

Abstract
Dual- or multi-target drugs have emerged as a promising alternative to combination therapies. Proteasome inhibitors (PIs) possess synergistic activity with histone deacetylase (HDAC) inhibitors due to the simultaneous blockage of the ubiquitin-degradation and aggresome pathways. Here, we present the design, synthesis, binding modes and anticancer properties of RTS-V5 as the first-in-class dual HDAC-proteasome ligand. The inhibition of both targets was confirmed by biochemical and cellular assays as well as X-ray crystal structures of the 20S proteasome and HDAC6 complexed with RTS-V5. Cytotoxicity assays with leukemia and multiple myeloma cell lines as well as therapy-refractory primary patient-derived leukemia cells demonstrated that RTSV5 possesses potent and selective anticancer activity. Our results will thus guide the structure-based optimization of dual HDAC-proteasome inhibitors for the treatment of hematological malignancies.
Graphical Abstract
INTRODUCTION
The approach “one drug multiple targets” or “multi-target drugs” is gaining major consideration in drug discovery and has been termed polypharmacology.1 Despite the highly significant therapeutic relevance of combination therapies, potential advantages of a targeted therapy based on a single drug acting through two or more independent modes of action include (a) a more predictable pharmacokinetic profile, (b) increased patient compliance, and (c) the simultaneous presence of the molecule in tissues where the active principles are intended to work.1
Histone deacetylases (HDACs) are clinically validated cancer targets and four inhibitors thereof (HDACi) have been approved by the FDA for cancer therapy.2 HDACi are characterized by a cap—linker—zinc-binding group pharmacophore model (Figure 1).3 Fortunately, the HDACi pharmacophore tolerates a variety of cap groups which allows scope for hybridization approaches.4 Consequently, the incorporation of a second pharmacophore in the cap region has been used to engineer several HDACi-based multi-target drugs.4 Notably, the dual kinase-HDAC inhibitors CUDC-101 and CUDC-907, the nitrogen mustard-HDACi hybrid tinostamustine, as well as the dual LSD1-HDAC inhibitor 4SC-202, are currently being investigated in clinical trials (Figure 1).4–5 In regards to combination therapy, the best investigated synergism of HDACi has been identified with proteasome inhibitors (PIs) leading to dual proteasome and aggresome blockage and apoptosis-induction due to the accumulation of misfolded proteins.6 However, to the best of our knowledge, no dual HDAC-proteasome inhibitor has been reported so far.
Figure 1.
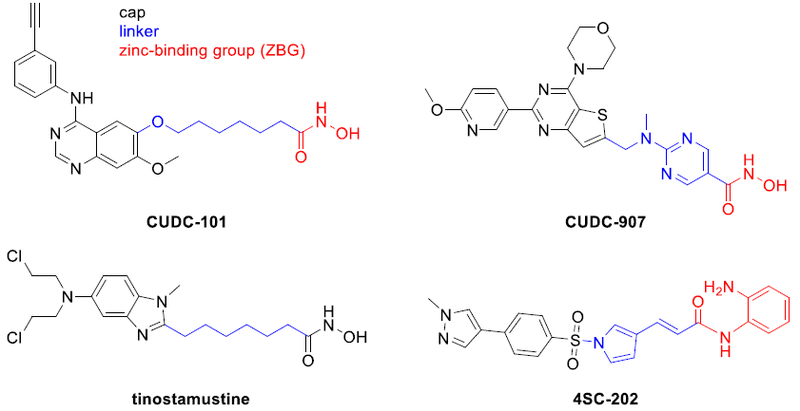
HDACi-based multi target drugs in clinical trials.
Herein, we present the design, synthesis, biological evaluation, and binding modes of RTS-V5 as the first-in-class dual HDAC-proteasome inhibitor.
RESULTS
Design and synthesis of RTS-V5.
PIs can be divided into covalent and non-covalent binders.7 We decided to focus on non-covalent scaffolds to suppress several drawbacks such as excessive reactivity, lack of specificity, and/or stability.8 Moreover, highly reactive electrophilic warheads might cause chemical incompatibilities with the typical HDACi zinc-binding groups (ZBGs) such as hydroxamic acids, aminoanilides or thioles. The first non-covalent acting PI was identified in the crystal structure of the yeast proteasome in complex with the natural product TMC-95A.9 In the following years, binding modes of TMC-95A derivatives10 as well as non-covalent linear peptide mimetics have been reported.11 In particular, a promising PI turned out to be compound ML16 (Figure 2) obtained from an elaborate study published by Blackburn and colleagues.11a The high affinity of ML16 and several analogs is primarily achieved by a P3-neopentyl-Asn residue (Figure 2). The comparison of currently available crystal structures of the proteasome in complex with peptidic ligands12 revealed that this bulky residue indeed represents a superb side chain to occupy the entire S3 specificity pocket of the chymotrypsin-like site of the 20S core particle. We, therefore, decided to use ML16 as a starting point for the design of dual HDAC-proteasome inhibitors. The S4 binding site does not resemble a pocket-like structure and a careful inspection of a series of X-ray structures of ML16 and its analogs indicated that the P4 residue is solvent exposed.7, 11a As a result, we aimed at the design of a HDAC-proteasome hybrid inhibitor by incorporating the HDACi part at the P4 position (Figure 2). The most obvious synergy between PIs and HDACi is derived from the inhibition of HDAC6.6, 13 Thus, we chose an N-hydroxybenzamide scaffold as HDACi part as this moiety provides HDAC6 selectivity.14
Figure 2.
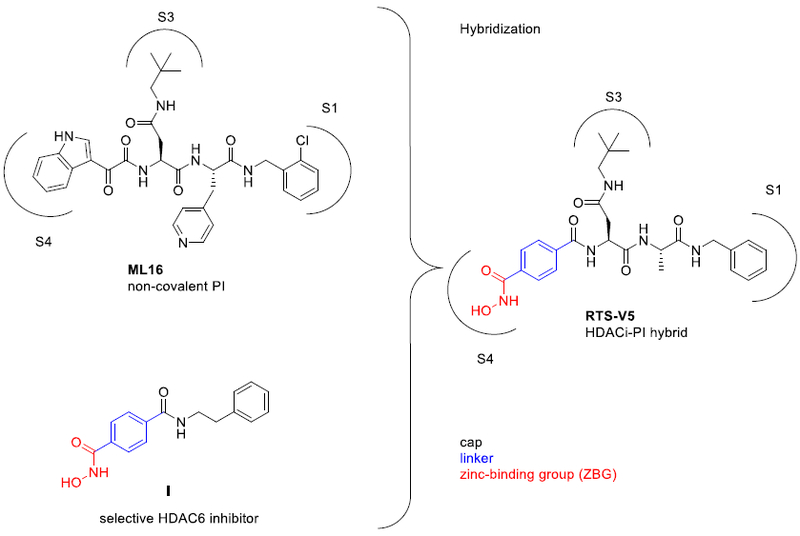
Design of RTS-V5 as the first-in-class dual HDAC-proteasome inhibitor.
Compound I (Figure 2) is a representative example of a selective HDAC6 inhibitor based on an N-hydroxybenzamide group. Furthermore, the solvent-exposed 4-picolyl group in the P2 position was replaced by a methyl group in order to reduce the molecular weight of the hybrid compound. Our hybridization strategy thus yielded the prototype HDAC-proteasome hybrid inhibitor RTSV5 (Figure 2).
RTS-V5 was synthesized as outlined in Scheme 1. The readily available building blocks 1 and 2 were combined by HATU-mediated coupling to generate dipeptide 3. Next, the deprotection of 3, followed by introduction of 4-((benzyloxy)carbamoyl)benzoic acid via another amide coupling reaction afforded the protected hydroxamic acid 5. Finally, catalytic hydrogenolysis of 5 provided the target compound RTS-V5.
Scheme 1.
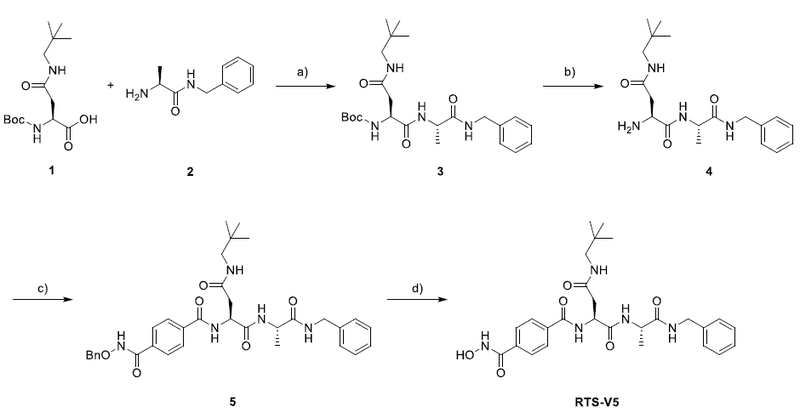
Synthesis of RTS-V5.a
a Reagents and conditions: a) HATU, DIPEA, DMF, rt, 16 h. b) TFA, CH2Cl2, rt, 4 h. c) 4-((Benzyloxy)carbamoyl)benzoic acid, HATU, DIPEA, DMF, rt, 24 h. d) Pd/C, H2, rt, 4 h.
