

Abstract
Metalloenzymes represent an important target space for drug discovery. A limitation to the early development of metalloenzyme inhibitors has been the lack of established structure-activity relationships (SARs) for molecules that bind the metal ion cofactor(s) of a metalloenzyme. Herein, we employed a bioinorganic perspective to develop an SAR for inhibition of the metalloenzyme influenza RNA polymerase PAN endonuclease. The identified trends highlight the importance of the electronics of the metal-binding pharmacophore (MBP), in addition to MBP sterics, for achieving improved inhibition and selectivity. By optimizing the MBPs for PAN endonuclease, a class of highly active and selective fragments were developed that display IC50 values <50 nM. This SAR led to structurally distinct molecules that also displayed IC50 values of ∼10 nM, illustrating the utility of a metal-centric development campaign in generating highly active and selective metalloenzyme inhibitors.
Graphical Abstract
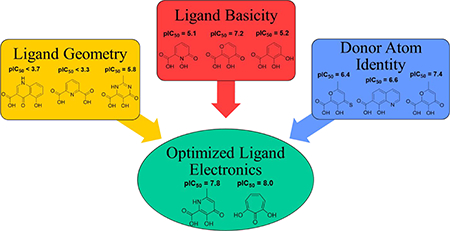
Introduction
Metalloenzymes comprise over one-third of all known enzymes, are ubiquitous across all domains of life, and are implicated in a wide variety of human diseases.1,2 As a result, metalloenzymes represent prime target space for drug discovery; however, the clinical development of metalloenzyme inhibitors is rather limited. In the past five years, only ∼9% of new molecular entities approved by the FDA target metalloenzymes, and <5% of all FDA approved drugs inhibit metalloenzymes.1,2 Compounds that are able to interact strongly with an active site metal center can effectively inhibit the catalytic activity of metalloenzymes, by disrupting substrate access to the active site and preventing metal-mediated catalysis.3 Metal binding inhibitors are reversible, but are capable of forming strong interactions due to the large bond enthalpy of metal-ligand dative or coordinate covalent bonds.
Within the context of metalloenzyme inhibitors, a shortcoming to the development of new inhibitors has been an over-reliance on a very limited number of metal-binding pharmacophores (MBPs).4,5 In addition, despite the importance of metal-ligand interactions in the development of metalloenzyme inhibitors, relatively little work has been focused on the development and optimization of MBPs, with a general lack of structural diversity in the MBP chemical space.6,7 Indeed, the only metalloenzyme targets where a substantial chemical diversity is present in terms of the MBPs are inhibitors of HIV integrase (HIV IN) and HIV reverse-transcriptase associated RNaseH (HIV RNaseH),8,9 with most of this structural diversity reported in the patent literature.10–12 However, despite the structural diversity in the patent literature against these targets, there is little analysis into the effects of varied MBP cores on metalloenzyme inhibition. Furthermore, these reports generally do not detail development of the MBP core nor efforts towards MBP optimization. To address these shortcomings, MBP libraries, consisting of fragment-like compounds designed to bind metal ion cofactors in metalloenzyme active sites, have been developed.13 These MBP libraries have been used in fragment-based drug discovery (FBDD) to identify novel inhibitors of several metalloenzymes, including the influenza RNA-dependent RNA polymerase PA subunit.13
The influenza polymerase complex is an attractive target for new antiviral therapies, particularly the polymerase PA endonuclease domain. This domain is both highly conserved across influenza strains and serotypes and is indispensable for the viral lifecycle.14 Crystallographic and biochemical studies have shown that the polymerase PA N-terminal endonuclease domain (PAN) contains a dinuclear metal active site which binds to two Mg2+ or Mn2+ cations.15,16 The metal cations reside in a pocket comprised of a histidine (His41), an isoleucine (Ile120), and a cluster of three acidic residues (Asp108, Glu80, Glu119) that all coordinate to the active site metal ions (Figure 1).15,17 These metal ions are essential for catalysis, and it has been shown that metal coordination by small molecules effectively inhibits endonuclease activity.13,18–22 Indeed, nearly all reported inhibitors of endonucleases have been shown by X-ray crystallography or modeling to coordinate to at least one active site metal center, including the polymerase PA inhibitor Baloxavir marboxil, developed by Roche and Shionogi, which is currently in Phase III clinical trials in the U.S. and has received regulatory approval in Japan.23
Figure 1.

Structure of the RNA-dependent RNA polymerase PA subunit active site (PDB ID: 5DES). The endonuclease active site employs two divalent metal cations to facilitate the hydrolytic cleavage of the phosphodiester backbone of RNA. Protein secondary structure elements are shown in cartoon representation (gray). Mn2+ cations are shown as purple spheres. Coordinating protein residues are colored by element and labeled and coordinating water/hydroxide molecules are shown as red spheres. All coordination bonds are displayed as dashed yellow bonds. This structure, as well as all other protein structures presented, were generated in PyMOL.24
The influenza virus RNA polymerase has no proofreading capability, which results in a high mutation rate of approximately one error per genome replication cycle.25 This results in each infected cell producing on average ∼10,000 new viral mutants during the course of infection.16 One primary advantage to a discovery campaign focused on metal binding is an intrinsic barrier to antiviral resistance. Any mutation to the PAN metal coordinating residues (with the exception of substituting Glu119 with Asp, which coordinates identically to Glu119) results in total loss of viral transcription activity and ultimately virulence.26,27 Hence, an inhibitor molecule that obtains significant binding energy from metal coordination may be less susceptible to antiviral resistance, as mutations that disfavor metal coordination will likewise disfavor substrate binding and/or catalytic activity.
Little work has been reported on the optimization of specific metal-ligand interactions for PAN inhibitors,19 despite some chemical diversity among the various reported PAN inhibitor MBPs both in terms of chemical structure and coordination motif. However, there are few clear structure-activity trends that can be derived from the different reported MBPs.28 This lack of consensus regarding preferred metal coordination interactions is not unique to PAN endonuclease, and has been a limitation to the development of inhibitors of many metalloenzymes.10 A more detailed understanding of the metal coordination preferences of the dinuclear metal center of PAN endonuclease would allow for optimization of MBP domains and produce highly active endonuclease inhibitors. To this end, a targeted FBDD screen was conducted employing a MBP library.13 This screen included MBPs with the ability to coordinate to both metals of the dinuclear metal active site simultaneously. Using a FRET-labeled DNA oligonucleotide substrate, the endonuclease activity of the PAN subunit of H1N1 influenza A polymerase was measured in the presence of MBPs to identify fragments that exhibit strong inhibition. Several MBP fragments were found to have IC50 values of <500 nM. SAR elucidated from the library screen and subsequent derivatization identified key chemical properties that were essential for tight binding to the metal cofactors in the PAN active site. In addition, good selectivity for PAN inhibition over other dinuclear metalloenzymes was also achieved. This strategy resulted in the identification of an optimized chemotype for MBP-based inhibition of PAN, with several MBPs displaying IC50 values of <50 nM. X-ray structural determination of MBPs in PAN were used to elucidate key binding interactions. Ultimately, the identification of key metal binding preferences allowed for the predictive development of other chemically distinct, highly active inhibitor chemotypes illustrating the value of a bioinorganic, coordination chemistry-based fragment discovery approach for metalloenzyme inhibitor development.
Results and Discussion
Fragments in the MBP library were screened for endonuclease inhibition against a truncated N-terminal (PAN) endonuclease construct employing a FRET-labeled DNA oligonucleotide, as previously reported.13,15,22 This construct is derived from an influenza A (A/California/04/2009) H1N1 clinical isolate that has been modified such that residues 52–64 of the N-terminal (PAN) endonuclease were replaced by a single glycine residue.18,29 This truncation removes a highly disordered protein loop that engages in protein-protein interactions with other subunits of the RNA polymerase complex. Loop removal was found to produce higher quality protein crystals and improve the solution stability of the isolated PAN domain.18,29 Note that several compounds used in this study were obtained from commercial suppliers (compounds 4–5, 7–14, 16, 19, 23, 31, and 34).
Compounds that exhibited inhibition of PAN endonuclease at a fragment concentration of 200μ;M and were predicted to simultaneously coordinate both metal centers in the endonuclease active site (i.e., possessed a suitable triad of donor atoms)30 were further analyzed with dose-response experiments to determine IC50 values. The activity of these MBPs is shown in Table 1. To ensure that the observed activity against endonuclease was not due to metal-stripping, all inhibition assays were performed with a large excess (2 mM) of both Mn2+ and Mg2+ metal cations in the assay media. From these results, a characteristic pharmacophore model with a common donor triad chemotype was identified that is shared by the most active inhibitors produced in this study. As shown in Figure 2, compounds sharing this chemotype were predicted to coordinate to the divalent metal centers as multicoordinate bridging ligands with the conserved phenolic oxygen atom replacing the bound hydroxide anion observed in the native protein. To elucidate the mode of binding, crystallographic studies were performed. The truncated PAN construct was co-crystalized or soaked with several ligands that share the active chemotype (Figure 2). All X-ray structures validate the multi-coordinate bridged coordination mode hypothesis. Compound 3 was observed to coordinate simultaneously to both metal centers with the carbonyl oxygen atom coordinating to Mn-1 at the open coordination site, the phenolic oxygen replacing the bound hydroxide anion as the bridging ligand, and the carboxylic acid group coordinating to Mn-2 (Figure 2). No steric clash was observed within the active site, and binding of compound 3 places each metal ion in an octahedral coordination geometry. From this co-crystal structure, it is inferred that the majority of the binding enthalpy for 3 is derived from metal coordination and not from other interactions with the protein active site. This is corroborated by the analogous binding mode and similar inhibitory activity of compound 1 (Figure 2).
