP, to be released less in the extracellular space. Intracellularly following endocytosis as the pH is reduced below 6.5 in the endosomes/lysosomes, VP can be released following demicellization to target the nucleus.
The cellular uptake of spherical and filamentous micelles by macrophages shows that at moderate doses, fVPM cellular uptake by macrophages is a bottleneck that is sensitive to fVPM concentration whereas for sVPM it is not a bottleneck and cellular uptake to macrophages is high even at reduced dosages. This difference is likely due to the high aspect ratio filamentous structures limiting the number of nanoparticles entering macrophages, via physically resisting macrophage engulfment.32
For cellular uptake by cancer cells, there is a significantly higher amount of sVPM entering cancer cells than fVPM as expected from the morphology-dependent cellular uptake effect, according to the normalized geometric mean fluorescence of cellular uptake. This observation translates into greater cell killing efficacy by sVPM compared with fVPM at several concentrations. Therefore, at the cellular level, spherical shape of VP-loaded micelles is shown to be more advantageous than filamentous shape. However, the capability of fVPM to avoid macrophage uptake is an advantageous feature at the systemic level. Prolonged circulation may lead to greater accumulation of the drug at the tumor site following systemic circulation through the enhanced permeability and retention effect, improving anti-cancer efficacy as well as reducing off-target effects in macrophages and other phagocytic cells.
The difference in dose response and kinetics of cell death between MDA-MB 231 and H446 cells can be due to a combination of factors. First, the level of total cellular uptake of sVPM, fVPM and free VP per cell, as indicated by the geometric mean fluorescence in cellular uptake, was different between two cancer cell types used in this study. This may potentially stem from a number of factors, including varying total endocytosis and exocytosis rates as well as differential cellular uptake pathways between different cells.33 Elucidation of the cellular uptake could allow more optimized and specific design of micelles tailored to a target cell type. Second, there could be greater drug resistance to VP by the human triple negative breast cancer cells compared to the human small cell lung cancer cells. Given the inherent molecular complexities and heterogeneity of these cell types, the expression pattern and levels of proto-oncogenes in cancer cells are rather diverse. Previous studies have indicated that cancer cells from different tissues of origins and disease grade/stage exhibit varying sensitivity to verteporfin-mediated cell death. In particular, recent studies have linked verteporfin’s cell killing potency to expression of wild-type p53.34 Thus, the stark different in their responses to the drug may stem from the presence of mutated vs. wild type p53 in MDA-MB 231 and H446 cells, respectively. Lastly, given that verteporfin disrupts the interaction between YAP and its cognate transcription factor, TEAD4, the levels and activity of YAP and its cognate transcription factor, TEAD4, may be different in the two cells types, which could affect verteporfin’s mechanism of action to disrupt the interaction between these two proteins. Thus, it is very likely that the unique molecular profiles of these cells may dictate their sensitivity to this drug, whereby the effective dosage may reflect the expression levels and activity of these oncogenic networks. This observation highlights that the same drug and delivery vehicle may require different optimization across different tumors to be most effective.
Since we were able to tune the triblock copolymer chemistry to observe a morphological shift from spherical micelles to filamentous micelles, a similar approach could be used to fabricate PEG-PBAE-PEG micelles with alternative aspect ratios and cargos depending on the application. We found dramatic efficacy of VP in killing human triple negative breast cancer cells and human small cell lung cancer cells without the need of a photodynamic trigger. This non-photodynamic approach to using VP therapeutically could obviate the many of the detrimental side effects of using a photodynamic therapy, such as skin photosensitivity or systemic toxicity.35 Moreover, VP acts on the Hippo pathway of tumor cells, hence would have minimal toxicity against normal cells. Alternatively, PEG-PBAE-PEG micelles for the delivery of VP could open up the possibility of combination therapies where the micelles could preferentially accumulate in the tumor compared to standard VP due to their shape properties and then a photodynamic trigger could further potentiate their efficacy. This work represents an important advancement in the design of anisotropic pH-sensitive PBAE delivery systems and the utility of VP as a chemotherapeutic agent.
