

Abstract
Cancers are complex diseases having several unique features, commonly described as ‘hallmarks of cancer’. Among them, altered signaling pathways are the common characteristic features that drive cancer progression; this is achieved due to mutations that lead to the activation of growth promoting(s) oncogenes and inactivation of tumor suppressors. As a result of which, cancer cells increase their glycolytic rate by consuming a large amount of glucose, and convert a majority of glucose to lactate even in the presence of oxygen known as the “Warburg effect”. Tumor cells like other cells are strictly dependent on energy for growth and survival; therefore, understanding energy metabolism will give us an idea to develop new effective anti-cancer therapies that target cancer energy production pathways. This review summarizes the roles of tumor suppressors and oncogenes and their products that provide metabolic advantages to cancer cells which in turn leads to the establishment of the “Warburg effect” and ultimately leads to cancer progression. Understanding cancer cell’s vulnerability will provide potential targets for its control.
Keywords: Glycolysis, HIF-1α, c-Myc, AMPK, p53
Introduction
Tumor cells, like other cells, are strictly dependent on an adequate supply of energy for their growth and survival. For a tumor to produce two daughter cells several anabolic processes are involved, all costly in term of energy, such as synthesis of nucleic acids, proteins, lipids. With regard to energy production, it is well established that tumor cells not only survive but thrive by increasing the rate of their cellular processes such as proliferation, migration and invasion as a results of selection of a particular metabolic pathways that are suitable for their needs to generate enough ATP and other metabolites even when underfed or hypoxic condition, a condition that is harmful to normal cells (Amoedo et al., 2013).
During the transition from normal to cancer cells, energy metabolism is the most affected processes, particularly glucose metabolism. In normal cells, glucose enters the cells via glucose transporters proteins (GLUTs). Once inside the cells, glucose is oxidized to pyruvate via a series of ten enzymatic steps (glycolysis). Pyruvate in the presence of O2 is further oxidized to CO2 and H2O in the mitochondria via TCA cycle, generating high energy molecules such as NADPH, FADH2 which are reduced in the inner mitochondria and creating energy in the form of ATP (Ward and Thompson, 2012). Typically 36/38 molecules of ATP are generated per molecule of glucose oxidized. Alternatively, in the absence of oxygen, pyruvate is converted into lactic acid via lactate dehydrogenase (anaerobic glycolysis) which is then transported to the liver via the Cori Cycle (Cox and Nelson, 2013).
In contrast to normal cells, tumor cells consume a large amount of glucose, maintain a much higher rate of glucose and convert majority of glucose to pyruvate even in the presence of oxygen, a phenomenon term as “aerobic glycolysis” or “Warburg Effect” as shown in Figure 1 (Koppenol et al., 2011). This increased aerobic glycolysis is considered as one of the hallmarks of cancer (Yeung, 2008), the other hallmarks being limitless replicative potential, self-sufficiency in growth signals, evading apoptosis, insensitivity to antigrowth signals, sustained angiogenesis, and tissue invasion and metastasis (Hanahan and Weinberg, 2000).
Figure 1.
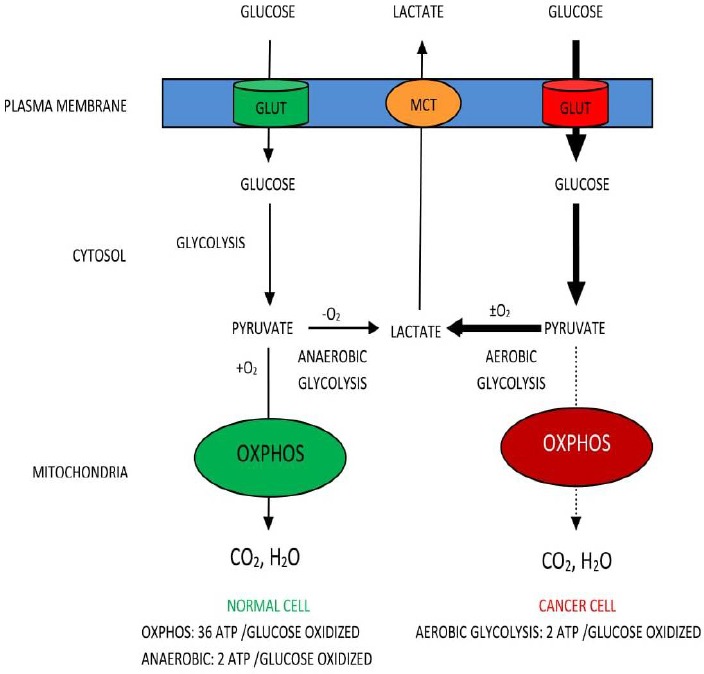
Utilizations of Glucose by Normal and Cancer Cell (The Warburg Effect)
Unlike normal cells, tumor cells preferentially used aerobic glycolysis over oxidative phosphorylation for glucose-dependent ATP production due to mitochondrial impairments (Zheng, 2012). Aerobic glycolysis yields 2 ATP whereas with mitochondria yields 36/38 ATP. Cancer cells have to compensate for this lower energy production to maintain their growth; part of this solution is to up-regulate glucose transporters as well as glycolytic enzymes (Phan et al., 2014). In fact, increase consumption of glucose by cancer cells has been well documented by using 18F-fluorodeoxyglucose (FDG-glucose) an analog of glucose (Burt et al., 2001). Once inside the cell, FDG-glucose is phosphorylated to FDG-glucose-6-phosphate by hexokinase but since 3’–OH group is occupied, it cannot be further oxidized as a result it accumulates and can be visualized by Positron Emission Tomography (PET) scan. This provides anatomical information about glucose intake on PET image (Dang, 2010; Palaskas et al., 2011). In addition, tumors remarkably elevated the expression of the majority of glycolytic enzymes in an insufficient p53-mediated control (Phan et al., 2014). Major oncogenes such as Ras, Myc, and HIF-1α are reported to be master inducers of cancer glycolysis through direct or indirect transactivation of cancer glycolytic genes, which is elaborated in Figure 2.
Figure 2.
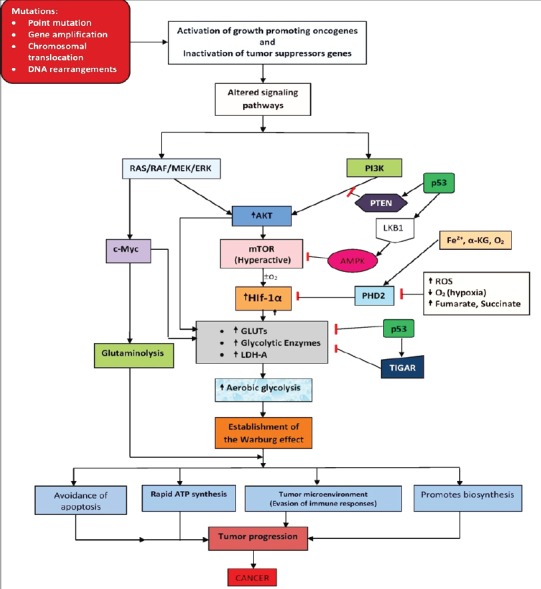
Altered Signaling Pathways and Their Contributions to the Warburg Effect in Cancer Glucose Metabolism. Up or -down arrows indicate an increase or decrease in activity, respectively.
