

Summary
Initial molecular details of cellular activation following αβT cell antigen receptor (TCR) ligation by peptide-major histocompatibility complexes (pMHC) remain unexplored. We determined the nuclear magnetic resonance (NMR) structure of the TCRα subunit transmembrane (TM) segment revealing a bipartite helix whose segmentation fosters dynamic movement. Positively charged TM residues Arg251 and Lys256 project from opposite faces of the helix, with Lys256 controlling immersion depth. Their modification caused step-wise reduction in TCR associations with CD3ζζ homodimers and CD3εγ plus CD3εδ heterodimers, respectively, leading to an activated transcriptome. Optical tweezers revealed that Arg251 and Lys256 mutations altered αβTCR-pMHC bond lifetimes, while mutations within interacting TCRα connecting peptide and CD3δ CxxC motif juxtamembrane elements selectively attenuated signal transduction. Our findings suggest that mechanical forces applied during pMHC ligation initiate T cell activation via a dissociative mechanism, shifting disposition of those basic sidechains to rearrange TCR complex membrane topology and weaken TCRαβ and CD3 associations.
Keywords: TCR, T cell activation, mechanoreceptor, NMR, EPR, optical tweezers, transmembrane domain, immunotherapy, CAR-T, transcriptome
Graphical abstract
In Brief
While TCR triggering is critical for adaptive immunity, how bioforces accompanying ligand binding transduce signals through the cell membrane is unknown. Brazin et al. define dynamic structural movements within the TCRα transmembrane domain linked to fundamental TCR complex mechanobiology, including subunit topological rearrangements that foster dissociation of CD3 dimers and cell activation.
Introduction
T lymphocytes are critical to the vertebrate adaptive immune system, performing wide-ranging immune surveillance to prevent or combat infections and cancerous transformations. The αβ T cell receptor complex (αβTCR), a mechanosensor displayed on the T cell surface, mediates recognition of cellular aberrations (Das et al., 2015; Das et al., 2016; Feng et al., 2017; Kim et al., 2009; Liu et al., 2014; Mallis et al., 2015). The αβTCR is a multi-subunit protein complex composed of a disulfide-linked TCRαβ heterodimer flanked by non-covalently associated dimeric CD3 subunits: CD3εγ, CD3εδ, and CD3ζζ [reviewed in (Rudolph et al., 2006; Wang and Reinherz, 2012)]. TCRα and β each contain an extracellular variable (V) and constant (C) domain, a membrane proximal connecting peptide (CP), a single transmembrane (TM) domain, and a short cytoplasmic tail. The TCR VαVβ extracellular domain module is most distal to the membrane, interacting directly with a peptide bound to a major histocompatibility complex molecule (pMHC) displayed on the surface of an antigen presenting cell (APC).
Following TCR-pMHC engagement and then force-driven conformational change and bond lifetime extension that tune antigen recognition (Das et al., 2015), there are orchestrated intracellular T cell signaling responses (Chakraborty and Weiss, 2014). The myriad of unique V modules form a repertoire of diverse T cells to interrogate the enormous immune peptidome of pathogen- and tumor-derived sequences displayed on the surface of altered cells. Each TCR specifically recognizes only one or a small number of pMHC ligands that it encounters on an APC. Since TCRα and β subunits have extremely short cytoplasmic tails lacking signaling sequences, CD3 molecules transfer cognate recognition event information into the cell (Sun et al., 2001; Sun et al., 2004). The CD3 molecules are invariant and thought to interact with the TCRα and β subunits through their extracellular and/or the TM domains to relay TCR-pMHC binding information to CD3, utilizing Immune Tyrosine Activation Motif (ITAM) regions within their cytoplasmic tails.
Formation of TCRαβ, CD3εγ and CD3εδ heterodimers is dependent on extracellular domain interactions, while for CD3ζζ, an interchain disulfide bond connects the two chains (Sun et al., 2001; Sun et al., 2004; Weissman et al., 1986). In contrast, TCRαβ association with CD3 subunits requires interactions in the membrane proximal and TM domains since only weak ectodomain-mediated interactions have been detected (Birnbaum et al., 2014; He et al., 2015; Natarajan et al., 2016). Each of the eight subunits comprising the αβTCR complex contains a single TM domain marked by the presence of highly conserved charged amino acids. A current hypothesis posits that the positions of these charged residues dictate charge-paired interactions between TCR α and β and CD3 subunits important for surface assembly, expression and signaling (Alcover et al., 1990; Blumberg et al., 1990; Call et al., 2002; Cosson et al., 1991; Manolios et al., 1990). The model suggests that basic amino acids within the TCRα and β subunits associate with acidic residues in each of the CD3ε, γ, and δ TM domains to generate trimeric TCRα-CD3εδ and TCRβ-CD3εγ sub-complexes. A similar electrostatic charged-based coupling role is proposed for TM residues in CD3ζζ and TCRα (Call et al., 2002; Call et al., 2006). There is no direct molecular evidence for this assembly hypothesis.
To elucidate the mechanistic underpinning of these TCR TM-based associations, we focused on the two basic amino acids in the TCRα TM. We determined the structure of its TM and cytoplasmic tail (TMC) in phospholipid micelles using nuclear magnetic resonance spectroscopy (NMR). In addition, we studied the structural features in liposomes using electron paramagnetic resonance spectroscopy (EPR), permitting membrane immersion depth and intra-helical distance measurements. Lastly, we assessed the biological importance of TCRαTMC features by engineering structurally-guided mutations with each variant assembled as a component of the TCR complex on the surface of T cells. We observed that the TCRαTMC consists of a conserved sequence in vertebrates forming a bipartite helical structure whose segmentation fosters dynamic movement in the lipid environment. Mechanical force operative during pMHC ligation of the TCR is capable of toggling this conformational switch, altering TM immersion topology to impact overall αβTCR subunit associations and T cell activation.
Results
The TCRαTMC segment adopts a bipartite L-shaped helix with a flexible C-terminal region.
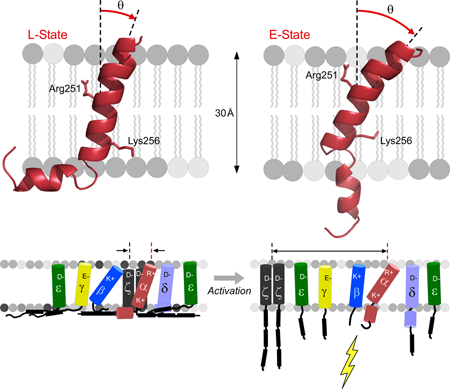
The 3D-structure of the TCRαTMC (residues 226–271) composed of the extracellular CP region, TM domain, and cytoplasmic tail was determined by NMR in phospholipid micelles (Figure 1A-1C) with structural restraints given in Figure S1A. The backbone RMSD was determined to be 0.38 Å for Helix 1, 0.46 Å for Helix 2, and 1.58 Å for both helices combined (Figure S1B). The N-terminal CP region (Asp226-Leu240) displays considerable flexibility and is disordered based on backbone chemical shifts. This agrees with PSIPRED prediction, and absence of the disulfide-linked connection to the TCRβ subunit. In contrast to the CP region, the TM domain (Asn241-Leu268) adopts an α-helical, L-shaped structure composed of two helices interrupted by a hinge (Figure 1B, bottom). Helix 1 (Asn241-Ala258) is of sufficient length, ~ 26–30 Å, to transverse a cellular membrane; however, the bend (Gly259-Asn261) and subsequent reorientation of Helix 2 (Leu262-Leu268) prevents the C-terminal residues from being solvent exposed outside the lipid environment (Figure 2A and2B). Two lines of evidence suggest the L-shaped helix is in conformational exchange with an extended helix. First, the doubling of spectral resonances observed in the 1H-15N HSQC spectrum indicates conformational exchange between a favored bent state and an extended state (Figure 1B and1C) at a rate slower than 5 s−1, an upper bound determined by the experimental time of the 1H-15N HSQC experiment. Second, alternate conformations of Helix 2 are observed in a subset of molecular models due to hinge flexibility. We postulate that the C-terminus becomes more solvent accessible when extended.
Figure 1. TCRα structure determination and sequence analysis.
A) Mouse TCRαTMC protein sequence studied by NMR. Numbering corresponds to the full-length sequence. The structural features displayed beneath the sequence were determined experimentally: CP (226–240), Helix 1 (241–258), Hinge (259–261), and Helix 2 (262–268). The TM domain residues (241–268) are highlighted in red. Met 264 is denoted with an asterisk, and starts the sequence defined by the blue bracket to mark those residues that gave rise to peak doublings in the NMR data in (C).
B)Ribbon representation of an average conformer of the TCRαTMC with side chains of key residues displayed. An ensemble of the 10 lowest energy conformers is shown below. The dynamic region undergoing conformational exchange is labeled and shaded to include both the bent (L)-state and extended (E)- state conformers. The flexible CP region has been removed for clarity, and every second residue is labeled. Residues undergoing conformational exchange are labeled in each main set of conformers.
C) 1H-15N HSQC spectrum of the TCRαTMC with complete backbone assignments. Residues exhibiting conformational exchange are labeled in two shades of red, the lighter shade designated with an asterisk represents the minor conformer.
D) TCRαTMC multiple sequence alignment from selected vertebrate species. Sequence numbering and secondary structure features depicted in the alignment correspond to the mouse TCRαTMC (residues 226–271). Conserved residues throughout all species are shown by grey shaded columns with the basic, acidic, and polar residues shown in cyan, red, and yellow font, respectively, and Trp is shown in white. A consensus secondary structure prediction (ssp α1) is shown at the bottom of the alignment while the average secondary structure calculated from the PDB conformers (PDB-SS) is shown at the top.
E) Cytoplasmic length analysis of TM-containing T cell proteins from the UNIPROT database plotted as the frequency of occurrence versus the cytoplasmic tail length in the histogram. The black bracketed region is expanded to show those proteins with cytoplasmic tails of 15 residues or less. The log-normal distribution of the cytoplasmic tail length is plotted in red over the frequency histogram ( also see Figure S1).
Figure 2. TCRα membrane depth analysis and generation of a straightened helix.
