

Abstract
This work reports the synthesis, and pharmacological and electrophysiological evaluation of new N-methyl-D-aspartic acid receptor (NMDAR) channel blocking antagonists featuring polycyclic scaffolds. Changes in the chemical structure modulate the potency and voltage dependence of inhibition. Two of the new antagonists display properties comparable to those of memantine, a clinically approved NMDAR antagonist.
Keywords: Alzheimer’s disease, electrophysiology, glutamate, NMDA receptor, memantine, polycyclic amines
TOC image
INTRODUCTION
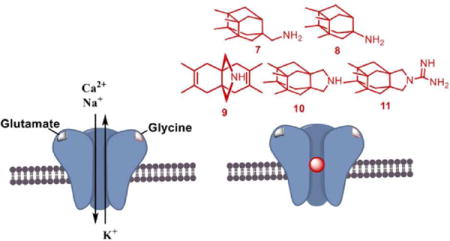
The amino acid L-glutamate1–2 is the main excitatory neurotransmitter in the central nervous system and activates a wide diversity of receptors comprising ionotropic (iGluRs) as well as metabotropic glutamate receptors. iGluRs, ligand-gated ion channels composed of four subunits, can be subdivided into three classes based on their subunit composition and their selective activation by the agonists (S)-2-amino-3-(3-hydroxy-5-methylisoxazol-4-yl)propionic acid (AMPA), kainate and N-methyl-D-aspartic acid (NMDA).3–5 Among iGluRs, the NMDA receptor (NMDAR) possesses unique properties including co-agonism, a high permeability for Ca2+ ions, and voltage-dependent channel blockade by Mg2+, which has to be relieved to allow ion flow through the channel.6–8
NMDARs are heterotetrameric complexes derived from three main types of subunits, namely GluN1, GluN2 and GluN3, of which GluN1 is obligatory.9–14 Usually a single NMDAR is composed of two glycine-binding GluN1 subunits plus two glutamate-binding GluN2 subunits. There are eight known splice variants of the GluN1 subunit, four GluN2 subunit subtypes (A-D), and two GluN3 subunit subtypes (A,B).8,15–17
NMDARs are expressed at nearly all vertebrate synapses and play key roles in neuronal development, plasticity, and survival. Ca2+ influx through NMDARs is a signal of paramount importance for synaptic plasticity, including long-term potentiation and long-term depression, physiological processes that are the cellular basis of many forms of learning and memory.18 However, NMDAR overstimulation triggers excessive Ca2+ influx and leads to excitotoxicity, which is the primary mediator of neuronal death following stroke and is believed to play a key role in the pathogenesis of neurodegenerative diseases, including Alzheimer’s disease (AD) and Parkinson’s disease (PD).9–20 Hence, NMDAR antagonists able to prevent overactivation of NMDARs are of interest as neuroprotective drugs.
Multiple types of NMDAR antagonists have been tested in clinical trials. Several competitive NMDAR antagonists failed trials for neurodegenerative disorders and related conditions, possibly because they blocked the physiological as well as the pathological effects of NMDARs, leading to severe adverse effects.21–23 NMDAR open channel blocking antagonists have also been tested as therapeutic agents. In contrast to competitive antagonists, NMDAR channel blockers bind at sites that overlap with the Mg2+ site and can only bind and unbind when the channel is open.24–26 Most NMDAR channel blockers also failed clinical trials, and several were found to be neurotoxic when administered at high doses to control animals,27 including dizocilpine (MK-801), phencyclidine, and ketamine (compounds 1, 2, and 3, respectively, in Figure 1). Nevertheless, two adamantane derivatives, amantadine and memantine (compounds 4 and 5, respectively, in Figure 1), which are low- (amantadine) and moderate- (memantine) affinity voltage-dependent NMDAR channel blockers, have been found to be moderately effective for treatment of PD and AD, respectively.28–32
Figure 1.
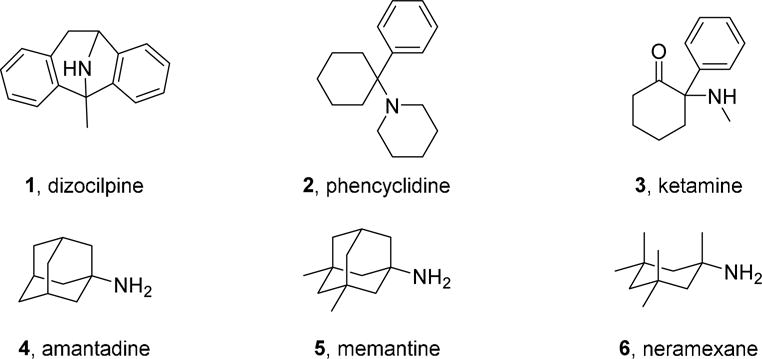
Structures of NMDAR channel blocking antagonists 1-6.
Several hypotheses have been proposed to explain the divergent clinical effects of NMDAR channel blockers. The kinetics of recovery from inhibition, which are much faster for memantine than dizocilpine, have been proposed to be a major determinant of clinical tolerability.29,33–34 An alternative hypothesis is that the utility of memantine may derive from an ability to preferentially inhibit extrasynaptic NMDARs, activation of which has been proposed to be especially neurotoxic.35–37 It is clear, however, that overactivation of synaptic NMDARs also can be neurotoxic.38,39 Another recent proposal is that clinical safety may be associated with preferential inhibition of NMDARs that undergo Ca2+3dependent desensitization following exposure to high intracellular Ca2+, a property exhibited by 5 but not 3.40
Although memantine is well-tolerated by AD patients, it possesses limited clinical efficacy.41 For this reason, new moderate-affinity NMDAR antagonists with similar but distinct pharmacological properties are of interest.29,33–34 Thus, we recently started a project aimed to design, synthesize, and characterize new polycyclic amines as analogues of 5 with improved pharmacological profiles.
Taking into account that carbocyclic amines other than 4 and 5 display similar affinity to NMDARs (e.g, neramexane, 6 in Figure 1),42–45 and that the methyl groups of memantine are critical for optimal potency at NMDARs (memantine is roughly fifty-fold more potent than amantadine),46–47 we envisaged the synthesis of polycyclic amine 7 (Figure 2) and a few selected analogues. These new analogues included amines 8, for considering the impact of the distance between the polcyclic cage and the amino group; 9 and 10, for assessing the effect of conformational freedom; and guanidine 11, for evaluating the effect of basicity.
Figure 2.
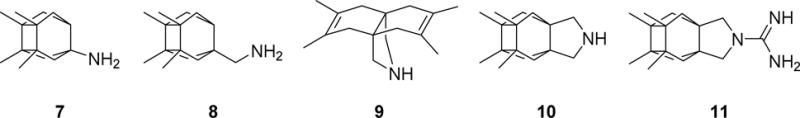
Chemical structures of new putative NMDAR antagonists 7-11.