RTS-V5 inhibits histone deacetylase and proteasomal activity.
RTS-V5 was evaluated for its ability to inhibit both histone deacetylase and proteasomal activity. First, we tested the compound in a biochemical assay for activity against recombinant HDAC6. The screening demonstrated potent submicromolar activity with an IC50 value of 0.27 μM (Figure 3a). To assess the selectivity of RTS-V5 for HDAC6, it was further tested for activity against all class I isoforms (HDACs 1, 2, 3, and 8, Figure 3a). Our analysis revealed that RTS-V5 has low activity against HDACs 1, 2, and 3. However, HDAC8 was blocked at submicromolar concentrations as well (HDAC8 IC50: 0.53 μM) which can be explained by the lowered rim of the catalytic channels of HDAC6 and HDAC8.15
Figure 3.
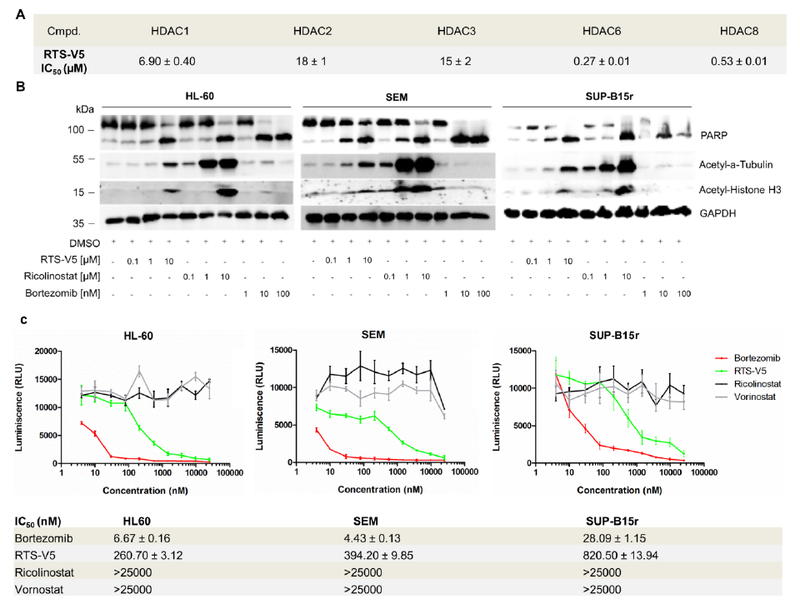
Functional specificity of RTS-V5 against HDAC6 and the proteasome. a) Inhibitory activities of compounds RTS-V5 against HDAC isoforms 1, 2, 3, 6, and 8. b), HL-60, SEM, and SUP-B15r cells were exposed to bortezomib, vorinostat, ricolinostat, and RTS-V5 at the indicated concentration for 24 h, after which lysates were immunoblotted with anti-acetyl-α-tubulin, anti-acetyl-Histone H3, poly (ADP-ribose) polymerase (PARP), and anti-glycerinaldehyd-3-phosphatdehydrogenase (GAPDH) antibodies. c) HL-60, SEM, and SUP-B15r cells were treated for 2 h with bortezomib, vorinostat, ricolinostat and RTS-V5 at concentrations ranging from 4 nM-25 μM. The proteasomal activity was measured after 2 h using the cell-based Proteasome-Glo Chymotrypsin-Like assay by taking Suc-LLVY-aminoluciferin (Succinyl-leucine-leucine-valinetyrosine-aminoluciferin) as a substrate. The compounds were printed on a 384-well plate using a randomization feature (n = 3).
In the following, we aimed to evaluate the inhibition of RTS-V5 against HDAC6 in a cellular environment. Therefore, we treated the acute myeloid leukemia (AML) cell line HL-60 as well as the B-cell precursor acute lymphoblastic leukemia (BCP-ALL) cell lines SEM and SUP-B15r (Tyrosine Kinase Inhibitor (TKI) - resistant)16 with RTS-V5, the preferential HDAC6 inhibitor ricolinostat and the FDA-approved proteasome inhibitor bortezomib for 24 h. Next, the cell lysates were immunoblotted with anti-acetyl-α-tubulin and acetyl-Histone H3 antibodies (Figure 3b). Compared to bortezomib, the treatment with RTS-V5 enhanced the expression of acetyl-α tubulin and acetyl-histone H3 in accordance to ricolinostat. Furthermore, RTS-V5 upregulated the expression of cleaved PARP, a marker of apoptosis, corresponding to ricolinostat and bortezomib (Figure 3b). The inhibition of proteasome activity by RTS-V5 was evaluated using a cell-based chymotrypsin-like Glo assay (Promega) by taking bortezomib as a positive control and ricolinostat (HDAC6i) or vorinostat (pan-HDACi) as a negative marker (Figure 3c). In all selected leukemic cell lines (HL-60, SEM and SUP-B15r), RTS-V5 blocked the chymotrypsin-like proteasome activity while vorinostat and ricolinostat were unable to inhibit the protease. Furthermore, it was shown that RTS-V5 acts specifically on the chymotrypsin-like activity, i.e. RTS-V5 was unable to inhibit the trypsin- and caspase-like proteasome activities (Figure S1a, S1b, Supporting Information). Thus, these results demonstrate that RTS-V5 is the first-in-class dual HDAC-proteasome inhibitor.
Co-crystal structures of RTS-V5 in complex with HDAC6 and the 20S proteasome.
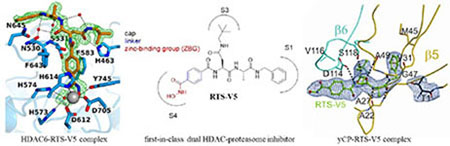
Encouraged by the functional specificity of RTS-V5 against HDAC6 and the proteasome, we set out to elucidate its binding modes in the vastly differing targets. First, the crystal structure of catalytic domain 2 (CD2) of Danio rerio (zebrafish) HDAC6 complexed with RTS-V5 was determined at 1.90 Å resolution (Rfree = 0.190, PDB ID: 6CW8, Table S1, Supporting Information). The crystal structures of zebrafish and human CD2 enzymes are identical,17a so zebrafish HDAC6 CD2 (henceforth simply “HDAC6”) serves as a more readily studied surrogate of the human enzyme. The crystal structure of the enzyme-inhibitor complex depicts no major conformational changes between the inhibitor-bound and unliganded states of the enzyme, and the root-mean-square (rms) deviation is 0.14 Å for 287 Cα atoms (unliganded HDAC6, PDB accession code 5EEM). Notably, there are two independent and essentially identical monomers in this crystal form (rms deviation = 0.15 Å for 299 Cα atoms). Electron density for RTS-V5 is generally well defined in both monomers (monomer A, Figure 4; monomer B, Figure S2, Supporting Information). Enzyme-inhibitor interactions are quite similar in both monomers, except for alternative interactions resulting from individual conformations of the benzyl-L-alanyl moiety in monomers A and B, respectively (Figure S3, Supporting Information).
Figure 4.
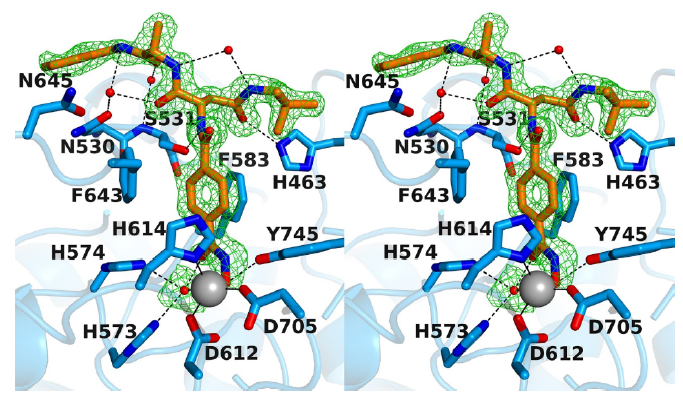
Stereoview of the Polder omit map of RTS-V5 bound to monomer A of HDAC6 (contoured at 3.0 σ) (PDB ID 6CW8). Atoms are color-coded as follows: C = orange (RTS-V5) or light blue (protein), N = blue, O = red, Zn2+ = gray sphere, solvent = red spheres. Metal coordination and hydrogen bond interactions are indicated by solid and dashed black lines, respectively. The Zn2+ coordination geometry is pentacoordinate square pyramidal.