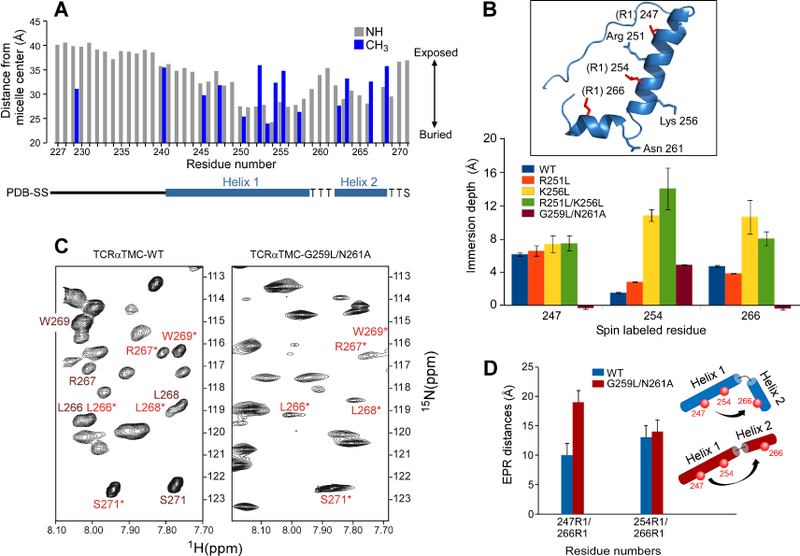
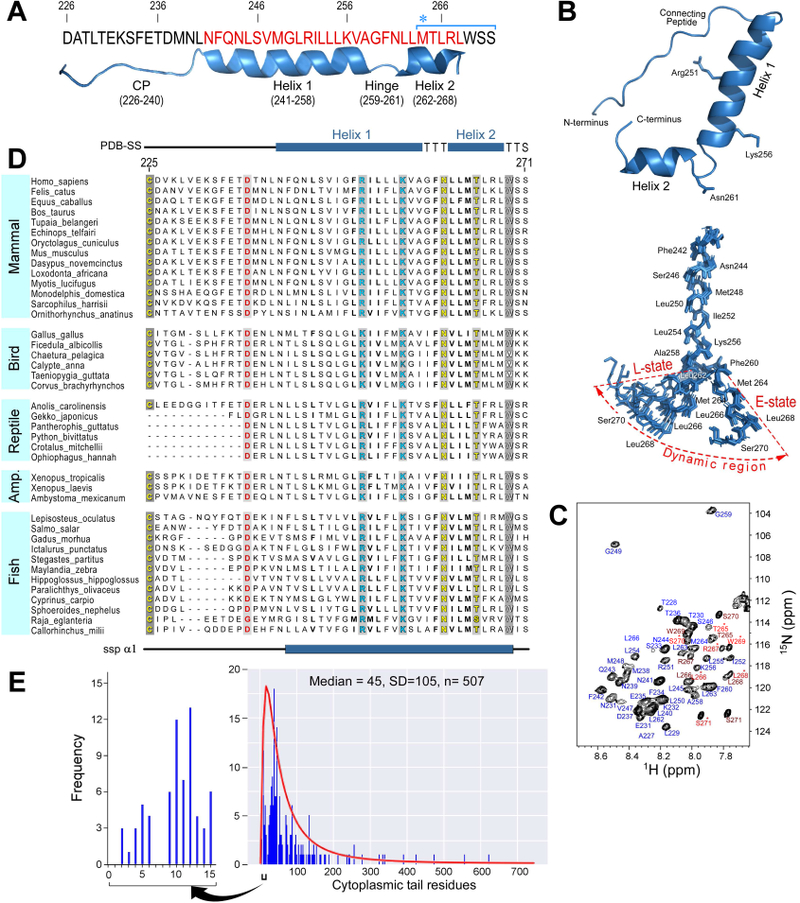
A) The 15N backbone resonances (grey) and 13C-methyl ILV resonances (blue) NMR determined membrane distance information of the TCRαTMC in LPPG micelles measured by a PRE-based analysis. A secondary structure diagram is shown below the x-axis. Residue 267 has been omitted due to spectral overlap.
B) EPR spin-labeled residues (R1) 247, 254, and 266 were used to determine the liposome immersion depths for singly labeled WT and mutant TCRα segments. Inset: Ribbon representation of the structurally determined TCRαTMC with spin-labeled residues (R1) in red and residues mutated for depth analysis highlighted in blue. Gly259 is not shown.
C) An expanded region of the 1H-15N HSQC spectra of the TCRαTMC WT (left) and the TCRαTMC G256L/N261A mutant (right). Resonances undergoing exchange in the WT segment are labeled in dark red and in light red with an asterisk. In the G256L/N261A mutant, those remaining peaks are labeled in light red with an asterisk.
D) The EPR measured distances calculated for the TCRαTMC WT (blue) and G259L/N261A mutant (red) segments in liposomes plotted as distance versus residue numbers of the spin labeled pairs. A cartoon illustrating the position of the spin labels (red spheres) and relative orientation of Helix 1 to Helix 2 is shown for the WT segment (blue) and G259L/N261L segment (red).
Sequence analysis of the TCRα segment shows that the hinge is highly conserved in mammals (Figure 1D). The CP region in mammals evolved to be ~14 amino acids long, containing a nearly 100% conserved FETDxxLN sequence, while both length and sequence conservation vary in non-mammalian vertebrates (Figure 1D and S2A). A pair of basic residues in the TCRαTMC segment, Arg251 and Lys256, are conserved in virtually all vertebrates. These same residues are conserved in an identical position in pTα, which pairs with TCRβ during thymic development to form the ligand binding heterodimer of the pre-TCR (von Boehmer et al., 2003) (Figure S2B). Arg251 and Lys256 in Helix 1 are positioned on opposite faces of the helix (Figure 1B). Helix 2 also displays high sequence conservation (Figure S2A) that continues through to the tail. The TCRα cytoplasmic tail is unusually short, predicted to comprise of only five residues. As noted below from the averaged paramagnetic relaxation enhancement (PRE) data, only two of the five residues are not membrane embedded. A survey of 507 type I integral membrane proteins (Figure 1E) on the surface of T cells in the UniProt database found that fewer than 5% have cytoplasmic tails of 10 residues or less, while fewer than 1% of 262 type I and type II cluster of differentiation (CD) proteins have cytoplasmic tails of 10 residues or less (Figure S2C and D). This short tail may facilitate a force-driven TCRα TM depth change to promote conformational change, dissociating the αβTCR complex subunits as described below.
Lys256 regulates the membrane depth of the TCRαTMC segment.
The immersion depth of TCRαTMC was investigated by NMR and EPR PRE-analyses to map the TM boundaries and geometry of the TMC segment relative to the lipid environment. Relaxation properties of solvent-exposed residues will be affected by addition of water-soluble paramagnetic ions, whereas membrane embedded residues will be largely unaffected (Figure 2A) (Respondek et al., 2007). As expected, the highly flexible extracellular CP region is exposed to solvent. Helix 1 in the TM domain and a portion of the Helix 2 appear to be micelle-embedded, but neither is deeply buried. The TCRαTMC becomes more solvent accessible at the hinge (residues 259–261) and more embedded in Helix 2, thereby reducing the solvent accessibility of the cytoplasmic tail (Figure 2A). The PRE data from the Ile, Leu and Val (ILV) methyl resonances also reveal that residue side chains in proximity to Arg251 and Lys256 are more exposed, perhaps due to the hydrophilic side chains pulling the helical TM domain towards solvent.
To corroborate observations on membrane insertion of TCRαTMC, we used EPR (Figure 2B) to measure immersion depth in DOPC:DOPG liposomes, a lipid bilayer environment distinct from detergent micelles utilized in our NMR studies. Single spin labels (R1; nitroxide spin label MTSL) were used at Val247 or Leu254 in Helix 1 or Leu266 in Helix2, within the liposome-embedded TCRαTMC to monitor responses to polar and nonpolar paramagnetic compounds thereby gauging the segment depth (Song et al., 2009). The targeted residues were immersed at relatively shallow depths in the acyl chain region (with head group defined as −5 Å to 0 Å and acyl chain region as >0 Å for the liposome) (Figure 2B). The WT TCRαTMC depth is shallow in both micelle and liposome membrane systems; insertion is not a product of the experimental system but a consequence of the TM amino acid composition.
We next examined the effect of the two basic amino acids (Arg251 and Lys256) on the positioning of the TCRα TM domain in the liposome via mutation to non-polar Leu residues, generating protein segments R251L, K256L, and R251L/K256L. Mutation of Arg251 had negligible effects on liposome immersion depth as assessed by EPR (Figure 2B). In contrast, mutation of Lys256 resulted in substantial depth change for residues within the TM domain compared to the WT or R251L segments (Figure 2B). For both the K256L and R251L/K256L segments, the spin label at position 247 remained relatively shallow. However, positions 254 and 266 were considerably more immersed in the liposome. The depth at position 254 increased substantially relative to WT for K256L and R251L/K256L. An increase in depth was also observed at position 266 relative to WT for K256L and R251L/K256L. Thus, Lys256 is essential for maintaining the shallow positioning of both helices in a membrane environment.
TCRαTM hinge mutations generate a straightened helix.
To investigate whether the TCRαTMC L-shaped configuration also contributes to the shallow depth, we engineered a straightened helix based on the NMR structure and mammalian sequence conservation in this region (Figure 1). The HSQC spectrum of the G259L/N261A mutant (Figure 2C) revealed elimination of resonance doubling; remaining chemical shifts were consistent with the minor population observed in Figure 1C. This result suggested that the minor population represented the extended conformer identified in our structural calculations. To confirm this observation, we used continuous wave (CW) EPR to measure whether the G259L/N261A mutant increased in helical length relative to the WT TCRαTMC (Figure 2D). Pairs of residues Val247 and Leu266 or Leu254 and Leu266 were R1 spin-labeled (Figure S2E). Measured distances ranged between 10–13 Å (Figure 2D) for the WT TCRαTMC segment, considerably shorter than the expected distance of approximately 20 Å for an ideal, unbroken helix. In comparison, distances measured for the G259L/N261A segment ranged between 14–19 Å, more consistent with an unbroken helix. The distance increase independently verifies our structural model of a split membrane-spanning helix for the TCRαTMC (Figure 1B). Moreover, membrane depth analysis of the straightened TM G259L/N261A mutant using single spin EPR depth measurements (Figure 2B) corroborated the distance measurements, as residues 247 and 266 were found to reside outside the liposome acyl region. The central residue 254 was slightly more immersed in the G259L/N261A liposome than in the WT TCRα liposome, indicating shallow positioning of the main body of the helix with the N-terminal end of Helix 1 and C-terminal end of Helix 2 extending from the acyl region into the headgroup for the G259L/N261A mutant. Although G259L/N261A mutation allowed the straightened segment to traverse the ~20 Å of lipid acyl chain, the L-shaped configuration alone did not drive the shallow membrane immersion depth of the TCRαTM, instead modulating TM boundaries.
Mutations within the TCRαTM affect αβ TCR surface assembly and function.
The impact of helix straightening and charged residue mutations of the TCRα subunit on signaling biology was investigated through cellular-based experiments. Stable T cell lines with full-length WT or mutated TCRα (R251L, K256L, R251L/K256L and G259L/N261L) were generated. Copies of surface αβTCR were comparable between the TCRα WT and all mutant cell lines relative to the negative control (Figure 3A). Anti-TCRβ clonotype and Vβ5-specific mAb binding confirmed a natively folded N15TCRαβ on the cell surface (Figure 3A and S3A). In contrast, the surface CD3 expression measured by staining with either anti-CD3ε or anti-CD3εγ mAbs, was less than 2% of WT for K256L and R251L/K256L cell lines (Figure 3A & S3A) and verified by cold blocking experiments to precisely assess background staining (Figure S3B). The TCRα association with CD3 was also investigated for the mutant cell lines using the anti-TCR Cα antibody H28 (Figure 3A). H28 only binds to surface TCRα in the absence of CD3εδ, as its epitope is occluded by CD3εδ and glycans attached to CD3δ. H28 surface staining was observed for the K256L and R251L/K256L cell lines but not for the WT or R251L cell line, confirming the presence of TCRα on the surface of the K256L cell lines lacking associated CD3 heterodimers. Hence, although the surface TCRαβ heterodimers were expressed at WT copy numbers in the K256L and R251/K256L cell lines, mutation of Lys256 eliminated both CD3εγ and CD3εδ from the cell surface. The flow cytometry data (Figure 3A and S3) suggest that K256 mutation dissociates TCRαβ from both CD3 heterodimers. Independently derived cell lines generated for each mutant corroborated the phenotypes.
Figure 3. Biochemical analysis and surface membrane expression of TCR complex subunits in T cells transfected with wildtype versus mutant TCRα.
