

Abstract
GLT-1 is the major glutamate transporter in the brain, and is expressed in astrocytes and in axon terminals in the hippocampus, cortex, and striatum. Neuronal GLT-1 accounts for only 5–10% of total brain GLT-1 protein, and its function is uncertain. In HD, synaptic dysfunction of the corticostriate synapse is well-established. Transcriptional dysregulation is a key feature of HD. We hypothesized that deletion of neuronal GLT-1, because it is expressed in axon terminals in the striatum, might produce a synaptopathy similar to that present in HD. If true, then some of the gene expression changes observed in HD might also be observed in the neuronal GLT-1 knockout. In situ hybridization using 33P labeled oligonucleotide probes was carried out to assess localization and expression of a panel of genes known to be altered in expression in HD. We found changes in the expression of cannabinoid receptors 1 and 2, preproenkaphalin, and PDE10A in the striatum of mice in which the GLT-1 gene was inactivated in neurons by expression of synapsin-Cre, compared to wild-type littermates. These changes in expression were observed at 12 weeks of age but not at 6 weeks of age. No changes in DARPP-32, PDE1B, NGFIA, or β-actin expression were observed. In addition, we found widespread alteration in expression of the dynamin 1 gene. The changes in expression in the neuronal GLT-1 knockout of genes thought to exemplify HD transcriptional dysregulation suggest an overlap in the synaptopathy caused by neuronal GLT-1 deletion and HD. These data further suggest that specific changes in expression of cannabinoid receptors, preproenkephalin, and PDE10A, considered to be the hallmark of HD transcriptional dysregulation, may be produced by an abnormality of glutamate homeostasis under the regulation of neuronal GLT-1, or a synaptic disturbance caused by that abnormality, independently of mutation in huntingtin.
Keywords: EAAT2, dynamin, cannabinoid, dopamine receptor, PDE10, preproenkephalin
Introduction1
The regulation of extracellular glutamate concentrations by glutamate transporters is essential for the normal functioning of excitatory synapses and for protection of the brain against excitotoxicity (Danbolt, 2001). Five Na+-dependent glutamate transporters have been identified : EAAT1 (GLAST, SLC1A3), EAAT2 (GLT-1, SLC1a2), EAAT3 (EAAC1, SLC1A1), EAAT4 and EAAT5 (Danbolt, 2001). Several studies have demonstrated that the majority of total glutamate reuptake activity occurs via GLT-1 (Danbolt et al., 1992; Haugeto et al., 1996; Holmseth et al., 2012; Otis and Kavanaugh, 2000; Tanaka et al., 1997). GLT-1 protein is abundant in astrocytes (Lehre et al., 1995) and is expressed at relatively low levels in hippocampal (Chen et al., 2004), cortical (Melone et al., 2009), and striatal axon terminals (Petr et al., 2013). Neuronal GLT-1 accounts for 5–10% of the GLT-1 protein expression in the hippocampus (Furness et al., 2008). Petr et al. (Petr et al., 2015) recently used astrocyte- and neuron-specific conditional GLT-1 knockout mice to demonstrate that neuronal GLT-1, but, remarkably, not astrocytic GLT-1, accounts for a substantial fraction of radioactive glutamate uptake into forebrain synaptosomes. Global (Tanaka et al., 1997) or astrocyte-specific conditional knockout of GLT-1 (Petr et al., 2015) results in a severe phenotype characterized by spontaneous seizures, lower body weight and early mortality. The functional consequences of neuronal glutamate transporter are largely unknown. We hypothesized that, because GLT-1 is expressed in axon terminals, deletion of GLT-1 in axon terminals produces a dysregulation of synaptic function. Since axon terminals in the striatum express GLT-1, we specifically hypothesized that corticostriate synapses might be dysfunctional.
Expression of the mutant huntingtin gene is the causative factor of Huntington’s disease (HD), a disease that is thought to produce dysregulation of the corticostriatal pathway (Bunner and Rebec, 2016; Cepeda et al., 2007; Raymond, 2016). Transcriptional dysregulation is a prominent feature of HD (Cha, 2000, 2007), and abnormalities in the expression of certain genes are found across multiple models of the disease and in human patients. For example, the levels of several transcripts, including the type 1 cannabinoid receptor (CB1) and the type 2 dopamine receptor (D2) are reduced, while levels of the type 2 cannabinoid receptor (CB2) are increased in the quinolinic acid lesion model of HD (Chiarlone et al., 2014). Similarly, CB1 and D2 transcript levels are reduced, while CB2 transcript levels are increased, in all studied transgenic mouse models of HD and in post-mortem tissue from HD patients reviewed in (Laprairie et al., 2015). Similarly, expression of the cyclic nucleotide phosphodiesterase 10A (PDE10A) was found to be altered in expression in R6/2 and R6/1 mice and in the caudate of autopsy HD brains (Hebb et al., 2004).
Based on the studies described above, we hypothesized that some of the changes in gene expression found in HD might be due to the corticostriate synaptopathy, rather than a direct effect of mutant huntingtin. Because GLT-1 is expressed in axon terminals in the striatum, we further hypothesized that mice in which GLT-1 is inactivated in neurons might have a corticostriate synaptopathy, and this synaptopathy might produce changes in gene expression. Further, these changes in gene expression might be similar to those seen in HD. If true, then such a result would suggest that the synaptopathy in HD might be similar to that in the neuronal GLT-1 knockout, and might provide a clue to the nature of the HD synaptopathy.
We chose to investigate expression levels of several transcripts--dopamine and cAMP-regulated protein phosphatase 32 kDA (DARPP-32), phosphodiesterases (PDE) 1B and 10A, CB1, CB2, D2, preproenkaphalin (ppENK), and nerve growth factor-induced clone A (NGFIA)-- that are known to be altered in HD (Cha et al., 1998; Hu et al., 2004; Laprairie et al., 2015; McCaw et al., 2004). The expression levels of dynamin (Dnm-1) and β-actin were also measured as controls because Dnm-1 and β-actin levels are not changed in HD (Gomez et al., 2006) and were not expected to be changed in neuronal GLT-1 knock-out mice. For these studies, we used in situ hybridization to assess differences in gene expression in the brains of mice in which the GLT-1 gene was conditionally deleted in neurons using synapsin-Cre compared with littermate controls (Petr et al., 2015). In situ hybridization has previously been used to document gene expression changes in mouse models of HD (Denovan-Wright et al., 1998) and is an especially valuable approach because of the anatomical information that it provides.
Materials and Methods
GLT-1 conditional knockout mice
The generation of neuronal GLT-1 knockout mice is described in detail elsewhere (Petr et al., 2015). These mice were obtained from the founder colony at Boston Children’s Hospital. This mouse strain is designated (Slc1A2tm1.1Pros ; MGI: 5752263). Briefly, neuronal knockout of GLT-1 was achieved by breeding male and female mice that were both homozygous for the conditional GLT-1 knockout allele; female mice only in the breeding pairs (Rempe et al., 2006) carried the synapsin I promoter-driven Cre recombinase transgene (syn/Cre with C57BL/6 background, JAX Stock No. 003966)(He et al., 2004; Zhu et al., 2001) which is integrated into an intronic region of chromosome 6, 1 Mb distant from the nearest gene (Cain-Hom et al., 2017). Female mice only were used to introduce synapsin-Cre because synapsin-Cre transgene expression in male mice produces germline recombination in the offspring (Rempe et al., 2006). Mice were genotyped by PCR using tail-derived DNA. Mice that demonstrated recombination in tail DNA, indicating recombination not specific to neurons (Rempe et al., 2006), were not used for experiments. Over a 1 year period in which 22 litters were produced, 5 animals were excluded for this reason. Mice had a mixed 126XC57L/6J background. The mice used for experiments had the genotypes GLT-1flox/flox; synapsin-Cre (synGLT-1 KO), and, their littermate controls, with normal GLT-1 function, GLT-1flox/flox.