In both monomers, the hydroxamate moiety of RTS-V5 coordinates to the active site Zn2+ ion in monodentate fashion, in a similar manner to that observed in complexes with other bulky phenylhydroxamate inhibitors such as HPOB, HPB, and ACY-1083.17 This binding mode is characterized by the coordination of the ionized hydroxamate hydroxyl group to Zn2+ (average Zn2+---O separation = 2.0 Å), while the hydroxamate carbonyl group accepts a hydrogen bond from a Zn2+-bound water molecule (average O---O separation = 2.8 Å). The Zn2+-bound N-O− group also accepts a hydrogen bond from Y745 (average O---O separation = 2.6 Å).
Beyond the Zn2+ coordination polyhedron, intermolecular interactions observed for RTS-V5 in both monomers contribute to inhibitor affinity and selectivity. The aromatic ring of the phenylhydroxamate is sandwiched between two fully conserved residues, F583 and F643. The para-substituted amide NH group forms a hydrogen bond with S531 on the L2 loop (average N---O separation = 3.0 Å). Notably, S531 is unique to HDAC6 and plays an important role in substrate binding.17a Thus, hydrogen bonds with S531 presumably contribute to HDAC6 inhibitor selectivity.
In monomer A, the carbonyl group of the neopentylamide moiety accepts a hydrogen bond from H463 in the L1 loop with an O---N separation of 2.9 Å; in monomer B, the O---N separation is 3.3 Å, which is slightly too long for, but perhaps within experimental error of, a hydrogen bond. Interestingly, H463 is unique to vertebrate HDAC6 isozymes, so this interaction may confer additional selectivity toward HDAC6.
Next, the crystal structure of the yeast 20S proteasome core particle (yCP) in complex with RTSV5 was determined to 2.5 Å resolution (Rfree = 0.217, PDB ID: 6H39, Table S2, Supporting Information). Intriguingly, the 2FO-FC electron density map displays the entire inhibitor molecule only bound to the chymotrypsin-like active site by adopting an antiparallel β-sheet structure (Figure 5a, b). Importantly, the HDACi hydroxamic acid ZBG of RTS-V5 is solvent exposed and thus, not in contact with protein residues. In agreement with our predictions, the complex structure depicts that the ligand acts non-covalently on the proteasome. Compared to standard inhibitors bound to the CP, such as the tripeptide aldehyde MG132,20 our study revealed that RTS-V5 is solely stabilized by Van der Waals interactions with its P1 benzene ring to Val31, Ala49 and predominantly Met45 of subunit β5, while its P3-neopentyl-Asn-moiety forms elaborate interactions with yβ5-Ala49 as well as Asp114, Val116 and Ser118 of yβ6 (Figure 5b, c). Notably, the electron density map uncovered the presence of an N-morpholino-ethane-sulfonic acid molecule (MES) in proximity to the inhibitor, which is derived from the crystallization buffer. Hereby, the sulfonate moiety of MES interacts with β5Gly47NH and hence, occupies the oxyanion hole, an area normally populated by the active residue of ligands such as i) the oxygen anion of the scissile peptide bond in its tetrahedral intermediate,21 or ii) functional groups of covalently bound inhibitors.22 Taken together, the crystallographic insights at the molecular resolution confirmed our structure-activity relationships demonstrating that the non-covalent proteasome inhibitor RTS-V5 fulfils elaborate interactions with the distinct specificity pockets of the chymotrypsin-like substrate binding channel, hereby generating target specificity.
Figure 5.
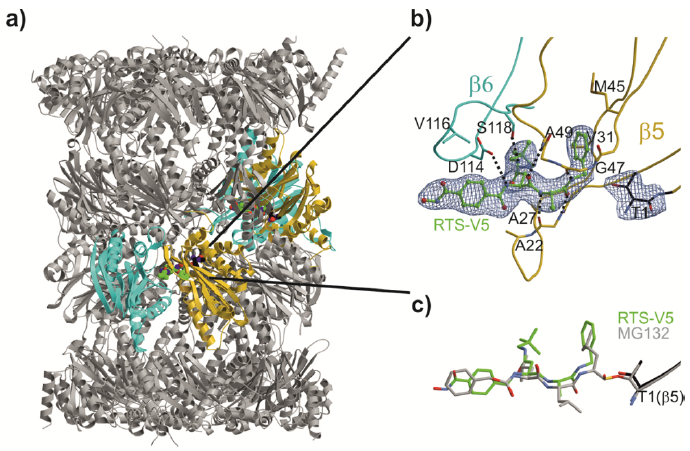
Yeast 20S proteasome in complex with RTS-V5. a) Cartoon representation of the yeast 20S proteasome core particle (yCP) in complex with RTS-V5 (PDB ID: 6H39). The decarboxylated ligand is presented as a sphere model, which is located at the intersection of the β–β′-rings. The molecule solely binds to the nonprimed substrate binding channel of the chymotrypsin-like active site, which is composed of subunits β5 (gold) and β6 (cyan), respectively. b) The 2FO-FC electron density map of the non-covalent inhibitor is illustrated as blue mesh and contoured to 1σ. Hydrogen bonds forming the antiparallel β-sheet between ligand and protein main chain residues are indicated by black dashed lines. RTS-V5 intensely interacts with the S1 and S3 sites, whereas the P2-Ala side chain is solvent-exposed. Amino acid numbering is according to Löwe et al.18 and Groll et al.19. c) Structural superposition of RTS-V5 with the covalently acting aldehyde inhibitor MG132 (PDB ID: 4NNN)20 depicts a uniform arrangement. The hemiacetal bond is highlighted in gold.
Specific cytotoxic activity of RTS-V5 against cancerous cells.
In order to investigate the anticancer properties of our dual HDAC-proteasome inhibitor, RTS-V5 was screened for cytotoxicity against a panel of leukemia and multiple myeloma cell lines using ricolinostat as a positive control (Table 1). Hereby, RTS-V5 showed comparable or higher cytotoxicity than ricolinostat with IC50 values in the single-digit micromolar to submicromolar concentration range. The highest activity of RTS-V5 was observed against the BCP-ALL cell line SEM (IC50: 0.89 μM). Our dual inhibitor was also active against TKI-resistant SUP-B15r and KCL-22r cells16 with IC50 values of 1.83 and 2.58 μM, respectively (Table 1). Due to its encouraging activity against chemosensitive and chemoresistant BCP-ALL cell lines, RTS-V5 was further tested for activity against primary BCP-ALL cells derived from four therapy-refractory patients (Patient 1 and 2 from initial diagnosis and Patient 3 and 4 from the relapse cohort) revealing IC50 values ranging from 1.51 to 5.23 μM (Table 1).
Table 1.
Cytotoxicity of RTS-V5 and ricolinostat against selected leukemia and multiple myeloma cell lines as well as patient-derived BCP-ALL cells.
| Cell line | Characteristic | RTS-V5 IC50[μM] |
Ricolinostat IC50[μM] |
|---|---|---|---|
| HL60 | AMLa | 1.55 ± 0.02 | 2.36 ± 0.07 |
| SEM | BCP-ALLb | 0.89 ± 0.01 | 1.61 ± 0.02 |
| SUP-B15 | BCP-ALLb | 1.77 ± 0.02 | 1.92 ± 0.07 |
| KCL-22 | CMLc | 3.14 ± 0.03 | 3.75 ± 0.09 |
| SUP-B15r | BCP-ALLb,d | 1.83 ± 0.03 | 3.54 ± 0.02 |
| KCL-22r | CMLc,d | 2.58 ± 0.04 | 3.38 ± 0.03 |
| RPMI-8226 | MMe | 1.75 ± 0.32 | 1.97 ± 0.12 |
| U266 | MMe | 2.04 ± 0.37 | 3.52 ± 0.38 |
| Patient 1 | BCP-ALLb | 2.06 ± 0.16 | 0.29 ± 0.01 |
| Patient 2 | BCP-ALLb | 1.84 ± 0.07 | 0.58 ± 0.04 |
| Patient 3 | BCP-ALLb | 5.23 ± 0.13 | 4.45 ± 0.14 |
| Patient 4 | BCP-ALLb | 1.51 ± 0.05 | 0.54 ± 0.01 |
Acute myeloid leukemia.