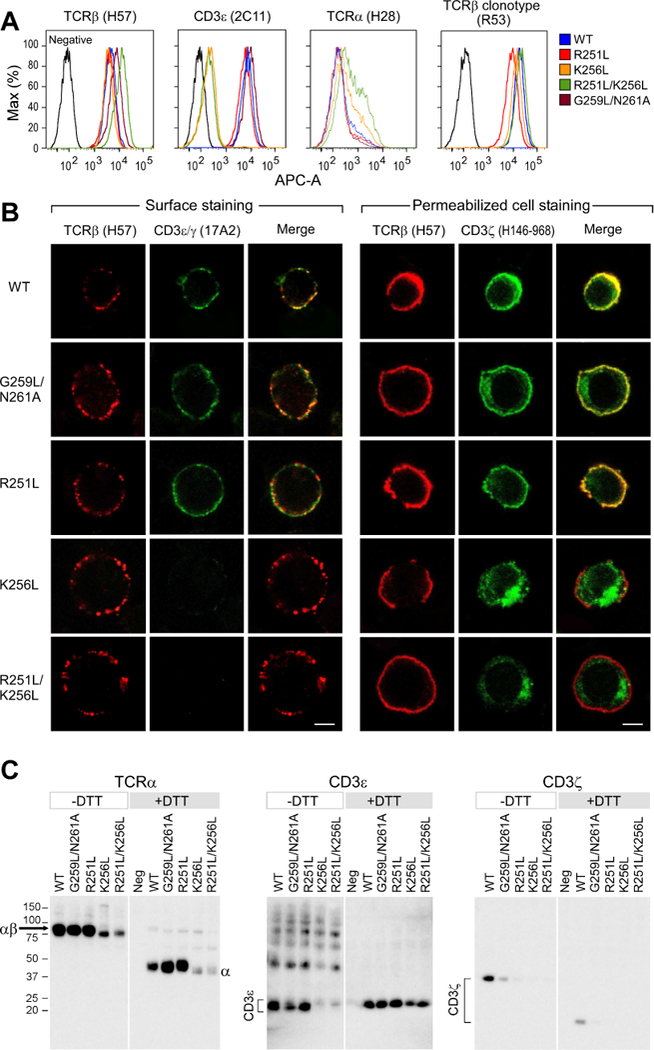
A) Flow cytometry analysis of the TCRβ, CD3ε, and TCRα cell surface expression using the indicated mAbs for each WT or variant TCRα chain containing cell line.
B) Confocal microscopy immunofluorescence analysis of the TCR components in the WT and mutant TCRα cell lines. A Z-plane representative slice is shown for each line with TCRβ, CD3εγ and a merge of the combined TCRβ (red) and CD3εγ (green) antibody cell surface staining results. Permeabilized cell staining results are shown for TCRβ (red) and CD3ζ (green) antibody intracellular staining of each cell line and a merge. Co-localization in merge appears as yellow in surface and intracellular staining. Bar = 5μm.
C) Immunoblot analysis of the TCRα, CD3ε, and CD3ζ co-immunoprecipitated with anti-TCRβ and run under reducing (+DTT) and non-reducing (-DTT) conditions.
TCRαβ and CD3 dimer cell surface assembly was further probed using immunofluorescence confocal microscopy (Figure 3B). Background surface staining of a negative control cell line is shown in Figure S3C. Surface expression of the TCRβ subunit and the CD3εγ heterodimer was determined in representative cells (Figure 3B left panel). Co-localization of fluorochrome staining is readily observable on the WT cell line (Figure 3B), consistent with co-association of these molecules in the normal αβTCR complex on a T cell surface. CD3ζ was also detected after permeabilization and staining with a CD3ζ cytoplasmic tail antibody (Figure 3B right panel). Co-localization of CD3ζ and TCRβ at the plasma membrane demonstrates a fully assembled TCR complex, given that CD3ζ is the last component to associate in the TCR complex (Weissman et al., 1986). Both the G259L/N261A and R251L cell lines had similar staining intensities of TCR and CD3ζ surface molecules as WT, despite straightening of the TM segment or loss of Arg251, the latter previously thought to be required for a CD3ζζ Asp-Asp residue interaction with the TCRα Arg251 (Call et al., 2006). The TCRαβ cell surface presentation and distribution on the Lys256 mutant cells lines (Figure 3B, left panel) were comparable with WT (Figure 3A). However, CD3 was completely absent on the surface of the Lys256 mutant, consistent with lack of CD3 cell surface staining by flow cytometry. CD3εγ intracellular staining revealed that the CD3 subunits were present as aggregates in lysosomal compartments (Figure S3D). Likewise, CD3ζ was observed in an intracellular locale distinct from the surface plasma membrane and unassociated with TCRβ (Figure 3B). Hence, the TCRα Lys256 mutation in K256L and R251L/K256L cell lines generates stable surface TCRαβ expression in the absence of all co-associated CD3 dimers.
Co-association of the TCR complex subunits for WT TCRα as well as the G259L/N261A, R251L, K256L and R251/K256L mutant cell lines was examined by immunoprecipitation and immunoblot analysis. TCRα and CD3ε band intensities were comparable for the WT, G259L/N261A, and R251L cell lines (Figure 3C, left two panels). However, both subunits were detected at a much lower intensity in the K256L and R251L/K256L cell lines. Despite displaying copy numbers of surface TCRαβ comparable to WT, the K256L and R251L/K256L TCRαβ subunits could not be readily immunoprecipitated from the cell lysates regardless of varying buffer conditions that included solubilizing additives such as SDS, positive or negatively charged detergents, or cyclodextrins, nor by using alternative TCRβ antibodies for immunoprecipitation. This finding indicates that the K256L and R251/K256L TCRs are localized in a detergent-resistant membrane fraction lacking CD3ζ or CD3ε subunits. Co-immunoprecipitation of CD3ζ was sensitive to loss of Arg251 and to TM helical straightening (Figure 3C, right panel). As cell lysis and immunoprecipitation disrupted CD3ζ association more readily with the R251L and G259L/N261A TCRα variants than in the WT cell line, even with the mild detergent digitonin, we conclude that Arg251 facilitates the association of CD3ζ to the TCR complex, as does the TM hinge. However, Arg251 and hinge residues G259 and N261 are not required for the TCR-CD3ζ interaction in vivo based on the observed WT IL-2 production and immunofluorescence cell staining (Figures 3B and 4A). Disruption of residues other than Arg251 in the TCRαTMC segment, including site-specific mutation of Leu245 or Phe260, mutation of the CP region, or deletion of Helix 2 have been previously observed to reduce or eliminate CD3ζ binding to the TCR complex (Bhatnagar et al., 2003) and references therein}, collectively implying that CD3ζ association with the TCRα TM domain is weak.
Figure 4. Activated T cell transcriptomes are differentially induced by modification of TCRα Lys256 and Arg251.
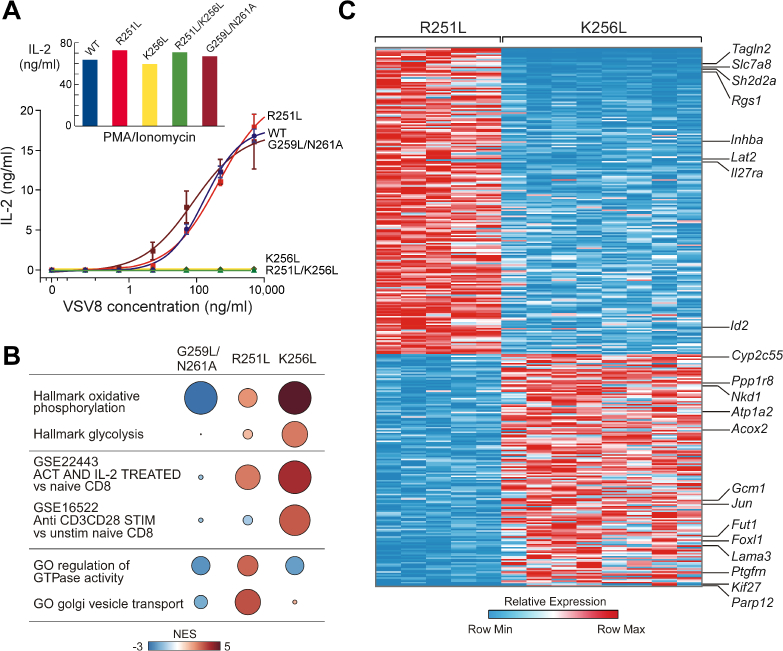
A) IL-2 ELISA results from a T cell stimulation assay using VSV8Kb for the TCRα WT and mutant TCRα cell lines. Inset: Measured IL-2 plotted vs. TCRα WT and mutant TCRα cell lines in response to PMA plus ionomycin stimulation.
B) GSEAPreranked results identifying MSigDB transcriptional signatures enriched and depleted in G259L/N261A, R251L, and K256L compared to WT. The color of each is the normalized enrichment score (NES) relative to WT, and the size is negative log10 false discovery rate (FDR). C) Heatmap of genes significantly differentially expressed between R251L and K256L (FDR-adjusted p-value < 0.01, log2(fold change) > 0.5).
Given uniform expression of cell surface TCRαβ in the absence of CD3 cell surface expression in Lys256 mutant cell lines (K256L and R251L/K256L), we posited that they would be unable to produce IL-2 upon TCR stimulation. However, we were uncertain as to whether weakening of the CD3ζ association revealed biochemically in R251L or G259L/N261A would impact TCR functionality. IL-2 response to pMHC stimulation was monitored as a sensitive bioassay of TCR complex signaling integrity (Figure 4A). No functional impact on IL-2 production was observed with the R251L variant, nor were defects observed in either surface expression or stimulation of IL-2 production in the G259L/N261A mutant (Figure 3A and Figure 4A). In contrast, IL-2 production was essentially undetectable for the K256L and R251L/K256L T cell lines, as expected. PMA plus ionomycin stimulation of K256L and R251L/K256L T cell lines yielded robust IL-2 production, confirming response to stimulation that bypasses the αβTCR complex (Figure 4A, inset).
Gene expression differences among WT, R251L and K256L αβTCR transfectants
Since basal activation states in the WT and mutant cells might differ more broadly, we performed transcriptome analysis on a minimum of two independently derived cell lines and several subclones from WT, G259L/N261A, R251L and K256L cells using RNA-Seq. The variance between the cell lines are shown in the principal component analysis (Figure S4). Gene set enrichment analysis (GSEA) (Figure 4B) revealed that relative to WT, K256L cells were substantially enriched for signatures of oxidative phosphorylation and glycolysis that signify an increase in cellular metabolism critical for supporting energy requirements of T cell activation. Additionally, patterns representative of stimulated T cells were observed in the K256L cells relative to the WT cells. The metabolic and transcriptional activity found in the K256L cells are characteristic of late stage T cell activation, whereas the R251L cells showed features of early stage T cell signaling. For example, the R251L cells displayed an elevation in GTPase activity, known to control cytoskeletal organization and transcriptional regulation, as well as an increase in Golgi vesicle transport necessary to maintain T cell signaling during activation. The G259L/N261A cells showed little variation in gene signatures from WT cells and suggesting that an additional conformational change may be required for gene activation.
To further analyze differences between R251L and K256L cells, differential expression analysis (DESeq2) was carried out between phenotypic groups with a comparison represented in the heatmap found in Figure 4C. A subset of genes is labeled in Figure 4C and all are listed in Supplemental Table 1. Relative to K256L, R251L exhibited upregulation of genes such as Tagln2 and Slc7a8, which stabilize cortical actin and participate in cellular transport of amino acids, respectively, both critical to promote immunological synapse formation and maintain T cell activation. In addition, Sh2d2a and Rgs1 were also upregulated in the R251L cell line, the former is an adaptor for Lck-signaling and the latter is involved in the regulation of GTPase activity at the cellular membrane. Other genes found to be up-regulated in R251L include those participating in TGFβ signaling (Inhba) and in innate and adaptive T cell defense mechanisms (Il27ra). In contrast to R251L cells, whose transcriptome implies genes involved in early T cell activation events, the K256L mutant cells displayed differentially regulated genes corresponding to broader cellular programs. For instance, upregulated cellular metabolism genes include Acox2, Atp1A2, and Cyp2c55 and genes upregulated and involved in transcription and/or translation include Jun, Foxl1 and Nkd1. Comparison of transcripts among mutants also revealed activation of Lama3 necessary for cell migration and Parp12 linked to the posttranslational modification of proteins. The transcriptome data show that R251L and K256L cell lines are in distinct states with R251L in an early stage of activation while K256L cells are in a later stage fostering broad transcription, translation and metabolic pathways linked to cell growth and proliferation. No gene signatures of immune exhaustion were evident to account for lack of K256L antigen responsiveness.