Table 1.
Structure and activity of a series of MBP derivatives.
| Number | Compound | IC50(μΜ) | pIC50a | LEb | Number | Compound | IC50(μΜ) | pIC50a | LEb |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 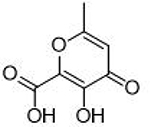 |
0.043 ± 0.009 μΜ | 7.4 | 0.84 | 10 | 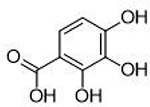 |
11.8 ± 2.1 μΜ | 4.9 | 0.56 |
| 2 | 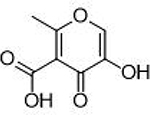 |
3.5 ± 0.6 μΜ | 5.5 | 0.63 | 11 | 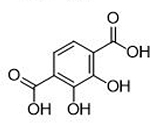 |
13.6 ± 0.9 μΜ | 4.9 | 0.48 |
| 3 | 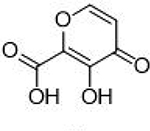 |
0.068 ± 0.011 μΜ | 7.2 | 0.89 | 12 | 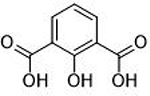 |
>500 μΜ | <3.3 | Ν/Α |
| 4 | 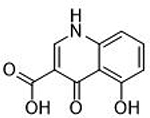 |
>200 μΜ | <3.7 | Ν/Α | 13 | 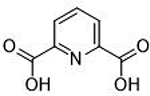 |
>500 μΜ | <3.3 | Ν/Α |
| 5 | 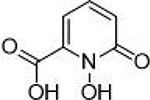 |
7.5 ± 1.2 μΜ | 5.1 | 0.64 | 14 | 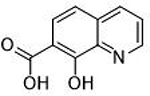 |
0.26 ± 0.04 μΜ | 6.6 | 0.65 |
| 6 | 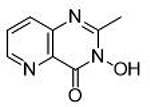 |
>100 μΜ | <4 | Ν/Α | 15 | 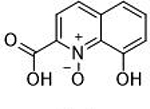 |
>100 μΜ | <4 | Ν/Α |
| 7 | 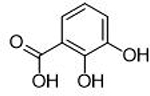 |
6.5 ± 0.9 μΜ | 5.2 | 0.65 | 16 | 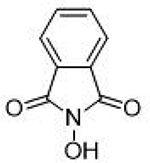 |
14.3 ± 1.7 μΜ | 4.8 | 0.55 |
| 8 | 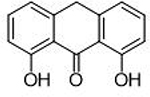 |
>100 μΜ | <4 | Ν/Α | Allomaltol | 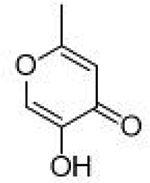 |
17.1 ±1.5 μΜ | 4.8 | 0.73 |
| 9 | 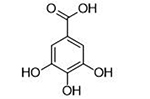 |
4.2 ± 1.0 μΜ | 5.4 | 0.61 | Pyromeconic Acid | 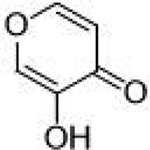 |
22.5 ±1.0 μΜ | 4.6 | 0.79 |
pIC50 is defined as pIC50 = −log(IC50) and is included to provide a linear comparison of relative inhibition activity.
Ligand efficiency (LE) provides a measure of binding energy per nonhydrogen atom in the fragment molecule.
Figure 2.

Top left: Representative chemotype shared by active MBP fragments of influenza PAN highlighting the coordination motif of these MBPs to the dinuclear active site metal ions. Top right: X-ray co-crystal structure of compound 3 (PDB ID: 6DCZ) in the PAN active site. Mn-2 is also coordinated by two water molecules at the open coordination sites (not shown). Bottom: X-ray co-crystal structure of compounds 1 (left; PDB ID: 6DZQ), 2 (middle; PDB ID: 6DCY), and 3 (right; PDB ID: 6DCZ) in the PAN active site. All ligands were found to coordinate in a similar manner. Compound 2 coordinates Mn-2 with the electron density (blue mesh is the 2FoFc map contoured to 2σ) corresponding to the carboxylic acid moiety found to be diffuse above the metal center. The poorer coordination ability of compound 2 is likely due to internal steric pressure exerted by the methyl group residing alpha to the carboxylic acid.
The inhibitory activity of compounds such as 1 is outstanding given the small molecular weight of the fragment (ligand efficiency of 0.84). Generally, hits identified in FBDD screening campaigns have IC50 values >100 μM, and not infrequently >1 mM.7,31 More striking is the comparison between 1 and the related MBPs allomaltol and pyromeconic acid (Table 1, IC50 = 17.1 μM and 22.5 μM, respectively), which were previously identified as PAN hits.13 It was observed that the addition of only three atoms (in the form of the coordinating carboxylate group) to these inhibitor scaffolds (to generate compounds 1 and 3, respectively) resulted in a ∼500-fold increase in activity.32 Interestingly, not every MBP that can present a donor atom triad to the PAN metal centers was found to display such tight binding affinities (e.g., compounds 4, 6, 8, Table 1), and suggests that the PAN metal centers have specific ligand preferences, which have not been previously identified for this metalloenzyme.
Based on the excellent activity observed with MBPs such as 1, other fragments were synthesized to obtain SAR to elucidate what aspects of these molecules were responsible for their tight binding affinity. Beyond probing simple steric effects or attempting to introduce new protein contacts, changes to the electronic character of the MBP ring system were explored. The effect of changes to donor atom identity, Lewis basicity, and isosteric replacement of coordinating moieties were also investigated. Multiple structural variants of the identified chemotype were ultimately designed and synthesized, and the effect of modulating each coordinating moiety was assessed by both biochemical and structural experiments to understand the observed trends.
Conversion of a pyrone to a pyridinone scaffold has been shown to improve activity in previously reported endonuclease inhibitors.13 Pyridinones have a greater aromatic character than pyrones, which can result in greater electron density on the oxygen donor atoms. This change in ligand electronics generally results in greater ligand basicity, which can lead to stronger coordination to the hard Lewis acidic metal centers found in endonucleases.33 To probe this effect, a series of analogs of 1 were synthesized and investigated (Table 2). As expected, conversion of a pyrone oxygen to a pyridinone nitrogen (17) resulted in an approximately 3-fold increase in activity relative to 1. In addition, no change in the activity of compounds 1 and 17 was observed as a function of the presence or absence of excess metal ions in solution (up to 25 mM). These results conclusively demonstrates that the high affinity of these inhibitor molecules is derived from a strong binding preference for the dinuclear Mn2+ PAN metal centers and not from any general affinity for free Mn2+ in solution.
Table 2.
Structure and activity of a series of MBP derivatives.
| Number | Compound | IC50 (nM) | pIC50 | Phenol pK aa |
|---|---|---|---|---|
| 1 | 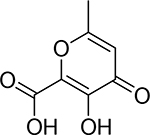 |
43 ±9 nM | 7.4 | 9.7 |
| 3 | 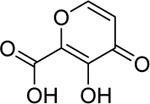 |
68 ± 11 nM | 7.2 | 9.6 |
| 5 | 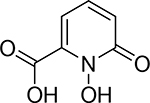 |
7500 ± 1200 nM | 5.1 | 7.0 |
| 7 | 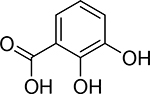 |
6400 ± 900 nM | 5.2 | 9.8; 14.3 |
| 17 | 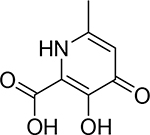 |
17 ± 3 nM | 7.8 | 11.2 |
| 18 | 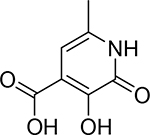 |
430 ± 20 nM | 6.4 | 9.2 |
| 19 | 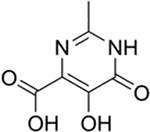 |
1600 ± 400 nM | 5.8 | 7.5 |
Calculated pKa values33
The position of the nitrogen atom within the ring system strongly effects inhibitory activity. While 3,4-hydroxypyridinones (17) did show an improvement in activity, 2,3-hydroxypyridinones (18) and 4,5-hydroxypyrimidinones (19) showed markedly less activity than the 3,4hydroxypyridinone or the parent hydroxypyrone fragments, with compounds 18 and 19 showing a 25-fold and 100-fold loss in activity, respectively (relative to 17). Furthermore, N-hydroxy-1,2hydroxypyridinones (5) were even less active, with a >400-fold loss in activity when compared to 17. Analysis of the pKa of the phenolic and N-hydroxy protons in these compounds revealed that the donor oxygen in 5 is less basic (pKa ∼7.0 calculated;33 pKa = 7.51 reported)34 than the equivalent donor oxygen in compounds 16 (pKa ∼9.5 calculated)33, 17 (pKa ∼11.3 calculated)33, 18 (pKa ∼9.2 calculated)33, or 19 (pKa ∼7.5 calculated)33. This change in ligand basicity is likely the electronic driving force behind the variation in inhibitory activity, with the most basic ligand 17 being the most active. Interestingly, the overall inhibitory activity (pIC50) of these fragments was found to be linearly correlated to the calculated pKa of the bridging oxygen atom (r = 0.96, n = 6, p < 2×10−3; Figure 3). It was also found that catechol-based fragments, which contained no endocyclic heteroatoms (e.g., 7), were far less active than the parent pyrone or pyridinone fragments in spite of their high ligand basicity. Compound 7 is likely too basic to fully deprotonate at physiological pH (pKa ∼9.8, 14.3 calculated;33 pKa = 10.30, 13.48 reported)35 and hence may be unable to efficiently coordinate to the metal centers in the PAN active site. Compound 7 also ultimately coordinates as a trianionic species, as opposed to the other MBPs in Table 2, which all coordinate as dianions.19 This difference in overall charge of the final coordination complex (−2 vs −1) is substantial, and the vast disparity in activity between compounds 7 and 1 may also be attributed to the formation of a highly anionic coordination center.