Dysregulation of glucose metabolism in cancer cells by oncogenes and tumor suppressors
It has been long proposed that altered expression or activities of glycolytic enzymes are regulated by oncogenes (i.e. the mutated version of proto-oncogenes) and tumor suppressor genes (DeBerardinis, 2008). Proto-oncogenes are groups of genes that encode proteins that are involved in cell division, cell differentiation, apoptosis and many another biological processes that are crucial for normal human development (Lodish et al., 2000). Mutation is the root cause of cancer and the mutated version of proto-oncogenes is called oncogenes. Various mutations that lead to inactivation of tumor suppressors and conversion of proto-oncogenes to oncogenes include point mutations, deletions, insertions, gene amplification, chromosomal translocations, DNA rearrangements (Lodish et al., 2000). Hyper-activation of oncogene leads to increased expression of their encoded proteins. In fact, many glycolytic enzymes are upregulated in tumors because of elevated HIF-1α and c-Myc transcriptional activity and insufficient p53-mediated control, which in turn altered the growth signaling pathways thus leading to uncontrolled cell division, inhibit apoptosis; altogether, drives cancer progression as summarized in Figure 2.
The PI3K/AKT/mTOR pathway reprograms glucose metabolism to fuel cell growth
In a higher organism, cells are exposed to a constant supply of nutrients. Therefore, a control system is required to prevent unwanted uptake of glucose because cells do not take glucose from the surrounding environment based on needs but based on instructions from growth factors through receptor tyrosine kinases (RTKs) (Ward and Thompson, 2012). Normal cells upon stimulation by growth factors activates PI3K and its downstream pathway AKT and mTOR, thereby promoting robust anabolic program involving increase glycolytic flux and FA synthesis that support cellular processes such as proliferation, growth, apoptosis and cytoskeleton rearrangement (Luo et al., 2010). In human cancer, this pathway is one of the most active and frequently altered growth signaling pathways, which not only provides a strong growth signal but also has a huge impact on glucose metabolism, the so-called “glucose addiction”(Long and Zierath, 2006). Aberrant activation of this pathway drives tumor progression independence on extrinsic growth factor stimulation (the hallmark of cancer) (Li et al., 2015). This is achieved through mutations in tumor suppressor genes, such as PTEN, mutations in the components of the PI3K complex itself or by hyperactivation of receptor tyrosine kinases (McCubrey et al., 2012).
The phosphoinositide-3 kinases (PI3K) are the family of lipid kinases that consist of three classes, of which Class I PI3K pathway is mainly associated with oncogenic cellular transformation and cancer (Cantley, 2002; Zhao and Vogt, 2008). Both the catalytic and a regulatory subunit of Class I PI3Ks are encoded by different genes. The catalytic subunit has four isoforms as p110α, p110β, p110γ and p110δ, whereas, the regulatory subunit consist of five different isoforms of p85 (p85α, p85β, p55α, p55γ and p50α). The catalytic subunit can form dimer with any of the five isoforms to generate an active kinase (Geering et al, 2007a; Geering et al, 2007b; Zhao and Vogt, 2008). Upon stimulation by growth factors such as HER2 (Human epidermal growth factor receptor 2), PDGFR (Platelet-derived growth factor receptor), EGFR (Epidermal growth factor receptor), insulin growth like factor 1 receptor (IGF1R), insulin receptor (IR) and G-protein coupled receptors, activates this pathway (Zhao and Vogt, 2008; Wong, 2010). Once activated, the p110 catalytic subunit phosphorylates phosphatidylinositol bisphosphate (PIP2) to generate phosphatidylinositol triphosphate (PIP3) which acts as a second cellular messenger. PIP3 then binds and recruit AKT1 kinase (protein kinase B) to the cell membrane for phosphorylation by phosphoinositide-dependent protein kinase 1 (PDK-1) at threonine 308 and serine 473 by PDK2 (Vivanco and Sawyers, 2002; Xing, 2010). Activated AKT1 stimulates glycolysis by phosphorylating key glycolytic enzymes, such as hexokinase and phosphofructokinase 2 (an allosteric activator of PFK1) (Robey and Hay, 2009). AKT1 also increase the expression and membrane translocation of glucose transporters, this increases the availability of glucose in the cytoplasm for oxidation of glucose to pyruvate (Barthel et al., 1999; Buzzai et al., 2005; DeBerardinis et al., 2007). Finally, AKT1 provide a strong signal toward its downstream element mTOR (mammalian Target of Rapamycin) by phosphorylating and inhibiting its negative regulator tuberous sclerosis 2 (TSC2). Activated mTOR directly stimulate anabolic processes and indirectly stimulated metabolism by activating key transcription factors such as cMyc, cyclin D, and HIF-1α even under normoxic condition (Cantor and Sabatini, 2012). This pathway is antagonized by the tumor suppressor protein (PTEN) which reverses the phosphorylation of PI3K products. Specifically, PTEN dephosphorylates PIP3, preventing Akt1 activation, thereby directly antagonizing the PI3K function and blocking, therefore the activation of downstream signaling events, including PDK1, (Akt) and Akt/mammalian target of rapamycin (mTOR) (Gingras et al., 2001). Loss of PTEN expression, which is often observed in several types of human cancers, specifically in glioblastoma, melanoma, endometrial, and prostate cancers (Hollander et al., 2011), promotes Akt activation which in turn increases glycolysis by promoting HK2 expression, furthermore Akt phosphorylate HK2 which directly interact with the mitochondrial pore to prevent the release of apoptotic proteins (Gottlob et al., 2001; Pastorino et al., 2002; Miyamoto et al., 2008). Recent studies show that silencing and deletion of HK2 in both human and mouse model of PTEN-deficient prostate tumors decrease cancer proliferation and increased cell death (Nogueira et al., 2018). Therefore, HK2 is considered as an attractive target for anticancer agents. In addition; Akt suppresses apoptosis through the activation of FOXO3a and promotes mitochondrial biogenesis to a growing cell to ensure cell survival (Dang, 2012). Thus, hyperactivation of mTOR or its upstream elements and loss of tumor suppressor PTEN induce aerobic glycolysis by direct stimulation of glucose transporter expression and stimulating hexokinase (HKII) and PFK1 activity through AKT, thereby enhance glucose oxidation to pyruvate and together with oncogenic proteins drives tumor progression (Cantor and Sabatini, 2012). Thus, increase and prolonged PI3K signaling pathway results in glucose addiction and promotes a glycolytic switch under normoxic conditions Cantor and Sabatini, (2012) as well as cell survival. Thus, cancer cells turn out to be dependent on aerobic glycolysis for their growth and survival under the AKT-mediated metabolic influence; consequently, activated AKT tumor cells undergo rapid cell death when shifted to low-dose glucose conditions (Elstrom et al., 2004). Small molecule inhibitors known as pan-inhibitor (pan-PI3K and pan-Akt inhibitors) that interfere with the PI3K/Akt signaling pathway have been developed in recent years which are currently being tested in clinical trial. However treatment with pan-inhibitors shows undesirable side effects such as such, hyperglycemia, hyperinsulinemia, and diabetes (Wang et al., 2017) (Nogueira et al., 2018), which indicates that development of selective inhibitors is crucially important.