Conclusion
PBAE polymers are excellent candidates for polymeric drug delivery systems due to their intrinsic biodegradability and pH-sensitivity, but their use for non-nucleic acid delivery has been limited. Through novel PBAE polymer design and modification, we synthesized a PEG-PBAE-PEG micellar system of two different morphologies capable of encapsulating and delivering a water-insoluble, newly emerging anti-cancer therapeutic drug, verteporfin. The spherical micelles displayed excellent stability in both human serum and PBS buffers, with size measuring sub-150 nm. The creation of PBAE-based filamentous micelles with an average aspect ratio of 20 enabled avoidance of off-target macrophage uptake. When treated to both human triple negative breast cancer cells and human small cell lung cancer cells, the micelles showed a larger therapeutic range when compared with free VP, being more effective at lower concentrations. This work demonstrates the first anisotropic PBAE-based self-assembled drug delivery system, and in addition, one of the first validations of VP nanomedicine as a direct anti-cancer approach without the need of external photodynamic therapy.
Supplementary Material
Figure S1 shows 1H NMR spectrum of B4S8m, PP1, B6S10m, and PP2 polymers as a confirmation of their structures and molecular weight following the described reactions. The figure also includes the theoretical partition coefficient of the two base polymers calculated by ChemBioDraw software.
Figure S2 shows the cell viability of murine macrophages that were treated with sVPM, fVPM, and free VP at concentrations tested for cellular uptake.
Figure S3 includes VP-induced cell death of all concentrations and all timepoints for both cancer cells, from which selective concentrations and timepoints were chosen as a representative data in Figure 6.
Acknowledgements
The authors acknowledge the NIH for support (CA195503, CA200399, CA183827, CA216855, NS070024, EB016721). AQH is also supported by the Mayo Clinician Investigator Award, Mayo Professorship, and the State of Florida. JJG thanks the Bloomberg~Kimmel Institute for Cancer Immunotherapy for support. JK thanks Samsung for scholarship support.
Abbreviations
- PBAE
poly(beta-amino ester)
- PEG
poly(ethylene glycol)
- VP
verteporfin
- sBM
spherical blank micelle
- sVPM
spherical VP-loaded micelle
- fBM
filamentous blank micelle
- fVPM
filamentous VP-loaded micelle
- CMC
critical micelle concentration
- DLS
dynamic light scattering
- PDI
polydispersity index
Footnotes
Associated content
Supporting information.
References
- (1).Kataoka K; Harada A; Nagasaki Y, Block copolymer micelles for drug delivery: Design, characterization and biological significance. Adv. Drug Deliv. Rev 2001, 47, (1), 113–31. [DOI] [PubMed] [Google Scholar]
- (2).Xu W; Ling P; Zhang T, Polymeric micelles, a promising drug delivery system to enhance bioavailability of poorly water-soluble drugs. J. Drug Deliv 2013, 2013, 340315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Williams HD; Trevaskis NL; Charman SA; Shanker RM; Charman WN; Pouton CW; Porter CJ, Strategies to address low drug solubility in discovery and development. Pharmacol. Rev 2013, 65, (1), 315–499. [DOI] [PubMed] [Google Scholar]
- (4).Kim J; Wilson DR; Zamboni CG; Green JJ, Targeted polymeric nanoparticles for cancer gene therapy. J. Drug. Target 2015, 23, (7–8), 627–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Ko J; Park K; Kim YS; Kim MS; Han JK; Kim K; Park RW; Kim IS; Song HK; Lee DS; Kwon IC, Tumoral acidic extracellular ph targeting of ph-responsive mpeg-poly(beta-amino ester) block copolymer micelles for cancer therapy. J. Control. Release 2007, 123, (2), 109–15. [DOI] [PubMed] [Google Scholar]