All animal experiments were carried out in accordance with ARRIVE (Kilkenny et al., 2010), and were approved by the Children’s Hospital Boston Institutional Animal Care and Use Committee. Both male and female adult mice were used in the experiments. For each particular experiment, animal age is indicated. The total number of animals used for in situ experiments and qRT-PCR was 48 (6 6-week wild-type males, 6 6-week old wild-type females, 6 12-week old wild-type males, 6 12-week-old wild-type females, 6 6-week synGLT-1 KO males, 6 6-week old synGLT-1 KO females, 6 12-week old synGLT-1 KO males, and 6 12-week-old synGLT-1 KO females). Tissue from each animal was divided and used for both in situ hybridization and qRT-PCR to reduce the number of animals used.
In situ hybridization
Synthetic, antisense oligonucleotide probes were obtained from Sigma-Aldrich (Oakville, ON) (Table 1). Ten pmol of oligonucleotide probe was radio-labelled at the 3′ end with [α-33P]dATP (Mandel Scientific, Guelph, ON) using the reagents and protocol provided in the 3′ end-labelling kit (Amersham Pharmacia Biotech). In situ hybridization was performed as described previously using 14 µm sagittal sections from male and female synGLT-1 KO and age-matched, littermate control mice (Denovan-Wright et al., 1998). The sections were exposed to Kodak MR film for 2 weeks at room temperature. Densitometric analysis of in situ autoradiographs was performed using ImageJ to determine the optical density (OD) of the radiolabel in the PFc, MCtx, Str, NAcc, thalamus (Th), CA1 and CA3 regions of the hippocampus, SNr, VTA, cerebellum (Cb), pons, and medulla (Fig. 1). Measurements were determined on 10 sections per target mRNA derived from 6 individual animals at each time point. Local background was subtracted from each OD value. In all cases multiple exposures were obtained to assure that signal was not saturated.
Table 1.
In situ hyribization oligonuvleotides.
| Target | Oligonucleotide Sequence(5’ – 3’) | Reference |
|---|---|---|
| DARPP-32 | TCCACTTGGTCCTCAGAGTTTCCATCTCTC | Gomez et al., 2006 |
| PDE1B | CATGTAGCGCAGCAGAGACCGTAGCTTAATCCACA | Hebb et al., 2004 |
| PDE10A | GACCAATGTCAAAGTGGAATAGCTCGATGTCCCGGC | Hebb et al., 2004 |
| CB1 | ATGTCTCCTTTGATATCTTCGTACTGAATGTCATTTG | McCaw et al., 2004 |
| ppENK | ATCTGCATCCTTCTTCATGAAACCGCCATACCTCTTGGCAAGGATCT | Rodriguez-Lebron et
al., 2005 |
| D2 | GGCAGGGTTGGCAATGATACACTCATTCTGGTCTGTATT | |
| NGFIA | CCGTTGCTCAGCAGCATCATCTCCTCCAGTTTGGGGTAGTTGTCC | |
| Dynamin | CACTGGCTTTCTCTTTGTCCCCAAGAGGCTC | |
| β-actin | GCCGATCCACACGGAGTACTTGCGCTCAGGAGGAGCAATGATCT |
Figure 1. Regions of interest identified for in situ hybridization analysis.

In situ hybridization was conducted to detect DARPP-32, PDE1B, PDE10A, CB1, ppENK, D2, NGFIA, Dnm-1, and β-actin mRNA in synGLT-1 KO and littermate control mice. DARPP-32, PDE1B and β-actin mRNA levels were not changed in synGLT-1 KO mice compared to littermate control mice. The brain regions of interest examined in this study are outlined and labeled in yellow in this representative autoradiogram of Dnm-1 probe hybridization in a 12 week-old, male, littermate control mouse.
Quantitative reverse transcriptase PCR
RNA was harvested from the tissue of male and female synGLT-1 KO and age-matched, littermate control mice using the Trizol® (Invitrogen, Burlington, ON) extraction method according to the manufacturer’s instruction. Reverse transcription reactions were carried out with SuperScript III® reverse transcriptase (Invitrogen), or without (-RT) as a negative control for use in subsequent PCR experiments according to the manufacturer’s instructions. Two micrograms of RNA were used per RT reaction. qRT-PCR was conducted using the LightCycler® system and software (Roche, Laval, QC). Reactions were composed 2 mM MgCl2, 0.5 µM each of forward and reverse primers CB2 forward 5’- GGATGCCGGGAGACAGAAGTGA-3’, reverse 5’- CCCATGAGCGGCAGGTAAGAAAT-3’; β-actin forward 5’- AAGGCCAACCGTGAAAAGAT-3’, reverse 5’- GTGGTACGACCAGAGGCATAC-3’), 2 µL of LightCycler® FastStart Reaction Mix SYBR Green I, and 1 µL cDNA to a final volume of 20µL with dH2O (Roche). The PCR program was: 95°C for 10 min, 50 cycles of 95°C 10 s, a primer-specific annealing temperature (57°C CB2, 59°C β-actin) for 5 s, and 72°C for 10 s. Experiments always included sample-matched –RT controls, a no-sample dH2O control, and a standard control containing product-specific cDNA of a known concentration. cDNA abundance was calculated using the ∆∆CT method and was in accordance with the MIQE guidelines (LightCycler Software version 4.1; Roche).
Synaptosomal glutamate uptake
The knockdown of GLT-1 expression in axonal terminals of striatum was verified in crude synaptosomes from a total of 6 male synGLT-1 KO and 6 male littermate control animals by determining sodium-dependent transport of L-[3H]glutamate as previously described (Petr et al., 2015). The isolated crude synaptosomes in 0.32 M sucrose were kept on ice and used immediately for the uptake assay. Glass tubes containing 450 µl of buffer [in mM: NaCl, 140 or choline chloride, 140; KCl, 2.5; CaCl2, 1.2; MgCl2, 1.2; K2HPO4, 1.2; glucose, 10; 2-amino-2-(hydroxymethyl)propane-1,3-diol (Tris base), 5; 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES), 10] with 10 µM L-glutamate [including 0.005 µM L-[3H]glutamate (PerkinElmer, Boston, MA, USA)] were preincubated for 5 min at 37°C. Glutamate uptake into synaptosomes was initiated by adding 50 µl of crude synaptosomes to each tube and incubating at 37°C for 30 seconds. To stop uptake activity, 2 ml of ice-cold choline buffer was added to the tube, which was then vortexed and plunged into an ice-water slurry. The samples were filtered through Whatman GF/C filter paper pre-wetted with 2ml choline buffer and the filters were washed three times with 2 ml ice-cold choline buffer. Radioactivity on the filters was measured by liquid scintillation counting (TRI-CARB2200CA, PACKARD; Long Island Scientific). The radioactivity taken up by the synaptosomes in the absence of sodium was subtracted from that taken up in the presence of sodium to determine the sodium-dependent component of transport. Glutamate uptake values were determined by normalizing the radioactivity count by protein concentration of the crude synaptosomes isolated from each brain region. Protein concentration was quantified using the Bio-Rad DC protein assay (Bio-Rad Laboratories, Hercules, CA, USA).