Association of TCRα with CD3δ is governed by TCRαCP region and CD3δ CxxC motif interaction.
Given the importance of the TCRα Lys256 residue in membrane positioning and TCR complex assembly, as well as prior publication purporting a critical charge pairing interaction of Lys256 in TCRαTM with Asp89 in CD3δ TM domain (Call et al., 2002), the TMC of TCRα and CD3δ was produced to study their binding interaction via NMR. Chemical shift and intensity changes of resonances occurred in the TCRα segment CP region upon addition of CD3δ (Figure 5A and B). The combined spectral changes corroborate a TCRα-CD3δ inter-subunit interaction involving the TCRαCP including residues in the FETDxxLN motif (aa234–241), referred to elsewhere as the TCRα CP motif (CPM) (Backström et al., 1998). Pointedly, chemical shift changes were not detected of similar magnitude in the TM segment nor in residue Lys256. Given undetected interactions between TCRα and CD3δ TM domains, we investigated possible interactions between TCRαTMC and CD3δTMC membrane proximal regions. CD3δTMC contains the highly conserved membrane proximal CxxC motif, CQNC, which creates an intramolecular disulfide bond in each CD3 heterodimer subunit (Brazin et al., 2014). When the CD3δ CxxC motif was mutated to SQNS (SxxS, Figure 5B) TCRα and CD3δ interaction was diminished. Thus, the dominant mode of interaction observed between TCRα and CD3δ segments is mediated by the TCRαCP region and the CD3δ CxxC motif.
Figure 5. The juxtamembrane TCRα CP region and the CD3δ CxxC motif mediate a key TCRα-CD3δ intermolecular interaction.
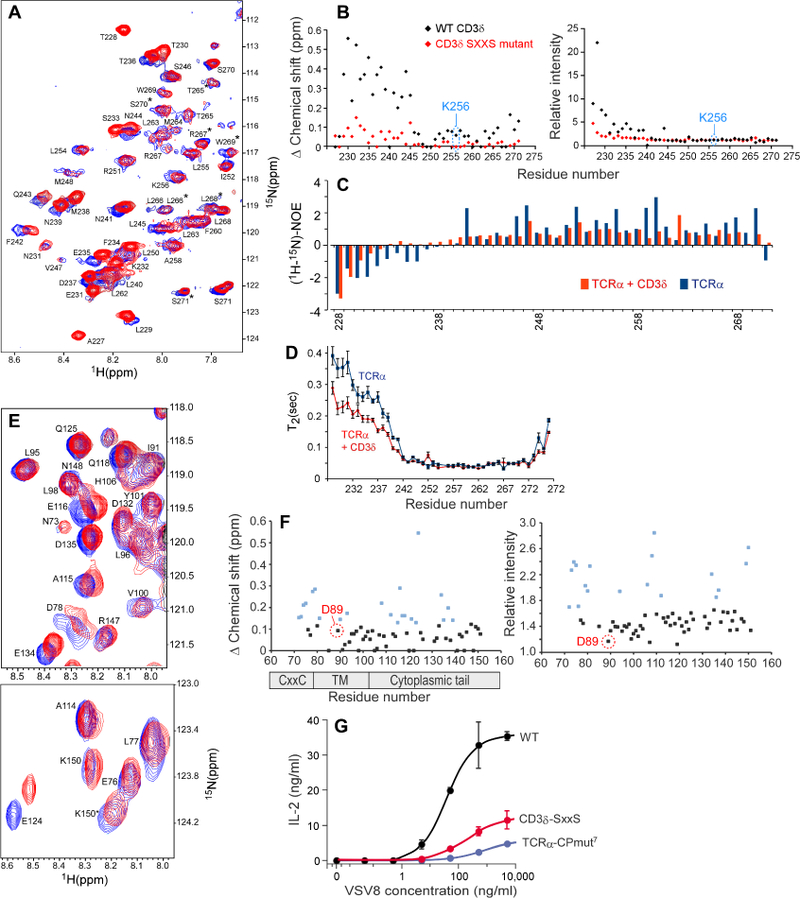
A) 1H-15N HSQC spectrum of 15N TCRα alone (blue) or in the presence of excess unlabeled CD3δ TMC (red). Residue specific backbone assignments are labeled and doubled resonances are denoted with an asterisk.
B) Chemical shift and intensity analysis. Left: Combined chemical shift changes plotted vs. residue number in TCRαTMC upon addition of WT CD3δTMC (black) or upon addition of CD3δ-SxxS TMC (red) are shown. Right: Relative intensity changes plotted vs. residue number in TCRαTMC upon addition of WT CD3δ TMC (black) or upon addition of CD3δ-SxxS TMC (red) are shown.
C) Residue specific heteronuclear NOEs were measured for the TCRαTMC segment alone (blue) or in the presence of excess CD3δ TMC (orange) and plotted as the 1H-15N NOE intensity versus residue number.
D) NMR determined T2 relaxation data of the 15N TCRαTMC segment alone (blue) and in the presence of excess unlabeled CD3δ TMC (orange). The T2 time is plotted vs. the residue number with SD.
E) Expanded regions of the 1H-15N TROSY-HSQC spectrum of CD3δTMC alone (blue) or in the presence of 7-fold excess unlabeled TCRαTMC (red). Residue numbers correspond to the full-length mouse sequence.
F) Chemical shift and intensity changes. Left: Combined chemical shift changes in 15N CD3δ TMC upon addition of unlabeled TCRαTMC plotted vs. residue number. Right: Relative intensity changes in 15N CD3δ TMC upon addition of unlabeled TCRαTMC plotted vs. residue number. The points highlighted in blue in each plot represent chemical shift changes and intensity changes in the top 30% with 0.13 ppm and 1.62 cut-off values for chemical shift and relative intensity, respectively.
G) IL-2 ELISA results from a T cell stimulation assay using VSV8 peptide for the TCRα WT, TCRα-CPmut7 and CD3δ-SxxS cell lines.
We also observed considerable changes in dynamics on TCRα as a result of CD3δ binding. Upon addition of CD3δ, the TCRα N-terminus and CP region heteronuclear 1H-15N-NOE values (Mandel et al., 1995) became considerably more positive and hence more structured with CD3δ binding, suggestive of convergence to a single conformational state (Figure 5C). The TM helices 1H-15N-NOE values became more invariant throughout in the presence of CD3δ, indicating a more uniform structure compared to the TCRα segment alone. The TCRαTMC dynamics were also monitored by measuring T2 relaxation times (Mandel et al., 1995). The addition of CD3δ to TCRαTMC resulted in a considerable decrease in the T2 time of the CP region indicative of a more structured state with negligible effects on the TM domain and the C-terminus. Combined, the T2 relaxation data and the 1H-15N-NOE data further support a CP interaction with CD3δ that results in an increase in structure within the TCRα CP region (Figure 5D).
Specific binding site information was then sought on the CD3δ segment. Spectral changes were observed in CD3δTMC in the presence of unlabeled TCRαTMC (Figure 5E). Several CD3δ residues changed upon addition of TCRαTMC, with the largest changes in the membrane proximal region (His67-Ser79) that encompasses the CxxC motif (Cys71-Cys74) and in the cytoplasmic tail, located near the ITAM motif (Tyr127-Leu141; Figure 5F). Extensive chemical shift perturbations are consistent with the structural effects observed previously following disruption of the CxxC motif (Brazin et al., 2014). As with TCRα, chemical shift changes were not observed in the TM domain of CD3δ. While we did observe chemical shift changes in L90 and T93, those residues may also be affected by conformational changes occurring at the CxxC region with TCRα binding, and not the result of a charge pairing interaction between TCRα Lys256 and CD3δ D89. The involvement of the CD3δ CxxC motif in the binding-site interaction is consistent with the observed disruption of the TCRα interaction when the two Cys were mutated to Ser (SxxS) (Figure 5B & S5A).
Mutations targeting the TCRα-CD3δ interaction site result in signaling defects.
To determine the functional importance of the TCRα and CD3δ interaction sites identified by NMR, TCRαCP mutants were generated in the full-length protein, mutating those residues having the largest spectral changes in the presence of CD3δ (Figures 5A and S5B). The IL-2 production of WT and mutant N15TCRα mutant constructs were quantitated with the largest effect observed in the mutant cell line CPmut7 (Figure 5G), constructed to enhance the WT TCRαCP flexibility and target key CD3δ binding residues without altering its length (EKSFETD to GGGSGSG). Effects of the TCRα CPmut7 were then compared with a CD3δ WT and mutant cell line, CD3δ-SxxS, in which the native CxxC motif was mutated to SxxS. The IL-2 response of the TCRα-CPmut7 cell line was substantially reduced (Figure 5G) with a calculated antigen EC50 of 860 ng/ml versus 38 ng/ml for the WT cells. CD3δ-SxxS also showed a reduction in IL-2 production with a calculated EC50 of 193 ng/ml. Given that surface TCR expression on mutants was equivalent to WT (Figure S5C) and with comparable response to PMA plus ionomycin stimulation, reduced cytokine production is a consequence of diminished TCR-pMHC ligand triggering sensitivity.
Deleterious functional outcomes in the above mutants might be due to disrupted structural connectivity at the TCRα and CD3δ juxtamembrane interface undermining force-dependent signal transduction. Optical tweezers (OT) were used in a single cell format (Feng et al., 2017) to characterize the mechanotransduction properties of the WT and mutant αβTCRs (Figure 6A and B). Concordant with the IL-2 functional results, OT analysis showed defects in Ca2+ triggering for both TCRα-CPmut7 and CD3δ-SxxS transduced cell lines relative to WT (Figure 6B, top panel). The mutant cell lines were unable to trigger Ca2+ flux at high pMHC copy number in the absence of force, in contrast to WT TCR expressing cells where internal actomyosin forces pulling on the TCR following pMHC ligation was sufficient. Only when substantial triggering force was applied (~21–27 pN for each mutant cell) were the mutant-transduced cell lines able to be activated by pMHC, an effect requiring a high ligand copy number (20,000 VSV8Kb per bead). Sensitivity of mutant receptor triggering was compromised compared to the WT TCR expressing cells where just 10 pN/TCR shear force and 2–29 molecules of VSV8Kb per bead readily induced activation. Thus, mutations targeting interaction sites identified by NMR produce defects in force-coupled signaling.
Figure 6. Functional consequences of mutations targeting the TCRα-CD3δ interaction site and TCRαTM as assessed by optical trap and fluorescence microscopy.