- (6).Green JJ; Langer R; Anderson DG, A combinatorial polymer library approach yields insight into nonviral gene delivery. Acc. Chem. Res 2008, 41, (6), 749–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Nguyen DN; Green JJ; Chan JM; Longer R; Anderson DG, Polymeric materials for gene delivery and DNA vaccination. Adv. Mater 2009, 21, (8), 847–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Kim J; Sunshine JC; Green JJ , Differential polymer structure tunes mechanism of cellular uptake and transfection routes of poly(beta-amino ester) polyplexes in human breast cancer cells. Bioconjug. Chem 2014, 25, (1), 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Eltoukhy AA; Chen D; Alabi CA; Langer R; Anderson DG, Degradable terpolymers with alkyl side chains demonstrate enhanced gene delivery potency and nanoparticle stability. Adv. Mater 2013, 25, (10), 1487–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Lombardo D; Kiselev MA; Magazu S; Calandra P, Amphiphiles self-assembly: Basic concepts and future perspectives of supramolecular approaches. Adv. Cond. Matter Phys 2015. [Google Scholar]
- (11).Geng Y; Dalhaimer P; Cai S; Tsai R; Tewari M; Minko T; Discher DE, Shape effects of filaments versus spherical particles in flow and drug delivery. Nat. Nanotechnol 2007, 2, (4), 249–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Lachelt U; Kos P; Mickler FM; Herrmann A; Salcher EE; Rodl W; Badgujar N; Brauchle C; Wagner E, Fine-tuning of proton sponges by precise diaminoethanes and histidines in pdna polyplexes. Nanomedicine 2014, 10, (1), 35–44. [DOI] [PubMed] [Google Scholar]
- (13).Holzer MP; Solomon KD; Vroman DT; Sandoval HP; Margaron P; Kasper TJ; Crosson CE, Photodynamic therapy with verteporfin in a rabbit model of corneal neovascularization. Invest. Ophthalmol. Vis. Sci 2003, 44, (7), 2954–8. [DOI] [PubMed] [Google Scholar]
- (14).Wilson BC; Patterson MS, The physics, biophysics and technology of photodynamic therapy. Phys. Med. Biol 2008, 53, (9), R61–109. [DOI] [PubMed] [Google Scholar]
- (15).Ichikawa K; Takeuchi Y; Yonezawa S; Hikita T; Kurohane K; Namba Y; Oku N, Antiangiogenic photodynamic therapy (pdt) using visudyne causes effective suppression of tumor growth. Cancer Lett. 2004, 205, (1), 39–48. [DOI] [PubMed] [Google Scholar]
- (16).Konan-Kouakou YN; Boch R; Gurny R; Allemann E, In vitro and in vivo activities of verteporfin-loaded nanoparticles. J. Control. Release 2005, 103, (1), 83–91. [DOI] [PubMed] [Google Scholar]
- (17).Brodowska K; Al-Moujahed A; Marmalidou A; Horste MMZ; Cichy J; Miller JW; Gragoudas E; Vavvas DG, The clinically used photosensitizer verteporfin (vp) inhibits yap-tead and human retinoblastoma cell growth in vitro without light activation. Exp. Eye Res 2014, 124, 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Martinez-Gutierrez JC R.-V. A; Shah SR; Riggins GJ; Quinones-Hinojosa A, Meningioma growth inhibition and radiosensitization by the small molecule yap inhibitor verteporfin. Neuro-Oncology 2015, 17, (suppl_5), v131. [Google Scholar]
- (19).Shah SR; David JM; Tippens ND; Mohyeldin A; Martinez-Gutierrez JC; Ganaha S; Schiapparelli P; Hamilton DH; Palena C; Levchenko A; Quinones-Hinojosa A, Brachyury-yap regulatory axis drives stemness and growth in cancer. Cell Rep 2017, 21, (2), 495–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Wei S; Wang J; Oyinlade O; Ma D; Wang S; Kratz L; Lal B; Xu Q; Liu S; Shah SR; Zhang H; Li Y; Quinones-Hinojosa A; Zhu H; Huang ZY; Cheng L; Qian J; Xia S, Heterozygous idh1 (r132h/wt) created by “single base editing” inhibits human astroglial cell growth by downregulating yap. Oncogene 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Liu-Chittenden Y; Huang B; Shim JS; Chen Q; Lee SJ; Anders RA; Liu JO; Pan D, Genetic and pharmacological disruption of the tead-yap complex suppresses the oncogenic activity of yap. Genes Dev 2012, 26, (12), 