Figure 3.

Inhibition values (pIC50) of MBP isologues of compound 1 plotted against the pKa of their bridging phenolic oxygen atom. A linear correlation (r = 0.96, n = 6, p < 2×10−3) was observed between pKa and pIC50, illustrating a ligand electronics-based justification for the observed disparity in inhibition values of otherwise structurally homologous inhibitors.
While hydroxypyridine and hydroxypyrimidine MBP scaffolds have been explored as inhibitors of endonucleases,13,19,21,22 no systematic study of the effects of their stereoelectronic differences on enzymatic inhibition has been performed previously. Surprisingly, the subtle differences in the electronic character of these structurally analogous MBPs has been shown to have a substantial impact on inhibitory activity. Similar relationships between small changes in ligand electronics and inhibitory activity have likewise not been well explored in other metalloenzyme systems;30 however, this new evidence would suggest that the exploration of such relationships would be extremely valuable in the development of metalloenzyme inhibitors.
Despite high structural similarity, a peculiar disparity in inhibitory activity was observed between several highly active MBPs (i.e., 1, 3, 17, 18) and compound 2. Compounds 1 and 2 are structurally nearly identical (the only difference is the position of the carboxylate moiety), but display a ∼100-fold difference in activity. A potential explanation is the steric pressure exerted on the carboxylic acid moiety by the α-methyl group of 2, which precludes an ideal coordination geometry. The difference in bridging atom type (phenolic vs. carbonyl oxygen) could also be an underlying reason for the disparity in activity, as has been previously suggested for related inhibitor molecules in HIV integrase (HIV IN).30 Structural determination using X-ray crystallography showed that 1 and 2 shared a similar mode of binding, except the electron density corresponding to the carboxylic acid moiety of compound 2 is diffuse above that of the Mn-2 metal ion (Figure 2). This suggests that the carboxylic acid does not coordinate to the metal center with a preferred orientation, is not co-planar with the aromatic ring, and only makes weak ionic interactions with the Mn-2 metal ion. To further evaluate this effect, compound 20 was synthesized (Table 3). Comparison of 17 and 20 shows a similar, ∼200-fold difference in activity as observed with compounds 1 and 2 giving evidence that steric pressure by a proximal methyl group is a central cause of the dramatic decrease in activity. To ensure that the disparity did not arise from a steric clash that was not observed crystallographically, compound 21 was synthesized. Compound 21 should exert the same steric pressure on any active site protein residue or water network as compound 20, but can also stabilize the carboxylic acid moiety in the proper orientation for metal binding through an intramolecular hydrogen bond. Compound 21 was found to exhibit activity similar to compounds 1 and 17. These data suggest that intramolecular steric pressure on coordinating moieties can cause dramatic differences in the ability of MBP ligands to effectively coordinate active site metal centers. Thus, care must be taken during structural elaboration of MBP leads that employ coordinating carboxylic acid moieties to prevent inadvertent perturbations to the desired modes of metal coordination. Despite the very strong binding enthalpy of coordination bonds, dative interactions are highly dependent on proper bond angle and orientation.36
Table 3.
Structure and activity of MBP inhibitor molecules.
| Number | Compound | IC50 (nM) | pIC50 | Number | Compound | IC50 (nM) | pIC50 | Number | Compound | IC50 (nM) | pIC50 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 20 | 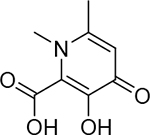 |
3900 ± 900 nM | 5.4 | 25 | 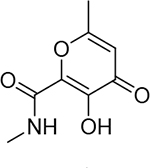 |
3800 ± 1100 nM | 5.4 | 30 | 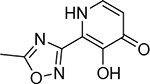 |
1200 ± 400 nM | 5.9 |
| 21 | 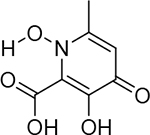 |
85 ± 17 nM | 7.1 | 26 | 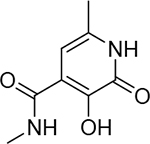 |
>50000 nM | 0 4.3 | 31 | 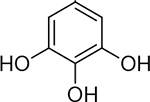 |
1700 ± 400 nM | 5.8 |
| 22 | 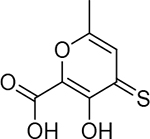 |
420 ± 60 nM | 6.4 | 27 | 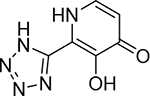 |
37 ± 8 nM | 7.4 | 32 |  |
11 ± 1.8 nM | 8.0 |
| 23 |  |
∼200000 nM | < 3.7 | 28 | 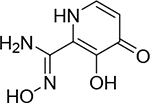 |
∼19000 nM | 4.7 | 33 | 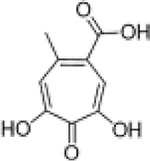 |
8.0 ± 1.1 nM | 8.1 |
| 24 | 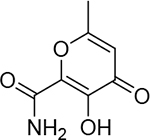 |
2700 ± 600 nM | 5.6 | 29 | 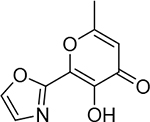 |
540 ± 80 nM | 6.3 |
Donor atom identity was also found to play a key role in MBP activity against PAN endonuclease. A small comparison of MBP fragments with similar structures, but different donor atom sets, is detailed in Table 3. It was found that oxygen donor atoms were preferred to sulfur donor atoms, with compound 22 displaying a 10-fold decrease in activity relative to 1. This trend is consistent with a hard-soft theory of Lewis acids and bases, where hard Mg2+ or Mn2+ Lewis acid metal centers are expected to form a more stable complexes with harder Lewis base oxygen donor atoms.37 Some MBPs containing nitrogen donor atoms were found to exhibit good activity against endonuclease (14), but only compounds containing borderline hard-soft Lewis basic pyridine-like aryl nitrogen atoms as the coordinating atom were found to be active. Compounds containing softer Lewis basic alkyl or aniline donor nitrogen atoms were found to have very little inhibitory activity against endonuclease. For example, 23 (IC50 >200μM, Table 3) was found to be a very poor inhibitor of PAN. While these trends are not unexpected based on principles of inorganic chemistry, such structural relationships are not often explored, optimized, or reported during the development of metalloenzyme inhibitors, despite the clear effect of incorporating the correct donor atom type on improving inhibition values.
Conversion of the coordinating carboxylic acid to N-H or N-alkyl amides was found to greatly impair inhibitory activity (24, 25) compared to the carboxylic acid (Table 3). This trend was observed with all acid-to-amide conversions, regardless of the activity of the parent compound (e.g., 18, 26). This transformation does not preclude metal coordination nor cause a conformational change in mode of binding; however, it does greatly alter the electronics of the coordinating moiety and the overall charge of the resulting coordination complex. This change to the electronic character of the coordinating carboxylate is presumed to be the underlying cause for the observed decrease in activity. This trend stands in contrast to that observed in HIV IN. Structurally related hydroxypyridinone and hydroxypyrimidinone inhibitors of HIV IN with good inhibitory activity have been reported that contain both carboxylic acid and amide metalcoordinating groups, and the inclusion of a metal-coordinating 4-fluorobenzyl amide is a characteristic motif found in many HIV IN inhibitors including the FDA-approved inhibitors raltegravir and dolutegravir.1,30 However, it should be noted that this 4-fluorobenzyl amide group also makes key interactions with active site nucleotides, and these favorable interactions may compensate for any potential loss in binding enthalpy from less favorable metal binding.
Isosteric replacement is a common practice in medicinal chemistry to improve activity or overcome pharmacokinetic liabilities of specific functional groups, such as carboxylic acids.38 The effects of isosteric replacement in metalloenzyme inhibitors have not been widely studied.39 To further probe the carboxylic acid moiety, a small series of hydroxypyrone and hydroxypyridinone core scaffolds containing carboxylic acid isosteres were synthesized.40 Compound 27 maintains an acidic tetrazole coordinating functional group and was found to have activity (IC50 = 37 nM) similar to that of 1. However, structurally similar compounds that were less acidic, such as amides 24 and 25, or azoles 29 and 30 (Table 3), were all found to be between 10- and 100-fold less active. Interestingly, the structurally similar but overall basic N-hydroxy amidine 28 (IC50 = 19 μM) was found to be substantially less active than all related isosteres, indicating a strong preference for acidic coordinating ligands in the PAN metal active site.