Statistical analyses
Previous studies have demonstrated a correlation between in situ hybridization and RNA abundance by northern blot and qRT-PCR for several isoforms of phosphodiesterase, affirming the utility of in situ hybridization as a technique that can by analyzed by parametric statistical tests (Hebb et al., 2004; Hu et al., 2004). Statistical analyses were conducted by two-way analysis of variance (ANOVA) and Bonferroni’s post-hoc test using GraphPad (version 5.0, Prism). Homogeneity of variance – and the correct application of parametric statistical analyses to these data--was confirmed using Bartlett’s test. All results are reported as the mean ± the standard error of the mean (SEM). Although both male and female mice were used in this study, no statistically significant differences in transcript levels were observed between male and female mice for the group sizes used in this study (data not shown). For synaptosomal uptake studies, unpaired t-test was performed to compare the means of two groups of data using Prism 5 (GraphPad Software, Inc. La Jolla, CA, USA). A factor was considered statistically significant if it had a p-value of <0.05.
Results
In situ hybridization of sagital sections was performed and the hybridizaton signal was quantified to determine the relative levels of DARPP-32, PDE1B, PDE10A, CB1, D2, ppENK, NGFIA mRNAs across several brain regions of 6 and 12 week-old synGLT-1 KO and littermate control mice (Table 1). These genes had been previously identified in animal models of HD as being dysregulated (Gomez et al., 2006). Hybridization of β-actin, and Dnm-1 specific probes were intended to provide control values. No changes in DARPP-32, PDE1B, NGFIA, or β-actin level were observed (data not shown).
Data presented in histograms herein represent all hybridization data collected for those genes. No exclusions were made. Representative images were chosen to provide reference for each transcript of interest. Hybridization of ppENK was visually distinct and quantified in the Str, VTA, NAcc, and PFc. No differences were observed in ppENK levels in any brain region analyzed for 6 week-old synGLT-1 KO mice compared to littermate control mice (data not shown). ppENK levels were lower in the Str of 12 week-old synGLT-1 KO mice compared to littermate control mice, but were not different in the VTA, NAcc, or PFc (Fig. 2A,B).
Figure 2. In situ hybridization detection of ppENK mRNA in 12 week-old wild-type (WT) and synGLT-1 KO mice.

A) Total Optical density (OD) was measured in the striatum, VTA, NAcc, and PFc (regions identifiable in the autoradiogram) of 12 week-old mice according to genotype. *P < 0.01 compared to WT (littermate control mice) within brain region. N = 6 per group (3 male and 3 female per genotype). B) Representative autoradiograms of ppENK mRNA hybridization.
CB1 was quantified in the Str, VTA, NAcc, PFc, and CA1 and CA3 regions of the hippocampus. No differences were observed in CB1 mRNA levels in any brain region analyzed for 6 week-old synGLT-1 KO mice compared to littermate control mice (data not shown). CB1 mRNA levels were lower in the Str of 12 week-old synGLT-1 KO mice compared to littermate control mice, but were not different in the other regions analyzed (Fig. 3A,B).
Figure 3. In situ hybridization detection of CB1 mRNA in 12 week-old wild-type (WT) and synGLT-1 KO mice.

A) Total Optical density (OD) was measured in the striatum, VTA, NAcc, PFc, CA1 and CA3 (regions identifiable in the autoradiogram) of 12 week-old mice according to genotype. *P < 0.01 compared to littermate control mice within brain region. N = 6 per group (3 male and 3 female per genotype). B) Representative autoradiograms of CB1 mRNA hybridization.
D2 was quantified in the Str, VTA, NAcc, and PFc. No differences were observed in D2 mRNA levels in any brain region analyzed for 6 week-old synGLT-1 KO mice compared to littermate control mice (data not shown). D2 mRNA levels were lower in the PFc of synGLT-1 KO mice compared to littermate control mice (Fig. 4A,B). They were not changed in the Str of 12 week-old synGLT-1 KO mice compared to littermate control mice.
Figure 4. In situ hybridization detection of D2 mRNA in 12 week-old wild-type (WT) and synGLT-1 KO mice.

A) Total Optical density (OD) was measured in the striatum, VTA, NAcc, and PFc (regions identifiable in the autoradiogram) of 12 week-old mice according to genotype. *P < 0.01 compared to littermate control mice within brain region. N = 6 per group (3 male and 3 female per genotype). B) Representative autoradiograms of D2 mRNA hybridization.
PDE10A was quantified in the Str, NAcc, and PFc. No overall differences were observed in PDE10A mRNA levels in any brain region analyzed for 6 week-old synGLT-1 KO mice compared to littermate control mice (data not shown). PDE10A mRNA levels were lower in the Str of 12 week-old synGLT-1 KO mice compared to littermate control mice, and were not different in the NAcc and PFc of synGLT-1 KO mice compared to littermate control mice (Fig. 5A,B).
Figure 5. In situ hybridization detection of PDE10A mRNA in 12 week-old wild-type (WT) and synGLT-1 KO mice.

A) Total Optical density (OD) was measured in the striatum, NAcc, and Pfc (regions identifiable in the autoradiogram) of 12 week-old mice according to genotype. *P < 0.01 compared to littermate control mice within brain region. N = 6 per group (3 male and 3 female per genotype). B) Representative autoradiograms of PDE10A mRNA hybridization.
Dnm-1 was quantified in the Str, VTA, NAcc, PFc, CA1, CA3, Cb, MCtx, pons, medulla, thalmus, and SNr using an in situ probe specific to the predominant Dnm-1 transcript variant 1, and not other dynamin isoforms or transcript variants. Dnm-1 mRNA levels were lower in the NAcc of 6 week-old synGLT-1 KO mice compared to littermate control mice (Fig. 6A,B). Dnm-1 mRNA levels were higher in the Str of 12 week-old synGLT-1 KO mice compared to littermate control mice, and were lower in the NAcc, PFc, CA3, pons and medulla of synGLT-1 KO mice compared to littermate control mice (Fig. 6C,D).
Figure 6. In situ hybridization detection of Dnm-1 mRNA in 6 and 12 week-old wild-type (WT) and synGLT-1 KO mice.

Total Optical density (OD) was measured in the striatum, VTA, NAcc, PFc, CA1, CA3, Cb, MCtx, pons, medulla, thalmus, and SNr of 6 (A,B) and 12 (C,D) week-old mice (regions identifiable in the autoradiogram) according to genotype. *P < 0.01 compared to littermate control mice within brain region. N = 6 per group (3 male and 3 female per age per genotype). B,D) Representative autoradiograms of Dnm-1 mRNA hybridization in 6 (B) and 12 (D) week-old mice.
Quantitative reverse transcriptase PCR analysis of CB2 expression
CB2 mRNA levels were measured because CB2 levels are increased in quinolinic acid lesion model of HD (Casteels et al., 2010; Chiarlone et al., 2014). Therefore, we hypothesized that CB2 levels might also be increased in synGLT-1 KO mice. Unfortunately, we were unable to develop a CB2-specific in situ probe (data not shown). Consequently, qRT-PCR was used to measure CB2 mRNA levels in synGLT-1 KO and littermate control mice. CB2 mRNA was quantified in total brain (Fig. 7A), and in the Str, VTA, NAcc, and PFc of 6 and 12 week-old mice (Fig. 7B). CB2 mRNA levels were higher in total brain from 12 week synGLT-1 KO mice compared to littermate control mice (Fig. 7A). CB2 mRNA levels were not different in any brain region in 6 week-old mice (Fig. 7B). CB2 mRNA levels were higher in the Str, NAcc, and PFc, but not VTA, of 12 week-old synGLT-1 KO mice compared to littermate control mice (Fig. 7C).