RESULTS AND DISCUSSION
Chemical synthesis
Compounds 9, 10 and 11 were prepared using procedures previously reported by our group.48–49 Primary amines 7 and 8 were synthesized following the sequence shown in Scheme 1, starting from known anhydride 12.50 Briefly, treatment of anhydride 12 with an excess of methanol at reflux furnished hemiester 13 in quantitative yield. Barton’s decarboxylation procedure led to ester 14, which upon hydrolysis yielded carboxylic acid 15. From this key intermediate, Curtius rearrangement led to primary amine 7. Finally, amine 8 was obtained by reduction of amide 16, in turn obtained from 15. Both target amines 7 and 8 were fully characterized as their corresponding hydrochlorides.
Scheme 1.
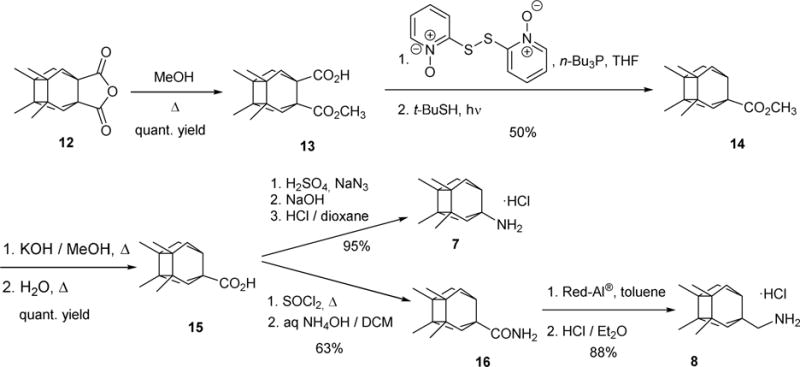
Synthesis of primary amines 7 and 8.
Pharmacology and Structure-Activity Relationships
To evaluate if the new compounds were able to antagonize NMDARs, we measured their effect on the increase in intracellular Ca2+ evoked by application of NMDA (100 μM, in the presence of 10 μM of glycine) to cultured rat cerebellar granule neurons.51 Pleasingly, inspection of the results shown in Table 1 reveals that all the new compounds were clearly more potent than amantadine (IC50 = 92 μM) with values of IC50 in the low micromolar range. Although differences are small, it seems that conformationally restricted secondary amines 9 (5.8 ± 1.0 μM) and 10 (5.1 ± 1.0 μM) are less potent than secondary amine 8 (2.8 ± 1.1 μM), while directly joining the polar amino group to the polycyclic ring slightly reduces the potency (compare 7vs8). Overall, guanidine 11 and primary amine 8 were the more potent compounds, with IC50 values (2.7 ± 0.4 and 2.8 ± 1.1 μM, respectively) only slightly higher than that of memantine (1.5 ± 0.1 μM).
Table 1.
IC50 (μM) values for amantadine, memantine and new analogs 7-11 as NMDAR antagonists.a
| Compound | NMDA (100 μM) IC50 (μM) |
|---|---|
| 4 | 92 ± 29 |
| 5 | 1.5 ± 0.1 |
| 7 | 4.1 ± 1.7 |
| 8 | 2.8 ± 1.1 |
| 9 | 5.8 ± 1.0 |
| 10 | 5.1 ± 1.0 |
| 11 | 2.7 ± 0.4 |
IC50 is the concentration of a compound that inhibits the measured response by 50%. Data were obtained from primary cultures of cerebellar granule neurons as described in Methods by measuring the intracellular Ca2+ concentration. Cells were exposed to 100 μM NMDA plus 10 μM glycine. Data shown are means ± SEM of at least three separate experiments carried out on three different batches of cultured cells.
Functional block of NMDARs by polycyclic compounds 7-11
We next evaluated electrophysiologically the functional ability of compounds 7-1 to block NMDARs. To carry out these studies, we performed whole-cell experiments on tsA201 cells transfected with expression plasmids codifying rat GluN1 and GluN2A subunits to measure the properties of the newly synthesized polycyclic amines. We clamped the cells at −60 mV and then evoked NMDAR currents by applying 100 μM NMDA plus 10 μM glycine. After the NMDAR-evoked current reached a steady state, we rapidly applied a given blocking compound by means of piezoelectric translation of a double-barreled theta glass tubing (<1 ms exchange between solutions). By doing so, we could compare the percentage of block for each tested compound. Figure 3 shows a typical example of an experiment for the compounds tested (5 and 7-1). Compound 5 at 10 μM blocked nearly 90% of the activated current while the percentage of block by the newly synthetized compounds varied amongst them. Compounds 7 and 8 at 10 μM displayed degree of NMDAR block similar to 5, i.e., 82.5 ± 3.2 % for 7 and 88.3 ± 3.7 % for 8vs 89.2 ± 1.0 % for 5 (Figure 4A). Compounds 9 and 10 at 10 μM induced less inhibition than 5, i.e., 71.9 ± 3.8 % for 9 and 70.4 ± 5.6 % for 10vs 89.2 ± 1.0 % for 5; (Figure 4A). On the other hand, 10 μM of compound 11 induced more inhibition than 5, although the difference was not significant, i.e., 97.9 ± 1.9 % for 11vs 89.2 ± 1.0 % for 5. Thus, compounds 7, 8 and 11 appeared to block NMDARs with potency similar to that of memantine.
Figure 3.
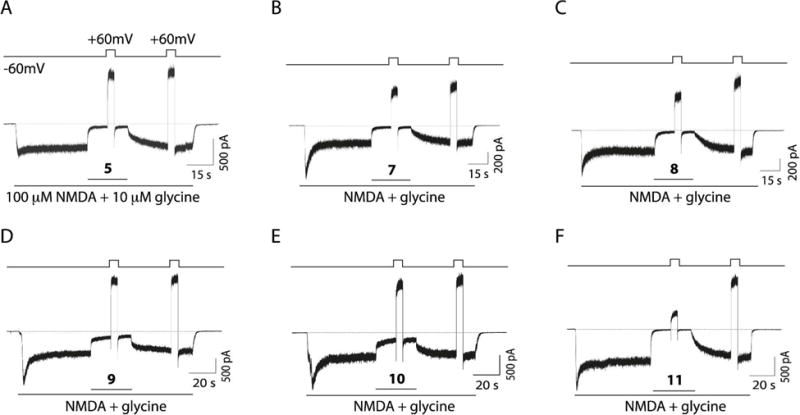
Block, unbinding, and voltage dependence of compounds 5, 7-11. A. Example recording showing the protocol used to study the degree of channel block, the voltage dependence and the unbinding percentage of compound 5. Whole-cell currents were evoked in tsA201 cells expressing GluN1/2A NMDARs by bath application of 100 μM NMDA plus 10 μM glycine. Compound 5 was rapidly applied at 10 μM. B-F. Example traces in the same conditions as described in A but for compounds 7, 8, 9, 10 and 11, respectively. All compounds were used at 10 μM.
Figure 4.