To illustrate the utility of a bioinorganic approach to SAR in the development of metalloenzyme inhibitors, we applied the obtained SAR to the development of other, chemically distinct PAN inhibitors. to highlight the applicability of a bioinorganic-guided inhibitor development paradigm beyond single targets or single chemotypes. The obtained SAR suggests that a tri-oxo, dianionic MBP that is aromatic and somewhat electron rich should be an ideal ligand for the PAN metal centers. With this information, we were able to synthesize an unrelated MBP, α-hydroxytropolone (32), that matches the proposed SAR, but is chemically distinct from 1 or 17. Indeed, 32 was found to inhibit PAN with an exceptional IC50 value of 11 nM (Table 3). Hydroxytropolone compounds 32 and 33 share similar ligand basicity as compound 17 despite belonging to a distinct chemotype. The calculated pKa of the phenolic oxygens of 32 are ∼10.1 and ∼12.7,33 respectively, and the calculated pKa of the phenolic oxygens of 33 are ∼9.8 and ∼11.9,33 respectively, compared to the phenolic oxygen of 17 (pKa = ∼11.2). The calculated pKa values of these compounds compared to their inhibitory activity are consistent with the trends observed with pyridinone carboxylate MPB inhibitors detailed in Table 2 and Figure 3, with relatively basic and electron rich donor oxygen atoms that result in exceptional inhibitory activity.
As compared to compounds 1 or 17, compound 32 is more rigid, in terms of metal-coordinating atoms; in compounds 1 or 17, the coordinating carboxylic acid moiety can rotate whereas in 32, the three coordinating oxygen atoms are geometrically constrained in the proper orientation for metal binding. The improved activity of 32 over 1 or 17 is likely due in part to the added rigidity of this MBP’s pre-organized binding pose for the two active site metal ions. The overall ligand geometry (i.e., two 5-membered chelate rings), which is likely mimetic of the shape and charge of the 5-membered phosphate ester transition state intermediate, may also explain the observed increase in inhibitory activity. It is also worth noting that α-hydroxytropolone derivatives have been identified as inhibitors of other dinuclear DNA/RNA processing metalloenzymes, such as HIV IN and HIV RNaseH.41 However, simple α-hydroxytropolone derivatives were shown to have 100- to 1000-fold less inhibitory activity against HIV IN or RNaseH than compound 32 displayed against PAN,41 highlighting that even similar metalloenzymes can display very specific ligand preferences and that ligand selectivity can be engineered into MBPs. Further analysis of several MBPs that share a similar chemotype with 32 indicate that the electronic, as opposed to steric, aspects of this compound are responsible for its very high activity. N-Hydroxyphthalamide (16), which possesses a similar donor triad, but is mono-anionic and more acidic, and pyrogallol (31), which also possesses a similar donor triad, but is trianionic and more basic, are both significantly less active than 32. Consistent with this SAR, gallic acid (9), was found to be much less active than the analogous α-hydroxytropolone compound 33. The structure of 33 bound to PAN (Figure S1) shows that the ligand binds to the active site as a bridging multidentate ligand, with the central carbonyl oxygen acting as a bridging atom and both hydroxyl moieties replacing the coordinating water molecules observed in the native structure. This binding mode is analogous to compounds such as 1. The carboxylic acid moiety of 33 was not observed to engage in any protein or water network interactions at the resolutions obtained, and the high inhibitory activity is attributed to the optimized metal coordination. The identification of this novel inhibitor scaffold demonstrates that metal-centric SAR is useful for predictive development of metalloenzyme inhibitors and that similar analysis can be broadly applicable for any metalloenzyme inhibitor development.
Finally, to validate the activity and selectivity of these highly active MBP inhibitors, cell-based antiviral activity assays and metalloenzyme cross-inhibition screens were performed.42 A subset of the MBP (compounds 1, 3, 17, 18, 22, 27, 32, and 33) were examined, which showed little antiviral activity in cell-based assays (Supporting Information; Figures S2 and S3), likely due to their high ionizability and low molecular weight.43,44 This result is not unexpected for these MBP fragments, which are intended as core scaffolds in FBDD for fragment growth efforts to achieve more complete inhibitors that can display cellular activity and drug-likeness. A crossinhibition screen of compound 1 was performed as a representative example of the series (Table S1). Compound 1 was screened against eight other relevant metalloenzymes, specifically: HIV IN, human carbonic anhydrase II, MMP-2, human glyoxalyase 1, NDM-1, HDAC-6, human arginase 1, and human methionine aminopeptidase 1. At a concentration of 200 μM, no substantial cross-inhibition was observed with 1, even against other dinuclear metalloenzymes, such as NDM1 (Zn2+), human arginase 1 (Mn2+), human methionine aminopeptidase 1 (Mn2+), and HIV integrase (Mg2+).30 Despite the small size of these fragment molecules, which increases the likelihood of off-target binding, optimized PAN inhibitors were found to display surprising good selectivity. The strong preference that this chemotype displays for PAN endonuclease over other metalloenzymes is evidence that target selectivity can be effectively engineered into MBP fragments by optimization for the specific coordination environment of a target metalloenzyme. This finding is corroborated by other work investigating metalloenzyme inhibitor selectivity,45,46 and stands in contrast to the widely held belief that MBP-containing compounds are intrinsically promiscuous. The data herein clearly suggest that the dogma that metal-coordinating metalloenzyme inhibitors may not be sufficiently selective over off-target metalloenzymes is not well substantiated.
To further illustrate that target selectivity can be engineered by MBP optimization, isologues of 1 were screened against the related dinuclear metalloenzymes human arginase 1 (Arg1), human methionine aminopeptidase 1 (MetAP1), and NDM-1 (Table 4). A total of 25 structurally related MBPs were screened against these three enzymes (200 μM MBP concentration) and PAN endonuclease (500 nM MBP concentration). Clear and distinct ligand preferences for each metalloenzyme are readily apparent (Table 4), despite the high degree of structural similarity between these fragments. This highlights the effects of varied ligand electronics on selectivity profiles in different metalloenzyme systems. Additional analysis shows that trends in donor atom preference remain, with compound 22 (which contains a sulfur donor atom) showing the expected preference for the soft Zn2+ metal centers of NDM-1 over the hard Mn2+ of Arg1 and MetAP1. Arg1 also clearly shows a preference for more highly basic MBP ligands, such as 7 and 28, which stands in contrast to the preferences displayed by PAN, despite both metalloenzymes employing dinuclear Mn2+ centers. These data further substantiate the hypothesis that activity and selectivity can both be achieved through proper optimization of metalligand interactions.
Table 4.
Single concentration cross-inhibition studies of MBPs against PAN endonuclease and three other dinuclear metalloenzymes. Final inhibitor concentration employed in assays is indicated in the table header for each enzyme. Percent inhibition is listed and colored from yellow to red with increasing inhibition values.
| Compound | PAN Endonuclease (500 nM) | Human Arginase 1 (200 μM) | Human MetAP1 Mn2+isoform (200 μM) | NDM-1 (200 μM) |
|---|---|---|---|---|
| 1 | 96 | 8 | 27 | 12 |
| 2 | 21 | 5 | 6 | 15 |
| 3 | 94 | 1 | 5 | 4 |
| 5 | 9 | 2 | 9 | 25 |
| 7 | 15 | 82 | 4 | 15 |
| 8 | 18 | 41 | 6 | 6 |
| 14 | 67 | 22 | 27 | 10 |
| 15 | 5 | 16 | 16 | 87 |
| 17 | 97 | 24 | 0 | 6 |
| 18 | 44 | 8 | 64 | 9 |
| 19 | 28 | 0 | 0 | 3 |
| 20 | 21 | 2 | 1 | 1 |
| 21 | 81 | 12 | 32 | 22 |
| 22 | 46 | 9 | 34 | 84 |
| 23 | 0 | 11 | 0 | 7 |
| 24 | 30 | 5 | 35 | 8 |
| 25 | 22 | 8 | 24 | 6 |
| 26 | 7 | 5 | 3 | 8 |
| 27 | 96 | 7 | 1 | 99 |
| 28 | 15 | 83 | 2 | 43 |
| 29 | 88 | 0 | 33 | 8 |
| 30 | 30 | 6 | 4 | 3 |
| 31 | 21 | 49 | 37 | 63 |
| 32 | 95 | 92 | 58 | 92 |
| 33 | 98 | 32 | 30 | 61 |
The results obtained in this SAR campaign indicate that both steric and electronic ligand effects must be explored to optimize MBP inhibitory activity for a given metalloenzyme microenvironment and that different metalloenzymes will exhibit different electronic and steric preferences at their active site. It was also found that small chemical modifications that might otherwise be considered innocuous in conventional inhibitor design (e.g., the position of a nonhydrogen bonding nitrogen atom in an aryl ring) were found to have a substantial effect on metal coordination and hence the overall activity of a MBP. These findings suggests that a more expanded view of which factors directly influence activity is absolutely necessary when developing and optimizing inhibitors of metalloenzymes as opposed to other classes of targets.