Figure 7. qRT-PCR quantification of CB2 mRNA in 6 and 12 week-old wild-type (WT) and synGLT-1 KO mice.
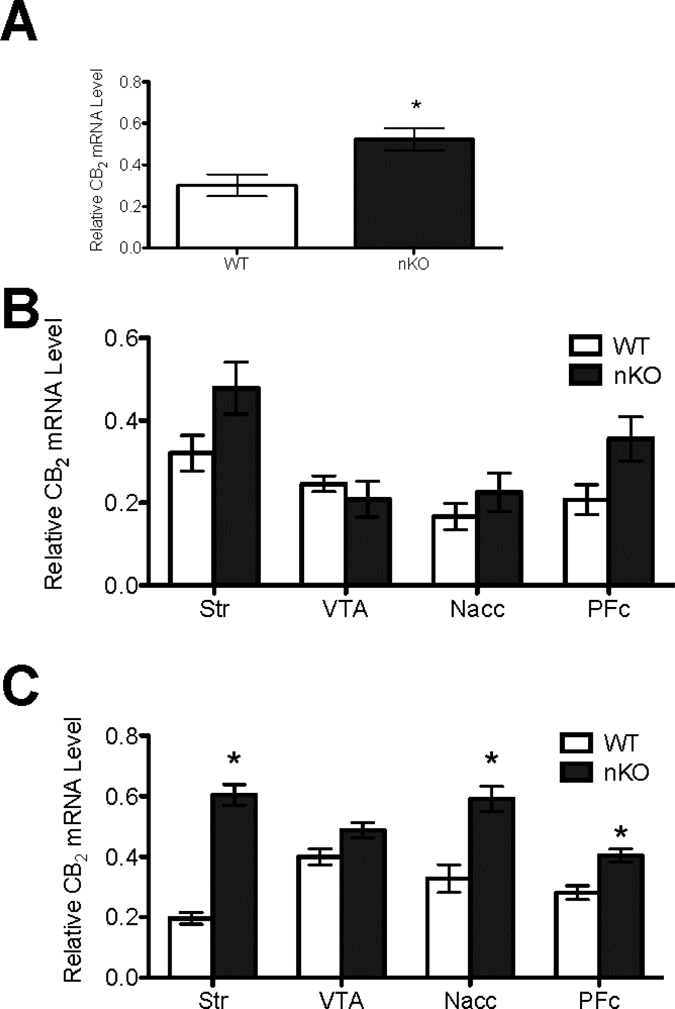
CB2 cDNA abundance was quantified relative to β-actin in the total brain (A) and in the striatum, VTA, NAcc, and PFc of 6 week-old (B) and 12 week old (C) mice according to genotype. *P < 0.01 compared to littermate controls within brain region. N = 6 per group (3 male and 3 female per age per genotype).
Validation of expression of GLT-1 in axon terminals in the striatum and efficacious knockdown in expression of GLT-1 in this region in the synGLT-1 KO
We previously demonstrated the expression of GLT-1 in axon terminals of the striatum (Petr et al., 2013) and, further, that the incidence of immunoreactive axon terminals in the striatum of the transgenic R6/2 mouse model of HD is not different from age matched controls (Petr et al., 2013). Using conditional knockout of GLT-1 restricted to neurons or astrocytes, we have shown that synaptosomal uptake of tritiated glutamate into a crude synaptosomal preparation from whole forebrain represents uptake mediated by neuronal GLT-1 but is not significantly affected by astrocytic GLT-1 (Petr et al., 2015; Rimmele and Rosenberg, 2016). Therefore, we reasoned that decrease in this parameter in mice in which neuronal GLT-1 is inactivated genetically can be used as an additional line of evidence for the expression of GLT-1 in axon terminals in any specific region.
The decrease in synaptosomal uptake with genetic deletion is dependent upon the efficacy of the Cre recombinase used to accomplish homologous recombination. Since most of the changes in gene expression we report here were observed in the striatum, we also wanted to verify that a decrease in synaptosomal glutamate uptake could be observed in the synGLT-1 KO in the striatum as evidence that the synapsin-Cre driver that we used produced effective deletion of GLT-1 in axon terminals in that region. We found reduced glutamate uptake into synaptosomes isolated from the dorsal striatum (45±3%, n=6; p<0.01) of synGLT-1 KO mice compared to littermate controls (Fig. 8).
Figure 8. Glutamate uptake into crude synaptosomes from the striatum was significantly reduced in the synGLT-1 KO.
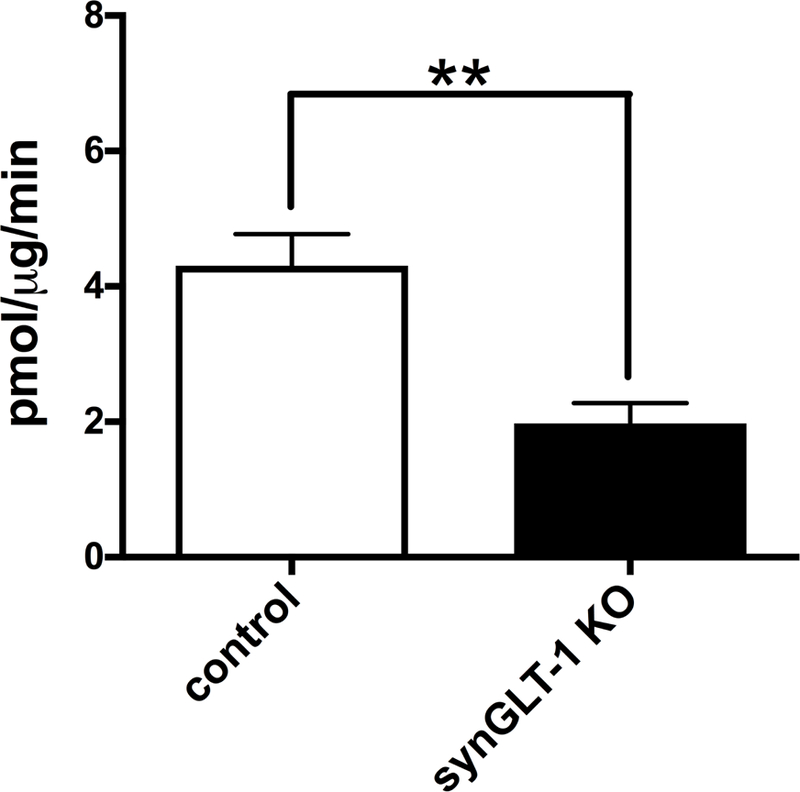
Glutamate uptake into crude synaptosomes from dorsal striatal tissue taken from the synGLT-1 KO and controls was determined using L-[3H]glutamate (N = 6 each genotype; all male). Glutamate uptake was significantly reduced by deletion of neuronal GLT-1 (*:p<0.01).
Discussion
We conducted a biased, hypothesis based survey of gene expression in synGLT-1 KO mice to test the idea that the synaptopathy produced by knockout of GLT-1 in neurons might be similar to the synaptopathy present in HD, and so produce similar changes in gene expression. Rather than conduct a microarray study (many of which demonstrate hundreds of changes that are not relevant to the striatum or synaptopathy (Cha, 2007; Thomas, 2006), a panel of genes was selected that are consistently observed as being dysregulated in both animal models and patient samples using different methodologies and are thought to be the hallmarks of the transcriptional dysregulation of the disease.