Hypoxia inducible factor
Like a normal cell, cancer cells must also adapt during hypoxia in order to maintain tumor growth. Since cancer cells divide rapidly, their proliferative rate will always exceed the rate of new blood vessel formation (Angiogenesis) (Dakubo, 2010). Therefore, all cancer cells, must, at some point experience a hypoxic condition which is coordinated by Hypoxia Inducible Factor 1 (HIF-1). HIF-1 is a heterodimeric transcription factor, composed of two subunits, the HIF-1α (O2 sensitive) which is ubiquitously expressed and HIF-1β subunit which is constitutively expressed (Wang et al., 1995). HIF-1α further consists of three isoforms; HIF-1α, HIF- 2α, and HIF-3α and they all possessed an oxygen-dependent degradation domain (ODDD) which is important in mediating O2 regulation stability (Bruick and McKnight, 2001). Unlike HIF-3α, HIF-2α shows 48%sequence similarities with HIF-1α and may form hetero-dimerization with HIF-1β and binding with HREs (Masoud and Lin, 2015). Interestingly, HIF-1α and HIF-2α show a different pattern of tissue distributions. HIF-1α is ubiquitously expressed in the body, which is primarily involved in transcriptional activation of many glycolytic genes (Masoud and Lin, 2015) whereas HIF-2α expression is stricter to specific tissues such as endothelial, lung, renal, and hepatic cells and does not involve in glucose metabolism directly, but drives cell cycle progression through the interaction with oncoprotein c-Myc (Ema et al., 1997; Wiesener et al., 2003; Gordan et al., 2007). Recent discovery show that both HIF-1α and HIF-2α are highly expressed in many human tumors (Lv et al., 2017), but HIF-3α is down-regulated in renal cell carcinomas (Maynard et al., 2015). Given the role of HIF-3α, it is not surprising that HIF-3α is down-regulated because of its ability to inhibit the transcriptional activity of HIF-1α (Pasanen et al., 2010), the main isoform which is responsible for activating transcriptional responses under hypoxia (Lv et al., 2017).
Under normoxia, HIF-1α undergo oxygen-dependent hydroxylation at proline residues 402 and 564 (human sequence) by prolylhydroxylases (PHD2) enzyme, resulting in their recognition by von Hippel–Lindau tumor suppressor (VHL), an E3 ubiquitin ligase, for poly-ubiquitylation and subsequent degradation by proteosomes (Hirsila et al., 2003). PHD2 is a Fe (II) and 2-oxoglutarate dependent dioxygenase which has an absolute requirement for molecular oxygen: because of its low affinity for oxygen (Km =250 μM, slightly above air pO2 (Semenza, 2000a). PHD2 is often described as an oxygen sensor. During hypoxia, PHD2 loses its ability to hydroxylate which allow the HIF-1α subunit to escape VHL-mediated destruction and accumulate to high levels. Then, HIF-1α dimerizes with the constitutively present HIF-1β subunit and accumulates in the nucleus. Subsequently, the HIF-1 dimer (i.e. the active complex of HIF-1α and HIF-1β) binds to the hypoxia response element of target genes (Semenza, 2000b; Maxwell and Pugh, 2001; Gordan et al., 2007; Semenza, 2012) resulting in their transcriptional activation. Initiation of transcription further requires the interaction of HIF-1 with the cofactors p300 and the DNA polymerase II complex to bind to hypoxia-responsive elements (HRE) of target genes and modulate genes expression (Porporato et al., 2011) such as those that are involved in the acceleration of glycolysis in response to HIF-1 activity (Semenza, 2000b; Maxwell and Pugh, 2001; Gordan et al., 2007; Semenza, 2010; Semenza, 2012) and those that are involved in vasculogenesis and/or neoangiogenesis (Baeriswyl and Christofori, 2009), such as vascular endothelial growth factor (VEGF), VEGF receptors FLT-1 and FLK, platelet-derived growth factor B (PDGF-B), matrix metalloproteinases (MMP-2 and MMP-9) plasminogen activator inhibitor-1 (PAI-1), the TIE-2 receptor, and angiopoietins (ANG-1 and ANG -2), which are required by tumor cells for growth and development (Hickey and Simon, 2006).
The HIF-1α protein level in a cell is also controlled by oncogenic signaling pathways. Activation of phosphatidylinositol-4, 5-bisphosphate-3-kinase (PI3K) and RAS/RAF/MEK/ERK kinase cascade can upregulate the HIF-1α protein translation (Conrad et al., 1999; Jiang et al., 2001; Semenza, 2002). Activated ERK, a downstream element of the RAS/RAF/MEK/ERK kinase cascade is not only involved in regulation of HIF-1α synthesis but also phosphorylates the co-activator CBP/p300 which further stabilized the binding of the active complex (i.e HIF-1α, HIF-1β and co-activator CBP/300) with RNA polymerase to positively modulates genes expression (Sang et al., 2003).
ROS generated from complexes II and III in the mitochondria also mediates HIF-1α stabilization (Guzy et al., 2005; Guzy et al., 2008; Klimova and Chandel, 2008). Neutralization of ROS is achieved through the oxidation of Fe2+ in the Fenton reaction to Fe3+. However, since Fe2+ and not Fe3+ is a cofactor for the enzyme prolyl hydroxylases (PHD2), therefore, a decreased in Fe2+ will decrease the activity of this enzyme (Dakubo, 2010). As a result of which HIF-1α escape hydroxylation by PHD2 for ubiquitination and degradation, leading to its stabilization and in turn modulate genes expression.
In addition, the TCA cycle metabolites fumarate and succinate and oncometabolite 2-hydroxyglutarate (2-HG) competitively inhibit HIF-1α hydroxylation, leading to the upregulation of HIF-1α (Dakubo, 2010). 2-HG competes with α-KG for the enzyme PHD2 as a result, the enzyme is inactivated which lead to HIF-1α stabilization (Dakubo, 2010). Mutations in several mitochondrial proteins are observed in tumors such as those involved in the TCA cycle enzymes i.e. succinate dehydrogenase and fumarate hydratase, making them unable to process succinate and fumarate as a result, these metabolites accumulate and promote HIF-1α stabilized (Gottlieb and Tomlinson, 2005; Pollard and Ratcliffe, 2009). In certain cases, the HIF-1α subunit is stabilized, even under normoxic conditions through a variety of mechanism including hyperactivation of mTOR, loss of vonHipel, accumulation of ROS and also the accumulation of TCA cycle metabolites such as succinate, fumarate, and 2-hydroxyglutarate (Soga, 2013). All these events have been attributed to a cancer-specific mutation in SDH and FH and IDH (Dakubo, 2010; Soga, 2013).
Hypoxia-Inducible Factor and glycolysis
HIF-1 positively regulates the transcription of over 100 genes, which involved in glucose metabolism, cell proliferation, migration and angiogenesis (Semenza, 2001). In a rapidly growing tumor tissue, HIF-1 helps hypoxic tumor cells to shift glucose metabolism from the more efficient oxidative phosphorylation to the less efficient glycolytic pathway in order to maintain their energy production (the Warburg effect) (Weinhouse et al., 1956). For this reason, hypoxic cells tend to consume more glucose in order to meet their energy needs (rapid ATP production). HIF-1 mediates this metabolic conversion through the induction of enzymes involved in the glycolysis pathway and over expression of glucose transporters (GLUT1 and GLUT3) which increase glucose import into tumor cells. This increases the availability of glucose within the cytoplasm for energy production (Denko, 2008). HIF-1 then facilitates the conversion of glucose to pyruvate by increasing the expression of glycolytic enzymes such as hexokinase 1/2 (HK I/II), pyruvate kinase M2 (PK-M2) (Levine and Puzio-Kuter, 2010). Besides catalyzing glucose breakdown, HK-II can also bind to mitochondria and interact with a 30 kDa pore protein, Voltage-Dependent Anion Channel (VDAC) that regulates the transport of metabolites in and out of the mitochondrial intermembrane space (Mathupala et al., 2006). The interaction of HK-II with VDAC interferes with the binding of the pro-apoptotic protein Bax to VDAC, thus preventing the formation of the channel through which cytochrome c can escape from mitochondria to trigger apoptosis (Pastorino et al., 2002). Therefore, over-expression of HK-II in cancer cells leads to a switch from HK-I to HK-II and offers a metabolic advantage by protecting cancer cells against apoptosis (the hallmark of cancer).