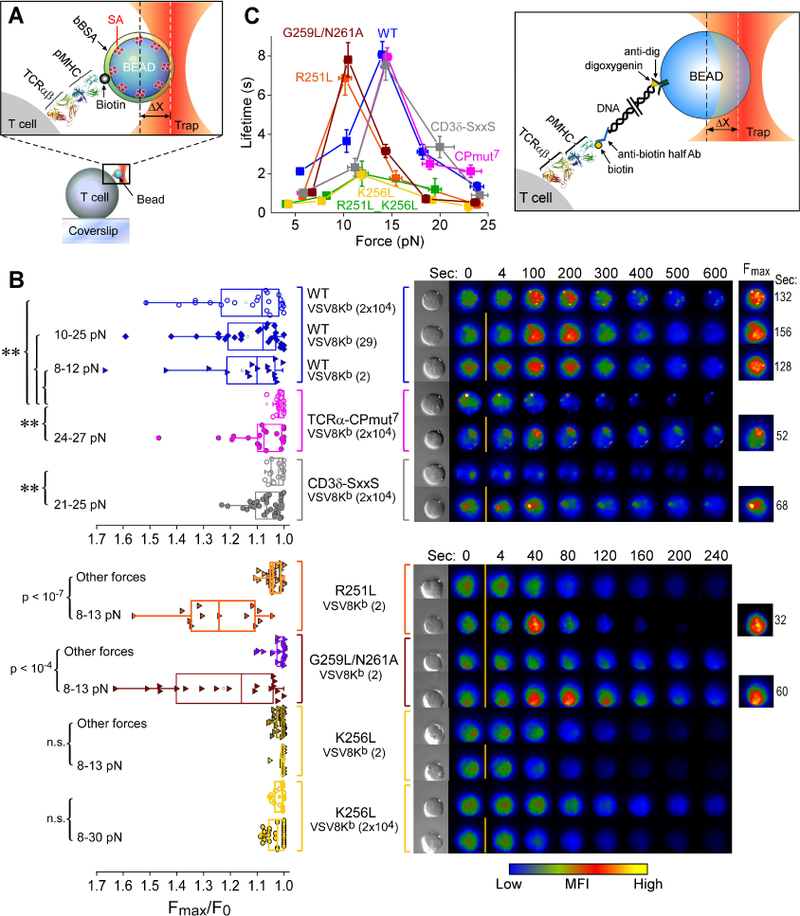
A) Diagram depicting the bead-cell contact in the optical trap used for applying force in the single-cell (SC) TCR triggering experiments. A streptavidin (SA)-coated polystyrene bead is bound with biotinylated pMHC, saturated with bBSA and brought into contact with an immobilized T cell. The trap force is calculated as the product of trap stiffness and displacement from the trap center (∆X). The molecular model depicts the TCRαβ heterodimer (1NFD) and pMHC (1KPU).
B) T cell activation SC analysis of the WT and mutant gene cell lines. Left: Ca2+ flux indicated by the ratio of maximum fluorescence intensity (Fmax) to initial fluorescence intensity (F0) was triggered at the indicated interfacial VSV8Kb copy numbers with applied trapping force (5–35 pN) or without force (no label) for the different cells. Each dot in the plot represents a single cell experiment. Width of the box plot illustrates a range from 25th to 75th percentiles in score distribution. Mean and median values are shown in the square and line, respectively. The (**) represents P values ≤ 0.01. Right: Intracellular Ca+2 flux measured over the experiment from representative individual cells. An increase inintracellular Ca+2 is shown by a colorization change through increases in red and yellow intensities over time. The top corresponds to the analysis of the WT, TCRαCPmut7 and CD3δ-SxxS over 0–600 seconds vs. the bottom representing that of R251L, G259L/N261A and K256L over 0–240 seconds. The initial fluorescence signal was recorded at time zero without trapping force application (0 s) and then shear force was loaded through the VSV8Kb coated bead between 0 s and 4 s, as indicated by the orange bar. The fluorescence images as well as the times exhibiting Fmax are shown on the right column.
C) The catch bond behavior of different TCR-expressing cell types under force is plotted as bond lifetime vs. force. Error bars represent SEM. The cartoon inset depicts SMSC tether assay for measurement of αβTCR-pMHC bond lifetimes.
Antigen recognition sensitivity is enhanced by R251L but abrogated by K256L: mutations associated with TCR-pMHC bond lifetime alterations
The effects of mutation at the hinge and TM residues R251 and K256 on mechanotransduction were even more striking (Figure 6B, bottom panel). With G259L/N261A and R251L cells just 2 molecules of VSV8Kb in the OT system led to a response more rapidly than with WT cells. The duration of calcium flux tested with 2 VSV8Kb molecules was sustained for 105 sec +/− 17 sec (11 cells, +/− SD) and 63 +/− 20 sec (10 cells, +/− SD) for the G259L/N261A and R251L N15 T cells, respectively, relative to the much longer response time of 288 +/− 119 sec (8 cells, +/− SD) for WT N15 T cells. By contrast, the K256L cells were unresponsive to mechanotransduction at any pMHC density and with any pN force applied. Given loss of CD3 dimers due to K256L mutation, it is not surprising that the surface membrane TCRαβ is signaling incompetent.
The OT system can also be used in single molecule, single cell (SMSC) format to measure individual TCR-pMHC bond lifetimes (Das et al., 2015). Force-bond lifetime dependence in SMSC assays was used to compare the WT and variant lines bearing TCRα mutations that alter the TCRα-CD3δ interaction site, the interconversion of the TM helicies between the L- and E-state, and the TCRαTMC membrane immersion depth and assembly (Figure 6C). The WT cell line showed similar force dependence of bond lifetimes as observed previously (Das et al., 2015; Das et al., 2016). The individual TCRs all manifest “catch” bonds, a counter-intuitive behavior in which bond strengthening occurs with application of force relative to 0 force and is manifested as longer bond lifetimes under load, with subsequent bond rupture occurring at higher forces exceeding that of the single bond strength (Das et al., 2015; Liu et al., 2014). Despite their impaired ability to facilitate T cell activation (Figure 6B), the CPmut7 and CD3δ-SxxS mutants display similar force maxima as WT for catch-bond formation, ~15 pN, and maximal lifetime, ~8s. In contrast, the R251L mutant showed a slightly reduced maximal lifetime, but the catch bond force maximum shifted downward from the WT to ~10pN, suggesting that a subunit assembly defect impacts antigen recognition function. We also observed a similar effect in the reduction of the maximal force bond lifetime for the G259L/N261A mutant, further supporting the conclusion that CD3ζζ dissociation accelerates T cell activation. Most strikingly, the two K256L bearing mutants showed both a large decrease of maximal bond lifetime and attenuation of catch bond formation. Given the normal protein fold of the mutant TCRs as determined by several anti-TCRβ mAbs (Figures 4A and S3A) the result suggests an inability of the force-transduction pathways to sustain necessary forces to gate ligand discrimination (Das et al., 2015). This could occur from attenuation of normal TM anchoring, modification of TM tilt geometry upon force-based ligation and/or CD3ζ-linked actin cytoskeletal associations necessary to transduce external force via the αβTCR complex.
Discussion
The TCRα subunit is a critical component of the αβTCR, essential for thymocyte development and mature T cell responsiveness to foreign antigens. We have determined the TCRαTMC structure using solution NMR and pursued in parallel biophysical and biological approaches that link the structural details to the fundamental understanding of TCR subunit organization and activation mechanism. Our structure differs from a model of a TCRαβ TM heterodimer that utilized a mutant TCRαTM segment where the vital Arg251 and Lys256 residues were mutated to Leu and disulfide crosslinking restraints were used to derive a tightly packed TCRαβ TM heterodimeric structural model through computations restricting the TMs each to a single uniform helix (Krshnan et al., 2016). By contrast, we observed that the WT TCRαTMC contains a mobile CP region, a membrane-spanning TM domain segmented into two helices by the hinge at Gly259-Asn261, and a short cytoplasmic tail. NMR and EPR analyses show that the TM domain exists in a dynamic bistable state exchanging between an L-shaped configuration (L-state) and a straightened configuration (E-state) facilitated by a flexible hinge. Moreover, TM residue sidechains in Helix 1, including Arg251 and Lys256, were found to have shallow immersion depths in a membrane-like environment.
The shallow position of the TCRαTM residues undoubtedly plays a regulatory role in modulating protein-protein interactions within the membrane while the L-shaped configuration of the TCRαTMC contributes to its disposition. Straightening of the helical segment induced a TM residue sidechain depth change as shown for the TCRαTMC G259L/N216A mutant. As a result, Lys256 and neighboring residues move deeper into the acyl chain region in the membrane. Conversion of a shallow L-shaped TM configuration to a more straightened TM that allows the Lys256 sidechain to adopt this more deeply buried membrane topology is a likely mechanism promoting TCR signaling. We propose that the straightened conformation is the primed state with force-mediated TCR-pMHC ligation inducing the switch.
Although Lys256 in the TCRαTMC segment is critical in regulating the membrane depth, we found no evidence of a charge-mediated interaction between TCRα and CD3δ. Instead, TCRα Lys256 is key in affecting the TM segment membrane disposition potentially through its positively charged sidechain amine and negatively charged lipid head groups. The mainchain likely remains embedded in the membrane while the charged sidechain is positioned in the polar lipid head group, a phenomenon is known as “snorkeling” (Strandberg and Killian, 2003). The TM domain TCRα Lys256 and, by extension, that of the TCRβ subunit Lys271 appear to be positioned to facilitate a charge-based association with the lipid head groups, mitigating energetic costs of charged sidechains within the hydrophobic environment. The TCRβ TM Lys271 residue may also play a critical role in maintaining αβTCR complex assembly and surface expression (Alcover et al., 1990). Although the TCRα Arg251 does not regulate TM depth, predicated on helix topology it may form an ionic interaction with the lipid headgroups and/or participate in an interaction with CD3ζζ in the αβTCR, either via a salt link or a polar or aromatic interface (Blázquez-Moreno et al., 2017; Call et al., 2006). As described in our dissociative model below, straightening of the TCRαTM would weaken or disrupt this Arg interaction with CD3ζζ, consistent with our results and with observations regarding the preTCR that readily activates early thymocytes. pTα subunit preserves the TM Arg and Lys residues but lacks a TM hinge, consistent with CD3ζζ absence in detergent lysates of preTCR complexes immunoprecipitated with anti-CD3ε (Shinkai et al., 1993).
Specific juxtamembrane interactions detailed by NMR on a residue-specific basis between the CP region of TCRα and the CxxC motif of CD3δ are consistent with the functional importance of this region shown here and in (Backström et al., 1998), establishing a mechanical link crucial to force-mediated T cell signaling without affecting TCR-pMHC bond lifetime. By contrast, the two TCRαTM charge residues control TCR-pMHC bond lifetime. Thus, the membrane proximal regions of these two subunits are involved in force relay while the TM segment residues control bond lifetime. Based on current structural and functional data, we propose a dissociative model (see graphical abstract and Figure S6) for pMHC-triggered αβTCR complex activation dependent on the distinct roles of TCRαTM Arg251 and Lys256 residues working in tandem as a part of force-driven mechanotransduction.
In the inactive state, rather loose association exists between TCRαβ and CD3 TM domains, as well as between TCRα and β subunit TM domains, with the CD3 cytoplasmic tails sequestered on the plasma membrane. The TCRα subunit exists in a predominantly L-shaped configuration with Lys256 snorkeling to associate with lipid head groups. The well-conserved hinge region assists in maintaining the inactive configuration. The CD3ζζ homodimer, including the paired negatively charged Asp residues, may be in contact with the TCRα Arg251 residue (Call et al., 2006). The heterodimeric CD3εδ and CD3εγ and their respective negatively charged TM residue pairs are potentially interacting with the lipid membrane positively charged choline or amino groups.
Force transduction through the membrane can initiate key changes causing the TCRαTM to alter its helical tilt, adopting a more straightened configuration, changing the position of the Arg251 sidechain and consequently disrupting an association with CD3ζζ. CD3ζζ then becomes displaced from the TCR complex resulting in the first stage of activation. Concurrent with load-dependent T cell activation, interchange of membrane lipid molecules causes the cytoplasmic tails to become released from the cell membrane and available for tyrosine phosphorylation and downstream signaling (Aivazian and Stern, 2000; Deford-Watts et al., 2009; Guo et al., 2017). With the subsequent change in TCRα membrane positioning, the Lys256 side chain becomes more embedded in the acyl chains in the membrane and the CD3 heterodimers then may dissociate in this later stage of activation. Perhaps force may transiently extrude the Lys256 sidechain into the cytosol for post-translational modification by acetylation or methylation leading to greater depth in the acyl chain region post-modification. The short TCRα cytoplasmic tail will facilitate such a movement of the straightened TCRαTM helix. The clear separation of ligand binding (TCRαβ) and signaling (CD3) components in the αβTCR complex affords a sophistication in receptor function that may be difficult to achieve with receptors that harbor both in one subunit, such as receptor tyrosine kinases.