Many of the transcriptional changes noted in HD have been observed in the striatum. The approach used here is contingent on the widespread expression of GLT-1 in neurons in the forebrain, and especially in axon terminals in the striatum. In fact, following the demonstration of GLT-1a expression in axon terminals in the hippocampus (Chen et al., 2004), that finding was confirmed (Furness et al., 2008) and extended to the cerebral cortex (Melone et al., 2009; Melone et al., 2011) and the striatum (Petr et al., 2013). The demonstration of a significant reduction in the synGLT-1 KO in glutamate uptake in crude synaptosomes derived from striatal tissue provides further evidence that GLT-1 is expressed in axon terminals in this region. A recent study specifically investigating the effect of neuronal GLT-1 knockout in different regions of the forebrain obtained a similar result, and extended the results presented here to include the cerebral cortex and thalamus (Zhou et al., 2018). This study also performed a detailed investigation of the expression of synapsin-Cre in the adult mouse brain, and found widespread although not universal expression in neurons that included heavy labeling of neurons of layers V and VI of the cerebral cortex which project to the striatum. Another recent study came to a similar conclusion (Taugher et al., 2017). Taken together, these published results and those presented herein further validate previous observations of GLT-1 expression in axon terminals in the striatum. They also validate the appropriateness of using synapsin 1 as a Cre-driver to inactivate the GLT-1 gene in neurons in the striatum and throughout the forebrain. Synapsin-Cre is widely used to produce conditional Cre-recombinase mediated inactivation of genes in neurons throughout the CNS (Gaveriaux-Ruff and Kieffer, 2007), but it is important to recognize that certain neuronal populations express little synapsin-Cre, and in most populations that express synapsin-Cre the expression is partial (Taugher et al., 2017; Zhou et al., 2018).
In the present study, we found transcriptional dysregulation of multiple transcripts in the central nervous system of synGLT-1 KO mice compared to littermate control controls (Fig. 9). The majority of changes in mRNA levels were observed in the Str, where ppENK, CB1, and PDE10A levels were lower, and Dnm-1 and CB2 levels were higher, in synGLT-1 KO mice compared to littermate control mice. Dnm-1 and CB2 mRNA levels were similarly dysregulated in the NAcc and PFc. D2 mRNA levels were also reduced in the PFc.
Figure 9. Summary of changes in gene expression observed in this study.

Overall changes in gene expression observed in synGLT-1 KO mice relative to littermate control mice are summarized for each of the brain regions studied. Neuronal projections between brain regions are shown as A) glutamatergic neurons, B) GABA-ergic neurons, and C) dopaminergic neurons. Lower mRNA levels in synGLT-1 KO mice relative to littermate control mice are indicated in red font. Higher mRNA levels in synGLT-1 KO mice relative to littermate control mice are indicated in green font.
Decreased GLT-1 transcript levels are observed in HD (Bunner and Rebec, 2016; Petr et al., 2013). Glutamate uptake is impaired in the YAC128 mouse model of HD due to reduced palmitoylation of GLT-1 (Huang et al., 2010). In YAC128 HD mice, reduced glutamate uptake via GLT-1 has been shown to lead to increased synaptic glutamate levels as early as 9 months prior to motor symptom onset, excitotoxicity, and eventual neuronal cell death (Huang et al., 2010). Cortical and striatal GLT-1 levels are decreased in the R6/2 mouse model of HD, however, the relative changes in astrocytic versus neuronal GLT-1 levels are not known. Decreased GLT-1 expression in itself does not exacerbate disease progression (Petr et al., 2013). This study tested the effect of putting the R6/2 mutation on a GLT-1 het null background, leaving 50% expression and function in both neurons and astrocytes. Petr et al. (2013) does not address the question of the impact of near total deletion of neuronal GLT-1 on disease progression in the R6/2 mouse model. We have observed the synGLT-1 KO mouse up to 1 year of age and have found no behavioral evidence of a neurodegenerative disorder. For example they do not show hindlimb clasping with tail suspension at that age (Lin et al., 2001). However, the point of the present study is not that the synGLT-1 KO is a model for HD, but rather that some of the transcriptional abnormalities that have been observed in HD may be due to disturbance of glutamate homeostasis in presynaptic terminals similar to that produced by neuronal GLT-1 knockout. The question then is whether HD produces alteration in the expression or function of neuronal GLT-1 itself or in the downstream pathways with which it is involved. If so, these alterations might be the cause of the transcriptional abnormalities, rather than a direct effect of mutant huntingtin on transcription, as has been proposed.
Although we found no change in expression of GLT-1 in axon terminals in the R6/2 transgenic mouse model of HD (Petr et al., 2013), this observation does not exclude the possibility that in other mouse models or in the human disease there is a decrease in expression of GLT-1 in neurons. Petr et al (2013) did find a significant decrease in total GLT-1 protein expression in the striatum assayed by western blot, but no change in tritiated glutamate uptake by crude striatal synaptosomes. These results are consistent with the conclusion drawn from prior studies (Petr et al., 2015; Rimmele and Rosenberg, 2016; Zhou et al., 2018) that synaptosomal uptake primarily reflects the activity of GLT-1 expressed in axon terminals and not astrocytes, and suggest that the decrease in GLT-1 protein that was observed in the R6/2 mouse was localized to astrocytes.
Neuronal GLT-1 may have roles other than glutamate clearance, and HD may disturb expression of GLT-1 itself or perturb mechanisms in the presynaptic terminals with which GLT-1 is involved. For example, it has been suggested that GLT-1 may be important for mitochondrial function and utilization of glutamate by mitochondria in astrocytes (Genda et al., 2011; Jackson and Robinson, 2017; Robinson and Jackson, 2016). HD may in some way interfere with GLT-1 dependent utilization of glutamate by mitochondria in axon terminals.
Several of the transcripts whose levels were dysregulated in this study, including ppENK, CB1, PDE10A, D2, and CB2 are also dysregulated in HD (Denovan-Wright and Robertson, 2000), while changes in other transcripts, such as Dnm-1, were unique to synGLT-1 KO mice. In HD, transcriptional dysregulation is thought to be due to direct effects on transcription by mutant huntingtin (Gomez et al., 2006; Hogel et al., 2012). However, the observations made here suggest that ppENK, CB1, PDE10A, and CB2 mRNA levels are highly sensitive to changes in glutamate homeostasis influenced by neuronal GLT-1, especially in the striatum (Fig. 8), and that a similar pattern of dysregulation to that observed in HD can be produced simply by alterations in glutamate homeostasis by inactivation of GLT-1 in neurons in the presence of normal huntingtin.
In the case of PDE10, observations concerning alteration in PDE10 expression in HD and on the functions of PDE10 in synaptic signalling (Beaumont et al., 2016; Kleiman et al., 2011) were the scientific justification for a clinical trial of the effect of a PDE10 inhibitor on HD progression. This study, which failed, is reported at the following website: https://www.clinicaltrialsregister.eu/ctr-search/trial/2014–001291-56/results. Our observation that a similar downregulation of PDE10 is observed in the GLT-1 neuronal knockout suggests that the alteration in PDE10 expression could be a distant consequence of mutant huntingtin expression. The change in PDE10 expression may be a consequence of the synaptopathy produced by mutant huntingtin, rather than a cause of the synaptopathy.
With the exception of Dnm-1, inactivation of GLT-1 in neurons did not affect transcript levels assessed at 6 weeks of age. CB1 limits neurotransmitter release and downregulation of CB1 levels may be a compensatory mechanism by neurons in response to circuit changes in the synGLT-1 KO (Ohno-Shosaku and Kano, 2014). Up-regulation of CB2 may be compensatory as well because higher astrocytic CB2 levels are observed following excitoxic lesions (Palazuelos et al., 2009) and in HD and Parkinson’s disease, where excitotoxicity may contribute to disease progression (Bisogno and Di Marzo, 2010; Laprairie et al., 2014). We did not determine whether changes in gene expression were astrocytic or neuronal in this study.