HIF-1α is also involved in the regulation of Fructose-2,6-bisphosphate (F-2,6-BP) a powerful allosteric regulator for controlling carbon flux through glycolysis by stimulating 6-phosphofructo-1-kinase (PFK1) to catalyzes the phosphorylation of fructose-6-phosphate (F6P) to fructose-1, 6-bisphosphate (Fru-1,6-BP) using MgATP as a phosphoryl donor (Lu et al., 2015). The synthesis and degradation of F-2,6-BP depend upon 6-phosphofructo 2-kinase/fructose-2,6- biphosphatase (PFK-2/F-2,6-BPase), which has both kinase and phosphatase activities. PFK-2/ F-2,6-BPase synthesis can be induced by mitogens, growth factors, and inflammatory cytokines, implicating its role in setting the glycolytic rate under multiple physiologic and pathologic conditions (Chesney et al., 1999). The family comprises four members among which PFKFB1, PFKFB2, and PFKFB4 display equal PFK2 and FBPase activities under basal conditions, whereas PFKFB3 has high PFK2 and almost no FBPase activity (Okarand Lange, 1999; Okar et al., 2001). The expression of PFK-2 genes in tumor cells is shown to be regulated by Ras and src (Yalcin et al., 2009) and that of PFKFB3has been demonstrated to be induced by HIF-1,c-Myc, ras, src, and loss of function of p53 (Minchenko et al., 2003). Among the four PFK-2 genes, PFKFB3 is the most significantly induced in response to hypoxia (Lu et al., 2015). Furthermore, HIF-1 activates PKM2 gene transcription (Zhang et al., 2008). In turn, PKM2 enhances HIF-1 binding to the hypoxia response element (HRE) and the recruitment of the p300 co-activator. This creates a positive feedback loop whereby PKM2 promotes HIF-1 transactivation (Luo et al., 2011; Tennant, 2011).
Besides promoting high glycolytic rate, HIF-1 activation also inhibits oxidative phosphorylation by up-regulating genes such as are pyruvate dehydrogenase kinase 1 (PDK1) and LDH-A (Kim et al., 2006; Papandreou et al., 2006) thus preventing pyruvate entrance into the TCA cycle (Papandreou et al., 2006). PDK1 inactivates pyruvate dehydrogenase (PDH) to inhibit the conversion of pyruvate into acetyl CoA (Kim et al., 2006). Making pyruvate available for conversion into lactate by LDH-A, as a result, the cancer cell produced a large amount of lactate (Papandreou et al., 2006). Lactate generated from LDH-A can exit cancer cells via the monocarboxylate transporters (MCTs) and cause acidification of the tumor microenvironment, thus facilitating cancer cell invasion. Furthermore, the HIF signaling pathway promotes fatty acid synthesis through activation of sterol regulatory-element binding protein (SREBP)-1 and up-regulation of fatty acid synthase (FASN) (Furuta et al., 2008). Overall, the HIF signaling pathway promotes glycolysis, fatty acid synthesis and angiogenesis which can be an attractive target for cancer therapy.
The c-Myc Oncogene and Glycolysis
c-Myc is another transcription factor that has huge influences on glucose metabolism (Osthus et al., 2000). c-Myc interact with its partner c-Myc-associated protein X (Max) which binds to DNA consensus core sequence, CACGTG or E-box (Blackwood et al., 1992; Meichle et al., 1992). In normal cells, c-Myc is induced upon growth factor stimulation, and involved in many biological processes, including proliferation, cell cycle progression, cell growth, metabolism, angiogenesis, differentiation, cell adhesion, and mobility (Chen and Russo, 2012). Therefore over-expression of c-Myc provides huge metabolic advantages to a tumor cell and needs to be tightly controlled. In several tumors, proto-oncogene myc is aberrantly activated by gene amplification, single nucleotide polymorphisms, and chromosomal translocations (Prendergast and Ziff, 1992). c-Myc expression promotes energy production and anabolic processes, which are required for rapid proliferation, independent of growth factor stimulation (Gordan et al., 2007; Yeung et al., 2008). Like HIF, c-Myc also has huge influences on glucose metabolism by up-regulating gene expression of many glycolytic enzymes from glucose transporters through pyruvate kinase as well as lactate dehydrogenase A, thereby allowing efflux of glucose-derived carbon as lactate (Shim et al., 1997; Osthus et al., 2000). Both HIF and c-Myc coordinate to promote the expression of key glycolytic enzymes such as HK-II, PFK1, TPI1, LDH-A, among others, in tumors (Dang et al.,1997;Dang, 2007; DeBerardinis et al., 2008a; DeBerardinis et al., 2008b;Denko, 2008; Vander and Cantley, 2009). In fact, Myc and HIF-1α contain consensus sequences present in the gene promoter of most glycolytic enzymes. Unlike HIF-1α which is mainly functional in hypoxia, c-Myc is well known to promote its glycolytic target genes expression in normoxia (Li et al., 2005). Many glycolytic enzymes are highly expressed in several cancers driven by HIF and c-MYC such as hexokinase II, glyceraldehyde-3-phosphate dehydrogenase, enolase 1, pyruvate kinase, and lactate dehydrogenase (Altenberg and Greulich, 2004; Mathupala et al., 2006). It has been shown that c-Myc can interact with genes-encoding glycolytic enzymes such as HK II, enolase, and lactate dehydrogenase A (Kim and Dang, 2005). Lactate dehydrogenase converts pyruvate to lactate, a metabolic pathway that is very active in glycolytic cancer cells (Dang et al., 2009). Lactate dehydrogenase expression is directly induced by oncogenes such, as c-Myc (Dang et al., 2009) and indirectly by activation of HIF-1α (Papandreou et al., 2006).
c-Myc has been found to promote preferential expression of PK-M2 over PK-M1 by modulating exon splicing (Wong et al., 2013). c-Myc upregulates the expression of heterogeneous nuclear ribonucleoproteins (hnRNPs) that bind to exon 9 of the PKmRNA, which suppress translation of the PK-M1 isoform, thereby allow preferential inclusion of exon 10 (David et al., 2009). Thus, is responsible for the predominant production of PK-M2. c-Myc also promotes the production of NADPH in order to match the increased ATP production and to support the rapidly dividing cells. c-Myc can collaborate with HIF to provide metabolic advantages to tumor cells (Phan et al., 2014). Hence, many glycolytic enzymes are upregulated in tumors because of elevated c-Myc and HIF-1α transcriptional activity and insufficient p53-mediated control. Indeed, c-Myc and HIF-1α are well recognized as two master inducers of glycolysis through direct or indirect transactivation of cancer glycolytic genes (Phan et al., 2014). This coordination allows tumors to continuously drive glycolysis for supporting their rapid proliferation and accelerated biosynthesis (Dang, 1999; Dang 2007; DeBerardinis et al., 2008b; Dang et al., 2009). Moreover, c-Myc drives anabolic pathways by promoting glutaminolysis (Dang, 1999). Oncogenic levels of c-Myc causes glutamine addiction and cells undergo apoptosis when deprived of glutamine (Wise et al., 2008; Cairns et al., 2011). Myc stimulates glutamine metabolism both directly and indirectly. As a transcription factor, c-Myc directly binds the promoters and stimulates expression of glutamine metabolism genes, such as the transporter Slc1a5 (Yuneva et al., 2007).c-Myc also promotes glutaminase activity indirectly by repressing expression of miR-23a/b, which targets Gls1 (Gao et al., 2009). Thus, excess Myc expression promotes glycolysis and several anabolic processes such as ribosomes biogenesis, protein translation that are required for growth and development. Therefore targeting Myc either directly or indirectly in Myc-driven cancer shows the promising direction for minimizing cancer progression (Chen et al., 2018).