B-cell precursor acute lymphoblastic leukemia.
Chronic myeloid leukemia.
Imatinib resistant.
Multiple myeloma.
Next, we evaluated the cell viability in peripheral blood derived mononuclear cells (PBMCs) from healthy individuals. Strikingly, RTS-V5 showed only marginal toxicity against PBMCs (IC50 >25 μM, Figure S4, Supporting Information). In contrast, the reference compounds ricolinostat, vorinostat and bortezomib caused significant cytotoxicity against PBMCs with IC50 values in the single-digit micromolar (ricolinostat, vorinostat) or even submicromolar concentration range (bortezomib) (Figure S4, Supporting Information). These data emphasize that RTS-V5 possesses promising anticancer properties against several leukemic and multiple myeloma cell lines as well as patient-derived BCP-ALL cells. Intriguingly, RTS-V5 acts in an encouraging therapeutic window.
Based on these promising results, the biological properties of RTS-V5 were analyzed in more detail using the BCP-ALL cell line SEM. RTS-V5 significantly inhibited the proliferation of SEM cells at its IC50 or 2x IC50 concentrations, comparable to ricolinostat (Figure S5, Supporting Information). RTS-V5 induced apoptosis in SEM cells as illustrated by Annexin V and PI staining with ~ 5-fold increase in the apoptotic (Annexin+PI+) cells upon 48 h treatment (Figure 6a). Comparable results were observed in a caspase 3/7 enzyme-dependent apoptosis assay with an induction of ~ 6-fold of apoptotic cells at its IC50 concentration (Figure 6b). SEM cells were dose dependently arrested in S phase and a reduction in G2/M phase was observed after exposure to RTS-V5 for 48 h (Figure 6c). The exposure of RTS-V5 induced early differentiation of SEM cells in the liquid medium marked by the expression of (CD14 and CD11b) myeloid markers (Figure S6, Supporting Information) and moreover, 48 h exposure of RTS-V5 to SEM cells significantly reduced their colony forming capacity (Figure 6d). In addition, exposure of RTS-V5 to SEM cells induces the heat shock response or HSR (marked by the overexpression of Grp94, HSP70, HSP40, and HSP27 proteins), unfolded protein response or UPR (marked by the overexpression of BIP, ATF4, ATF6, and pJNK proteins) in response to combat the proteotoxic stress and autophagy (marked by the overexpression of LC3B and p62 proteins) (Figure 6e).
Figure 6.
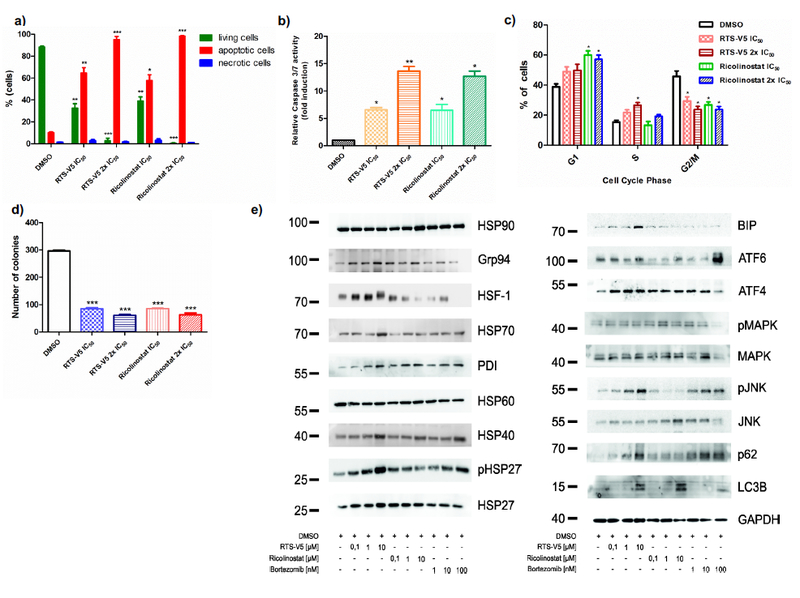
RTS-V5 as a potent inhibitor in a leukemic cell line. a) SEM cells were treated with RTS-V5 and ricolinostat at IC50 or at 2x IC50 concentration for 48 h. Subsequently dual staining was performed with annexin V/PI and measured by FACS. Viable cells (-ve for annexin V/PI) were analyzed if they are necrotic (+ve for PI) and are either in an early (+ve for annexin V) or in a late (+ve for both annexin/PI) apoptotic stage. The bar graph is depicting the percentage of living, apoptotic and necrotic cells after 48 h exposure to RTS-V5 or ricolinostat. b) SEM cells were treated with RTS-V5 for 48 h followed by determining the enzymatic activity of caspase 3/7 by applying a Glo assay (absorbance at 405 nm) to record the induction of apoptosis. c) SEM cells were treated with RTS-V5 for 48 h, and after propidium iodide staining cell cycle analysis was carried out by FACS. d) SEM cells were seeded in a semisolid methylcellulose-based medium after 48 h treatment with RTS-V5 or controls. Next, the impact of RTS-V5 on the differentiation ability of leukemic cells was evaluated. The bar-graphs depict the colonies counted after 14 days. e) SEM cells were treated with bortezomib, ricolinostat, and RTS-V5 at the indicated concentration for 18 h, after which the expression of proteins involved in the HSR, UPR and autophagy were analyzed by western blot analyses. The achieved values depicted in the Figure 6 are plotted as a bar graph. Columns depict the mean of 3 independent experiments (n = 3). Significance analyses of normally distributed data with variance similar between groups used paired, two-tailed student’s t-test. * p < 0.05, ** p < 0.005, *** p < 0.001.
Aggresomes are inclusion bodies produced in response to inhibition of the ubiquitin-proteasome machinery. HDAC6 together with the motor protein dynein is required to recruit cytotoxic, ubiquitylated proteins to aggresomes. The effect of RTS-V5 on the aggresome accumulation was studied using fluorescence microscopy and FACS upon staining with an aggresome dye (Figure 7a, b). The well-known proteasome inhibitors MG132 and bortezomib were used as positive controls whereas ricolinostat served as negative control. RTS-V5 significantly blocked aggresome accumulation at its inhibitory concentration as opposed to bortezomib and MG132, but in accordance with ricolinostat.
Figure 7.
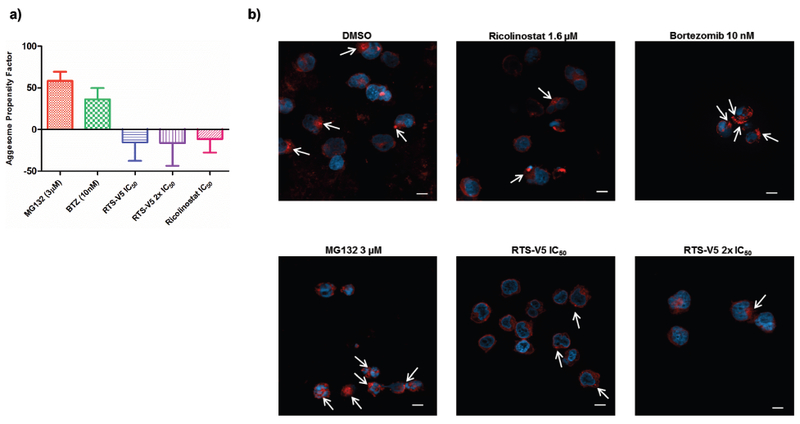
Effect of RTS-V5 on the aggresome accumulation. a) SEM cells were treated with the respective compounds at their indicated concentration for 18 h. In the following, the Enzo proteostat aggresome detection kit was used to stain the cells along with their DNA. Pictures were taken with a 63X objective using fluorescence microscopy. Scale = 10 μm. b) Treated SEM cells were analyzed by FACS upon staining with aggresome dye to determine the aggresome propensity factor according to the relative mean fluorescence intensity (MFI).
These results led us to conclude that RTS-V5 induces apoptosis and blocks proliferation, cell cycle, colony formation, and aggresome accumulation in the SEM cell line. Furthermore, the exposure of RTS-V5 leads to the activation of HSR and UPR. Hence, our findings together with the crystallographic and biochemical data demonstrate that RTS-V5 eradicates cancer cells by dual blockage of the aggresome-proteasome pathway.