Conclusions
Based on the results described herein, SAR for metalloenzyme inhibitor development is useful for inhibitors that directly interact with active site metal ions. As opposed to probing primarily steric effects when preforming SAR analysis, the effects of modulating ligand electronics on metal coordination must also be considered. Upon metal binding, an MBP forms a coordination complex, which will display preferences for certain ligand interactions (like any inorganic coordination complex).47–49 In the field of metalloenzyme inhibitor development, this scenario has often been overlooked, due in part to a long-standing focus on perceived ‘privileged’ scaffolds such as hydroxamic acids,4 but also because principles of inorganic chemistry are often not considered during development. Nevertheless, these design principles can be readily applied to the development of metalloenzyme inhibitors to afford selectivity and activity, even at the fragment level (i.e., before hit-to-lead development). In this report, we have demonstrated that consideration of SAR derived from MBP electronic effects can have substantial impact on optimizing both activity and selectivity of metalloenzyme inhibitor fragments. As shown by this MBP-FBDD approach, expanding the toolkit used by chemists working on metalloenzyme inhibitor development will allow for improved progress toward highly active and selective inhibitors and may help to overcome the barriers that have limited the clinical success of this class of drug target.
Experimental Section:
General Experimental Details.
All reagents and solvents were obtained from commercial sources and used without further purification. All reactions, unless otherwise stated, were done under nitrogen atmosphere. Reactions were monitored using glass-backed silica TLC plates impregnated with a fluorescent indicator, absorbing at 254 nm. Silica gel column chromatography was performed on a CombiFlash Rf Teledyne ISCO system using hexane, ethyl acetate, methylene chloride, or methanol as eluent. Reverse phase column chromatography (C18 column) was performed on the same instrument using 0.1% formic acid in methanol, acetonitrile, or water as eluent. Separations were monitored by mass spectrometry via a Teledyne ISCO RF+ PurIon ESIMS or APCI-MS detector with 1 Da resolution. 1H and NMR spectra were obtained on a Varian (400 MHz) spectrometers in the Department of Chemistry and Biochemistry at U.C. San Diego. The purity of all compounds used in assays was determined to be ≥95% by HPLC-MS analysis. Standard resolution MS was performed either at U.C. San Diego Molecular Mass Spectrometry Facility or on the aforementioned Teledyne ISCO RF+ PurIon MS. HRMS analysis was performed using an Agilent 6230 Accurate-Mass LC-TOFMS located at the U.C. San Diego Molecular Mass Spectrometry Facility. Compounds 4–5, 7–14, 16, 19, 23, 31, and 34 were obtained from commercial suppliers. Compounds 1,50 2,51 3,6 15,6 32,52 and 3353 were previously reported. Xray diffraction data for co-crystal structures were collected either on an in-house Bruker X8 Proteum diffractometer at 100 K, using a Bruker Microfocus Rotating Anode (MicroSar FR-592) X-ray generator with a Bruker APEX II CCD detector at wavelength 1.54178 Ǻ (compound 2), or ALS 5.0.1 beamline using a single-crystal, cylindrically bent, Si(220) monochromator set to a wavelength of 0.977 Ǻ with a Pilatus 6M 25 Hz detector (compounds 1, 33), or at ALS 5.0.2 beamline using a double-crystal Si(111) monochromator set to a wavelength of 1.00 Ǻ with a Pilatus3 6M 25 Hz detector (compound 2). Statistics for data collection and refinement of the Xray crystal structures are listed in Table S2 and discussed in the supporting information.
Synthesis of Compounds 1, 17, and 33.
The chemical synthesis of the most active reported pyrone, pyridinone, and α-hydroxytropolone MBP molecules is described in Scheme 1. Full synthetic details of all other reported molecules is described in the Supporting Information.
Scheme 1.

aReagents and conditions: (a) thionyl chloride, CH2Cl2, rt, 6–8 h; (b) zinc dust, HCl, water, 70 °C, 4–6 h; (c) NaOH, formaldehyde, water/MeOH, 0–20 ºC, 18 h; (d) benzyl bromide, TBACl, 70 °C, 3 h; (e) NaCO3H, KI, TEMPO, TBACl, NaOCl, CH2Cl2/water, 5 °C, 2.5 h; (f) 5:5:1 HOAc/HCl/TFA, 40 °C, 18 h. (g) NH4OH, MeOH/water, 75 °C, 18–24 h; (h) methyl triflate, DCM/chloroform, reflux, 18 h; (i) ethyl propiolate, TEA, chloroform, microwave 100 ºC, 25 min; (j) BCl3, CH2Cl2, 0–20 °C, 30 min; (k) HBr/HOAc, neat, 95 °C, 6–8 h.
Allomaltol.
Kojic chloride (35) (10 g, 62.3 mmol) and water (75 mL) were stirred at 40 °C. Zinc dust (8.14 g, 125 mmol) was added and the reaction was stirred vigorously and heated to 60 °C. Concentrated HCl (15.4 mL, 187 mmol) was added dropwise via an addition funnel over ∼30 min. Effervescing hydrogen gas was observed. The slurry was left to stir for 2–3 h after HCl addition was complete, at which time the excess zinc was removed from the pale green reaction by hot filtration. The filtrate was adjusted to pH 1 and extracted with CH2Cl2 3×50 mL. The extracts were dried over magnesium sulfate and concentrated under vacuum, and the product was isolated by silica chromatography in 85% yield. 1H NMR (400 MHz, Acetone-d6): δ 7.82 (s, 1H), 6.21 (s, 1H), 2.28 (d, J = 0.5 Hz, 3H). ESI-MS Experimental: 125.22. Calculated for [C6H5O3]-: 125.03
3-Hydroxy-6-methyl-4-oxo-4H-pyran-2-carboxylic acid (1).
Compound 37 was taken up in a 5:5:1 mixture of concentrated HCl:HOAc:TFA and was stirred at room temperature for 48 h. After this time, acids were removed under high vacuum, the resultant solids were co-evaporated several times with methanol. The remaining solids were then purified by C18 chromatography eluting in a water/methanol system to yield the target compound as a white solid in 93 % yield. 1H NMR (400 MHz, DMSO-d6): δ 6.34 (s, 1H), 2.28 (s, 3H). 13C NMR (126 MHz, DMSO-d6): δ 174.66 (s), 166.15 (s), 164.07 (s), 149.36 (s), 135.69 (s), 112.39 (s), 20.07 (d, J = 4.3 Hz). HR-ESI-MS Experimental: 169.0141. Calculated for [C7H5O5]-: 169.0142. Δ = −0.6 ppm.
3-Hydroxy-6-methyl-4-oxo-1,4-dihydropyridine-2-carboxylic acid (17).
Compound 38 was taken up in a 5:5:1 mixture of concentrated HCl:HOAc:TFA and was stirred at room temperature for 48 h. Acids were then removed under high vacuum, and the resultant solids were co-evaporated several times with methanol. The remaining solids were then purified by C18 chromatography eluting in a water/methanol system to yield the target compound as a white solid in 74 % yield. 1H NMR (400 MHz, DMSO-d6): δ 6.92 (s, 1H), 2.48 – 2.38 (m, 3H). 13C NMR (126 MHz, DMSO-d6) δ 174.66 (s), 166.15 (s), 164.07 (s), 149.36 (s), 135.69 (s), 112.39 (s), 20.07 (d, J = 4.3 Hz). HR-ESI-MS Experimental: 168.0302. Calculated for [C7H6NO4]-: 168.0302. Δ = 0.0 ppm.
5-Methyl-2,7-dihydroxytropolone-4-carboxylic acid (33).
Compound 41 (200 mg, 0.84 mmol) was taken up in a 1:1 mixture of acetic acid and 50% HBr (15 mL) and was heated to 95 °C for 6–8 h. After this time, solvent was removed under high vacuum and the residual solids were purified by reverse phase chromatography to afford 33 in 56% yield. 1H NMR (400 MHz, CD3OD): δ 7.62 (s, 1H), 7.44 (s, 1H), 2.53 (s, 3H). 13C NMR (126 MHz, DMSO-d6): δ 170.69 (s), 169.02 (s), 160.40 (s), 157.70 (s), 138.08 (s), 132.49 (s), 124.22 (s), 119.76 (s), 25.11 (s). HR-ESI-MS Experimental: 195.0298. Calculated for [C9H7O5]-: 195.0299. Δ = −0.5 ppm.
Kojic chloride (35).
To a rapidly stirring suspension of kojic acid (34) (10 g, 70.4 mmol) in DCM (250 ml) at room temperature was added thionyl chloride (5.9 mL, 81 mmol), dropwise over the course of 25 min. Throughout the addition, the suspension tends to clump. When clumping occurred, the addition was paused to allow the solution to return to homogeneity. After 4 h of stirring, the suspension was filtered, and the solids were recrystallized from ethanol to afford kojic chloride as white needles in 91% yield. 1H NMR (400 MHz, DMSO-d6): δ 8.12 (s, 1H), 6.56 (s, 1H), 4.65 (s, 2H). ESI-MS Experimental: 159.14. Calculated for [C6H6ClO3]+: 159.99
3-(Benzyloxy)-2-(hydroxymethyl)-6-methyl-4H-pyran-4-one (36).