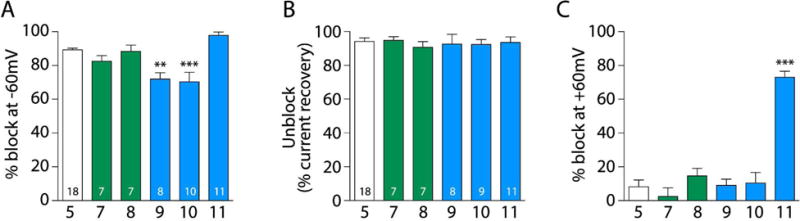
Quantification of block, unbinding and percentage of block at +60mV of compounds 5, 7-11. A. Summary of the blocking percentage at the holding potential of −60 mV for the different compounds tested. Asterisks identify mean values with statistically significant differences. The number of asterisks indicates the magnitude of the p-value, the probability of measuring by chance a difference equal to or greater than the observed difference between indicated mean values. **p<0.01 and ***p<0.001 vs compound 5; one-way ANOVA with Tukey post hoc analysis. Numbers inside bars denote the number of experiments. B. Degree of unbinding (“Unblock”), measured as the percentage of current recovery after removal of the blocker; no differences between compounds was observed (p>0.05 for all compounds compared with compound 5). Numbers inside bars denote the number of experiments. C. Percentage of block at +60 mV for the studied compounds. ***p<0.0001 vs compounds 5, 7, 8, 9, and 10; one-way ANOVA with Tukey post hoc analysis; n=17, 7, 7, 8, 10 and 10 for compounds 5, 7, 8, 9, 10 and 11, respectively.
We also evaluated the ability of the newly synthesized blockers to unbind from the channel pore upon drug removal. Unbinding was measured by rapidly removing the blocker in the continuous presence of agonists (100 μM NMDA plus 10 μM glycine). Unbinding (“Unblock”, Fig. 4B) was calculated as the percentage of current recovery after a 30-s application of agonists without blocker, when the current was at or very near steady state. All tested compounds showed similar abilities to unbind from the pore compared with 5, i.e., 94.9 ± 2.2 %, 90.6 ± 3.5 %, 92.5 ± 5.9, 92.3 ± 3.0 % and 93.5 ± 3.4 % for compounds 7, 8, 9, 10 and 11, respectively, vs 94.2 ± 2.1 % for 5 (Figure 4B).
Finally we assessed the voltage dependence of channel block by the compounds. During the recordings we applied two positive pulses to +60 mV for 0.5 s during the sustained NMDA- and glycine-evoked current. The first pulse was applied in the presence of the blocking compound at 10 μM and a second +60 mV pulse was applied in the absence of the blocker. Hence, we could extract the percentage block at +60 mV. Compound 11, which appeared to be the most potent compound when tested at −60 mV, also displayed the greatest inhibition at +60 mV, i.e., 73.0 ± 3.6 % block for 11vs 8.1 ± 4.0 % for 5 (Figure 4C). Compounds 7-10 had similar blocking percentages at +60 mV to 5, i.e., 2.4 ± 5.1 %, 14.7 ± 4.5 %, 9.0 ± 3.6 % and 10.4 ± 6.4 % for compounds 7, 8, 9, and 10, respectively; (Figure 4C).
Concentration and voltage dependence of NMDAR inhibition by 5, 7, 8, and 11
Whole-cell patch-clamp recordings from tsA201 cells expressing GluN1/2A receptors were used to further assess the pharmacological properties of three promising derivatives, primary amines 7 and 8, and guanidine 11. Experiments measuring the IC50 and voltage-dependence of block by compound 5 were performed for comparison. In cells held at −65 mV, inhibition by each drug was measured at increasing drug concentrations (Figures 5–8, A) and used to calculate the IC50 and Hill coefficient (nH, which reflects the steepness of the concentration-inhibition curve; see Equation 2). The IC50 value and Hill coefficient measured for 5 (Fig. 5B) are similar to previously-reported values measured under the same conditions.42 Compounds 7, 8, and 11 were found to have moderate IC50 values (Figures 6–8, B). The IC50s of compounds 8 (1.01 ± 0.13 μM) and 11 (0.48 ± 0.09 μM) were significantly lower than the IC50s of 5 (1.84 ± 0.39 μM) or 7 (2.78 ± 0.25 μM), and the IC50 of 5 was significantly lower than the IC50 of 7 (Fig. 9A). There were no significant differences between the nH of the drugs, i.e., 1.07 ± 0.27, 0.98 ± 0.08, 1.01 ± 0.11, and 1.00 ± 0.03 for compounds 5, 7, 8, and 11, respectively. Modest differences between IC50s measured using intracellular Ca2+ measurements from cerebellar granule neurons (Table 1) and patch-clamp recordings from tsA201 cells expressing GluN1/2A receptors (Figs. 5 – 9) were observed. The differences may have resulted from expression of GluN2 subunits other than GluN2A in cerebellar granule neurons, and from differences in recording technique.
Figure 5.
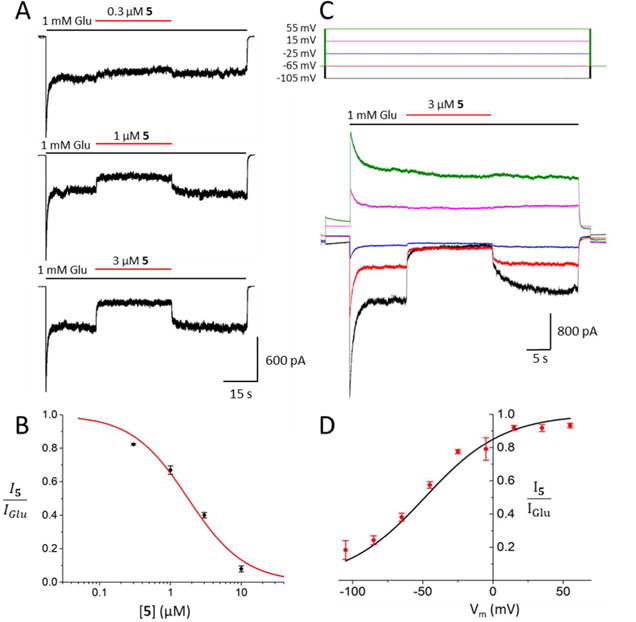
Concentration and voltage dependence of NMDAR inhibition by 5. A. Representative current traces from one cell depicting effect of 5 on GluN1/2A receptor currents. Application of 1 mM Glu (black bars) elicited an inward current that was antagonized by application of 5 (red bars). B. Concentration-inhibition relation for 5. Line shows best fit of Equation 2 (IC50 = 1.84 ± 0.39 μM, nH = 1.07 ± 0.27; n=5). C. Representative voltage (Vm; top) and current (bottom) traces depicting effect of membrane potential upon inhibition by 3 μM 5. Traces from 5 of the 9 membrane potentials tested are displayed for clarity. D. Current-voltage relation of inhibition by 5. Line shows best fit of Equation 3 (V0 = 28.0 ± 2.2; n=5). Points in B and D represent mean fractional currents measured at each concentration (B) or voltage (D); error bars represent SEM and are sometimes smaller than symbols. Comparison of the concentration and voltage dependence of NMDAR inhibition by compounds 5, 7, 8, and 11 is shown in Figure 9.
Figure 8.