AMP-activated protein kinase(AMPK)
External signals such as growth factors, hormonal signals, and availability of nutrients activate the PI3K pathway thereby promoting glucose transport, aerobic glycolysis, and anabolic synthesis of macromolecules (Luo et al., 2010). However, in the absence of an abundant supply of nutrients such as glucose, this pathway is ineffective and the cell must rely on other sources of ATP generation such as fatty acid oxidation (Buzzai et al., 2005). Energy (AMP/ATP ratio) status in the cell is sensed by AMP kinase. In case of energetic stress, such as that triggered by nutrient deprivation or hypoxia, AMP levels increases and in turn activates AMPK (Hawley et al., 2003). AMP kinase is a serine/threonine kinase consisting of two subunits, catalytic (α) and two regulatory subunits (β and γ) (Hardie, 2007). During period of energy stress (high AMP/ATP ratio) the AMPK become activated which in turn controlled by three upstream kinases, the tumor suppressor LKB1 (Hawley et al., 2003), calmodulin-dependent protein kinase kinase β (CamKKβ) (Hawley et al., 2005; Hurley et al., 2005) and transforming growth factor-β (TGF-β)-activated kinase-1 (TAK1)(Woods et al., 2005; Momcilovic et al., 2006). LKB, like p53 act as a tumor suppressor that is required for AMPK activation which suppresses anabolic processes under conditions of bioenergetic stress including glucose withdrawal (Hawley et al., 2003; Woods et al., 2003; Xie et al., 2006).
Once activated the AMPK inhibits mTORC1, a downstream element of the PI3K pathway thereby interfere with the rapidly dividing cell (Inoki et al. 2003; Gwinn et al., 2008). Consequently, AMPK suppress anabolic processes by blocking protein translation and fatty acid (FA) synthesis (Jones et al., 2005; Kuhajda, 2008; Shackelford and Shaw, 2009) and is responsible for shifting cell to an oxidative phosphorylation (Shaw et al., 2004; Jones et al., 2005; Kuhajda, 2008; Shackelford and Shaw, 2009). Given the role of AMPK mentioned above, it is clear that AMPK plays a major role in decreasing the growth of a rapidly diving cell. Therefore, a mutation in the components of the AMPK signaling pathway allows the tumor to divide under abnormal nutrient conditions (Shackelford and Shaw, 2009). Thus, a mutation in the tumor suppressor such as LKb1as in Peutz–Jeghers syndrome (Jenne, 1998) resulting in a decrease in AMPK signaling and loss of mTOR inhibition (Shackelford and Shaw, 2009). LKb1 is also frequently mutated in sporadic cases of non-small-cell lung cancer (Ji, 2007) and cervical carcinoma (Wingo, 2009). Thus abnormalities in the AMPK signaling allow the activation of mTOR and HIF1, which indirectly support the Warburg effect and promote tumor progression.
p53
The p53 transcription factor serves as a master regulator and defenders, either directly or indirectly during cellular stress such as DNA damage or any other events that can compromise the genome stability and promote tumorigenesis. p53 is involved in many biological processes including DNA repair, cell-cycle arrest, and apoptosis (Vousden and Ryan, 2009) and has been considered as the protector of the genome. In an unstressed cell, wild-type p53 is associated with an E3 ubiquitin ligase enzyme MDM2 that target p53 for degradation thus maintained the basal expression of p53 (Marine et al., 2006). In response to DNA damage or oncogenic stress, wild-type p53 undergo post-translational modification such as phosphorylation, which prevents its association with MDM2, thus leading to its stabilization (Kruse et al., 2010; Wade et al., 2010). P53 induce cell cycle arrest allowing DNA to repair in order to maintain the genome stability and cell survival (Kim et al., 2009; Guo et al., 2015). If the damage is unrepairable, p53 induced cell death through the expression of apoptotic proteins such as PUMA, NOXA or BAX (Villunger et al., 2003; Chipuk et al., 2004), thus preventing cellular transformation and tumor development. Besides its involvement in DNA damage, repairs and apoptosis, p53 also play major roles in metabolism by decreasing the glycolytic rate (Vousden and Ryan, 2009). p53 has a negative impact on the Warburg effect by suppressing glucose consumption and modulating glycolysis at multiple levels (Tran et al., 2016). Glucose enters the cell via transporters (Medina and Owen, 2002). Among transporters, GLUT1 and GLUT3 have a high affinity for glucose and are expressed in most mammalian cells (Medina and Owen, 2002). The GLUT3 expression is stimulated by the IKK/nuclear factor-κB pathway, p53 inhibit this pathway and indirectly reduces GLUT3 expression (Kawauchi et al., 2008). Besides GLUT3, GLUT1 and GLUT4 (insulin-regulated glucose transporter) expression are also suppressed by p53 (Schwartzenberg-Bar-Yoseph et al., 2004). Similarly, wild type p53 also repressed the expression of hexokinase 2 (HK-II) by interacting with the promoter genes coding for HK-II, which control the production of glucose-6-phosphate (G6P) (Mathupala et al., 1997). Thus p53 indirectly control the level of glucose-6-phosphate (G6P) through glucose transporters and HK, which is a central metabolite that is directed into glycolysis, glycogen synthesis and the pentose phosphate pathway (PPP).
Phosphofructokinase 1 (PFK1) an enzyme which catalyzed the rate limiting steps in glycolysis converts fructose 6-phosphate to fructose 1,6-bisphosphate, is the most important enzyme controlling the glycolytic flux, PFK1 is inhibited by ATP and citrate and activated by AMP (Alfarouk et al., 2014). Fructose 2,6-bisphosphate is another important activator of PFK1, whose levels is in turn controlled by the bi-functional enzyme 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFK2/FBPase) (Alfarouket al., 2014). In response to DNA damage, p53 indirectly reduced the level of fructose 2,6-bisphosphate by increasing the expression of TP53-induced glycolysis and apoptosis regulator (TIGAR), which exhibits sequence similarity to the bisphosphatase2 domain of PFK2/FBPase and is able to reduce the glycolytic flux (Bensaad et al., 2006) diverting glucose-6-phosphate to the pentose phosphate pathway. Pentose phosphate pathway activation increases the levels of NADPH, which increases the levels of reduced glutathione to scavenge ROS (Bensaad et al., 2006). Therefore, in a cell harboring normal TIGAR, ROS level declined (Bensaad et al., 2006). Tumor cell with mutated p53 and loss of TIGAR expression causes increased glycolysis at the expense of the pentose phosphate pathway, as well as increased ROS production (Bensaad et al., 2006). TIGAR can also be expressed independently of p53, which occurs in a number of tumor cells, and it plays a role in tumorigenesis (Cheung et al., 2013). p53 also reduce the levels of phosphoglycerate mutase (PGM) which catalyzes the conversion of 3-phosphoglycerate to 2-phosphoglycerate (Kondoh et al., 2005). Downregulation of PGM by p53, 3PG accumulates, which in turn inhibit 6-phosphogluconate dehydrogenase (6PGD), the second NADPH producing enzyme of the PPP (Hitosugi et al., 2012). However overexpression of PGM in p53-deficient cells leads to overproduction of 2PG, which is in turn allosterically activate phosphoglycerate dehydrogenase (PHGDH), the first enzyme of the serine biosynthesis pathway, thus promoting serine biosynthesis (Hitosugi et al., 2012). Furthermore, serine promote the expression of PKM2, which has low affinity toward pyruvate as compared to PKM1, this results in accumulation of glycolytic intermediates that can be used for serine biosynthesis (Chaneton et al., 2012; Ye et al., 2012) and in the PPP pathway (Anastasiou et al., 2011). Thus serine availability plays a crucial role in tumor metabolism.