DISCUSSION AND CONCLUSIONS
The ‘one-disease-one-drug’ paradigm has dominated drug development strategies for decades.23 However, the so-called magic bullets, molecules that exhibit high selectivity and potency for one target, are often not effective to treat multifactorial diseases such as cancer or neurological disorders.4a Consequently, combination therapy is a cornerstone of cancer therapy: the combination of anti-cancer drugs enhances efficacy compared to the mono-therapy approach because it modulates key pathways in an additive or even synergistic manner.24 Bortezomib is often given in combination with the pan-HDACi panobinostat. The combination of HDAC6i and proteasome inhibitors leads to increased α-tubulin acetylation as well as to accumulation of misfolded proteins.25 Misfolded proteins accumulate because both clearance routes, the proteasome and the aggresome pathway, are blocked; in turn, this leads to apoptosis of the cell.26 Thus, the simultaneous inhibition of both pathways could be of high clinical importance to combat hematological malignancies.
A phase I/II trial conducted for patients with relapsed or refractory multiple myeloma showed that therapy with ricolinostat as a single agent resulted in neither significant toxicity nor clinical responses.27 However, combination therapy with the proteasome inhibitor bortezomib and dexamethasone achieved a response rate of 37%.27 Similar results were reported in a study using a combination therapy including the proteasome inhibitor MG132 and vorinostat, which induced synergistic cytotoxicity in leukemia cells by downregulating BCR-ABL1 expression and by inducing intracellular ROS levels.28
In recent years, multi-target drugs have emerged as a powerful alternative to combination chemotherapy. Although several HDACi-based multi-target drugs have been described before, it is surprising that no dual HDAC-proteasome inhibitors have been reported to date. In this work, we have designed and synthesized RTS-V5 as a first-in-class dual HDAC-proteasome inhibitor. We have shown that this compound inhibits both HDAC6 and the chymotrypsin-like proteasome activity. RTS-V5 induces apoptosis, HSR, UPR and autophagy in the SEM cell line. Furthermore, it blocks cell cycle, colony formation, and aggresome accumulation. It is an encouraging finding that RTS-V5 displayed potent anticancer activity against a panel of chemosensitive, chemoresistant leukemic and multiple myeloma cell lines, as well as against therapy-refractory primary patient-derived leukemia cells without imposing toxicity against PBMC cells from healthy volunteers. In future studies the efficacy and toxicity of RTS-V5 or improved analogues will be investigated in in vivo models in comparison to a combination treatment with a HDAC6 inhibitor and a proteasome inhibitor (e.g. ricolinostat combined with bortezomib) in order to further evaluate the therapeutic potential of this promising new class of multi-target ligands.
To the best of our knowledge, this is also the first report of a dual target binder with accompanying co-crystal structures of complexes with both protein targets. The X-ray structures confirmed several important features that might lead to fewer side effects. The non-covalent and selective inhibition of chymotrypsin-like proteasome activity may explain the selective toxicity profile of RTS-V5 compared to covalent proteasome inhibitors such as bortezomib.12 Selective HDAC6 inhibition is clinically preferable since there is growing evidence that there are intrinsic toxic side effects associated with inhibition of HDAC1–3.29 The monodentate zinc-binding observed for RTS-V5 can be exploited by bulky phenylhydroxamate-based HDACi. However, binding of these inhibitors in the sterically constricted active site of HDAC1–3 would be disfavored.17b Thus, the monodentate zinc-binding mode is believed to contribute to the significantly reduced inhibition of HDAC1–3 and low toxicity of RTS-V5 compared to the pan-inhibitor vorinostat. Moreover, our determined crystal structures of RTS-V5 in complex with HDAC6 and the 20S proteasome will ultimately pave the way for the structure-based optimization of dual HDAC-proteasome inhibitors for advanced preclinical studies.
EXPERIMENTAL SECTION
Chemistry
General.
All reagents and solvents were purchased from commercial sources and used without further purification. Thin layer chromatography was carried out using Macherey-Nagel pre-coated aluminium foil sheets which were visualised using UV light (254 nm) and, in the case of hydroxamic acids, stained with a 1% solution of iron(III) chloride in methanol. 1H-NMR and 13CNMR spectra were recorded at room temperature on Bruker Avance III HD (400 MHz), Bruker Avance III (600 MHz), Bruker Avance DRX (500 MHz) and Varian/Agilent Mercury-plus (300 MHz) spectrometers. Chemical shifts (δ) are quoted in parts per million (ppm). All spectra were standardised in accordance with the signals of the deuterated solvent (DMSO-d6: δH = 2.50 ppm; δC = 39.5 ppm). Coupling constants (J) are reported in Hertz (Hz). Mass-spectra were measured by the Leipzig University Mass Spectrometry Service, using electrospray ionisation (ESI) on a Bruker Daltonics ESI-TOF micrOTOF. The uncorrected melting points were determined using a Barnstead Electrothermal 9100 apparatus. Analytical HPLC analysis were carried out using a Knauer Azura P 6.1L system equipped with P 6.1L (pumps), a Smartline UV detector 2600 and a Phenomenex Luna 5u C18(2) 1.8 μm particle (250 mm × 4.6 mm) column, supported by Phenomenex Security Guard Cartridge Kit C18 (4.0 mm × 3.0 mm). UV absorption was detected at 254 nm with a linear gradient of 10% B to 100% B within 20 min. HPLC-grade water (solvent A) and HPLC-grade acetonitrile (solvent B) were used for elution at a flow rate of 1 mL/min. Both solvents were enriched with 0.1% TFA. The purity of the final compound was at least 95%. The synthesis of 4-((benzyloxy)carbamoyl)benzoic acid is described in the Supporting Information.
tert-Butyl ((S)-1-(((S)-1-(benzylamino)-1-oxopropan-2-yl)amino)-4-(neopentylamino)-1,4-dioxobutan-2-yl)carbamate (3).
tert-Butyl (S)-(1-benzylamino)-1-oxopropan-2-yl)carbamate (6.64 g, 23.85 mmol, 1 eq) was dissolved in a mixture of trifluoroacetic acid/CH2Cl2 (1:2, 30 mL) and stirred at room temperature for 4 h. After completion of the reaction, the solution was basified (pH ≈ 9) using sat. sodium carbonate solution. The mixture was extracted with CH2Cl2 (3 × 20 mL) and washed with 1M sodium hydroxide solution (3 × 20 mL) and brine (3 × 20 mL). Subsequently, the collected organics were dried over sodium sulfate and the solvent was removed under reduced pressure to yield compound 2 as a crude product. The crude product 2 (1.9 g, 10.66 mmol, 1.1 eq) was added to a mixture of 1 (2.93 g, 9.69 mmol, 1 eq), diisopropylethylamine (1.65 mL, 9.69 mmol, 1 eq), and HATU (3.68 g, 9.69 mmol, 1 eq) in 10 mL DMF and stirred at room temperature for 16 h. The mixture was extracted with CH2Cl2 (3 × 15 mL) and washed with H2O (3 × 15 mL), citric acid (3 × 15 mL) and sat. sodium carbonate solution (3 × 15 mL). The combined organics were dried over sodium sulfate and the solvent was removed under reduced pressure. Product 3 was crystallized from n-hexane and ethyl acetate. White solid; 73% yield; mp. 143–145 °C; 1H-NMR (400 MHz, DMSO-d6) δ 8.45 (t, J = 5.4 Hz, 1H, NH), 8.02 (d, J = 7.4 Hz, 1H, NH), 7.83 – 7.67 (t, J = 5.4 Hz, 1H, NH), 7.37 – 7.22 (m, 5H, arom. H), 6.91 (d, J = 6.9 Hz, NH), 4.36 – 4.19 (m, 4H, CH, CH, CH2), 2.93 – 2.76 (m, 2H, CH2), 2.75 – 2.57(m, 2H, CH2), 1.36 (s, 9H, tBu), 1.24 (d, J = 7.0 Hz, 3H, CH3), 0.80 (s, 9H, tBu) ppm; 13C-NMR (101 MHz, DMSO-d6) δ 172.0, 171.0, 169.6, 155.0, 139.3, 128.2, 127.0, 126.7, 78.2, 51.5, 49.7, 48.4, 42.0, 37.7, 31.9, 28.1, 27.2, 18.2 ppm; HRMS (m/z): M– calcd. for C24H37N4O5, 461.2769; found, 461.2755.
(S)-2-Amino-N1-((S)-1-(benzylamino)-1-oxopropan-2-yl)-N4-neopentylsuccinamide (4).