Dnm-1 mRNA levels were lower in both 6 and 12 week-old synGLT-1 KO mice compared to littermate control mice in all brain regions examined that receive glutamatergic input (Fig. 8), suggesting that Dnm-1 mRNA regulation is particularly sensitive to changes in glutamate homeostasis, at least that component of glutamate homeostasis that is regulated by GLT-1 expressed in axon terminals. Because Dnm-1 regulates vesicular neurotransmitter release, Dnm-1 levels may be lower in glutamatergic neurons in synGLT-1 KO mice because of a compensatory change in order to reduce glutamate release in response to lower glutamate uptake (Chen-Hwang et al., 2002). In contrast, Dnm-1 levels may be higher in GABAergic neurons, such as those in the Str, to inhibit the activity of glutatamergic neurons onto which they synapse (Chen-Hwang et al., 2002). Further investigation is required to determine whether dysregulation of Dnm-1 affects mitochondrial function, vesicle shuttling, and neurotransmitter release in synGLT-1 KO mice (Chen-Hwang et al., 2002).
Enkephalin is co-packaged with glutamate in several regions of the brain (Van Bockstaele et al., 2000). If glutamate release was reduced to compensate for reduced glutamate uptake, it is possible that enkephalin – and thus ppENK mRNA levels – was also decreased in glutamatergic neurons (Van Bockstaele et al., 2000). It is noteworthy that changes in mRNA levels in synGLT-1 KO were observed – for the majority of changes – at 12 weeks of age but not at 6 weeks of age, suggesting an important role for processes active during adolescence in governing the transcriptional response to the lack of GLT-1 in neurons. Future studies are required to determine if excess extracellular glutamate per se contributes to the changes in gene expression seen in the synGLT-1 KO mice.
These observations suggest that neuronal GLT-1 plays an important physiological role in glutamate uptake and transcriptional regulation throughout the brain, and particularly in the striatum and prefrontal cortex, which are critical targets of dopaminergic signaling (Berger et al., 2005; Pavese et al., 2003). Beyond its potential relevance to understanding neurodegenerative disorders, our findings suggest that GLT-1 expressed in neurons may regulate dopamine-dependent reward, motivation, and addiction processes. Interestingly, higher CB2 levels produced by overexpression have been shown to reduce addiction and cocaine self-administration in mice (Aracil-Fernández et al., 2012). It has recently been reported that synGLT-1 KO mice show decreased locomotor response to acute administration of another psychostimiulant, amphetamine (Fischer et al., 2018). Whether this phenotype is related to increased expression of CB2 receptors in these mice is unknown. As with CB2, PDE10A activity is thought to have important effects on dopaminergic signaling in the striatum (Wilson and Brandon, 2015). PDE10A inhibition has been shown to improve corticostriate pathway function in Huntington’s disease models (Beaumont et al., 2016) and has been considered as a therapeutic modality in schizophrenia as well as in HD (Geerts et al., 2016; Harada et al., 2017). Future research should determine the specific alterations in glutamate homeostasis and synaptic function produced by inactivation of GLT-1 in neurons, how these alterations produce the transcriptional dysregulation observed here, and whether and how the functions of GLT-1 expressed in neurons affects dopaminergic signaling. Most intriguing is the possibility that the synaptopathy in HD might be similar to that in the neuronal GLT-1 knockout, and that understanding the latter might provide a clue to the nature and origin of the HD synaptopathy.
Highlights.
Transcriptional dysregulation was assessed in a knockout of GLT-1 confined to neurons
A panel of genes was assayed known to be altered in Huntington’s disease
Similar changes in certain genes were observed in the neuronal GLT-1 KO as in HD
Expression changes in HD may be due a synaptopathy caused by glutamate dyshomeostasis
Acknowledgements
This work was supported by the Hereditary Disease Foundation, Children’s Hospital Intellectual and Developmental Disabilities Research Center (IDDRC) core grant HD 018655, NIH RO1NS066019, NIH R21MH104318, a partnership grant from Canadian Institutes of Health Research (CIHR), Nova Scotia Health Research Foundation (NSHRF), and the Huntington Society of Canada (HSC) (ROP-97185). RBL was supported by studentships from CIHR, HSC, Killam Trusts, NSHRF, and Canadian Consortium for the Investigation of Cannabinoids. The authors also wish to acknowledge Kathleen Murphy from Dalhousie University, for her technical assistance with this study.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors have no conflict of interest to report.
Abbreviations:
Cb, cerebellum;
CB1, type 1 cannabinoid receptor;
CB2, type 2 cannbinoid receptor;
D2, type 2 dopamine receptor;
DARPP-32, dopamine and cAMP-regulated protein phosphatase 32 kDA;
Dnm1, dynamin-1;
EAAT1 (GLAST), Excitatory amino acid tranporter 1;
EEAT2 (GLT-1 slc1a2), Excitatory amino acid transporter 2;
EAAT3 (EAAC1), Excitatory amino acid transporter 3;
EAAT4, Excitatory amino acid transporter 4;
EAAT5, Excitatory amino acid transporter 5;
HD, Huntington’s disease;
MCtx, motor cortex;
NAcc, nucleus accumbens;
NGFIA, nerve growth factor-induced clone A;
nGLT-1−/− (synGLT-1 KO), conditional neuronal GLT-1 knockout;
OD, optical density;
PDE1B, phosphodiesterase 1B;
PDE10A, phosphodiesterase 10A;
PFc, prefrontal cortex;
ppENK, preproenkephalin;
RT, reverse transcriptase;
SNr, substantia nigra;
Str, striatum;
Th, thalmus;
VTA, ventral tagmental area;
and WT, wild-type
References
- Aracil-Fernández A, Trigo JM, García-Gutiérrez MS, Ortega-Álvaro A, Ternianov A, Navarro D, Robledo P, Berbel P, Maldonado R, Manzanares J, 2012. Decreased cocaine motor sensitization and self-administration in mice overexpressing cannabinoid CB₂ receptors. . Neuropsychopharmacology 37, 1749–1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaumont V, Zhong S, Lin H, Xu W, Bradaia A, Steidl E, Gleyzes M, Wadel K, Buisson B, Padovan-Neto FE, Chakroborty S, Ward KM, Harms JF, Beltran J, Kwan M, Ghavami A, Haggkvist J, Toth M, Halldin C, Varrone A, Schaab C, Dybowski JN, Elschenbroich S, Lehtimaki K, Heikkinen T, Park L, Rosinski J, Mrzljak L, Lavery D, West AR, Schmidt CJ, Zaleska MM, Munoz-Sanjuan I, 2016. Phosphodiesterase 10A Inhibition Improves Cortico-Basal Ganglia Function in Huntington’s Disease Models. Neuron 92, 1220–1237. [DOI] [PubMed] [Google Scholar]
- Berger UV, DeSilva TM, Chen W, Rosenberg PA, 2005. Cellular and subcellular mRNA localization of glutamate transporter isoforms GLT1a and GLT1b in rat brain by in situ hybridization. . J Comp Neurol 492, 78–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisogno T, Di Marzo V, 2010. Cannabinoid receptors and endocannabinoids: role in neuroinflammatory and neurodegenerative disorders. . CNS Neurol Disord Drug Targets 9, 564–573. [DOI] [PubMed] [Google Scholar]