Besides its involvement in the cytosol, p53 also plays a major role in the mitochondria by increasing the synthesis of cytochrome c oxidase 2 genes, which is required for the assembly of the cytochrome oxidase complex (COX) of the electron transport chain, thereby enhanced oxidative phosphorylation (Matoba et al., 2006). Loss of p53 leads to loss of cytochrome c oxidase expression and improper COX assembly which in turn reduced the oxidative phosphorylation thus forcing cells to depend on glycolysis for energy production (Matoba et al., 2006). Furthermore wild-type p53 represses pyruvate dehydrogenase kinase-2 (PDK2), which phosphorylates and inhibits the pyruvate dehydrogenase (PDH) activity (Contractor et al., 2012) thereby promoting the conversion of pyruvate to acetyl CoA for entry in the TCA cycle. Thus, overall wild-type p53 favors mitochondrial respiration for energy production over aerobic glycolysis (the Warburg Effect) exhibited by many cancer cells. Wild-type p53 also repress the expression of the Lactate/proton symporter monocarboxylate transporter 1 (MCT1) (Boidot et al., 2012). Therefore in a p53-deficient cell, the PDH activity is repressed and the majority of pyruvate is converted to lactate via LDH (Warburg effect). Lactate/proton is then transported via a symporter MCT1 that contribute to the acidity of the tumor microenvironment, which is one of the strategies that tumor cell avoid immune responses (Susana et al., 2016).
Furthermore, wild-type p53 promotes the expression of phosphate and tensin homolog deleted on chromosome 10 (PTEN), which inhibits the phosphatidylinositol-3-kinase (PI3K) pathway and in turn lower the expression of its downstream elements that drives glycolysis (Figure 2) (Cairns et al., 2011). Several mutations resulting in loss of PTEN function occurs in human cancers through various genetic alterations including point mutations, large chromosomal deletions and epigenetic mechanisms such as hypermethylation of the PTEN promoter region (Mutter, 2001; Sansal and Sellers, 2004; Chow and Baker, 2006). In addition, PTEN could be inactivated by other non-structural alterations which compromise the protein’s integrity (Chalhoub and Baker, 2009; Zhang and Yu, 2010; Song et al., 2012).
Glutaminase-2 (GLS-2) enzyme catalyzes the hydrolysis of glutamine to glutamate is another p53 target gene that encodes a mitochondrial GLS (Hu et al., 2010). Glutamate can be further deaminated to form α-KG, which can enter the TCA cycle for energy metabolism (Figure 2). Glutamate also preserves total reduced glutathione after oxidative stress (Suzuki et al., 2010). However, mutation or suppression of p53, a frequent occurrence in cancer, results in the loss of control of its metabolic regulation (Johnson and Perkins, 2012). Another oncogene, K-ras, has been found to be frequently mutated in some type of cancer (Normanno et al., 2009). Mutated K-ras upregulate glucose transporter protein 1 (GLUT1) expression leading to an increase in aerobic glycolysis by cancer cells (Figure 2) (Annibaldi and Widmann, 2011). Interestingly, in a cell harboring mutated K-ras, the integrity of mitochondrial functions and oxidative phosphorylation still remain intact, which provide an advantage to the K-ras mutant cells during glucose deprivation (Annibaldi and Widmann, 2011).
Therefore, inactivation of p53 provides huge advantages to tumor cells and their progression. Recently, it has been found that with approximately 50% of all human cancers harboring either a mutation or deletion in the TP53 encoding gene (Lee et al., 2010) and another scenario where p53 not only lose its tumor suppressive function but gain a new oncogenic function that is independent of wild-type p53, designated as mutp53 (Zhang et al., 2013). In mutp53 the structural integrity is compromised, thus reducing its ability to bind to the promoter of target genes (Wong et al., 1999). Furthermore, an E3 ubiquitin ligase MDM2 protein cannot target mutp53 for degradation leading to its stabilization (Kubbutat et al., 1997) and can exert additional promoting functions (Muller et al., 2013). Hence, cancer cells with mutant p53 promote glycolysis in several ways, including expression of GLUT (GLUT 1, GLUT4 and GLUT3) (Zhang et al., 2013), which causes the cancer cell to consume more glucose as compared to the normal cell, the so-called ‘glucose addiction’. Low energy in the cell is sensed by AMPK, which in turn decrease the anabolic activities in the cell. However expression of mutp53, inhibit AMPK activation, allowing cancer cells to proliferate even under energy stress (Zhou et al., 2014). Unlike wild-type p53, mutp53 is a potent activator of the c-Myc promoter which regulates its expression in cancer cells (Frazier et al., 1998). Therefore, in p53- deficient cell or cell harboring mutp53, HIF and cMyc promotes the expression of several glycolytic enzymes (HK-II, PFK1, PGM) (Dang et al., 1997; Dang, 2007; DeBerardinis et al., 2008a; DeBerardinis et al., 2008b; Denko, 2008; Vander and Cantley, 2009) HK-II increases the production G-6P in the cytosol, which is a central metabolite for anabolic process (PPP) and catabolic process (glycolysis) for energy production. HK-II also promotes cancer cell survival by reducing the production of cytochrome c oxidase 2 to trigger apoptosis (Pastorino et al., 2002). Overexpression of PFK1 and PGM drives cancer glycolysis not only for energy production but also provides metabolites for the anabolic process. Besides energy, rapidly dividing cells also required raw materials for their growth and development. PPP play a major role in providing raw materials such as ribose phosphate and NADPH, which is frequently altered in many tumors (Deberardinis et al., 2008; Riganti et al., 2012). Wild-type p53 directly control the activity of the PPP by interacting with the rate-limiting enzyme G6PD, which prevent its active dimer formation, leading to a decrease in enzyme activity (Jiang et al., 2011). Thus, in a cell harboring mutp53, the activity of G-6PD, a major PPP enzyme, increases in proliferating cancer cells (Jones et al., 1992). PPP not only provide ribose-phosphate for NT biosynthesis but also NADPH that protects cancer cell from oxidative stress and facilitating DNA damage repair (Riganti et al., 2012). Furthermore, mutp53 also promotes activation of AKT and HIF, which are effectors downstream of PI3K (Figure 2). Thus, the interaction among p53, c-Myc, and HIF-1α has a decisive impact on the status of cancer glycolysis. Therefore, activation of oncogenes and loss of tumor suppressors are believed to underlie the metabolic switch in cancer cells (Warburg effect) which in turn drives cancer progression.