Concentration and voltage dependence of NMDAR inhibition by 11. A, B. Same as Figure 5 A, B, except concentration-inhibition measurements made using 11. Line in B shows best fit of Equation 2 (IC50 = 0.48 ± 0.09 μM, nH = 1.00 ± 0.03; n=4). C, D. Same as Figures 5 C, D, except measurements of voltage-dependence made using 1 μM 11. Line in D shows best fit of Equation 3 (V0 = 33.6 ± 1.5; n=4). Comparison of the concentration and voltage dependence of NMDAR inhibition by compounds 5, 7, 8, and 11 is shown in Figure 9.
Figure 6.

Concentration and voltage dependence of NMDAR inhibition by 7. A, B. Same as Figure 5A, B, except concentration-inhibition measurements made using 7. Line in B shows best fit of Equation 2 (IC50 = 2.78 ± 0.25 μM, nH = 0.98 ± 0.08; n=7). C, D. Same as Figure 5C, D, except measurements of voltage-dependence made using 5 μM 7. Line in D shows best fit of Equation 3 (V0 = 26.5 ± 1.8; n=7). Comparison of the concentration and voltage dependence of NMDAR inhibition by compounds 5, 7, 8, and 11 is shown in Figure 9.
Figure 7.
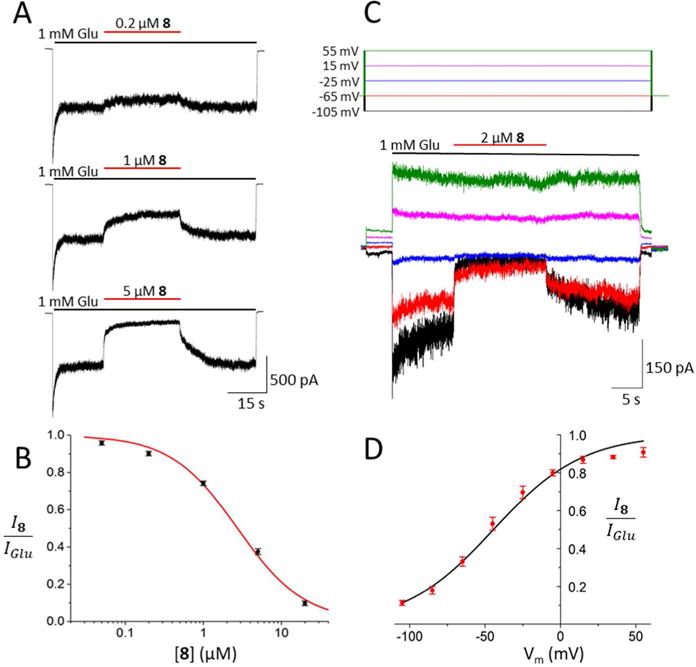
Concentration and voltage dependence of NMDAR inhibition by 8. A, B. Same as Figure 5A, B, except concentration-inhibition measurements made using 8. Line in B shows best fit of Equation 2 (IC50 = 1.01 ± 0.13 μM, nH = 1.01 ± 0.11; n=7). C, D. Same as Figure 5C, D, except measurements of voltage-dependence made using 2 μM 8. Line in D shows best fit of Equation 3 (V0 = 29.9 ± 1.9; n=8). Comparison of the concentration and voltage dependence of NMDAR inhibition by compounds 5, 7, 8, and 11 is shown in Figure 9.
Figure 9.

Comparison of NMDAR channel blocker properties. Sample size denoted by number inside column. A. Comparison of blocker IC50 values measured at −65 mV. B. Comparison of voltage dependence of inhibition by the blockers. All comparisons made by one-way ANOVA with Tukey post hoc analysis; *p<0.01, **p<0.001, ***p<0.0001.
To measure voltage dependence of inhibition by 5, 7, 8, and 11, inhibition elicited by roughly twice the IC50 of each drug was measured at 9 different voltages (examples from 5 voltages are shown in Figures 5–8, C). The inhibition produced by the drugs decreased as voltage was depolarized (Figures 5–8, C and D), as expected of positively charged channel blockers. Fitting of Equation 3 to current-voltage data was used to quantify V0, the change in voltage (in mV) that results in an e-fold change in the IC50 of a drug. Equation 4 was used to calculate δ, an estimate of the fraction of the total transmembrane voltage field felt by the blocker at its binding site.52 The value of δ is calculated from the value of V0 (Equation 4); strong voltage dependence is reflected by a large δ and a small V0. All compounds displayed strongly voltage-dependent block, i.e., for 5, V0 = 28.0 ± 2.2 mV and δ = 0.91 ± 0.08; for 7, V0 = 26.5 ± 1.8 mV and δ = 0.99 ± 0.05; for 8, V0 = 29.9 ± 1.9 mV and δ = 0.87 ± 0.05; for 11, V0 = 33.6 ± 1.5 mV and δ = 0.76 ± 0.03 (Figures 5–8, D). The voltage dependence of inhibition was found to be significantly weaker for 11 than for either 5 or 7 (Fig. 9B).
CONCLUSIONS
We have described the synthesis, pharmacological evaluation and electrophysiology of a novel, family of N-methyl-D-aspartic acid receptor (NMDAR) channel blockers. Despite profound, structural modifications, e.g., compare diene 9 with the other compounds, or different pKas, e.g., compare amine 10 with guanidine 11, all the inhibitors showed similar potency as NMDAR, antagonists. However, more subtle changes in the chemical structure modulated both the degree, of inhibition and voltage-dependence of inhibition by the inhibitors, e.g., compare primary amine, 8 with secondary amine 10 or amine 10 with guanidine 11 (Figures 3 and 4). Primary amines 7, and 8 displayed voltage-dependence and potency comparable to memantine (5).