Compound 3 (340 mg, 0.73 mmol, 1 eq) was dissolved in a mixture of trifluoroacetic acid/CH2Cl2 (1:2.25, 13 mL) and stirred at room temperature for 4 h. After completion of the reaction, the mixture was basified (pH ≈ 9) using sat. sodium carbonate solution. The resulting solution was extracted with CH2Cl2 (3 × 20 mL) and washed with brine (1 × 10 mL). The collected organics were dried over magnesium sulfate and the solvent was removed under reduced pressure. The crude product was recrystallized from methanol and diethyl ether to yield compound 4. White solid; 75% yield; mp. 160–163 °C; 1H-NMR (400 MHz, DMSO-d6) δ 8.49 (t, J = 5.8 Hz, 1H, NH), 8.32 (d, J = 6.5 Hz, 1H, NH), 7.97 (t, J = 5.6 Hz, 1H, NH), 7.36 – 7.15 (m, 5H, arom. H), 4.35 – 4.11 (m, 3H, CH, CH2), 3.70 – 3.61 (m, 1H, CH), 2.84 (d, J = 6.2 Hz, 2H, CH2), 2.68 – 2.53 (m, 2H, CH2), 1.26 (d, J = 7.1 Hz, 3H, CH3), 0.81 (s, 9H, tBu) ppm; 13 C-NMR (75 MHz, DMSO-d6) δ 172.0, 171.9, 170.1, 169.9, 139.3, 128.2, 127.0, 126.7, 51.1, 49.7, 48.3, 42.0, 31.8, 27.2, 18.3 ppm; HRMS (m/z): MNa+ calcd. for C19H30N4NaO3, 385.2210; found, 385.2210.
N1-((S)-1-(((S)-1-(Benzylamino)-1-oxopropan-2-yl)amino)-4-(neopentylamino)-1,4-dioxobutan-2-yl)-N4-(benzyloxy)terephthalamide (5).
A mixture of compound 4 (95 mg, 0.26 mmol, 1 eq), ((benzyloxy)carbamoyl)benzoic acid (84 mg, 0.31 mmol, 1.2 eq) and HATU (118 mg, 0.31 mmol, 1.2 eq) was suspended in DMF (3 mL) and diisopropylethylamine (53 μL, 0.31 mmol, 1.2 eq) was added. The resulting solution was stirred at room temperature for 24 h. After completion of the reaction, the solvent was removed under reduced pressure and the remaining solid was washed with sat. sodium bicarbonate solution (2 × 15 mL), 10% HCl (2 × 15 mL) and diethyl ether (2 × 15 mL). The crude product was recrystallized from methanol and diethyl ether to yield compound 5. White solid; 65% yield; mp. 232–236 °C; 1H-NMR (400 MHz, DMSO-d6): δ 11.90 (s, 1H, NH), 8.70 (d, J = 7.6 Hz, 1H, NH), 8.43 (t, J = 6.1 Hz, 1H, NH), 8.25 (d, J = 7.4 Hz, 1H, NH), 7.94 – 7.87 (m, 2H, arom. H), 7.84 – 7.78 (m, 2H, arom. H), 7.48 – 7.17 (m, 10H, arom. H), 4.94 (s, 1H, OCH2), 4.82 – 4.73 (m, 1H, CH), 4.34 – 4.19 (m, 3H, CH2, CH), 2.89 – 2.63 (m, 4H, CH2, CH2), 1.26 (d, J = 7.1 Hz, 3H, CH3), 0.78 (s, 9H, tBu) ppm; 13C-NMR (101 MHz, DMSO-d6): δ 172.0, 170.7, 169.7, 165.4, 163.7, 139.4, 136.5, 135.8, 134.7, 129.0, 128.4, 128.2, 127.6, 127.1, 127.0, 126.7, 77.1, 50.9, 49.7, 48.6, 42.0, 37.5, 31.9, 27.2, 18.0 ppm; HRMS (m/z): M– calcd. for C34H40N5O6, 614.2984; found, 614.2979.
N1-((S)-1-(((S)-1-(Benzylamino)-1-oxopropan-2-yl)amino)-4-(neopentylamino)-1,4-dioxobutan-2-yl)-N4-hydroxyterephthalamide (RTS-V5).
Compound 5 (50 mg, 0.08 mmol, 1 eq) was dissolved in 5 mL MeOH and Pd(C) (5 mg, 10% wt, 4.70 μmol, 0.06 eq) was added. The mixture was stirred under hydrogen atmosphere at room temperature for 4 h. After completion of the reaction, the mixture was filtered over celite. The solvent was removed under reduced pressure and the product RTS-V5 was crystallized from n-hexane and ethyl acetate. White solid; 98% yield; mp. 220 °C (decomp.); tR: 10.67 min, purity: 95%; 1H-NMR (500 MHz, DMSO-d6): δ 11.31 (bs, 1H, OH), 9.10 (bs, 1H, NH), 8.67 (d, J = 7.6 Hz, 1H, NH), 8.40 (t, J = 5.8 Hz, 1H, NH), 8.20 (d, J = 7.3 Hz, 1H, NH), 7.91 – 7.87 (m, 2H, arom. H), 7.84 – 7.80 (m, 2H, arom. H), 7.32 – 7.19 (m, 5H, arom. H), 4.83 – 4.72 (m, 1H, CH), 4.34 – 4.21 (m, 3H, CH2, CH), 2.91 – 2.79 (m, 2H, CH2), 2.77 – 2.63 (m, 2H, CH2), 1.26 (d, J = 7.1 Hz, 3H, CH3), 0.78 (s, 9H, tBu) ppm; 13C-NMR (126 MHz, DMSO-d6): δ 171.9, 170.5, 169.6, 165.5, 139.2, 136.0, 135.2, 128.1, 127.3, 126.9, 126.7, 126.6, 50.9, 49.7, 48.5, 42.0, 37.4, 31.7, 27.1, 17.9 ppm; HRMS (m/z): MH+ calcd. for C27H36N5O6, 526.2660; found, 526.2669.
Biological evaluation
Cell culture.
SEM, HL60, KCL22, K562 leukemic cell lines were cultured in RPMI1640 supplemented with 10% FCS and maintained at 37°C with 5% CO2, except for SUP-B15 (BCRABL1) BCP-ALL cell line which was cultured in McCoy’s 5A supplemented with 20% of FCS (DSMZ, Braunschweig, Germany). Mononuclear cells (MNC) were isolated by Ficoll density gradient centrifugation using standard procedures and later cultured in Mononuclear Cell Medium (PromoCell, Heidelberg, Germany). CD34+ cells were later sorted from these MNC using MACS (Miltenyi Biotec, Bergisch Gladbach, Germany). Primary patient samples were obtained from newly diagnosed patients or from relapse after informed consent approval of the local ethics committee and were cultured either in Stemline® II Hematopoietic Stem Cell Expansion Medium (Sigma-Aldrich) or in Mononuclear Cell Medium (PromoCell)
CellTiter-Glo® luminescent cell viability assay.
CellTiter-Glo® Luminescent Cell Viability Assay (Promega, Madison, USA) was performed to determine the IC50 values for every cell line. Inhibitors were printed on white 384-well plates (Thermo Fisher Scientific, Waltham, USA) with increasing concentrations (50 nM-25 μM) by using a digital dispenser (D300e, Tecan, Männedorf, Switzerland). Cell viability was monitored after 72 h using CellTitre-Glo luminescent assay using a Microplate reader (Spark®, Tecan). IC50 for the compounds were determined by plotting raw data (normalized to controls) using sigmoid dose curve and non-linear regression (GraphPad Prism Inc., San Diego, CA).
Proteasome activity assay.
To analyze if treatment with RTS-V5 leads to decreased proteasome activity, chymotrypsin-like, trypsin-like, and caspase-like protease activities associated with the proteasome complex were measured. Therefore, Cell-Based Proteasome-Glo™ assay kits (Promega, Madison, USA) were used which contain luminogenic substrates that are recognized and cleaved by the proteasome into aminoluciferin. Aminoluciferin is consumed by Ultra-Glo™ luciferase, producing a luminescent signal correlating to proteasome activity. Luminescence was measured afterwards using a Microplate reader (Spark®, Tecan).
Caspase assay.
Caspase-Glo® 3/7 assay was used to show the impact of the inhibitors on the activity of caspase-3 and −7 in the cells. Caspase-3 and −7 are key players in apoptosis. The kit contains a luminogenic substrate of caspase-3 and −7 with a DEVD sequence. This sequence can be recognized and cleaved by caspase-3 and −7. The cleavage results in a luminescent signal which is proportional to the amount of caspase activity. Cells were seeded and treated with the IC50 or 2x IC50 concentrations of the inhibitors for 48 h and later the luminescence was measured with a Microplate reader (Spark®, Tecan).