In conclusion, this review highlights the cumulative findings of the roles of oncogenes and tumor suppressors in cancer, glucose metabolism and their contribution to the Warburg Effect as summarized in Figure 2 and provides a potential direction towards the possible path to control cancer progression. Alteration of metabolic pathways has been considered as one of the hallmarks of cancer. Cancer being a complicated disease with multiples abnormalities, have one thing in common i.e. they need glucose for energy production as well as for biosynthesis of cell’s raw material such as lipids, proteins, nucleotides and anti-oxidant glutathione which are required by rapidly diving cells. Therefore, starving cancer cells by exploiting their energy production pathways is clearly a very promising direction. It is becoming clear that metabolic reprogramming is induced by HIF-1, c-Myc, p53, mTOR, PK-M2, IDH, and other molecules that act both independently and in concert with each other as mentioned previously. Increase glycolytic rate in cancer cells is under the master command of the transcription factor HIF-1α which, in collaboration with other oncogenic signaling pathways including c-Myc, AMPK, and mTOR, promotes the expression of most glycolytic enzymes and transporters. Therefore, targeting their activities has a potential to reduce cancer’s progression. Many compounds have been developed that interfere with the metabolic pathway by inhibiting metabolic enzymes that are important for tumors (Porporato et al., 2011). However, normal cells also have the same metabolic requirements as a cancer cell, therefore one of the most effective anti-cancer metabolism strategies is by selectively and effectively inhibits metabolic enzymes that are mostly expressed or used in tumor cells, and without harming the normal cells. This therapeutic strategy by targeting proteins that are highly active or over-expressed in cancer would minimize the effect of anti-cancer drugs in the normal cell.
Many proteins involved in glycolysis are overexpressed in cancer cell but not all can be safely targeting without harming the normal cell. HKII has been well known as a facilitator of cancer glycolysis and as a repressor of apoptosis in several types of cancer (Pastorino et al., 2002). High expression of HK2 isoform in cancer cells is induced by several mechanisms, whereas its expression in most mammalian tissues is low (Hay, 2016). Therefore, overexpression of HKII isoform over HKI in some type of cancer provides a window to target HKII without harming normal cell that expressed other isoforms. Since HKII is specifically required by a cancer cell, therefore inhibiting HKII forces other HK isoforms re-expression which can interfere with cancer progression. Beside 2-DG and lonidamine, many efforts have thus been made to identify specific inhibitors (Floridi et al., 1981). The current leading compound is 3-bromopyruvate (3BP), an HK inhibitor which is shown to be toxic to the cancer cell (Ko et al., 2004; Mathupala et al., 2009). Another target is PFKFB3 which is highly expressed in cancer under the influence of HIF-1, c-Myc, ras, src (Minchenko et al., 2003). PFKFB3 stimulate the expression of Fructose-2,6-bisphosphate (F-2,6-BP), an important driver of glycolysis. Therefore, inhibition of PFKFB3 reduces the glycolytic rate which can slow down cancer progression. 3PO (3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one) is the only inhibitor of PFKFB3 identified so far (Clem et al., 2008). Preferential expression of PKM2 over PKM1 in most cancer can be therapeutically beneficial. Inhibitions of PKM2 forces the expression of PKM1 which is less active as compared to PKM2 may help divert glucose metabolites for oxidative phosphorylation in cancer cells. Peptide aptamers that promote the expression of PKM1 over PKM2 have been shown to cause energy stress and cell death (Spoden et al., 2008). DCA is another compound which has been known to inhibit PDK1 (Whitehouse and Ran, 1973) whose expression is under the influence of HIF- 1 (Kim et al., 2006). PDK1 prevent the entry of pyruvate into the TCA cycle by inhibiting PDH. Thus, inhibition of PDK1 leads to PDH activation, allowing pyruvate entry in the TCA cycle and decreased lactate production. Because lactate is exported out of the cell, blocking its production or transportation can also be beneficial in antagonizing the Warburg effect. With every lactate formed there is a rise in H+ concentration, and since lactate/proton is transported via a symporter MCT. Blocking lactate transfer decrease the pH inside the cancer cell and since PFK 1 (a major regulatory enzyme in glycolysis) is a pH-sensitive enzyme, therefore, as pH decreases, the activity of PFK1 decreases and in turn reduce the glycolytic rate (Berg et al., 2002). In fact, inhibiting MCT1/MCT2by inhibitors induce cell starvation and subsequent apoptosis (Sonveaux et al., 2008).
Another important strategy to safely target tumor cell without harming normal cell is by restoring p53 function. Since p53 is involved in a wide range of metabolic activities as previously discussed, restoring its function should provide a major therapeutic benefit. One strategy is by restoring wild-type p53 that have lost its expression due to an increases expression or amplification of MDM2 protein. One such compound is Nutlin (RG7112), which interfere with the p53 binding domain of the MDM2 protein, leading to stabilization of the p53 protein (Vassilev et al., 2004). Another strategy is to restore the structural integrity of mutant p53 by a compound that acts as a chaperone (Bykov et al., 2002). PRIMA-1MET (APR-246) acts as such a compound and has been shown to inhibit tumor harboring mutant p53 (Zache et al, 2008). Despite the success of targeting enzyme involved in glucose metabolism, cancer cells may bypass certain inhibitions mediated by therapeutic agents because of their metabolic plasticity and heterogeneity (Dang, 1999, 2007; Yeung et al., 2008; Hanahan and Weinberg, 2011; Dang, 2012; Hu et al., 2013) to rewire their metabolic pathway in order to ensure their own survival. Therefore, a better understanding of how cancer rewires their metabolism is needed for effective therapy development. Although there is still much to study and discover about the role of oncogenes and tumor suppressors in cancer metabolism, a recent discovery has unveiled exciting therapeutic windows to target cancer metabolism and bioenergetics. Thus, exploiting their energy production pathway for cancer treatment is clearly a promising proposition.