METHODS
Chemistry
(3,4,8,9-Tetramethyltetracydo[4.4.0.03,9.04,8]dec-1-yl)amme hydrochloride (7)
A solution of the acid 15 (182 mg, 1.35 mmol) in DCM (5 mL) was prepared in a two-neck round-bottom flask equipped with a condenser, a gas outlet and a magnetic stirring. Concentrated H2SO4 (0.43 mL) was added and the reaction was heated to 50 °C. Then, NaN3 (189 mg, 2.90 mmol) was carefully added portionwise. The reaction was kept at 50 °C for 1.5 h. The resulting mixture was cooled with an ice bath. Crushed ice (2 g) was added to the reaction and the aqueous layer was basified with 2 N NaOH to basic pH. The layers were separated, and the aqueous layer was extracted with warmed CH2Cl2 due to the low solubility of the product (6 × 20 mL). The combined organics were dried over anhydrous Na2SO4 and filtered. An excess of HCl in 1,4-dioxane was added to the residue and the suspension was concentrated under reduced pressure to give the 7·HCl as a brown solid (182 mg, 95% yield). The analytical sample was obtained by crystallization from hot CH2Cl2, mp 200 °C (sublimation). IR (ATR) ν: 628, 685, 716, 1010, 1041, 1062, 1090, 1114, 1147, 1163, 1188, 1232, 1294, 1310, 1341, 1369, 1385, 1452, 1460, 1483, 1501, 1524, 1547, 1620, 1646, 2072, 2583, 2697, 2790, 2873, 2899, 2961, 3395, 3426 cm−1. 1H-NMR (400 MHz, CD3OD) δ: 0.90 [dd, J = 11.4 Hz, J’ = 2.4 Hz, 2H, 5(7)-Ha], 0.98-1.02 [d, J = 10.8 Hz, 2H, 2(10)-Ha], 1.01 [s, 6H, 3(9)-CH3or 4(8)-CH3], 103 [s, 6H, 4(8)-CH3 or 3(9)-CH3], 1.94 [dd, J = 11.4 Hz, J’ = 1.6 Hz, 2H, 5(7)-Hb], 1.99 [d, J = 10.8 Hz, 2H, 2(10)-Hb], 2.25 (m, 1H, 6-H). 13C-NMR (100.5 MHz, CD3OD) δ: 15.4 [CH3, C3(9)-CH3 or C4(8)-CH3], 15.6 [CH3, C4(8)-CH3 or C3(9)-CH3], 38.9 [CH2, C5(7)], 40.8 (CH, C6), 42.0 [CH2, C2(10)], 45.8 [C, C3(9) or C4(8)], 46.2 [C, C3(9) or C4(8)], 58.1 (C, C1). HRMS-ESI+ m/z [M+H]+ calcd for [C14H23N+H]+: 206.1903, found: 206.1907. Anal. Calcd for C14H23N·HCl·0.66H2O: C 66.24, H 10.06, N 5.52. Found: C 66.07, H 9.59, N 5.40.
(3,4,8,9-Tetramethyltetracydo[4.4.0.03,9.04,8]dec-1-yl)methylamine hydrochloride (8)
A solution of 16 (96 mg, 0.41 mmol) in anhydrous toluene (10 mL) was cooled to 0 °C and Red-Al® (0.7 mL, 2.05 mmol) was added dropwise. The reaction was heated to reflux overnight. The resulting mixture was cooled with an ice bath and aqueous 10 N NaOH solution was added dropwise to basic pH. Then, the reaction was stirred for 10 min. The layers were separated and the aqueous layer extracted with DCM (3 × 20 mL). The combined organics were dried over anhydrous Na2SO4, filtered and HCl/Et2O was added. After concentration under reduced pressure, 8·HCl was obtained as a white solid (92 mg, 88% yield). The analytical sample was obtained by washing the solid with cooled Et2O, mp 240 °C (sublimation). IR (ATR) ν: 715, 844, 933, 968, 986, 998, 1016, 1031, 1064, 1097, 1117, 1130, 1163, 1178, 1223, 1297, 1350, 1367, 1383, 1451, 1461, 1479, 1501, 1514, 1613, 2860, 2916, 2941, 3012 cm−1. 1H-NMR (400 MHz, CD3OD) δ: 0.63 [d, J = 11.2 Hz, 2H, 2(10)-Ha], 0.79 [broad d, J = 11.2 Hz, 2H, 5(7)-Ha], 0.995 [s, 6H, 3(9)-CH3 or 4(8)-CH3], 0.999 [s, 6H, 4(8)-CH3 or 3(9)-CH3], 1.76 [d, J = 11.2 Hz, 2H, 5(7)-Hb], 1.82 [d, J = 11.2 Hz, 2H, 2(10)-Hb], 2.12 (m, 1H, 6-H), 3.01 (s, 2H, CCH2NH2). 13C-NMR (100.5 MHz, CD3OD) δ: 15.8 [CH3, C3(9)-CH3 or C4(8)-CH3], 16.0 [CH3, C4(8)-CH3 or C3(9)-CH3], 38.2 (CH, C6), 39.3 [CH2, C5(7)], 42.1 [CH2, C2(10)], 42.5 (C, C1), 45.9 [C, C3(9) or C4(8)], 47.1 [C, C3(9) or C4(8)], 48.1 (CH2, CH2NH2). HRMS-ESI+ m/z [M+H]+ calcd for [C15H25N+H]+: 220.2060, found: 220.2050. Anal. Calcd for C15H25N·HCl·0.25H2O: C 69.21, H 10.26, N 5.38. Found: C 69.15, H 10.01, N 5.19.
Intracellular Ca2+ measurements
The functional assay of antagonist activity at NMDA receptors was performed using primary cultures of rat cerebellar granule neurons that were prepared according to established protocols.51 Cells were grown on 10 mm poly-L-lysine coated round glass cover slips and used for the experiments after 6-9 days in vitro. Cells were loaded with 6 μM Fura-2 AM (Invitrogen-Molecular Probes) for 30 min. Afterwards a coverslip was mounted on a quartz cuvette containing a Mg2+ -free Locke-HEPES buffer using a special holder. Measurements were performed using a PerkinElmer LS-55 fluorescence spectrometer equipped with a fast-filter accessory, under mild agitation and at 37 °C. Analysis from each sample was recorded real-time during 1600 s. After stimulation with NMDA (100 μM, in the presence of 10 μM glycine), increasing cumulative concentrations of the compound to be tested were added. The percentages of inhibition at every tested concentration were analyzed using a non-linear regression curve fitting (variable slope) using the software Prism 5.04 (GraphPad Software Inc.).
Cell culture, transfection, and recordings for electrophysiology experiments
All electrophysiological experiments were performed at room temperature using the tsA201 cell line (European Collection of Authenticated Cell Cultures) transiently cotransfected with mammalian expression plasmids containing cDNAs encoding the rat GluN1-1a and GluN2A subunits.
For Figures 3–4, cells were maintained as previously described53 in DMEM:F12 supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin (Sigma). Cells were plated at 0.1-0.2 × 105 cells/dish in onto 10 mm glass coverslips treated with poly D-lysine. 12–24 hours after plating, the cells were transiently transfected using PEI transfection reagent (1 mg/ml) in a 3:1 ratio (PEI:DNA). Culture medium was supplemented with the competitive NMDAR antagonist D, L-2-amino-5-phosphonopentanoate (dl-APV, Sigma, 500 μM) at the time of transfection to prevent NMDAR-mediated cell death. Whole-cell voltage-clamp recordings were performed 48 hours after transfection. Pipettes were pulled from borosilicate capillary tubing (OD = 1.5 mm, ID = 0.86 mm) using a PC-10 vertical puller (Narishige Instruments) and subsequently fire-polished to a resistance of 3-5 MΩ using an MF-830 forge (Narishige). Intracellular pipette solution contained (in mM): 140 CsCl, 10 HEPES, 5 EGTA, 4 Na2ATP and 0.1 Na3GTP with pH balanced to 7.25 with CsOH. Extracellular recording solution contained (in mM): 140 NaCl, 5 KCl, 1 CaCl2, 10 HEPES and 10 glucose, balanced to pH 7.2 ± 0.05 with NaOH. Drugs were diluted from concentrated stock solutions (5 stock = 10 mM in dH2O; 7-11 stocks = 10 mM in 75% HEPES buffer and 25% ethanol) in extracellular solution each day of experiments. Whole-cell currents were recorded using an Axopatch 200B patch-clamp amplifier (Molecular Devices). Current signal was low-pass filtered at 1 kHz and sampled at 2 kHz in pClamp 10 (Molecular Devices). Series resistance was 10-15 MΩ. Solutions containing agonists (100 μM NMDA and 10 μM glycine) or agonists and 10 μM blocker were applied by piezoelectric translation (P-601.30; Physik Instrumente) of a theta-barrel application tool made from borosilicate glass (1.5 mm o.d.; Sutter Instruments).