Our dissociative hypothesis is consistent with observations made years ago showing that anti-clonotypic antibodies which bind to the VαVβ-module recognition surface, akin to pMHC, dissociate the CD3 dimers from the TCRαβ heterodimer in detergent lysates whereas anti-CD3ε antibodies preserve the integrity of the entire αβTCR complex (Meuer et al., 1983). Subsequent studies have shown uncoupling of CD3 homodimers and CD3 heterodimers upon antigen exposure (Kishimoto et al., 1995; La Gruta et al., 2004). Several lines of evidence suggest that the CD3ζζ dimer can readily dissociate from the αβTCR complex. First, loss of CD3ζζ proteins from human T cells has been observed in areas of immune inflammation, particularly in associations with cancer, among other alterations, [reviewed in (Baniyash et al., 2014; Koneru et al., 2005)]. Second, weakening of CD3ζζ association described above augments the rapidity of mechanotransduction-linked calcium flux. Third, in addition to R251L, other TCRαTM mutations facilitate CD3ζζ dissociation, underscoring its weak basal interaction with the TCR subunits in the membrane environment. Fourth, early T cell transcriptional activation is a consequence of this weakening.
Chimeric antigen receptors (CARs) are recombinant proteins transduced into T cells with ligand binding activity distinct from the TCR, often employing a scFv ectodomain, a CP linker and TM segment unrelated to αβTCR components and a cytoplasmic tail consisting of an amalgam of CD3ζζ and one or more costimulatory receptor domains (Hartmann et al., 2017). In these constructs CD3ζζ is separated from TCR subunits. Nuancing of the molecular design of CARs based on evolving principles of mechanotransduction and dissociative αβTCR complex activation may extend the number of targets amenable to effective immunotherapy.
STAR Methods
Recombinant protein expression and purification
Complete methods for protein expression and purification of the 15N- labeled TCRα and CD3δ NMR samples have been described previously (Brazin et al., 2014). Briefly, N-terminal His tag, GB1 domain linked TCRα and CD3δ TMC constructs were produced and isotopically expressed in M9 minimal media containing 15N ammonium chloride as the sole nitrogen source. For expression of ILV-2H, 12C/13C, 15N TCRα samples the precursors alpha-ketoisovaleric acid (125 mg/L) and alpha-ketobutyric acid (100 mg/L) were added to the expression media 1 hour prior to IPTG induction. Expressed proteins were solubilized with SDS lysis buffer, and the CD3δ Cys residues were oxidized and/or modified, and the protein segments were affinity purified and His-GB1 was removed by TEV digestion. The TMC segments were purified to homogeneity by size exclusion chromatography and then were TCA-precipitated and then dried. The samples were then dissolved in 30 mM Tris buffer, pH 7.0, containing 100 mM LPPG, 10% D2O, and 0.02% NaN3 for NMR analysis. Deuterated LPPG (d31-LPPG obtained from fbreagents.com) was used in the solubilization buffer for the TCRαTMC sample during NMR acquisition of backbone data.
NMR spectroscopy
NMR spectra were acquired on Bruker 500, 600 and 750 or Varian 600 and 700 MHzspectrometers equipped with a 5-mm cryogenic probe. 1H,15N heteronuclear single quantum correlation (HSQC) NMR experiments and transverse relaxation optimized spectroscopy (TROSY)-enhanced 1H,15N HSQC spectra for the TCRα TMC, CD3δ TMC, and CD3δ-SxxS TMC were performed at 310K. Standard three-dimensional triple resonance backbone experiments were recorded on the TCRαTMC segment for completion of the backbone resonance assignments. The sidechain assignments were completed using HCCONH and CCONH experiments and Nuclear Overhauser Enhancement (NOE) distance constraints were obtained from ILV 15N- and 13C- dispersed NOE experiments with a mixing time of 200 milliseconds for both experiments. Data were processed with NMRPipe and all the data were analyzed with CARA and CcpNmr software.
Residue specific PREs were determined through the acquisition of successive T1 delay modulated 1H-15N HSQC and 1H-13C HSQC spectra recorded on a ILV-2H, 12C, 15N TCRα sample over the course of a titration from 0 to 10mM with Gd+3 (Gd(DTPA-BMA) purchased as Gadodiamide from Toronto Research Chemicals, Toronto, Canada. Relaxation delays of 0.15, 0.25, 0.35, 0.5, 0.75, 1, 2, 5 seconds were used in each experiment, with repeats at 0.5 and 2 seconds. Peak intensities were measured for each 15N or 13C-ILV resonance throughout the titration to calculate the relaxation effect (R1) at each concentration of Gd+3, and the R1 was then used to calculate the PRE value (Franzmann et al., 2009). The calculated PRE-derived distance constraints were weighted at 30% with respect to NOEs in the structure calculation, and +/−3 Å was used for the upper and lower boundaries.
Chemical shift assignments and NOE assignments were completed using CARA and CcpNmr, respectively. The ARIA program was also used to determine the NOE integrals for an independent assessment of the NOE peak heights and volumes. An ensemble of folded TCRα TMC structures was generated using the following NMR determined restraints: 220 NOE restraints, 13 hydrogen bond restraints, and 68 dihedral angle restraints (obtained from the chemical shift data using TALOS. 100 structures were calculated using the simulated annealing protocol in CYANA and the 10 structures with the lowest energy target functions were chosen for deposition. PyMOL was used for structure visualization. Structural quality was assessed with PSVS and iCING.
The T2 and heteronuclear NOE (15N[1H] NOE) measurements were conducted as described (Roberts, 1993). The relaxation delays of: 16, 32, 49, 65, 81, 97, 114, ms, were used to calculate the T2 relaxation times, with repeats on the 49 and 81 ms delays. The peak heights were integrated with CcpNmr, and T2 times were calculated within CcpNmr. The interleaved (15N[1H] NOE) experiments were recorded with a 3-s saturation delay and the 15N[1H] NOE values were calculated within CcpNmr.
Chemical shift mapping experiments were performed by the solubilization of 15N-TCRα TMC, 15N-CD3δ TMC, or 15N-CD3δ TMC-SxxS at a concentration of 0.5 mM for TCRα and CD3δ-SxxS TMC, and 0.4 mM for CD3δ-TMC. Samples were dissolved in 100mM LPPG, 30mM Tris pH 7.0, 0.1 mM EDTA, 0.02 % NaN3 as described (Brazin et al., 2014). Spectra were acquired on the 15N protein samples alone and then in the presence of 14N protein samples at 310K using the 1H-15N HSQC pulse sequence for 15N TCRα samples and 1H-15N TROSY-HSQC for 15N CD3δ samples. Solubilized 14N samples were then added to the 15N samples as follows: 14N- CD3δ TMC or 14N- CD3δ TMC-SxxS was added in 8x excess to 15N-TCRα TMC; 14N-TCRαTMC was added in 7x excess to 15N-CD3δ TMC; and 14N-TCRα synthetic CP peptide (sequence: DATLTEKSFETDMNLNFQN) was added in 8x excess to 15N- CD3δ-SxxS TMC. Chemical shift and relative intensity changes were plotted vs. residue number in TCRαTMC upon addition of WT CD3δ TMC or upon addition of CD3δ-SxxS TMC and the median +/− standard deviation (SD) are 0.08 +/− 0.16 and 1.20 +/− 3.49 for the chemical shift and intensity changes, respectively. Combined chemical shift and intensity changes in 15N CD3δ TMC upon addition of unlabeled TCRαTMC were plotted vs. residue number with the median +/− SD for the chemical shift and intensity changes were calculated to be 0.089 +/− 0.091 and 1.487 +/− 0.381, respectively.
TCRα multiple sequence alignment and cytoplasmic tail analysis
The multiple sequence alignment of the TCRα transmembrane region from selected vertebrate species was generated using MUSCLE from a selection of TCRα orthologues identified in the ENSEMBL and PANTHER databases. Partial reptile TCRα sequences in the alignment were obtained after BLAST searches with Anolis carolinensis against high throughput genome sequences at NCBI. TCRα secondary structure definition was obtained from the relevant 3D-coordinates of the NMR models using DSSP or predicted from sequence analysis using PHISPRED.
Cytoplasmic tail residues analysis was completed on two sets of proteins obtained from the UNIPROT knowledgebase database. One set consisted of 507 single-pass type I membrane proteins expressed in T cells from mammalians. The other set included 247 Human Cluster of Differentiation (CD) proteins, consisting of 218 type I and 35 type II single-pass membrane proteins. A complete list of CD proteins is available at http://www.uniprot.org/docs/cdlist. We used PERL scripts to retrieve the complete records of the selected proteins from UNIPROT and processed them to identify the topology and the number of cytoplasmic residues of each protein. The size distribution of the cytoplasmic tail (number of cytoplasmic residues) of the proteins was analyzed under the Jupyter Notebook environment and fitted the density distribution to cytoplasmic tail residues to a lognormal equation (Eq.1):
| Eq.1 |
where µ is the average size of the cytosolic tails, is the standard deviation and is the number PI
EPR experiments
Large unilamellar vesicles (LUVs) were prepared as described previously (Hope et al., 1985; Szoka et al., 1980). Lipids (DOPC/DOPG, 4:1 wt/wt) in chloroform were mixed in a glass tube and dried as thin films under a stream of nitrogen gas, and were further dried using a vacuum pump for ~16 h to remove residual solvent. The lipids were resuspended in an HK buffer (20 mM HEPES, 150 mM KCl, pH 7), vortexed for 1–2 min and then subjected to 10–15 freeze-and-thaw cycles. The lipid suspension was extruded 10–15 times through a mini-extruder with a 100 nm polycarbonate membrane (Avanti Polar Lipids). Synthetic TCRα WT and mutant peptides spin labeled with MTSL (S-(1-oxyl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl) methyl methanesulfonothioate) (Toronto Research Chemicals) were obtained from the Biopolymers & Proteomics Core of Koch Institute at MIT. The peptides were dissolved in methanol (100 μM) and subsequently co-dried along with the lipids during the liposome preparation.
The EPR measurements were carried out on a Bruker E680 EPR spectrometer (Billerica, MA) in X-band at the National High Magnetic Field Laboratory (NHMFL). Spin-spin distance data were recorded on a Bruker high-sensitivity resonator (4119HS) at 200 K. Spectra were collected at 2 mW microwave power with a field modulation frequency of 100 kHz, a modulation amplitude of one Gauss (G), and a 200 G sweep width. EPR spectra were analyzed with a Monte-Carlo/Simplex Gaussian convolution method to extract spin-spin distance (Fajer et al., 2007). For depth measurements, EPR power saturation experiments were performed on a loop-gap resonator (Molecular Specialties, Milwaukee, WI). LUV samples were loaded in gas permeable TPX capillary tubes (Molecular Specialties, Milwaukee, WI) and purged using either a stream of air or N2 gas. EPR spectra were collected at microwave powers ranging from 0.4 to 100 mW with a modulation field of 2 G and a modulating frequency of 100 kHz. Immersion depth was calculated by the ratio of the accessibility values of O2 to 50 mM nickel (II) ethylenediaminediacetic acid (NiEDDA). Depth standard curves were determined using lipid vesicles containing trace amounts of spin-labeled lipids (1:500 by weight) as described (Song et al., 2009).