Allomaltol (1.26 g, 10 mmol) was added to a solution of NaOH (0.44 g, 11 mmol) in water (60 mL) and the mixture was cooled to 0 °C. Formaldehyde (37%, 0.83 mL, 11.1 mmol) was added dropwise maintaining the solution temperature at ∼0 °C. The mixture was stirred and allowed to warm to room temperature after addition of formaldehyde. A solid began to appear after 1.5 h, and methanol (30 mL) was added to solubilize the solids and the mixture was left stirring 18 h. The reaction was then heated to 40 °C and benzyl bromide (1.3 ml, 10.9 mmol) and tetra-N-butylammonium chloride (TABCl) (0.069 g, 0.25 mmol) were added and the reaction was heated to reflux for 3.5 h. The reaction mixture was then cooled and the pH of the solution adjusted to pH=1. A solution of sodium hydroxide (0.25 g) in 10 mL water was added. Benzyl bromide (0.200 mL) was added and the mixture was heated to reflux for 1.5 h and was then cooled to room temperature and methanol was removed by evaporation under vacuum. The mixture was then extracted with CH2Cl2 3×100 mL. The CH2Cl2 layer was washed with brine and dried over magnesium sulfate and evaporated yielding a red oil, which was purified by column chromatography to isolate the product in 61% yield. 1H NMR (400 MHz, DMSO-d6): δ 7.48 – 7.19 (m, 5H), 6.24 (s, 1H), 5.43 (t, J = 6.0 Hz, 1H), 4.98 (s, 2H), 4.23 (d, J = 6.0 Hz, 2H), 2.24 (s, 3H). ESI-MS Experimental: 247.27. Calculated for [C14H15O4]+: 247.09
3-(Benzyloxy)-6-methyl-4-oxo-4H-pyran-2-carboxylic acid (37).
3-(Benzyloxy)-2-(hydroxymethyl)-6-methyl-4H-pyran-4-one (36) (1.25 g, 5.08 mmol) was dissolved in CH2Cl2 (60 ml) and stirred. A solution of sodium bicarbonate (1.83 g, 21.8 mmol) and KI (0.084 g, 0.508 mmol) in water (60 mL) was added and the mixture was cooled in an ice bath to 0 °C. TEMPO (0.012 g, 0.076 mmol) and TABCl (0.056 g, 0.203 mmol) were added. Sodium hypochlorite (14.7 mL, 11.2 mmol) was added dropwise, in three portions, maintaining the internal temperature of the reaction below 7° C. After each portion, the pH was measured and 10% NaCO3H solution was added after each portion to maintain basic pH. Before the final portion of hypochlorite was added, a half weight of TEMPO and KI were added, followed by CH2Cl2 (15 mL). The reaction was stirred at ice bath temperatures for one hour (3 h total reaction time). The reaction mixture was then filtered and the filtrate was transferred to a separatory funnel where the resultant two layers were separated. The aqueous layer was mixed with a sodium thiosulfate solution to remove excess hypochlorite and was placed under reduced pressure for 5 min to remove residual organic solvent. The solution was then cooled in an ice-bath with rapid stirring, and concentrated HCl was added until the pH reached ∼1. An insoluble white solid appeared and was isolated by filtration to afford the product in 73% yield. 1H NMR (400 MHz, DMSO-d6): δ 7.42 (d, J = 7.3 Hz, 2H), 7.33 (ddd, J = 9.4, 7.5, 3.7 Hz, 3H), 6.39 (s, 1H), 5.08 (s, 2H), 2.27 (s, 3H). ESI-MS Experimental: 259.48. Calculated for [C14H11O5]−: 259.07.
3-(Benzyloxy)-6-methyl-4-oxo-1,4-dihydropyridine-2-carboxylic acid (38).
3-(Benzyloxy)-6-methyl-4-oxo-4H-pyran-2-carboxylic acid (37) (200 mg, 0.77 mmol) was taken up in a 1:1 mixture of water (10 mL) and methanol (10 mL) in a sealable vessel. Ammonium hydroxide (30%) (328 μL, 2.3 mmol) was added and the vessel was sealed and heated to 75 °C for 18–24 h. The reaction was then cooled and concentrated under vacuum, diluted with 1M HCl, and concentrated under vacuum to remove ammonium salts, and the resultant solids were purified by C18 column chromatography utilizing a MeOH/water system to afford 3-(benzyloxy)-6-methyl-4-oxo-1,4dihydropyridine-2-carboxylic acid (122 mg, 0.47 mmol, 69% yield) as a white powder. 1H NMR (400 MHz, CD3OD): δ 7.45 (t, J = 5.6 Hz, 4H), 7.31 (dd, J = 15.9, 6.3 Hz, 8H), 5.18 (s, 4H), 2.62 (s, 5H). ESI-MS Experimental: 259.66 Calculated for [C14H14NO4]+: 259.27.
5-Hydroxy-4-methoxy-2-methylpyrylium trifluoromethanesulfonate (39).
Allomaltol (5.4 g, 42.8 mmol) was stirred in a mixture of CH2Cl2 (10 ml) and chloroform (30.0 ml). To the mixture was slowly added fresh methyl trifluoromethansulfonate (7.1 mL, 64.2 mmol). The reaction mixture was then heated to reflux and vigorously stirred. Note: the reaction mixture may form two layers at lower temperatures. The mixture was heated to reflux for 18 h and the reaction progress was monitored by TLC (5% MeOH in CH2Cl2) and stained with Iron chloride. Iron test alone can give false negatives for reaction progress as some residual allomaltol always remains unreacted and the free phenol of the product stains at high concentrations. After ∼18 h the reaction mixture was cooled and evaporated to dryness under high vacuum to remove solvent and residual methyl triflate. The resultant dark oil was then taken up in minimal ethyl acetate and was heated to dissolve residual solids. The mixture was then cooled to −20 °C for 4 h to allow for crystallization. White needle-like crystals were isolated by filtration and a second crop of crystals were isolated by the same method from concentrated mother liquor. Drying the isolate white crystals afforded 5-hydroxy-4-methoxy-2-methylpyrylium triflate (9.4 g, 32.4 mmol, 76% yield). 1H NMR (400 MHz, DMSO-d6): δ 8.99 (s, 1H), 7.91 (s, 1H), 4.25 (s, 3H), 2.73 (s, 3H). ESI-MS Experimental: 141.45. Calculated for [C7H9O3]+: 141.05.
Ethyl 3-methoxy-5-methyl-2-oxo-8-oxabicyclo[3.2.1]octa-3,6-diene-6-carboxylate (40).
Compound 39 (250 mg, 0.861 mmol) was added to chloroform (1 mL) in a 10 mL microwave vessel. To the mixture was added ethyl propiolate (0.61 mL, 6.03 mmol) followed by TEA (0.181 mL, 1.03 mmol). The mixture was stirred briefly, sealed, and then heated in the microwave reactor at 125 °C for 25 min with stirring. Upon completion, the crude mixture was evaporated under high vacuum to remove ethyl propiolate and purified by silica chromatography employing a stepwise gradient from 6% to 12% ethyl acetate in hexanes, affording the titled product in 67% yield (130 mg, 0.546 mmol) as a light-yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.08 (d, J = 2.5 Hz, 1H), 6.08 (s, 1H), 5.01 (dd, J = 2.5, 0.5 Hz, 1H), 4.25 (qd, J = 7.1, 6.6 Hz, 2H), 3.55 (s, 3H), 1.77 (s, 3H), 1.32 (td, J = 7.1, 0.6 Hz, 3H). ESI-MS Experimental: 239.34. Calculated for [C12H15O5]+: 239.08.
Ethyl 7-methoxy-5-methyl-2-hydroxytropolone carboxylate (41).
Compound 40 (1.5 g, 6.30 mmol) was taken up in CH2Cl2 (20 mL) and was cooled to 0–5 °C on an ice bath with stirring. To the cooled solution was added boron trichloride solution (1M, 12.6 mL, 12.6 mmol) dropwise. After addition, the reaction mixture was removed from the ice bath and was allowed to warm to room temperature for 30 min, when the reaction was quenched by the addition of MeOH. Solvent was removed under high vacuum and the solids were co-evaporated with methanol three additional times and the resultant solids were purified by silica chromatography to afford 41 in 85% yield. 1H NMR (400 MHz, CD3OD): δ 7.49 (s, 1H), 7.08 (s, 1H), 4.35 (q, J = 7.1 Hz, 2H), 3.98 (s, 3H), 2.50 (s, 3H), 1.36 (t, J = 7.1 Hz, 3H). ESI-MS Experimental: 239.61. Calculated for [C12H15O5]+: 239.08.
Endonuclease Activity Assay.