For Figures 5–9, cells were maintained as previously described54 in DMEM supplemented with 10% fetal bovine serum, 1% GlutaMAX (Thermo Fisher Scientific), and for some experiments 1% penicillin/streptomycin (Sigma). Cells were plated at 1 × 105 cells/dish in 35 mm petri dishes with three 15 mm glass coverslips treated with poly D-lysine (0.1 mg/ml) and rat-tail collagen (0.1 mg/ml, BD Biosciences). 12-24 hours after plating, the cells were transfected using FuGENE 6 Transfection Reagent (Promega) as previously described.54 Culture medium was supplemented with 200 μM dl-APV at the time of transfection to prevent NMDAR-mediated cell death. Whole-cell voltage-clamp recordings were performed 18-30 hours after transfection. Pipettes were pulled from borosilicate capillary tubing (OD = 1.5 mm, ID = 0.86 mm) using a Flaming Brown P-97 electrode puller (Sutter Instruments) and subsequently fire-polished to a resistance of 2.5 – 4.5 MΩ using an in-house fabricated microforge. Intracellular pipette solution contained (in mM): 130 CsCl, 10 HEPES, 10 BAPTA, and 4 MgATP with pH balanced to 7.2 ± 0.05 with CsOH and an osmolality of 280 ± 10 mOsm. Extracellular recording solution contained (in mM) 140 NaCl, 2.8 KCl, 1 CaCl2, 10 HEPES, 0.01 EDTA, and 0.1 glycine, and was balanced to pH 7.2 ± 0.05 with NaOH and to osmolality 290 ± 10 mOsm with sucrose. Drugs were diluted from concentrated stock solutions (5 stock = 10 mM in dH2O; 7, 8, and 11 stocks = 40 mM in 100% DMSO) in extracellular solution each day of experiments. Whole-cell currents were recorded using either an Axopatch 1D or Axopatch 200A patch-clamp amplifier (Molecular Devices). The current signal was low-pass filtered at 5 kHz and sampled at 20 kHz in pClamp 10 (Molecular Devices). Series resistance was compensated 80-90% in all experiments. A −6 mV liquid junction potential between the intracellular pipette solution and extracellular solution was corrected in all experiments. Glutamate and drug solutions were delivered to the cell via an in-house fabricated ten-barreled gravity-fed fast perfusion system.40,54
Data Analysis
The percentage of channel block, unbinding, and recovery shown in Figures 3 – 4 were measured with the following protocol: at a holding potential of −60 mV, NMDA (100 μM) and glycine (10 μM) were applied until current reached a clear steady-state (about 90 s). Then compounds 5, 7, 8, 9, 10 or 11 were rapidly applied by piezo control (1 ms solution exchange) for 30 seconds as described.55 During the application of the blocker, a 5-s voltage step to +60 mV was applied in order to study the voltage dependence of block. Blockers were then removed in the presence of agonists to allow recovery of the current. During this period (around 1 min) a second voltage step (5 s duration) to +60 mV was applied. Finally, agonists were removed. Percentage of block was calculated by dividing steady state current in the presence of the blocker by steady state current in the absence of the blocker. Percentage of unbinding (“Unblock”) was calculated by dividing the steady state current after blocker removal by the steady state current before blocker application. Finally, the voltage dependence (% of block at +60 mV) was calculated by using Equation 1:
where I+60(Drug) is the steady state current at the holding voltage of +60 mV in the presence of agonists and the blocker, and I+60(Agonist) is the steady state current at +60 mV in the presence of agonists but absence of the blocker.
Concentration-inhibition relations were measured using the protocol shown in Figures 5–8, A and B. Glutamate (1 mM) was applied until current reached steady-state (20 s), then 5, 7, 8, or 11 at the plotted concentration was applied in the presence of glutamate until a new steady-state current level was reached (30 s). Glutamate in the absence of drug was then reapplied for 30 s to allow drug unbinding and recovery from inhibition. Cells in which recovery from inhibition did not reach 90% of steady-state current during initial glutamate application were excluded from analysis. IC50 and nH (Hill coefficient) were estimated by fitting Equation 2 to concentration-inhibition data:
where IDmg/IGlu was calculated as the mean current over the final 1 s of drug application (IDrug) divided by the average of the mean steady state currents (final 1 s) elicited by glutamate before and after drug application (IGlu). IC50 and nH were free parameters during fitting.
Voltage dependence of block by 5, 7, 8, and 11 was measured using the protocol shown in Figures 5–8, C and D. Cells were subjected to voltage jumps from −65 mV to nine voltages ranging from −105 to +55 mV. The protocol at each voltage consisted of: a 4-s wait in extracellular solution following the voltage step; application of 1 mM glutamate for 10 s; application of drug with 1 mM glutamate for 15 s; application of 1 mM glutamate for 15 s to allow drug unbinding; application of extracellular solution for 2 s. Voltage was then returned to −65 mV for 4 s before the next voltage jump was made. ~2 times the IC50 of each drug was used in voltage dependence experiments. Voltage dependence of block was calculated using Equation 3:
where lC50 (−65 mV) is the IC50 at −65 mV calculated in concentration-inhibition experiments, and V0 represents the change in voltage (in mV) that results in an e-fold change in the IC50 of the drug. IDrug/IGlu was calculated as described for concentration-inhibition data. V0 was the only free parameter during fitting. An estimate of the fraction of the total membrane voltage field felt by the blocker at its binding site (δ)52 was calculated using Equation 4:
Where R, T, z and F have their usual meanings. Note that, although δ is useful for comparing voltage dependence of blockers, voltage dependence of NMDAR channel block is influenced by permeant ions.56 Therefore, δ should be used only as a rough estimate of binding site location in the voltage field.
Supplementary Material
Acknowledgments
This study was supported by the Ministerio de Economía Industria y Competitividad and Fondo Europeo de Desarrollo Regional (MINECO-FEDER) (Projects SAF2014-57094-R and BFU2014-57562-P) and by National Institutes of Health grant R01MH045817. The authors thank the Spanish Ministerio de Educacion, Cultura y Deporte (FPU fellowships to R. L. and A. L. T.).
ABBREVIATIONS
- AD
Alzheimer’s disease
- AMPA
(S)-2-amino-3-(3-hydroxy-5-methylisoxazol-4-yl)propionic acid
- iGluRs
ionotropic glutamate receptors
- NMDA
N-methyl-D-aspartic acid
- NMDAR
NMDA receptor
- PD
Parkinson’s disease
Footnotes
The following files are available free of charge on the ACS Publications website at DOI: General methods for the chemical synthesis. Description of the synthesis and characterization of intermediates 13-16. (PDF).