Generation of the BW5147 T cell line
The BW5147 cell line and plasmids CD3δγεζWT pMIY and TCRαβ pMIG were a gift from the Vignali lab (St. Jude Children’s Research Hospital, Memphis, Tennessee). Cells were maintained in high glucose DMEM medium (Sigma-Aldrich) supplemented with 20 % (v/v) fetal bovine serum (FBS) (Sigma-Aldrich), 100 U/ml penicillin, 100 U/ml streptomycin (Life technologies), 2 mM glutamine (Life technologies). The WT mouse CD3 and N15 TCR genes in each plasmid were constructed using the viral 2A-linked system to generate multicistronic vectors for co-transfection of the CD3 and TCR genes. The N15 αβTCR is specific for the vesicular stomatitis virus nucleoprotein octapeptide (VSV8: RGYVYQGL) bound to H-2Kb (Mallis et al., 2015). The CD8αβ gene was sub-cloned into the pcDNA3.1 vector and transfected into the BW5147 cell line. The N15 TCRαβ expressing cell line was generated by transfection of the CD3δγεWT and N15 TCRαβ plasmids into Pheonix-Eco packing cells (ATCC) for CD3 and TCR retrovirus production. The viral supernatants were harvested and then used to retrovirally transduce the BW5147-CD8αβ cells to incorporate the CD3 and TCR genes in that order. TCRα mutants were produced using the QuikChange II mutagenesis kit (Agilent Technologies) and plasmids were then transfected in the Pheonix-Eco packing cells for TCR retrovirus production containing a mutant TCRα gene.
Immunoprecipitation experiments
For each cell line, 2 0 × 107 cells were washed 3× in TBS and lysed in lysis buffer containing: 1 % Triton X-100, 50 mM Tris pH 8.0, 150 mM NaCl, 2mM NEM, and protease inhibitor (Roche cOmplete cocktail). Cell lysis was carried-out for 30 min on ice, lysates were then centrifuged to remove insoluble material and pre-cleared with protein G beads (GE Healthcare Life Sciences, GammaBind Plus). Anti-TCRβ (H57) coupled beads were then added to the lysates and incubated at 4°C for 2 hours with rotation. The beads were washed 4× with cell lysis buffer and then resuspended in buffer containing 10 mM NEM, 1× NUPAGE buffer, and +/− 50mM DTT. Samples were separated on 4–12% Bis-Tris NuPAGE gels (Invitrogen), transferred to PVDF membranes, and detected with the indicated antibodies. The following antibodies were used for western blotting: TCR Cα (H28), CD3ζ (H146–968, Sigma), CD3ε (gift from J.E. Coligan).
Flow cytometry
The following list of antibodies were used in FACS analysis: anti-TCRβ-APC (H57–597) BD Pharmingen, anti-CD3ε-APC (145–2C11) eBioscience, anti-CD3εγ-APC (17A2) BD Biosciences, anti-TCRαβ clonotype (R53), anti-TCR Vβ5 (MR9.4), Isotype Control (HTK888) BioLegend, anti-TCR Cα (H28), anti-mouse IgG-APC (poly4053) BioLegend, anti-hamster IgG-PE (polyclonal) eBioscience, anti-rat IgG-APC (poly4054) BioLegend, anti-CD3ε unconjugated (2C11).
The BW5147 cells were surface stained with H57-APC and sorted for equivalent surface expressing cells on the Aria II SORP machine. APC was detected using a 660/20 band pass filter, excited with a 633nM laser and sorted with a 70 μM nozzle. FACS analysis samples were surface stained with the indicated APC- or PE-conjugated antibodies and analyzed on the J-Fortessa machine. Dana-Farber Cancer Institute Jimmy Fund Flow Cytometry Facility.
Confocal microscopy immunofluorescence
1.0 × 105 TCR-transfected or un-transfected cells were washed twice with 3% FCS-PBS and then surfaced stained with PE-conjugated anti-TCRβ (H57–597, BD Biosciences) and Alexa Fluor® 647-conjugated anti-CD3 (17A2, BD Biosciences) simultaneously for 60 min on ice. After removal of excess antibody, the PE-TCRβ was amplified by rabbit anti-Phycoerythrin-R/R-PE antibody (Thermo Fisher Scientific) for 60 min on ice and then further labeled by Alexa Fluor® 594-conjugated F(ab’)2 fragment of goat anti-rabbit IgG (H+L) (Thermo Fisher Scientific) for 60 min on ice. Cells were then washed and filtered through 7μm cell strainer to remove cell aggregates before introduction via flow into a fabricated flow cell chamber. To this end, the flow cell chamber consisted of glass slide (Fisherbrand) and cover slip (FisherBrand) coated with poly-L-lysine (PLL) solution (Sigma P2890, 3% solution in ethanol). Double-sided tape was put on each edge of the glass slide to create a channel. The PLL coated cover slip was adhered to the tape face down. Cells were introduced into the channel via a pipette and kept on ice in a polystyrene tube right before microscope observation to avoid surface molecule internalization.
For CD3ζ subunit staining, TCRβ staining was carried-out first using Alexa Fluor® 594-conjugated anti-TCRβ (H57, Biolegend) for 60 min on ice. Cells were then washed with 3% FCS-PBS and permeabilized with eBioscience FoxP3 Transcription Factor Staining Buffer Set (Thermo Fisher Scientific) according to the manufacturer’s protocol. Permeabilized cells were blocked with Armenian Hamster IgG (Abcam) for 10 min on ice and then stained with FITC-conjugated anti-CD3ζ (H146–968, Abcam) for 30 min at room temperature and then washed. The FITC-labeled antibody was then amplified with Alexa Fluor® 488-conjugated anti-FITC antibody (Jackson ImmunoResearch Laboratories Inc.) for 30 min on ice and then washed. In some experiments, CD3εγ staining was carried out simultaneously with lysosome staining using FITC-conjugated anti-mouse CD107a (LAMP-1, BD Biosciences, clone 1D4B, 1:100 dilution) plus FITC-conjugated anti-mouse CD107b (LAMP-2, BD Biosciences, clone M3/84, 1:100 dilution) overnight at 4 °C, then labeled with Alexa Fluor® 488-conjugated mouse anti-FITC secondary antibody for 1h at 4 °C and washed.
The Leica SP5X laser scanning confocal microscope equipped with an acousto-optical beam splitter (AOBS) system (Leica Camera AG, Wetzlar, Germany) with a 40× oil objective (PL APO, NA1.25) was used for image acquisition with LAS AF software. Image processing and analyses were carried out using Fiji/ImageJ (http://rsbweb.nih.gov/ij/).
T cell activation assays and IL-2 ELISA
The T cell activation assay was carried-out in triplicate in a 96 well plate using 2 × 105 R8 cells irradiated at 3000 rads prior to use with 2 ×105 N15 TCRαβ BW cells in each well. Concentrations of the stimulatory peptide VSV8 were added to each well ranging from 50 pg/ml to 5 μg/ml. A negative control lacked VSV8 peptide while PMA plus ionomycin was used as a positive control. The VSV8 stimulated cells and the controls were incubated for 16–18 hours overnight in a 37°C incubator, the ce ll supernatants were then harvested for an IL-2 ELISA assay.
The IL-2 ELISA assay was completed using the mouse IL-2 DuoSet and ancillary reagent kit 2 (R&D systems). T cell supernatants were diluted in media such that the O.D. 450 nm readings fall within the standard curve for the assay. The assay was then carried out following the kit instructions. Negative control values were subtracted from each sample point and concentrations in pg/ml were calculated from the standard curve. Measured IL-2 was plotted vs. the concentration of VSV8 peptide and fit to a 4-parameter logistic model.
RNA-Seq
A minimum of two independently derived cell lines of each mutant or WT, as well as additional subclones derived at different culture times, were used for analysis. RNA was extracted from approximately 0.25–1 × 106 cells using the QIAGEN Rnaeasy kit with QIAshredder treatment. Total RNA was quantified using the Qubit RNA Assay Kit (Life Tech) and RNA quality was determined on the Bioanalyzer using the RNA Pico Kit (Agilent). The NuGen Ovation Human RNA-Seq Multiplex system (NuGen, part 0341) Prep Kit, was used to target deletion of unwanted high abundance transcripts and ribosomal RNA. More than 100ng of total RNA was converted into each DNA library following the manufacturer’s protocol without modification. Following library construction, DNA libraries were quantified using the Qubit High Sensitivity DNA Kit (Life Tech) and library size was determined using the Bioanalyzer High Sensitivity Chip Kit (Agilent). Finally, qPCR was carried-out on the libraries using the Universal Library Quantification Kit for Illumina (Kapa Biosystems) and run on the 7900HT Fast qPCR machine (ABI). Libraries passing quality control were diluted to 2nM in sterile water and then sequenced on the NextSeq500 (Illumina) at a final concentration of 12pM, following all manufacturer protocols. RNA-seq data was collected at the DFCI Center for Cancer Computational Biology.
Sequencing reads were aligned to the mm10 genome using the STAR aligner and quantified as integer counts using the featureCounts summarization program of the Rsubread R package (Bioconductor). Batch correction was performed using the ComBat algorithm. DESeq2 was used to normalize the counts matrix and perform differential expression analysis between all phenotypic groups. GSEAPreranked was performed between phenotypic groups following differential expression analysis using the ranking metric k, where k = −1*log10(q) and q is the DESeq2 FDR-adjusted p-value for a gene in a given phenotypic comparison. All GSEAPreranked analyses were performed using canonical pathway gene-sets from the MSigDB database.
SMSC analysis, force-bond lifetime measurements, and induced T cell activation by optical traps
In the SMSC assay, bond lifetime measurements were carried-out using tethers constructed from half anti-biotin antibody functionalized DNA to bridge the anti-digoxygenin–coated polystyrene beads (1.0 μm in diameter, Spherotech Inc.) at one end and biotin-labeled VSV8Kb at the other end. After washing with PBST buffer (1X PBS + 0.02% (v/v) Tween-20) two times, the bead slurry was diluted 200-fold with 5 mg/mL Bovine serum albumin (BSA) (Sigma-Aldrich) in colorless DMEM medium (Sigma-Aldrich) for bond lifetime measurements. Cells were washed once with colorless DMEM medium and re-suspended to 2 × 106 cells/mL. 20 μL of the cell suspension was transferred into the flow chamber, and cells were bound to a coverslip, after 30 mins incubation at 37°C and 5% CO2 the coverslip surface was further blocked by 5 mg/mL BSA in colorless DMEM medium. Following a 10-min incubation at 37°C and 5% CO 2, the pMHC-DNA bead slurry (~20 μL) was introduced into the same chamber. The tether-functionalized bead was trapped and brought into the cell’s vicinity to form a stable tether. Detailed bead preparation procedures and methods for measuring the bond lifetime were elaborated in the previous work (Das et al., 2015).
The T cell activation SC assay was carried-out by staining the cells with Quest Rhod-4, AM (AAT Bioquest, Inc.) to observe the intracellular calcium flux during T cell triggering and then VSV8Kb coated beads with three different interfacial copy numbers (2×10 4, 29 and 2) were used to test the triggering capacity for the different T cell lines. Procedures of VSV8Kb bead preparation and characterization, Ca2+ dye loading and cell triggering with optical trapped beads are available in previously published methods (Feng et al., 2017)
Supplementary Material
Highlights.
The TCRα transmembrane (TM) domain is a bipartite helix separated by a dynamic hinge.
Lys256 controls the TCRαTM depth and CD3 homo- and heterodimer associations.
The TCRα- and CD3δ-juxtamembrane elements mediate a specific TCRα-CD3δ interaction.
Key residues within TCRαTM or juxtamembrane domains govern αβTCR mechanotransduction.