- Bunner KD, Rebec GV, 2016. Corticostriatal Dysfunction in Huntington’s Disease: The Basics. Front Hum Neurosci 10, 317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cain-Hom C, Splinter E, van Min M, Simonis M, van de Heijning M, Martinez M, Asghari V, Cox JC, Warming S, 2017. Efficient mapping of transgene integration sites and local structural changes in Cre transgenic mice using targeted locus amplification. Nucleic Acids Res 45, e62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casteels C, Martinez E, Bormans G, Camon L, de Vera N, Baekelandt V, Planas AM, Van Laere K, 2010. Type 1 cannabinoid receptor mapping with [18F]MK-9470 PET in the rat brain after quinolinic acid lesion: a comparison to dopamine receptors and glucose metabolism. . European Journal of Nuclear Medicine and Molecular Imaging 37, 2354–2363. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Wu N, Andre VM, Cummings DM, Levine MS, 2007. The corticostriatal pathway in Huntington’s disease. Prog Neurobiol 81, 253–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha JH, 2000. Transcriptional dysregulation in Huntington’s disease. Trends Neurosci 23, 387–392. [DOI] [PubMed] [Google Scholar]
- Cha JH, 2007. Transcriptional signatures in Huntington’s disease. Prog Neurobiol 83, 228–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha JH, Kosinski CM, Kerner JA, Alsdorf SA, Mangiarini L, Davies SW, Penney JB, Bates GP, Young AB, 1998. Altered brain neurotransmitter receptors in transgenic mice expressing a portion of an abnormal human huntington disease gene. Proc Natl Acad Sci U S A 95, 6480–6485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Mahadomrongkul V, Berger UV, Bassan M, DeSilva T, Tanaka K, Irwin N, Aoki C, Rosenberg PA, 2004. The glutamate transporter GLT1a is expressed in excitatory axon terminals of mature hippocampal neurons. J Neurosci 24, 1136–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen-Hwang MC, Chen HR, Elzinga M, Hwang YW, 2002. Dynamin is a minibrain kinase/dual specificity Yak-1 related kinase 1A substrate. Journal of Biological Chemistry 277, 17597–17604. [DOI] [PubMed] [Google Scholar]
- Chiarlone A, Bellocchio L, Blázquez C, Resel E, Soria-Gómez E, Cannich A, Ferrero JJ, Sagredo O, Benito C, Romero J, Sánchez-Prieto J, Lutz B, Fernández-Ruiz J, GalveRoperh I, Guzmán M, 2014. A restricted population of CB1 cannabinoid receptors with neuroprotective activity. . Proceedings of the National Academy of Science USA 111, 8257–8262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danbolt NC, 2001. Glutamate uptake. Progress in Neurobiology 65, 1–105. [DOI] [PubMed] [Google Scholar]
- Danbolt NC, Storm-Mathiesen J, Kanner BI, 1992. An [Na+ K+] coupled L-glutamate transporter purified from rat brain is located in glial cell processes. . Neuroscience 51, 295–301. [DOI] [PubMed] [Google Scholar]
- Denovan-Wright EM, Newton RA, Armstrong JN, Babity JM, Robertson HA, 1998. Acute administration of cocaine, but not amphetamine, increases the level of synaptotagmin IV mRNA in the dorsal striatum of rat. Brain Res Mol Brain Res 55, 350–354. [DOI] [PubMed] [Google Scholar]
- Denovan-Wright EM, Robertson HA, 2000. Cannabinoid receptor messenger RNA levels decrease in a subset of neurons of the lateral striatum, cortex and hippocampus of transgenic Huntington’s disease mice. Neuroscience 98, 705–713. [DOI] [PubMed] [Google Scholar]
- Fischer KD, Houston ACW, Desai RI, Doyle MR, Bergman J, Mian M, Mannix R, Sulzer DL, Choi SJ, Mosharov EV, Hodgson NW, Bechtholt A, Miczek KA, Rosenberg PA, 2018. Behavioral phenotyping and dopamine dynamics in mice with conditional deletion of the glutamate transporter GLT-1 in neurons: resistance to the acute locomotor effects of amphetamine. Psychopharmacology [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furness DN, Dehnes Y, Akhtar AQ, Rossi DJ, Hamann M, Grutle NJ, Gundersen V, Holmseth S, Lehre KP, Ullensvang K, Wojewodzic M, Zhou Y, Atwell D, Danbolt NC, 2008. A quantitative assessment of glutamate uptake into hippocampal synaptic terminals and astrocytes: new insights into a neuronal role for excitatory amino acid transporter 2 (EAAT2). Neuroscience 157, 80–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaveriaux-Ruff C, Kieffer BL, 2007. Conditional gene targeting in the mouse nervous system: Insights into brain function and diseases. Pharmacology and Therapeutics 113, 619–634. [DOI] [PubMed] [Google Scholar]
- Geerts H, Spiros A, Roberts P, 2016. Phosphodiesterase 10 inhibitors in clinical development for CNS disorders. Expert Rev Neurother, 1–8. [DOI] [PubMed] [Google Scholar]
- Genda EN, Jackson JG, Sheldon AL, Locke SF, Greco TM, O’Donnell JC, Spruce LA, Xiao R, Guo W, Putt M, Seeholzer S, Ischiropoulos H, Robinson MB, 2011. Co-compartmentalization of the astroglial glutamate transporter, GLT-1, with glycolytic enzymes and mitochondria. Journal of Neuroscience 31, 18275–18288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez GT, Hu H, McCaw EA, Denovan-Wright EM, 2006. Brain-specific factors in combination with mutant huntingtin induce gene-specifc transcriptional dysregulation. . Molecular Cellular Neuroscience 31, 661–675. [DOI] [PubMed] [Google Scholar]
- Harada A, Suzuki K, Kimura H, 2017. TAK-063, a Novel Phosphodiesterase 10A Inhibitor, Protects from Striatal Neurodegeneration and Ameliorates Behavioral Deficits in the R6/2 Mouse Model of Huntington’s Disease. J Pharmacol Exp Ther 360, 75–83. [DOI] [PubMed] [Google Scholar]
- Haugeto Ø, Ullensvang K, Levy LM, Chaudry FA, Honoré T, Neilsen M, Lehre KP, Danbolt NC, 1996. Brain glutamate transporter proteins form homomultimers. Journal of Biological Chemistry 271, 2715–2772. [DOI] [PubMed] [Google Scholar]
- He XP, Kotloski R, Nef S, Luikart BW, Parada LF, McNamara JO, 2004. Conditional deletion of TrkB but not BDNF prevents epileptogenesis in the kindling model. Neuron 43, 31–42. [DOI] [PubMed] [Google Scholar]
- Hebb AL, Robertson HA, Denovan-Wright EM, 2004. Striatal phosphodiesterase mRNA and protein levels are reduced in Huntington’s disease transgenic mice prior to the onset of motor symptoms. Neuroscience 123, 967–981. [DOI] [PubMed] [Google Scholar]
- Hogel M, Laprairie RB, Denovan-Wright EM, 2012. Promoters are differentially sensitive to N-terminal mutant huntingtin-mediated transcriptional repression. PLoS One 7, e41152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmseth S, Dehnes Y, Huang YH, Follin-Arbelet VV, Grutle NJ, Mylonakou MN, Plachez C, Zhou Y, Furness DN, Bergles DE, Lehre KP, Danbolt NC, 2012. The density of EAAC1 (EAAT3) glutamate transporters expressed by neurons in the mammalian CNS. Journal of Neuroscience 32, 6000–6013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, McCaw EA, Hebb AL, Gomez GT, Denovan-Wright EM, 2004. Mutant huntingtin affects the rate of transcription of striatum-specific isoforms of phosphodiesterase 10A. Eur J Neurosci 20, 3351–3363. [DOI] [PubMed] [Google Scholar]