Notes
The authors declare no competing financial interest.
References
- 1.Parsons CG, Danysz W, Quack G. Glutamate in CNS disorders as a target for drug development: an update. Drug News & Perspectives. 1998;11:523–569. doi: 10.1358/dnp.1998.11.9.863689. [DOI] [PubMed] [Google Scholar]
- 2.Zhou Y, Danbolt NC. Glutamate as a neurotransmitter in the healthy brain. J Neural Transm. 2014;121:799–817. doi: 10.1007/s00702-014-1180-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- 4.Mayer ML, Armstrong N. Structure and function of glutamate receptor ion channels. Ann Rev Physiol. 2004;66:161–181. doi: 10.1146/annurev.physiol.66.050802.084104. [DOI] [PubMed] [Google Scholar]
- 5.Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev. 2010;62:405–496. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McBain CJ, Mayer ML. N-methyl-D-aspartic acid receptor structure and function. Physiol Rev. 1994;74:723–760. doi: 10.1152/physrev.1994.74.3.723. [DOI] [PubMed] [Google Scholar]
- 7.Danysz W, Parsons CG. Glycine and N-methyl-D-aspartate receptors: physiological significance and possible therapeutic applications. Pharmacol Rev. 1998;50:597–664. [PubMed] [Google Scholar]
- 8.Iacobucci GJ, Popescu GK. NMDA receptors: linking physiological output to biophysical operation. Nat Rev Neurosci. 2017;18:236–249. doi: 10.1038/nrn.2017.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karakas E, Furukawa H. Crystal structure of a heterotetrameric NMDA receptor ion channel. Science. 2014;344:992–997. doi: 10.1126/science.1251915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee CH, Lü W, Michel JC, Goehring A, Du J, Song X, Gouaux E. NMDA receptor structures reveal subunit arrangement and pore architecture. Nature. 2014;511:191–197. doi: 10.1038/nature13548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu S, Stein RA, Yoshioka C, Lee CH, Goehring A, Mchaourab HS, Gouaux E. Mechanism of NMDA receptor inhibition and activation. Cell. 2016;165:704–714. doi: 10.1016/j.cell.2016.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou Q, Sheng M. NMDA receptors in nervous system diseases. Neuropharmacology. 2013;74:69–75. doi: 10.1016/j.neuropharm.2013.03.030. [DOI] [PubMed] [Google Scholar]
- 13.Tajima N, Karakas E, Grant T, Simorowski N, Díaz-Avalos R, Grigorieff N, Furukawa H. Activation of NMDA receptors and the mechanism of inhibition by ifenprodil. Nature. 2016;534:63–68. doi: 10.1038/nature17679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lü W, Du J, Goehring A, Gouaux E. Cryo-EM structures of the triheteromeric NMDA receptor and its allosteric modulation. Science. 2017;355:eaal3729. doi: 10.1126/science.aal3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cull-Candy S, Brickley S, Farrant M. NMDA receptor subunits: diversity, development and disease. Curr Opin Neurobiol. 2001;11:327–335. doi: 10.1016/s0959-4388(00)00215-4. [DOI] [PubMed] [Google Scholar]
- 16.Paoletti P, Bellone C, Zhou Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci. 2013;14:383–400. doi: 10.1038/nrn3504. [DOI] [PubMed] [Google Scholar]
- 17.Glasgow NG, Siegler Retchless B, Johnson JW. Molecular bases of NMDA receptor subtype-dependent properties. J Physiol. 2015;593:83–95. doi: 10.1113/jphysiol.2014.273763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morris RG. NMDA receptors and memoring encoding. Neuropharmacology. 2013;74:32–40. doi: 10.1016/j.neuropharm.2013.04.014. [DOI] [PubMed] [Google Scholar]
- 19.Kalia LV, Kalia SK, Salter MW. NMDA receptors in clinical neurology: excitatory times ahead. Lancet Neurol. 2008;7:742–755. doi: 10.1016/S1474-4422(08)70165-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mota SI, Ferreira IL, Rego C. Dysfunctional synapse in Alzhimer’s disease – A focus on NMDA receptors. Neuropharmacology. 2014;76:16–26. doi: 10.1016/j.neuropharm.2013.08.013. [DOI] [PubMed] [Google Scholar]
- 21.Ikonomidou C, Turski L. Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet Neurol. 2002;1:383–386. doi: 10.1016/s1474-4422(02)00164-3. [DOI] [PubMed] [Google Scholar]
- 22.Lipton SA. Failures and successes of NMDA receptor antagonists: molecular basis for the use of open-channel blockers like memantine in the treatment of acute and chronic neurologic insults. NeuroRx. 2004;1:101–110. doi: 10.1602/neurorx.1.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muir KW. Glutamate-based therapeutic approaches: clinical trials with NMDA antagonists. Curr Opin Pharmacol. 2006;6:53–60. doi: 10.1016/j.coph.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 24.Blanpied TA, Clarke RJ, Johnson JW. Amantadine inhibits NMDA receptors by accelerating channel closure during channel block. J Neurosci. 2005;25:3312–3322. doi: 10.1523/JNEUROSCI.4262-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson JW, Kotermanski SE. Mechanism of action of memantine. Curr Opin Pharmacol. 2006;6:61–67. doi: 10.1016/j.coph.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 26.Johnson JW, Glasgow NG, Povysheva NV. Recent insights into the mode of action of memantine and ketamine. Curr Opin Pharmacol. 2015;20:54–63. doi: 10.1016/j.coph.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Olney JW, Labruyere J, Price MT. Pathological Changes Induced in Cerebrocortical Neurons by Phencyclidine and Related Drugs. Science. 1989;244:1360–1362. doi: 10.1126/science.2660263. [DOI] [PubMed] [Google Scholar]
- 28.Danysz W, Parsons CG, Kornhuber J, Schmidt WJ, Quack G. Aminoadamantanes as NMDA receptor antagonists and antiparkinsonian agents— preclinical studies. Neurosci Biobehav Rev. 1997;21:455–468. doi: 10.1016/s0149-7634(96)00037-1. [DOI] [PubMed] [Google Scholar]
- 29.Lipton SA. Paradigm shift in neuroprotection by NMDA receptor blockade: memantine and beyond. Nat Rev Drug Discovery. 2006;5:160–170. doi: 10.1038/nrd1958. [DOI] [PubMed] [Google Scholar]
- 30.Hubsher G, Haider M, Okun MS. Amantadine: the journey from fighting flu to treating Parkinson disease. Neurology. 2012;78:1096–1099. doi: 10.1212/WNL.0b013e31824e8f0d. [DOI] [PubMed] [Google Scholar]