Proliferation assay.
To investigate the influence of the inhibitors on the proliferation, cells were counted after every 24 h interval through trypan exclusion method using automated cell counter (Vi-CELL™ XR -Beckman Coulter).
Annexin V staining.
For evaluating apoptosis, cells treated with respective compounds or control for 48 h were stained with Annexin V and PI and later subjected to FACS, following supplier’s guidelines (Invitrogen, Carlsbad, CA, USA).
Cell cycle analysis.
To investigate if cells treated with the inhibitors show differences in the cell cycle progression as compared to untreated cells, cell cycle analysis was performed. Therefore, cells were permeabilized and DNA was stained with PI which binds stoichiometric, i.e. proportional to the amount of DNA present in the cell. Fluorescence was measured by flow cytometry with FACSCalibur (Becton Dickinson, Heidelberg, Germany).
Western blotting.
Cell lysates were generated after 24 h treatment with the respective inhibitors and later immunoblotted using anti-PARP (# 9542), anti-Acetyl-α-tubulin (# 5335), anti-Histone H3 (# 9677), anti-HSP90 (# 4877), anti-Grp94 (# 2104), anti-HSF-1 (# 4356), anti-HSP70 (# 4872), anti-PDI (# 2446), anti-HSP60 (# 12165), anti-HSP40 (# 4871), anti-pHSP27 (# 9709), anti-HSP27 (# 2402), anti-BIP (# 3177), anti-ATF6 (# 65880), anti-ATF4 (# 11815), anti-pMAPK (# 4370), anti-MAPK (# 4695), anti-pJNK (# 4668), anti-JNK (# 9252), anti-p62 (# 5114), anti-LC3B (# 3868) and anti-GAPDH (# 2118) (Cell Signaling Technology, Danvers, MA).
Differentiation assay.
Healthy cells differentiate into specialized cells when they mature. In contrast, cancer cells remain undifferentiated or poorly differentiated to maintain their ability of fast replication. Differentiation assay was performed to analyze if treatment with the inhibitors leads to increased differentiation of the cells. Therefore, antibodies against specific surface molecules that cells express depending on their stage of differentiation were used as differentiation markers. FACS was performed on FACSCalibur (Becton Dickinson, Heidelberg, Germany) by using fluorochrome coupled monoclonal antibodies (mAbs) along with matched isotype controls: anti-CD11b (Bear1) and anti-CD14 (RMO52) (Beckman Coulter).
CFU assay.
Colony forming unit assays (CFU) were performed initially treating the cells in the liquid medium for 24 h and later the treated cells were seeded in the semisolid methylcellulose-based medium containing respective compounds or control. Colonies were counted after 14 days.
Aggresome detection assay.
As RTS-V5 simultaneously blocks the 20S proteasome and HDAC6 we planned to evaluate its effect on aggresome production using a proteostat aggresome detection kit (Enzo Life Sciences). Cells were labeled using an aggresome detection kit following the manufacturer’s guidelines. After 18 h treatment with the respective compounds the aggresomes were detected using either fluorescence microscopy or FACS.
HDAC IC50 profiling.
The in vitro inhibitory activity of RTS-V5 against five human HDAC isoforms (1, 2, 3, 6, and 8) were determined at Reaction Biology Corp. (Malvern, PA) with a fluorescence-based assay according to the company’s standard operating procedure using RHKK(Ac)AMC (HDACs 1, 2, 3, and 6) or RHK(Ac)K(Ac)AMC (HDAC8) as substrates. The IC50 values were determined in duplicate using 10 different concentrations with 3-fold serial dilution starting at 100 μM. TSA (HDAC1 IC50: 9.4 nM, HDAC2 IC50: 26.7 nM, HDAC3 IC50: 12.7 nM, HDAC6 IC50: 8.5 nM, and HDAC8 IC50: 609 nM) was used as reference compound.
X-ray crystallography
Experimental details for the X-ray crystal structure determination of HDAC6 and the yeast 20S proteasome in complex with RTS-V5 can be found in the Supporting Information.
Supplementary Material
ACKNOWLEDGMENT
We thank Nicholas Porter for helpful scientific discussions. Additionally, we thank Alexei Soares and the beamline staff at the National Synchrotron Light Source II (NSLS-II) for assistance with data collection using AMX beamline 17-ID-1. NSLS-II is a U.S. Department of Energy (DOE) Office of Science User Facility operated by Brookhaven National Laboratory under Contract No. DE-SC0012704. We are grateful to the staff of the beamline X06SA at the Paul-Scherrer-Institute, Swiss Light Source, Villigen (Switzerland), for assistance during data collection of the proteasome complex. Richard Feicht is acknowledged for the purification and crystallization of the yeast 20S core particle. Experimental support from Alexandra Herrlich is gratefully appreciated. This research was financially supported by NIH Grant GM49758 (to D.W.C) and by the Deutsche Forschungsgemeinschaft (SFB1035, to M.G.). F.K.H. acknowledges financial support from the Fonds der Chemischen Industrie (FCI). J.H. has been supported by the German Children’s Cancer Foundation (Project 110997 and Translational Oncology Program 70112951), the German Carreras Foundation (DJCLS 02R/2016), the Kinderkrebsstiftung (2016/17) and the Elterninitiative Kinderkrebsklinik e.V. Düsseldorf. S.B. acknowledges the financial support by Forschungskommission, HHU (2018–04).
ABBREVIATIONS USED
- CFU
colony forming assay
- DMSO
dimethylsulfoxide
- HDAC
histone deacetylase
- HDAC6 CD2
histone deacetylase 6 catalytic domain 2
- PI
proteasome inhibitor
- AML
acute myeloid leukemia
- BCP-ALL
B-cell precursor acute lymphoblastic leukemia
- CML
chronic myeloid leukemia
- MM
multiple myeloma
- TKI
tyrosine kinase inhibitor
- PARP
poly(ADP-ribose) polymerase
- PBMCs
peripheral blood derived mononuclear cells
- HSR
heat shock response
- UPR
unfolded protein response
Footnotes
Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Figures and Tables, experimental procedures, compound characterization data, X-ray crystallography, HPLC traces and NMR spectra of newly synthesized compounds.
Molecular Formula Strings and some data (CSV).
Accession codes. Protein Data Bank (PDB): HDAC6–RTS-V5 complex, 6CW8; Proteasome–RTS-V5 complex, 6H39. Authors will release the atomic coordinates and experimental data upon article publication.
The authors declare no competing financial interest.