Acknowledgments
We gratefully acknowledge Dr. Derin Keskin, Zahra Hayati, and Dr. Vlado Gelev for technical assistance and Dr. Jia-huai Wang for scientific discussions. This work is supported by NIH grants PO1 GM047467 to GW and ELR, AI037581 to GW, and R56 AI138489 and R01AI100643 to ELR and MJL. Funding is also provided by the FWF project J3872-B21 to AB, and NSF DMR 1157490 to the NHMFL, and the NHMFL UCGP 5080 grant to LS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Aivazian D, and Stern LJ (2000). Phosphorylation of T cell receptor zeta is regulated by a lipid dependent folding transition. Nat Struc Biol 7, 1023–1026. [DOI] [PubMed] [Google Scholar]
- Alcover A, Mariuzza RA, Ermonval M, and Acuto O (1990). Lysine 271 in the transmembrane domain of the T-cell antigen receptor beta chain is necessary for its assembly with the CD3 complex but not for alpha/beta dimerization. J Biol Chem 265, 4131–4135. [PubMed] [Google Scholar]
- Backström BT, Muller U, Hausmann B, and Palm er E (1998). Positive selection through a motif in the alphabeta T cell receptor. Science 281, 835–838. [DOI] [PubMed] [Google Scholar]
- Baniyash M, Sade-Feldman M, and Kanterman J (2014). Chronic inflammation and cancer: suppressing the suppressors. Cancer Immunol Immunother 63, 11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatnagar A, Gülland S, Bascand M, Palmer E , Gardner TG, Kearse KP, and Bäckström BT (2003). Mutational analysis of conserved amino acids in the T cell receptor alpha-chain transmembrane region: a critical role of leucine 112 and phenylalanine 127 for assembly and surface expression. Mol Immunol 39, 953–963. [DOI] [PubMed] [Google Scholar]
- Birnbaum ME, Berry R, Hsiao YS, Chen Z, Shingu-Vazquez MA, Yu X, Waghray D, Fischer S, McCluskey J, Rossjohn J, et al. (2014). Molecular architecture of the αβ T cell receptor-CD3 complex. Proc Natl Acad Sci USA 111, 17576–17581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blázquez-Moreno A, Park S, Im W, Call MJ, Call ME, and Reyburn HT (2017). Transmembrane features governing Fc receptor CD16A assembly with CD16A signaling adaptor molecules. Proc Natl Acad Sci USA 114, E5645–E5654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumberg RS, Alarcon B, Sancho J, McDermott FV, Lopez P, Breitmeyer J, and Terhorst C (1990). Assembly and function of the T cell antigen receptor. Requirement of either the lysine or arginine residues in the transmembrane region of the alpha chain. J Biol Chem 265, 14036–14043. [PubMed] [Google Scholar]
- Brazin KN, Mallis RJ, Li C, Keskin DB, Arthanari H, Gao Y, Wu S-L, Karger BL, Wagner G, and Reinherz EL (2014). Constitutively oxidized CxxC motifs within the CD3 heterodimeric ectodomains of the T cell receptor complex enforce the conformation of juxtaposed segments. J Biol Chem 289, 18880–18892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Call ME, Pyrdol J, Wiedmann M, and Wucherpfennig KW (2002). The organizing principle in the formation of the T cell receptor-CD3 complex. Cell 111, 967–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Call ME, Schnell JR, Xu C, Lutz RA, Chou JJ, and Wucherpfennig KW (2006). The structure of the zetazeta transmembrane dimer reveals features essential for its assembly with the T cell receptor. Cell 127, 355–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty AK, and Weiss A (2014). Insights into the initiation of TCR signaling. Nat Immunol 15, 798–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosson P, Lankford SP, Bonifacino JS, and Klausner RD (1991). Membrane protein association by potential intramembrane charge pairs. Nature 351, 414–416. [DOI] [PubMed] [Google Scholar]
- Das DK, Feng Y, Mallis RJ, Li X, Keskin DB, Hussey RE, Brady SK, Wang J-H, Wagner G, Reinherz EL, et al. (2015). Force-dependent transition in the T-cell receptor β-subunit allosterically regulates peptide discrimination and pMHC bond lifetime. Proc Natl Acad Sci USA 112, 1517–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das DK, Mallis RJ, Duke-Cohan JS, Hussey RE, Tetteh PW, Hilton M, Wagner G, Lang MJ, and Reinherz EL (2016). Pre-T cell receptors (pre-TCRs) leverage Vβ complementarity determining regions (CDRs) and hydrophobic patch in mechanosensing thymic self-ligands. J Biol Chem 291, 25292–25305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deford-Watts LM, Tassin TC, Becker AM, Medeiros JJ, Albanesi JP, Love PE, Wülfing C, and van Oers NSC (2009). The cytoplasmic tail of the T cell receptor CD3 epsilon subunit contains a phospholipid-binding motif that regulates T cell functions. J Immunol 183, 1055–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fajer PG, Brown LJ, and Song L (2007). Practical Pulsed Dipolar ESR (DEER), Chapter 4. In ESR Spectroscopy in Membrane Biophysics (SBN), pp. 95–128.
- Feng Y, Brazin KN, Kobayashi E, Mallis RJ, Reinherz EL, and Lang MJ (2017). Mechanosensing drives acuity of αβ T-cell recognition. Proc Natl Acad Sci USA 114, E8204–E8213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzmann M, Otzen D, and Wimmer R (2009). Quantitative use of paramagnetic relaxation enhancements for determining orientations and insertion depths of peptides in micelles. Chembiochem 10, 2339–2347. [DOI] [PubMed] [Google Scholar]
- Guo X, Yan C, Li H, Huang W, Shi X, Huang M, Wang Y, Pan W, Cai M, Li L, et al. (2017). Lipid-dependent conformational dynamics underlie the functional versatility of T-cell receptor. Cell Res 27, 505–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann J, Schüßler-Len M, Bondanza A, and B uchholz C,J (2017). Clinical development of CAR T cells-challenges and opportunities in translating innovative treatment concepts. EMBO Mol Med 9, 1183–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Rangarajan S, Kerzic M, Luo M, Chen Y, Wang Q, Yin Y, Workman CJ, Vignali KM, Vignali DA, et al. (2015). Identification of the docking site for CD3 on the T cell receptor β chain by solution NMR. J Biol Chem 290, 19796–19805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hope MJ, Bally MB, Webb G, and Cullis PR (1985). Production of large unilamellar vesicles by a rapid extrusion procedure: characterization of size distribution, trapped volume and ability to maintain a membrane potential. Biochim Biophys Acta 812, 55–65. [DOI] [PubMed] [Google Scholar]
- Kim ST, Takeuchi K, Sun ZY, Touma M, Castro CE, Fahmy A, Lang MJ, Wagner G, and Reinherz EL (2009). The alphabeta T cell receptor is an anisotropic mechanosensor. J Biol Chem 284, 31028–31037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto H, Kubo RT, Yorifuji H, Nakayama T, Asano Y, and Tada T (1995). Physical dissociation of the TCR-CD3 complex accompanies receptor ligation. J Exp Med 182, 1997–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koneru M, Schaer D, Monu N, Ayala A, and Frey AB (2005). Defective proximal TCR signaling inhibits CD8+ tumor-infiltrating lymphocyte lytic function. J Immunol 174, 1830–1840. [DOI] [PubMed] [Google Scholar]
- Krshnan L, Park S, Im W, Call MJ, and Call ME (2016). A conserved αβ transmembrane interface forms the core of a compact T-cell receptor-CD3 structure within the membrane. Proc Natl Acad Sci USA 113, E6649–E6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Gruta NL, Liu H, Dilioglou S, Rhodes M, Wiest DL, and Vignali DA (2004). Architectural changes in the TCR:CD3 complex induced by MHC:peptide ligation. J Immunol 172, 3662–3669. [DOI] [PubMed] [Google Scholar]
- Liu B, Chen W, Evavold BD, and Zhu C (2014). Accumulation of dynamic catch bonds between TCR and agonist peptide-MHC triggers T cell signaling. Cell 157, 357–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallis RJ, Bai K, Arthanari H, Hussey RE, Handley M, Li Z, Chingozha L, Duke-Cohan JS, Lu H, Wang J-H, et al. (2015). Pre-T cell receptor ligand binding impacts thymocyte development prior to αβTCR expression. Proc Natl Acad Sci USA 112, 8373–8378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandel AM, Akke M, and Palmer A.G.r. (1995). Backbone dynamics of Escherichia coli ribonuclease HI: correlations with structure and function in an active enzyme. J Mol Biol 246, 144–163. [DOI] [PubMed] [Google Scholar]
- Manolios N, Bonifacino JS, and Klausner RD (1990). Transmembrane helical interactions and the assembly of the T cell receptor complex. Science 249, 274–247. [DOI] [PubMed] [Google Scholar]
- Meuer SC, Cooper DA, Hodgdon JC, Hussey RE, Fitzgerald KA, Schlossman SF, and Reinherz EL (1983). Identification of the antigen and major histocompatibility complex receptor on human inducer T lymphocytes. Science 222, 1239–1242. [DOI] [PubMed] [Google Scholar]
- Natarajan A, Nadarajah V, Felsovalyi K, Wang W, Jeyachandran VR, Wasson RA, Cardozo T, Bracken C, and Krogsgaard M (2016). Structural model of the extracellular assembly of the TCR-CD3 complex. Cell Rep 14, 2833–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Respondek M, Madl T, Göbl C, Golser R, and Zangger K (2007). Mapping the orientation of helices in micelle-bound peptides by paramagnetic relaxation waves. J Am Chem Soc 129, 5228–5234. [DOI] [PubMed] [Google Scholar]
- Roberts GCK (1993). NMR of Macromolecules:A Practical Approach (Oxford UK: Oxford University; ). [Google Scholar]
- Rudolph MG, Stanfield RL, and Wilson IA (2006). How TCRs bind MHCs, peptides, and coreceptors. Annu Rev Immunol 24, 419–466. [DOI] [PubMed] [Google Scholar]
- Shinkai Y, Koyasu S, Nakayama K, Murphy KM, Loh D, Reinherz EL, and Alt FW (1993). Restoration of T cell development in RAG-2-deficient mice by functional TCR transgenes. Science 259, 822–825. [DOI] [PubMed] [Google Scholar]
- Song L, Sun ZY, Coleman KE, Zwick MB, Gach JS, Wang JH, Reinherz EL, Wagner G, and Kim M (2009). Broadly neutralizing anti-HIV-1 antibodies disrupt a hinge-related function of gp41 at the membrane interface. Proc Natl Acad Sci USA 106, 9057–9062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strandberg E, and Killian JA (2003). Snorkeling of lysine side chains in transmembrane helices: how easy can it get? FEBS Lett 544, 69–73. [DOI] [PubMed] [Google Scholar]
- Sun Z-YJ, Kim KS, Wagner G, and Reinherz EL (2001). Mechanisms contributing to T cell receptor signaling and assembly revealed by the solution structure of an ectodomain fragment of the CD3εγ heterodimer. Cell 105, 913–923. [DOI] [PubMed] [Google Scholar]
- Sun Z-YJ, Kim ST, Kim IC, Fahmy A, Reinherz EL, and Wagner G (2004). Solution structure of the CD3episilondelta ectodomain and comparison with CD3episilongamma as a basis for modeling T cell receptor topology and signaling. Proc Natl Acad Sci USA 101, 16867–16872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szoka F, Olson F, Heath T, Vail W, Mayhew E, and Papahadjopoulos D (1980). Preparation of unilamellar liposomes of intermediate size (0.1–0.2 mumol) by a combination of reverse phase evaporation and extrusion through polycarbonate membranes. Biochim Biophys Acta 601, 559–571. [DOI] [PubMed] [Google Scholar]
- von Boehmer H, Aifantis I, Gounari F, Azogui O, Haughn L, Apostolou I, Jaeckel E, Grassi F, and Klein L (2003). Thymic selection revisited: how essential is it? Immunol Rev 191, 62–78. [DOI] [PubMed] [Google Scholar]
- Wang JH, and Reinherz EL (2012). The structural basis of αβ T-lineage immune recognition: TCR docking topologies, mechanotransduction, and co-receptor function. Immunol Rev 250, 102–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissman AM, Samelson LE, and Klausner RD (1986). A new subunit of the human T-cell antigen receptor complex. Nature 324, 480–482. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.