- Huang K, Kang MH, Askew C, Kang R, Sanders SS, Wan J, Davis NG, Hayden MR, 2010. Palmitoylation and function of glial glutamate transporter-1 is reduced in the YAC128 mouse model of Huntington disease. Neurobiology of Disease 40, 207–215. [DOI] [PubMed] [Google Scholar]
- Jackson JG, Robinson MB, 2017. Regulation of mitochondrial dynamics in astrocytes: Mechanisms, consequences, and unknowns. Glia [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG, 2010. Improving Bioscience Research Reporting: The ARRIVE Guidelines for Reporting Animal Research. PLoS Biology 8, e1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiman RJ, Kimmel LH, Bove SE, Lanz TA, Harms JF, Romegialli A, Miller KS, Willis A, des Etages S, Kuhn M, Schmidt CJ, 2011. Chronic suppression of phosphodiesterase 10A alters striatal expression of genes responsible for neurotransmitter synthesis, neurotransmission, and signaling pathways implicated in Huntington’s disease. Journal of Pharmacology and Experimental Therapeutics 336, 64–76. [DOI] [PubMed] [Google Scholar]
- Laprairie RB, Bagher AM, Precious SV, Denovan-Wright EM, 2015. Components of the endocannabinoid and dopamine systems are dysregulated in Huntington’s disease: analysis of publicly available microarray datasets. Pharmacology Research and Perspectives 3, e00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laprairie RB, Warford JR, Hutchings S, Robertson GS, Kelly ME, Denovan-Wright EM, 2014. The cytokine and endocannabinoid systems are co-regulated by NF-κB p65/RelA in cell culture and transgenic mouse models of Huntington’s disease and in striatal tissue from Huntington’s disease patients. Journal of Neuroimmunology 267, 61–72. [DOI] [PubMed] [Google Scholar]
- Lehre KP, Levy LM, Ottersen OP, Storm-Mathiesen J, Danbolt NC, 1995. Differential expression of two glial glutamate transporters in the rat brain: quantitative and immunocytochemical observations. . Journal of Neuroscience 15, 1835–1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CH, Tallaksen-Greene S, Chien WM, Cearley JA, Jackson WS, Crouse AB, Ren S, Li XJ, Albin RL, Detloff PJ, 2001. Neurological abnormalities in a knock-in mouse model of Huntington’s disease. Human Molecular Genetics 10, 137–144. [DOI] [PubMed] [Google Scholar]
- McCaw EA, Hu H, Gomez GT, Hebb AL, Kelly ME, Denovan-Wright EM, 2004. Structure, expression and regulation of the cannabinoid receptor gene (CB1) in Huntington’s disease transgenic mice. Eur J Biochem 271, 4909–4920. [DOI] [PubMed] [Google Scholar]
- Melone M, Bellesi M, Conti F, 2009. Synaptic localization of GLT-1a in the rat somatic sensory cortex. Glia 57, 108–117. [DOI] [PubMed] [Google Scholar]
- Melone M, Bellesi M, Ducati A, Iacoangeli M, Conti F, 2011. Cellular and Synaptic Localization of EAAT2a in Human Cerebral Cortex. Front Neuroanat 4, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Kano M, 2014. Endocannabinoid-mediated retrograde modulation of synaptic transmission. Current Opinion in Neurobiology 29C, 1–8. [DOI] [PubMed] [Google Scholar]
- Otis TS, Kavanaugh MP, 2000. Isolation of current components and partial reaction cycles in the glial glutamate transporter EAAT2. Journal of Neuroscience 20, 2749–2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palazuelos J, Aguado T, Pazos MR, Julien B, Carrasco C, Resel E, Sagredo O, Benito C, Romero J, Azcoitia I, Fernández-Ruiz J, Guzmán M, Galve-Roperh I, 2009. Microglial CB2 cannabinoid receptors are neuroprotective in Huntington’s disease excitotoxicity. . Brain 132, 3152–3164. [DOI] [PubMed] [Google Scholar]
- Pavese N, Andrews TC, Brooks DJ, Ho AK, Rosser AE, Barker RA, Robbins TW, Sahakian BJ, Dunnett SB, Piccini P, 2003. Progressive striatal and cortical dopamine receptor dysfunction in Huntington’s disease: a PET study. . Brain 126, 1127–1135. [DOI] [PubMed] [Google Scholar]
- Petr GT, Schultheis LA, Hussey KC, Sun Y, Dubinsky JM, Aoki CJ, Rosenberg PA, 2013. Decreased expression of GLT-1 in the R6/2 model of Huntington’s disease does not worsen disease progression. European Journal of Neuroscience 38, 2477–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petr GT, Sun Y, Frederick NM, Zhou Y, Dhamne SC, Hameed MQ, Miranda C, Bedoya EA, Fischer KD, Armsen W, Wang J, Danbolt NC, Rotenberg A, Aoki CJ, Rosenberg PA, 2015. Conditional deletion of the glutamate transporter GLT-1 reveals that astrocytic GLT-1 protects against fatal epilepsy while neuronal GLT-1 contributes significantly to glutamate uptake into synaptosomes. J Neurosci 35, 5187–5201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond LA, 2016. Striatal synaptic dysfunction and altered calcium regulation in Huntington disease. Biochem Biophys Res Commun [DOI] [PubMed] [Google Scholar]
- Rempe D, Vangeison G, Hamilton J, Li Y, Jepson M, Federoff HJ, 2006. Synapsin I Cre transgene expression in male mice produces germline recombination in progeny. Genesis 44, 44–49. [DOI] [PubMed] [Google Scholar]
- Rimmele TS, Rosenberg PA, 2016. GLT-1: The elusive presynaptic glutamate transporter. Neurochemistry International 98, 19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MB, Jackson JG, 2016. Astroglial glutamate transporters coordinate excitatory signaling and brain energetics. Neurochemistry International 98, 56–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, Wada K, 1997. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 276, 1699–1702. [DOI] [PubMed] [Google Scholar]
- Taugher RJ, Lu Y, Fan R, Ghobbeh A, Kreple CJ, Faraci FM, Wemmie JA, 2017. ASIC1A in neurons is critical for fear-related behaviors. Genes Brain Behav 16, 745–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas EA, 2006. Striatal specificity of gene expression dysregulation in Huntington’s disease. J Neurosci Res 84, 1151–1164. [DOI] [PubMed] [Google Scholar]
- Van Bockstaele EJ, Saunders A, Commons KG, Liu X-B, Peoples J, 2000. Evidence for coexistence of enkephalin and glutamate in axon terminals and cellular sites for functional interactions of their receptors in the rat locus coeruleus. Journal of Comparative Neurology 417, 103–114. [DOI] [PubMed] [Google Scholar]
- Wilson LS, Brandon NJ, 2015. Emerging biology of PDE10A. Curr Pharm Des 21, 378–388. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Hassel B, Eid T, Danbolt NC, 2018. Axon-terminals expressing EAAT2 (GLT-1; Slc1a2) are common in the forebrain and not limited to the hippocampus. Neurochemistry International [DOI] [PubMed] [Google Scholar]
- Zhu Y, Romero MI, Ghosh P, Ye Z, Charnay P, Rushing EJ, Marth JD, Parada LF, 2001. Ablation of NF1 function in neurons induces abnormal development of cerebral cortex and reactive gliosis in the brain. Genes Dev 15, 859–876. [DOI] [PMC free article] [PubMed] [Google Scholar]