Endonuclease activity assays were carried out in Black Costar 96well plates. Each well contained a total volume of 100 μL comprised of: buffer (20 mM Tris, 150 mM NaCl, 2 mM MnCl2, 10 mM β-mercaptoethanol, 0.2% Triton-X100, pH=8.0), influenza PA endonuclease (4 nM), inhibitor (various concentrations) in buffer, and fluorescent ssDNA-oligo substrate (200 nM). A single-stranded, 17-mer DNA substrate labeled with a 5′-FAM fluorophore and a 3′-TAMRA quencher ([6-FAM]AATCGCAGGCAGCACTC[TAM]) synthesized by Sigma-Aldrich was employed to measure endonucleic cleavage. Upon addition of the substrate, the change in fluorescence was measured over 45 min at 37 ºC (excitation: 485 nm; emission 528 nm). The gain was set to 100 and the first 10 min of data were excluded from the activity calculations. L-742,001 (IC50 value of 430 nM against Endo) was obtained from Sigma (catalog number SML1010) and utilized as a positive control. Dose-response curves were generated, fitted, and analyzed using Origin16 graphing software.
Other Protein Activity Assays.
A cross inhibition screen was performed on compound 1 against a battery of eight different metalloenzymes, including structurally related dinuclear metalloenzymes (Figure S1). Experiments were carried out according to literature protocols or were previously reported. Compound 1 was screened at a concentration of 200 μM against each enzyme. Percent inhibition, relative to positive and negative controls, is as follows: HIV IN 3′processing activity, no inhibition;30 HIV IN strand transfer activity, 37% inhibition;30 human carbonic anhydrase II (hCAII),36 no inhibition; MMP-2, 11% inhibition;6 human glyoxylase 1 (Glo-1), 11%;54 NDM-1, 13% inhibition;55 HDAC-6, no inhibition;45 human arginase 1, 8% inhibition;56 human methionine aminopeptidase 1, 27% inhibition.57
Supplementary Material
Acknowledgements:
The authors acknowledge Dr. Yongxuan Su (U.C. San Diego, Molecular Mass Spectrometry Facility) for aid with HR-MS and HPLC compound purity analysis. We also thank Dr. Curtis Moore, Dr. Milan Gembicky, the UC San Diego X-ray Facility, the J. P. Noel lab at the Salk Institute, and the staff at beam lines 5.0.1 and 5.0.2 at the Advanced Light Source. This work was supported by grants from the National Institutes of Health (R01 GM098435 to S.M.C.; R56 AI127371 to I.A.W.; F32GM125233 to C.N.M.), and by the University of California President’s Postdoctoral Fellowship (to C.N.M.). S.M.C. is a co-founder, has an equity interest, and receives income as member of the Scientific Advisory Board for Cleave Biosciences and is a co-founder, has an equity interest, and a member of the Scientific Advisory Board for Forge Therapeutics. Both companies may potentially benefit from the research results of certain projects in the laboratory of S.M.C. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies.
Abbreviations Used:
- SAR
structure-activity relationship
- MBP
metal-binding pharmacophore
- FBDD
fragment-based drug discovery
- PAN
Influenza RNA-dependent polymerase PA N-terminal endonuclease domain
- FRET
Forster resonance energy transfer
- LE
ligand efficiency
- HIV IN
human immunodeficiency virus integrase
- MMP-2
matrix-metalloprotease 2
- NDM-1
New Delhi metallo-β-lactamase 1
- Arg1
human arginase 1
- MetAP1
human methionine aminopeptidase 1
- TBACl
tetra-N-butylammonium chloride
- TEA
triethyl amine
- TEMPO
2,2,6,6-tetramethylpiperidine-1-oxyl
Footnotes
Ancillary Information:
Full descriptions of the chemical synthesis of reported compounds, protein expression, purification, and crystallography, and biochemical activity assays are given in the Supporting Information. Tables of Molecular Formula Strings for all reported compounds are also available as a CSV file. Statistics for data collection and refinement of the X-ray crystal structures are compiled in Supplemental Table S2 (Supporting Information). Coordinates and structure factors have been deposited in the PDB under the accession codes: 6DZQ (compound 1), 6DCY (compound 2), 6DCZ (compound 3), and 6E0Q (compound 33). Authors will release the atomic coordinates and structure factors immediately upon article publication.
References
- (1).Yang Y; Hu XQ; Li QS; Zhang XX; Ruan BF; Xu J; Liao CZ Metalloprotein Inhibitors for the Treatment of Human Diseases. Curr. Top. Med. Chem 2016, 16, 384–396. [DOI] [PubMed] [Google Scholar]
- (2).Chen AY; Adamek RN; Dick BL; Credille CV; Morrison CN; Cohen SM Targeting Metalloenzymes for Therapeutic Intervention. Chem. Rev 2018, 118, ASAP contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).White RJ; Margolis PS; Trias J; Yuan ZY Targeting Metalloenzymes: A Strategy that Works. Curr. Opin. Pharmacol 2003, 3, 502–507. [DOI] [PubMed] [Google Scholar]
- (4).Cohen SM A Bioinorganic Approach to Fragment-Based Drug Discovery Targeting Metalloenzymes. Acc. Chem. Res 2017, 50, 2007–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Whittaker M; Floyd CD; Brown P; Gearing AJH Design and Therapeutic Application of Matrix Metalloproteinase Inhibitors. Chem. Rev 1999, 99, 2735–2776. [DOI] [PubMed] [Google Scholar]
- (6).Jacobsen JA; Fullagar JL; Miller MT; Cohen SM Identifying Chelators for Metalloprotein Inhibitors Using a Fragment-Based Approach. J. Med. Chem 2011, 54, 591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Agrawal A; Johnson SL; Jacobsen JA; Miller MT; Chen LH; Pellecchia M; Cohen SM Chelator Fragment Libraries for Targeting Metalloproteinases. ChemMedChem 2010, 5, 195–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Wang X; Gao P; Menendez-Arias L; Liu X; Zhan P Update on Recent Developments in Small Molecular HIV-1 RNase H Inhibitors (2013–2016): Opportunities and Challenges. Curr. Med. Chem 2018, 25, 1682–1702. [DOI] [PubMed] [Google Scholar]
- (9).Cao L; Song W; DeClercq E; Zhan P; Liu X Recent progress in the research of small molecule HIV-1 RNase H inhibitors. Curr. Med. Chem 2014, 21, 1956–1967. [DOI] [PubMed] [Google Scholar]
- (10).Rogolino D; Carcelli M; Sechi M; Neamati N Viral Enzymes Containing Magnesium: Metal Binding as a Successful Strategy in Drug Design. Coord. Chem. Rev 2012, 256, 3063–3086. [Google Scholar]
- (11).Dayam R; Detig FX; Neamati N HIV-1 integrase inhibitors: 2003–2004 update. Medicinal Research Reviews 2006, 26, 271–309. [DOI] [PubMed] [Google Scholar]
- (12).Cotelle P Patented HIV-1 integrase inhibitors (1998–2005). Recent Pat Antiinfect Drug Discov 2006, 1, 1–15. [DOI] [PubMed] [Google Scholar]
- (13).Credille CV; Chen Y; Cohen SM Fragment-Based Identification of Influenza Endonuclease Inhibitors. J. Med. Chem 2016, 59, 6444–6454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Monod A; Swale C; Tarus B; Tissot A; Delmas B; Ruigrok RW; Crepin T; Slama-Schwok A Learning from Structure-Based Drug Design and New Antivirals Targeting the Ribonucleoprotein Complex for the Treatment of Influenza. Expert Opin. Drug Discov 2015, 10, 345–371. [DOI] [PubMed] [Google Scholar]
- (15).Dias A; Bouvier D; Crepin T; McCarthy AA; Hart DJ; Baudin F; Cusack S; Ruigrok RW The Cap-Snatching Endonuclease of Influenza Virus Polymerase Resides in the PA Subunit. Nature 2009, 458, 914–918. [DOI] [PubMed] [Google Scholar]
- (16).Boivin S; Cusack S; Ruigrok RWH; Hart DJ Influenza A Virus Polymerase: Structural Insights into Replication and Host Adaptation Mechanisms. J. Biol. Chem 2010, 285, 28411–28417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Rao P; Yuan WM; Krug RM Crucial Role of CA Cleavage Sites in the Cap-Snatching Mechanism for Initiating Viral mRNA Synthesis. Embo J.2003, 22, 1188–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Kowalinski E; Zubieta C; Wolkerstorfer A; Szolar OHJ; Ruigrok RWH; Cusack S Structural Analysis of Specific Metal Chelating Inhibitor Binding to the Endonuclease Domain of Influenza pH1N1 (2009) Polymerase. PLoS Pathog 2012, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).DuBois RM; Slavish PJ; Baughman BM; Yun MK; Bao J; Webby RJ; Webb TR; White SW Structural and Biochemical Basis for Development of Influenza Virus Inhibitors Targeting the PA Endonuclease. PLoS Pathog.2012, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Nakazawa M; Kadowaki SE; Watanabe I; Kadowaki Y; Takei M; Fukuda H PA Subunit of RNA Polymerase as a Promising Target for Anti-Influenza Virus Agents. Antiviral Res.2008, 78, 194–201. [DOI] [PubMed] [Google Scholar]
- (21).Sagong HY; Bauman JD; Patel D; Das K; Arnold E; LaVoie EJ Phenyl Substituted 4-Hydroxypyridazin-3(2H)-ones and 5-Hydroxypyrimidin-4(3H)-ones: Inhibitors of Influenza A Endonuclease. J. Med. Chem 2014, 57, 8086–8098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Bauman JD; Patel D; Baker SF; Vijayan RSK; Xiang A; Parhi AK; Martinez-Sobrido L; LaVoie EJ; Das K; Arnold E Crystallographic Fragment Screening and Structure-Based Optimization Yields a New Class of Influenza Endonuclease Inhibitors. ACS Chem. Biol 2013, 8, 2501–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Heo YA Baloxavir: First Global Approval. Drugs 2018, 78, 693–697. [DOI] [PubMed] [Google Scholar]
- (24).Schrodinger L; Version 1.8 ed. 2015.