- 31.Danysz W, Parsons CG. Alzheimer’s disease, β-amyloid, glutamate, NMDA receptors and memantine – searching for the connections. British J Pharmacol. 2012;167:324–352. doi: 10.1111/j.1476-5381.2012.02057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alam S, Lingenfelter KS, Bender AM, Lindsley CW. Classics in chemical neuroscience: memantine. ACS Chem Neurosci. 2017;8:1823–1829. doi: 10.1021/acschemneuro.7b00270. [DOI] [PubMed] [Google Scholar]
- 33.Chen HSV, Lipton SA. The chemical biology of clinically tolerated NMDA receptor antagonists. J Neurochem. 2006;97:1611–1626. doi: 10.1111/j.1471-4159.2006.03991.x. [DOI] [PubMed] [Google Scholar]
- 34.Lipton SA. Pathologically activated therapeutics for neuroprotection. Nat Rev Neurosci. 2007;8:803–808. doi: 10.1038/nrn2229. [DOI] [PubMed] [Google Scholar]
- 35.Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signaling: implications for neurodegenerative disorders. Nat Rev Neurosci. 2010;11:682–696. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gladding CM, Raymond LA. Mechanisms underlying NMDA receptor synaptic/extrasynaptic distribution and function. Mol Cell Neurosci. 2011;48:308–320. doi: 10.1016/j.mcn.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 37.Parsons MP, Raymond LA. Extrasynaptic NMDA receptor involvement in central nervous system disorders. Neuron. 2014;82:279–293. doi: 10.1016/j.neuron.2014.03.030. [DOI] [PubMed] [Google Scholar]
- 38.Wroge CM, Hogins J, Eisenman L, Mennerick S. Synaptic NMDA receptors mediate hypoxic excitotoxic death. J Neurosci. 2012;32(19):6732–6742. doi: 10.1523/JNEUROSCI.6371-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou X, Hollern D, Liao J, Andrechek E, Wang H. NMDA receptor-mediated excitotoxicity depends on the coactivation of synaptic and extrasynaptic receptors. Cell Death Dis. 2013;4:e560. doi: 10.1038/cddis.2013.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Glasgow NG, Povysheva NV, Azofeifa AM, Johnson JW. Memantine and ketamine differentially alter NMDA receptor desensitization. J Neurosci. 2017;37:9686–9704. doi: 10.1523/JNEUROSCI.1173-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matsunaga S, Kishi T, Iwata N. Memantine monotherapy for Alzheimer’s Disease: a systematic review and meta-analysis. PLoS One. 2015;10:e0123289. doi: 10.1371/journal.pone.0123289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Glling K, Jatzke C, Wollenburg C, Vanejevs M, Kauss V, Jirgensons A, Parsons CG. A novel class of amino-alkylcyclohexanes as uncompetitive, fast, voltage-dependent, N-methyl-D-aspartate (NMDA) receptor antagonists – in vitro characterization. J Neural Transm. 2007;114:1529–1537. doi: 10.1007/s00702-007-0792-7. [DOI] [PubMed] [Google Scholar]
- 43.Rammes G. Neramexane: a moderate-affinity NMDA receptor channel blocker: new prospects and indications. Expert Rev Clin Pharmacol. 2009;2:231–238. doi: 10.1586/ecp.09.7. [DOI] [PubMed] [Google Scholar]
- 44.Camps P, Duque MD, Vázquez S, Naesens L, DeClercq E, Sureda FX, López-Querol M, Camins A, Pallàs M, Prathalingam SR, Kelly JM, Romero V, Ivorra D, Cortés D. Synthesis and pharmacological evaluation of several ring-contracted amantadine analogs. Bioorg Med Chem. 2008;16:9925–9936. doi: 10.1016/j.bmc.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Valverde E, Sureda FX, Vázquez S. Novel benzopolycyclic amines with NMDA receptor antagonist activity. Bioorg Med Chem. 2014;22:2678–2683. doi: 10.1016/j.bmc.2014.03.025. [DOI] [PubMed] [Google Scholar]
- 46.Blanpied TA, Boeckman FA, Aizenman E, Johnson JW. Trapping channel block of NMDA-activated responses by amantadine and memantine. J Neurophysiol. 1997;77:309–323. doi: 10.1152/jn.1997.77.1.309. [DOI] [PubMed] [Google Scholar]
- 47.Limapichat W, Yu WY, Branigan E, Lester HA, Dougherty DA. Key biding interactions for memantine in the NMDA receptor. ACS Chem Neurosci. 2013;4:255–260. doi: 10.1021/cn300180a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Torres E, Leiva R, Gazzarrini S, Rey-Carrizo M, Frigolé-Vivas M, Moroni A, Naesens L, Vázquez S. Azapropellanes with anti-influenza A virus activity. ACS Med Chem Lett. 2014;5:831–836. doi: 10.1021/ml500108s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rey-Carrizo M, Barniol-Xicota M, Ma C, Frigolé-Vivas M, Torres E, Naesens L, Llabrés S, Juárez-Jiménez J, Luque FJ, DeGrado WF, Lamb RA, Pinto LH, Vázquez S. Easily accessible polycyclic amines that inhibit the wild-type and amantadine-resistant mutants of the M2 channel of influenza A virus. J Med Chem. 2014;57:5738–5747. doi: 10.1021/jm5005804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Avila WB, Silva RA. 3,4,8,9-Tetramethyltetracyclo[4.4.0.03,9.04,8]decane-1,6-dioic anhydride. J Chem Soc D. 1970:94–95. [Google Scholar]
- 51.Canudas AM, Pubill D, Sureda FX, Verdaguer E, Camps P, Muñoz-Torrero D, Jiménez A, Camins A, Pallàs M. Neuroprotective effects of (+/−)-huprine Y on in vitro and in vivo models of excitotoxicity damage. Exp Neurol. 2003;180:123–130. doi: 10.1016/s0014-4886(02)00029-8. [DOI] [PubMed] [Google Scholar]
- 52.Woodhull AM. Ionic Blockage of Sodium Channels in Nerve. J Gen Physiol. 1973;61:687–708. doi: 10.1085/jgp.61.6.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gratacos-Batlle E, Yefimenko N, Cascos-García H, Soto D. AMPAR interacting protein CPT1C enhances surface expression of GluA1-containing receptors. Front Cell Neurosci. 2014;8:469. doi: 10.3389/fncel.2014.00469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Glasgow NG, Johnson JW. Whole-Cell Patch-Clamp Analysis of Recombinant NMDA Receptor Pharmacology Using Brief Glutamate Applications. In: Martina M, Taverna S, editors. Patch-Clamp Methods and Protocols. Springer New York; New York, NY: 2014. pp. 23–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Soto D, Olivella M, Grau C, Armstrong J, Alcon C, Gasull X, Gómez de Salazar M, Gratacòs-Batlle E, Ramos-Vicente D, Fernández-Dueñas V, Ciruela F, Bayés À, Sindreu C, López-Sala A, García-Cazorla À, Altafaj X. Rett-like severe encephalopathy caused by a de novo GRIN2B mutation is attenuated by D-serine dietary supplement. Biol Psychiatry. 2018;83:160–172. doi: 10.1016/j.biopsych.2017.05.028. [DOI] [PubMed] [Google Scholar]
- 56.Antonov SM, Gmiro VE, Johnson JW. Binding sites for permeant ions in the channel of NMDA receptors and their effects on channel block. Nature Neurosci. 1998;1:451–461. doi: 10.1038/2167. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.