REFERENCES
- (1).Anighoro A; Bajorath J; Rastelli G Polypharmacology: challenges and opportunities in drug discovery. J. Med. Chem 2014, 57, 7874–7887. [DOI] [PubMed] [Google Scholar]
- (2).Roche J; Bertrand P Inside HDACs with more selective HDAC inhibitors. Eur. J. Med. Chem 2016, 121, 451–483. [DOI] [PubMed] [Google Scholar]
- (3).Maolanon AR; Kristensen HM; Leman LJ; Ghadiri MR; Olsen CA Natural and synthetic macrocyclic inhibitors of the histone deacetylase enzymes. ChemBioChem 2017, 18, 5–49. [DOI] [PubMed] [Google Scholar]
- (4).(a) Ganesan A Multitarget Drugs: an Epigenetic Epiphany. ChemMedChem 2016, 11, 1227–1241; [DOI] [PubMed] [Google Scholar]; (b) de Lera AR; Ganesan A Epigenetic polypharmacology: from combination therapy to multitargeted drugs. Clin. Epigenetics 2016, 8, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Millard CJ; Watson PJ; Fairall L; Schwabe JWR Targeting class I histone deacetylases in a “complex” environment. Trends Pharmacol. Sci 2017, 38, 363–377; [DOI] [PubMed] [Google Scholar]; (b) Schobert R; Biersack B Multimodal HDAC inhibitors with improved anticancer activity. Curr. Cancer Drug Targets 2018, 18, 39–56. [DOI] [PubMed] [Google Scholar]
- (6).Hideshima T; Richardson PG; Anderson KC Mechanism of action of proteasome inhibitors and deacetylase inhibitors and the biological basis of synergy in multiple myeloma. Mol. Cancer Ther 2011, 10, 2034–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Beck P; Dubiella C; Groll M Covalent and non-covalent reversible proteasome inhibition. Biol. Chem 2012, 393, 1101–1120. [DOI] [PubMed] [Google Scholar]
- (8).Gallastegui N; Beck P; Arciniega M; Huber R; Hillebrand S; Groll M Hydroxyureas as noncovalent proteasome inhibitors. Angew. Chem. Int. Ed 2012, 51, 247–249. [DOI] [PubMed] [Google Scholar]
- (9).Groll M; Koguchi Y; Huber R; Kohno J Crystal structure of the 20 S proteasome:TMC-95A complex: a non-covalent proteasome inhibitor. J. Mol. Biol 2001, 311, 543–548. [DOI] [PubMed] [Google Scholar]
- (10).(a) Kaiser M; Groll M; Siciliano C; Assfalg-Machleidt I; Weyher E; Kohno J; Milbradt AG; Renner C; Huber R; Moroder L Binding mode of TMC-95A analogues to eukaryotic 20S proteasome. ChemBioChem 2004, 5, 1256–1266; [DOI] [PubMed] [Google Scholar]; (b) Groll M; Gotz M; Kaiser M; Weyher E; Moroder L TMC-95-based inhibitor design provides evidence for the catalytic versatility of the proteasome. Chemistry & biology 2006, 13, 607–614; [DOI] [PubMed] [Google Scholar]; (c) Desvergne A; Genin E; Marechal X; Gallastegui N; Dufau L; Richy N; Groll M; Vidal J; Reboud-Ravaux M Dimerized linear mimics of a natural cyclopeptide (TMC-95A) are potent noncovalent inhibitors of the eukaryotic 20S proteasome. J. Med. Chem 2013, 56, 3367–3378. [DOI] [PubMed] [Google Scholar]
- (11).(a) Blackburn C; Gigstad KM; Hales P; Garcia K; Jones M; Bruzzese FJ; Barrett C; Liu JX; Soucy TA; Sappal DS; Bump N; Olhava EJ; Fleming P; Dick LR; Tsu C; Sintchak MD; Blank JL Characterization of a new series of non-covalent proteasome inhibitors with exquisite potency and selectivity for the 20S β5-subunit Biochem. J 2010, 430, 461–476; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Groll M; Gallastegui N; Marechal X; Le Ravalec V; Basse N; Richy N; Genin E; Huber R; Moroder L; Vidal J; Reboud-Ravaux M 20S proteasome inhibition: designing noncovalent linear peptide mimics of the natural product TMC-95A. ChemMedChem 2010, 5, 1701–1705. [DOI] [PubMed] [Google Scholar]
- (12).Huber EM; Groll M Inhibitors for the immuno- and constitutive proteasome: current and future trends in drug development. Angew. Chem. Int. Ed 2012, 51, 8708–8720. [DOI] [PubMed] [Google Scholar]
- (13).Hideshima T; Qi J; Paranal RM; Tang W; Greenberg E; West N; Colling ME; Estiu G; Mazitschek R; Perry JA; Ohguchi H; Cottini F; Mimura N; Gorgun G; Tai YT; Richardson PG; Carrasco RD; Wiest O; Schreiber SL; Anderson KC; Bradner JE Discovery of selective small-molecule HDAC6 inhibitor for overcoming proteasome inhibitor resistance in multiple myeloma. Proc. Natl. Acad. Sci. USA 2016, 113, 13162–13167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).(a) Olson DE; Wagner FF; Kaya T; Gale JP; Aidoud N; Davoine EL; Lazzaro F; Weiwer M; Zhang YL; Holson EB Discovery of the first histone deacetylase 6/8 dual inhibitors. J. Med. Chem 2013, 56, 4816–4820; [DOI] [PubMed] [Google Scholar]; (b) Wagner FF; Olson DE; Gale JP; Kaya T; Weïwer M; Aidoud N; Thomas M; Davoine EL; Lemercier BC; Zhang Y-L; Holson EB Potent and selective inhibition of histone deacetylase 6 (HDAC6) does not require a surface-binding motif. J. Med. Chem 2013, 56 (4), 1772–1776. [DOI] [PubMed] [Google Scholar]
- (15).Miyake Y; Keusch JJ; Wang L; Saito M; Hess D; Wang X; Melancon BJ; Helquist P; Gut H; Matthias P Structural insights into HDAC6 tubulin deacetylation and its selective inhibition. Nat. Chem. Biol 2016, 12, 748–754. [DOI] [PubMed] [Google Scholar]
- (16).Bhatia S; Diedrich D; Frieg B; Ahlert H; Stein S; Bopp B; Lang F; Zang T; Kroger T; Ernst T; Kogler G; Krieg A; Ludeke S; Kunkel H; Rodrigues Moita AJ; Kassack MU; Marquardt V; Opitz FV; Oldenburg M; Remke M; Babor F; Grez M; Hochhaus A; Borkhardt A; Groth G; Nagel-Steger L; Jose J; Kurz T; Gohlke H; Hansen FK; Hauer J Targeting HSP90 dimerization via the C-terminus is effective in imatinib resistant CML and lacks the heat shock response. Blood 2018, 132, 307–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).(a) Hai Y; Christianson DW Histone deacetylase 6 structure and molecular basis of catalysis and inhibition. Nat. Chem. Biol 2016, 12, 741–747; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Porter NJ; Mahendran A; Breslow R; Christianson DW Unusual zinc-binding mode of HDAC6-selective hydroxamate inhibitors. Proc. Natl. Acad. Sci. USA 2017, 114, 13459–13464; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mackwitz MKW; Hamacher A; Osko JD; Held J; Schöler A; Christianson DW; Kassack MU; Hansen FK Multicomponent Synthesis and binding mode of imidazo[1,2-a]pyridine-capped selective HDAC6 inhibitors. Org. Lett 2018, 20, 3255–3258. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Porter NJ; Osko JD; Diedrich D; Kurz T; Hooker JM; Hansen FK; Christianson DW Histone deacetylase 6-selective inhibitors and the influence of capping groups on hydroxamate-zinc denticity. J. Med. Chem 2018, 61, 8054–8060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Löwe J; Stock D; Jap B; Zwickl P; Baumeister W; Huber R Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 A resolution. Science 1995, 268, 533–539. [DOI] [PubMed] [Google Scholar]
- (19).Groll M; Ditzel L; Lowe J; Stock D; Bochtler M; Bartunik HD; Huber R Structure of 20S proteasome from yeast at 2.4 A resolution. Nature 1997, 386, 463–471. [DOI] [PubMed] [Google Scholar]
- (20).Stein ML; Cui H; Beck P; Dubiella C; Voss C; Kruger A; Schmidt B; Groll M Systematic comparison of peptidic proteasome inhibitors highlights the alpha-ketoamide electrophile as an auspicious reversible lead motif. Angew. Chem. Int. Ed 2014, 53, 1679–1683. [DOI] [PubMed] [Google Scholar]
- (21).Huber EM; Heinemeyer W; Li X; Arendt CS; Hochstrasser M; Groll M A unified mechanism for proteolysis and autocatalytic activation in the 20S proteasome. Nature Commun 2016, 7, 10900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Borissenko L; Groll M 20S proteasome and its inhibitors: crystallographic knowledge for drug development. Chem. Rev 2007, 107, 687–717. [DOI] [PubMed] [Google Scholar]
- (23).Morphy R; Kay C; Rankovic Z From magic bullets to designed multiple ligands. Drug Discov. Today 2004, 9, 641–651. [DOI] [PubMed] [Google Scholar]
- (24).Bayat Mokhtari R; Homayouni TS; Baluch N; Morgatskaya E; Kumar S; Das B; Yeger H Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Hideshima T; Bradner JE; Wong J; Chauhan D; Richardson P; Schreiber SL; Anderson KC Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc. Natl. Acad. Sci. USA 2005, 102, 8567–8572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Delic J; Morange M; Magdelenat H Ubiquitin pathway involvement in human lymphocyte gamma-irradiation-induced apoptosis. Mol. Cell. Biol 1993, 13, 4875–4883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Vogl DT; Raje N; Jagannath S; Richardson P; Hari P; Orlowski R; Supko JG; Tamang D; Yang M; Jones SS; Wheeler C; Markelewicz RJ; Lonial S Ricolinostat, the first selective histone deacetylase 6 inhibitor, in combination with bortezomib and dexamethasone for relapsed or refractory multiple myeloma. Clin. Cancer Res 2017, 23, 3307–3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Zhou W; Zhu W; Ma L; Xiao F; Qian W Proteasome inhibitor MG-132 enhances histone deacetylase inhibitor SAHA-induced cell death of chronic myeloid leukemia cells by an ROS-mediated mechanism and downregulation of the Bcr-Abl fusion protein. Oncol. Lett 2015, 10, 2899–2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Kalin JH; Bergman JA Development and therapeutic implications of selective histone deacetylase 6 inhibitors. J. Med. Chem 2013, 56, 6297–6313. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.