- (25).Drake JW Rates of Spontaneous Mutation Among RNA Viruses. Proc. Natl. Acad. Sci. U. S. A 1993, 90, 4171–4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Crepin T; Dias A; Palencia A; Swale C; Cusack S; Ruigrok RWH Mutational and Metal Binding Analysis of the Endonuclease Domain of the Influenza Virus Polymerase PA Subunit. J. Virol 2010, 84, 9096–9104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Song MS; Kumar G; Shadrick WR; Zhou W; Jeevan T; Li ZM; Slavish PJ; Fabrizio TP; Yoon SW; Webb TR; Webby RJ; White SW Identification and Characterization of Influenza Variants Resistant to a Viral Endonuclease Inhibitor. P. Natl. Acad. Sci. USA 2016, 113, 3669–3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Ju H; Zhang J; Huang BS; Kang DW; Huang B; Liu XY; Zhan P Inhibitors of Influenza Virus Polymerase Acidic (PA) Endonuclease: Contemporary Developments and Perspectives. J. Med. Chem 2017, 60, 3533–3551. [DOI] [PubMed] [Google Scholar]
- (29).Iwai Y; Takahashi H; Hatakeyama D; Motoshima K; Ishikawa M; Sugita K; Hashimoto Y; Harada Y; Itamura S; Odagiri T; Tashiro M; Sei Y; Yamaguchi K; Kuzuhara T Anti-Influenza Activity of Phenethylphenylphthalimide Analogs Derived From Thalidomide. Bioorg. Med. Chem 2010, 18, 5379–5390. [DOI] [PubMed] [Google Scholar]
- (30).Agrawal A; DeSoto J; Fullagar JL; Maddali K; Rostami S; Richman DD; Pommier Y; Cohen SM Probing Chelation Motifs in HIV Integrase Inhibitors. P. Natl. Acad. Sci. USA 2012, 109, 2251–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Scott DE; Coyne AG; Hudson SA; Abell C Fragment-Based Approaches in Drug Discovery and Chemical Biology. Biochemistry 2012, 51, 4990–5003. [DOI] [PubMed] [Google Scholar]
- (32).Martin DP; Blachly PG; McCammon JA; Cohen SM Exploring the Influence of the Protein Environment on Metal-Binding Pharmacophores. J. Med. Chem 2014, 57, 7126–7135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Sanna D; Buglyo P; Biro L; Micera G; Garribba E Coordinating Properties of Pyrone and Pyridinone Derivatives, Tropolone and Catechol toward the VO2+ Ion: An Experimental and Computational Approach. Eur. J. Inorg. Chem 2012, 1079–1092. [Google Scholar]
- (34).Thipyapong K; Ruangpornvisuti V Isomers of Various Species of 1-hydroxy-2-pyridinone6-carboxylic Acid, their Proton Dissociation and Complexes with Cr(III) and Zn(II). J. Mol. Struct 2008, 891, 1–10. [Google Scholar]
- (35).Evanko CR; Dzombak DA Influence of Structural Features on Sorption of NOM-Analogue Organic Acids to Goethite. Environ. Sci. Technol 1998, 32, 2846–2855. [Google Scholar]
- (36).Dick BL; Patel A; McCammon JA; Cohen SM Effect of Donor Atom Identity on Metal-Binding Pharmacophore Coordination. J. Biol. Inorg. Chem 2017, 22, 605–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Pearson RG Hard and Soft Acids and Bases. J. Am. Chem. Soc 1963, 85, 3533. [Google Scholar]
- (38).Ballatore C; Huryn DM; Smith AB Carboxylic Acid (Bio)Isosteres in Drug Design. ChemMedChem 2013, 8, 385–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Dowell RI; Hales NH; Tucker H Novel Inhibitors of Prolyl 4-Hydroxylase .4. Pyridine-2-Carboxylic Acid Analogs with Alternative 2-Substituents. Eur. J. Med. Chem 1993, 28, 513–516. [Google Scholar]
- (40).Lassalas P; Gay B; Lasfargeas C; James MJ; Tran V; Vijayendran KG; Brunden KR; Kozlowski MC; Thomas CJ; Smith AB 3rd; Huryn DM; Ballatore C Structure Property Relationships of Carboxylic Acid Isosteres. J. Med. Chem 2016, 59, 3183–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Didierjean J; Isel C; Querre F; Mouscadet JF; Aubertin AM; Valnot JY; Piettre SR; Marquet R Inhibition of human immunodeficiency virus type 1 reverse transcriptase, RNase H, and integrase activities by hydroxytropolones. Antimicrobial Agents and Chemotherapy 2005, 49, 4884–4894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Wu NC; Grande G; Turner HL; Ward AB; Xie J; Lerner RA; Wilson IA In vitro evolution of an influenza broadly neutralizing antibody is modulated by hemagglutinin receptor specificity. Nature Communications 2017, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Hu Y; Gupta-Ostermann D; Bajorath J EXPLORING COMPOUND PROMISCUITY PATTERNS AND MULTI-TARGET ACTIVITY SPACES. Computational and Structural Biotechnology Journal 2014, 9, e201401003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Sturm N; Desaphy J; Quinn RJ; Rognan D; Kellenberger E Structural Insights into the Molecular Basis of the Ligand Promiscuity. Journal of Chemical Information and Modeling 2012, 52, 2410–2421. [DOI] [PubMed] [Google Scholar]
- (45).Chen Y; Cohen SM Investigating the Selectivity of Metalloenzyme Inhibitors in the Presence of Competing Metalloproteins. ChemMedChem 2015, 10, 1733–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Day JA; Cohen SM Investigating the Selectivity of Metalloenzyme Inhibitors. J. Med. Chem 2013, 56, 7997–8007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Haas KL; Franz KJ Application of Metal Coordination Chemistry to Explore and Manipulate Cell Biology. Chem. Rev 2009, 109, 4921–4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Szunyogh D; Gyurcsik B; Larsen FH; Stachura M; Thulstrup PW; Hemmingsen L; Jancso A Zn(II) and Hg(II) Binding to a Designed Peptide that Accommodates Different Coordination Geometries. Dalton Trans. 2015, 44, 12576–12588. [DOI] [PubMed] [Google Scholar]
- (49).Comba P; Schiek W Fit and Misfit Between Ligands and Metal Ions. Coord. Chem. Rev 2003, 238–239, 21–29. [Google Scholar]
- (50).Puerta DT; Mongan J; Tran BL; McCammon JA; Cohen SM Potent, Selective Pyrone-Based Inhibitors of Stromelysin-1. J. Am. Chem. Soc 2005, 127, 14148–14149. [DOI] [PubMed] [Google Scholar]
- (51).Yan YL; Cohen SM Efficient Synthesis of 5-amido-3-hydroxy-4-pyrones as Inhibitors of Matrix Metalloproteinases. Org. Lett 2007, 9, 2517–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Takeshita H; Mori A; Kusaba T An Improved Synthesis of 2,7-Dihydroxytropone (3-Hydroxytropolone). Synthesis-Stuttgart 1986, 578–579. [Google Scholar]
- (53).Meck C; Mohd N; Murelli RP An Oxidopyrylium Cyclization/Ring-Opening Route to Polysubstituted alpha-Hydroxytropolones. Org. Lett 2012, 14, 5988–5991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Perez C; Li J; Parlati F; Rouffet M; Ma Y; Mackinnon AL; Chou TF; Deshaies RJ; Cohen SM Discovery of an Inhibitor of the Proteasome Subunit Rpn11. J. Med. Chem 2017, 60, 1343–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Chen ALY; Thomas PW; Stewart AC; Bergstrom A; Cheng ZS; Miller C; Bethel CR; Marshal SH; Credille CV; Riley CL; Page RC; Bonomo RA; Crowder MW; Tierney DL; Fast W; Cohen SM Dipicolinic Acid Derivatives as Inhibitors of New Delhi Metallo-beta-lactamase-1. J. Med. Chem 2017, 60, 7267–7283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Di Costanzo L; Moulin M; Haertlein M; Meilleur F; Christianson DW Expression, Purification, Assay, and Crystal Structure of Perdeuterated Human Arginase I. Arch. Biochem. Biophys 2007, 465, 82–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Huang M; Xie SX; Ma ZQ; Huang QQ; Nan FJ; Ye QZ Inhibition of Monometalated Methionine Aminopeptidase: Inhibitor Discovery and Crystallographic Analysis. J. Med. Chem 2007, 50, 5735–5742. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.