1300–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Chang HI; Yeh MK, Clinical development of liposome-based drugs: Formulation, characterization, and therapeutic efficacy. Int. J. Nanomedicine 2012, 7, 49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Kim J; Kang Y; Tzeng SY; Green JJ, Synthesis and application of poly(ethylene glycol)-co-poly(beta-amino ester) copolymers for small cell lung cancer gene therapy. Acta. Biomater 2016, 41, 293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Kobayashi H; Watanabe R; Choyke PL, Improving conventional enhanced permeability and retention (epr) effects; what is the appropriate target? Theranostics 2013, 4, (1), 81–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Sunshine JC; Peng DY; Green JJ, Uptake and transfection with polymeric nanoparticles are dependent on polymer end-group structure, but largely independent of nanoparticle physical and chemical properties. Mol. Pharm 2012, 9, (11), 3375–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Hassan S; Prakash G; Ozturk A; Saghazadeh S; Sohail MF; Seo J; Dockmeci M; Zhang YS; Khademhosseini A, Evolution and clinical translation of drug delivery nanomaterials. Nano Today 2017, 15, 91–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Meyer RA; Sunshine JC; Perica K; Kosmides AK; Aje K; Schneck JP; Green JJ, Biodegradable nanoellipsoidal artificial antigen presenting cells for antigen specific t-cell activation. Small 2015, 11, (13), 1519–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Martinez Rivas CJ; Tarhini M; Badri W; Miladi K; Greige-Gerges H; Nazari QA; Galindo Rodriguez SA; Roman RA; Fessi H; Elaissari A, Nanoprecipitation process: From encapsulation to drug delivery. Int J Pharm 2017, 532, (1), 66–81. [DOI] [PubMed] [Google Scholar]
- (29).Bae YH; Park K, Targeted drug delivery to tumors: Myths, reality and possibility. J. Control. Release 2011, 153, (3), 198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Zhang JX; Hansen CB; Allen TM; Boey A; Boch R, Lipid-derivatized poly(ethylene glycol) micellar formulations of benzoporphyrin derivatives. J. Control. Release 2003, 86, (2–3), 323–38. [DOI] [PubMed] [Google Scholar]
- (31).Tzeng SY; Green JJ, Subtle changes to polymer structure and degradation mechanism enable highly effective nanoparticles for sirna and DNA delivery to human brain cancer. Adv. Healthc. Mater 2013, 2, (3), 468–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Champion JA; Mitragotri S, Role of target geometry in phagocytosis. Proc. Natl. Acad. Sci. USA 2006, 103, (13), 4930–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Oh N; Park JH, Endocytosis and exocytosis of nanoparticles in mammalian cells. Int. J. Nanomedicine 2014, 9 Suppl 1, 51–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Wang C; Zhu X; Feng W; Yu Y; Jeong K; Guo W; Lu Y; Mills GB, Verteporfin inhibits yap function through up-regulating 14–3-3sigma sequestering yap in the cytoplasm. Am J Cancer Res 2016, 6, (1), 27–37. [PMC free article] [PubMed] [Google Scholar]
- (35).Lucky SS; Soo KC; Zhang Y, Nanoparticles in photodynamic therapy. Chem. Rev 2015, 115, (4), 1990–2042. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 shows 1H NMR spectrum of B4S8m, PP1, B6S10m, and PP2 polymers as a confirmation of their structures and molecular weight following the described reactions. The figure also includes the theoretical partition coefficient of the two base polymers calculated by ChemBioDraw software.
Figure S2 shows the cell viability of murine macrophages that were treated with sVPM, fVPM, and free VP at concentrations tested for cellular uptake.
Figure S3 includes VP-induced cell death of all concentrations and all timepoints for both cancer cells, from which selective concentrations and timepoints were chosen as a representative data in Figure 6.