References
- Alfarouk KO, Verduzco D, Rauch C, et al. Glycolysis, tumor metabolism, cancer growth and dissemination. A new pH-based etiopathogenic perspective and therapeutic approach to an old cancer question. Oncol Sci. 2014;1:777–91. doi: 10.18632/oncoscience.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altenberg B, Greulich KO. Genes of glycolysis are ubiquitously over expressed in cancer classes. Genomics. 2004;84:1014–20. doi: 10.1016/j.ygeno.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Amoedo ND, Valencia JP, Rodrigues MF, Galina A, Rumjanek FD. How does the metabolism of tumor cells differ from that of normal cells? Biosci Rep. 2013;33:865–871. doi: 10.1042/BSR20130066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastasiou D, Poulogiannis G, Asara JM, et al. Inhibition of pyruvate kinase m2 by reactive oxygen species contributes to cellular antioxidant responses. Science. 2011;334:1278–83. doi: 10.1126/science.1211485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annibaldi A, Widmann C. Glucose metabolism in cancer cells. Curr Opin Clin Nutr Metab Care. 2011;13:466–70. doi: 10.1097/MCO.0b013e32833a5577. [DOI] [PubMed] [Google Scholar]
- Baeriswyl V, Christofori G. The angiogenic switch in carcinogenesis. Semin Cancer Biol. 2009;19:329–37. doi: 10.1016/j.semcancer.2009.05.003. [DOI] [PubMed] [Google Scholar]
- Barthel A, Okino ST, Liao J, et al. Regulation of GLUT1 gene transcription by the serine/threonine kinaseakt1. J Biol Chem. 1999;274:20281–86. doi: 10.1074/jbc.274.29.20281. [DOI] [PubMed] [Google Scholar]
- Bensaad K, Tsuruta A, Selak MA, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126:107–20. doi: 10.1016/j.cell.2006.05.036. [DOI] [PubMed] [Google Scholar]
- Berg JM, Tymoczko Jl, Stryer L Biochemistry. Glycolysis and Gluconeogenesis. 5th Edition. Chapter 16:2. New York: W H Freeman; 2002. pp. 634–5. [Google Scholar]
- Blackwood EM, Kretzner L, Eisenman RN. Myc and Max function as a nucleoprotein complex. Curr Opin Gen Dev. 1992;2:227–35. doi: 10.1016/s0959-437x(05)80278-3. [DOI] [PubMed] [Google Scholar]
- Boidot R, Vegran F, Meulle A, et al. Regulation of monocarboxylate transporter mct1 expression by p53 mediates inward and outward lactate fluxes in tumors. Cancer Res. 2012;72:939–48. doi: 10.1158/0008-5472.CAN-11-2474. [DOI] [PubMed] [Google Scholar]
- Bruick RK, McKnight SL. A conserved family of prolyl-4- hydroxylases that modify HIF. Science. 2001;294:133–740. doi: 10.1126/science.1066373. [DOI] [PubMed] [Google Scholar]
- Burt MB, Hummy JL, Kooby DA, et al. Using positron emission tomography with [18F] FDG to predict tumor behavior in experimental colorectal cancer 1. Neo, 3. 2001:189–95. doi: 10.1038/sj.neo.7900147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzzai M, Bauer DE, Jones RG, et al. The glucose dependence of Akt-transformed cells can be reversed by pharmacologic activation of fatty acid b-oxidation. Oncogene. 2005;24:4165–73. doi: 10.1038/sj.onc.1208622. [DOI] [PubMed] [Google Scholar]
- Bykov VJ, Issaeva N, Shilov A, et al. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat Med. 2002;8:282–8. doi: 10.1038/nm0302-282. [DOI] [PubMed] [Google Scholar]
- Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11:85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–57. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- Chalhoub N, Baker SJ. PTEN and the PI3-kinase pathway in cancer. Annu Rev Pathol. 2009;4:127–500. doi: 10.1146/annurev.pathol.4.110807.092311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaneton B, Hillmann P, Zheng L, et al. Serine is a natural ligand and allosteric activator of pyruvate kinase m2. Nat. 2012;491:458–462. doi: 10.1038/nature11540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Liu H, Qing G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct Target Ther. 2018;3:5. doi: 10.1038/s41392-018-0008-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JQ, Russo J. Dysregulation of glucose transport, glycolysis, TCA cycle and glutaminolysis by oncogenes and tumor suppressors in cancer cells. Biochim Biophys Acta. 2012;1826:370–84. doi: 10.1016/j.bbcan.2012.06.004. [DOI] [PubMed] [Google Scholar]
- Chesney J, Mitchell R, Benigni F, et al. An inducible gene product for 6-phosphofructo-2-kinase with an AU-rich instability element:role in tumor cell glycolysis and the Warburg effect. Proc Natl Acad Sci U S A. 1999;96:3047–52. doi: 10.1073/pnas.96.6.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung EC, Athineos D, Lee P, et al. TIGAR is required for efficient intestinal regeneration and tumorigenesis. Dev Cell. 2013;25:463–77. doi: 10.1016/j.devcel.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipuk JE, Kuwana T, Bouchier-Hayes L, et al. Direct activation of bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science. 2004;303:1010–4. doi: 10.1126/science.1092734. [DOI] [PubMed] [Google Scholar]
- Chow LM, Baker SJ. PTEN function in normal and neoplastic growth. Cancer Lit. 2006;241:184–196. doi: 10.1016/j.canlet.2005.11.042. [DOI] [PubMed] [Google Scholar]
- Clem B, Telang S, Clem A, et al. Small-molecule inhibition of 6-phosphofructo-2-kinase activity suppresses glycolytic flux and tumor growth. Mol Cancer Ther. 2008;7:110–20. doi: 10.1158/1535-7163.MCT-07-0482. [DOI] [PubMed] [Google Scholar]
- Conrad PW, Freeman TL, Beitner Johnson D, Millhorn DE. EPAS1 trans-activation during hypoxia requires 42/p44MAPK. J Biol Chem. 1999;274:337–913. doi: 10.1074/jbc.274.47.33709. [DOI] [PubMed] [Google Scholar]
- Contractor T, Harris CR. P53 negatively regulates transcription of the pyruvate dehydrogenase kinase pdk2. Cancer Res. 2012;72:560–67. doi: 10.1158/0008-5472.CAN-11-1215. [DOI] [PubMed] [Google Scholar]
- Cox MM, Nelson DL. In “Gluconeogenesis”. 6th Edition. New York: W.H. Freeman and Company; 2013. Lehninger principle of biochemistry; p. 543. [Google Scholar]
- Dakubo GD. The warburg phenomenon and other metabolic Alterations of Cancer Cells. Mitochon Genet Cancer. 2010;1:39–66. [Google Scholar]
- Dang CV. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol. 1999;19:1–11. doi: 10.1128/mcb.19.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV. The interplay between MYC and HIF in the Warburg effect. Ernst Schering Found Symp Proc. 2007;4:35–53. doi: 10.1007/2789_2008_088. [DOI] [PubMed] [Google Scholar]
- Dang CV. Rethinking the warburg effect with Myc micromanaging glutamine metabolism. Cancer Res. 2010;70:859–62. doi: 10.1158/0008-5472.CAN-09-3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV. Links between metabolism and cancer. Genes Dev. 2012;26:877–90. doi: 10.1101/gad.189365.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV, Le A, Gao P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin Cancer Res. 2009;15:6479–83. doi: 10.1158/1078-0432.CCR-09-0889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV, Lewis BC, Dolde C, Dang G, Shim H. Oncogenes in tumor metabolism, tumorigenesis, and apoptosis. J Bioenerg Biomembr. 1997;29:345–54. doi: 10.1023/a:1022446730452. [DOI] [PubMed] [Google Scholar]
- Dang CV, Semenza GL. Oncogenic alterations of metabolism. Trends Biochem Sci. 1999;24:68–72. doi: 10.1016/s0968-0004(98)01344-9. [DOI] [PubMed] [Google Scholar]
- David CJ, Chen M, Assanah M, Canol P, Manley JL. hnRNP proteins controlled by c Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature. 2009;463:364–8. doi: 10.1038/nature08697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ. Is cancer a disease of abnormal cellular metabolism? Genet Med. 2008;10:767–77. doi: 10.1097/GIM.0b013e31818b0d9b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer:metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008b;7:11–20. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- DeBerardinis RJ, Mancuso A, Daikhin E, et al. Beyond aerobic glycolysis:transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A. 2007;104:19345–50. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deberardinis RJ, Sayed N, Ditsworth D, Thompson CB. Brick by brick:metabolism and tumor cell growth. Curr Opin Genet Dev. 2008a;18:54–61. doi: 10.1016/j.gde.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denko NC. Hypoxia, HIF1 and glucose metabolism in the solid tumor. Nat Rev Cancer. 2008;8:705–13. doi: 10.1038/nrc2468. [DOI] [PubMed] [Google Scholar]
- Elstrom RL, Bauer DE, Buzzai M, et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004;64:3892–9. doi: 10.1158/0008-5472.CAN-03-2904. [DOI] [PubMed] [Google Scholar]
- Ema M, Taya S, Yokotani N, et al. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1αregulates the VEGF expression and is potentially involved in lung and vascular development. Proc Natl Acad Sci U S A. 1997;94:738–42. doi: 10.1073/pnas.94.9.4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floridi A, Paggi MG, Marcante ML, et al. Lonidamine, a selective inhibitor of aerobic glycolysis of murine tumor cells. J Natl Cancer Inst. 1981;66:497–9. [PubMed] [Google Scholar]
- Frazier MW, He X, Wang J, et al. Activation of c-myc gene expression by tumor-derived p53mutants requires a discrete C-terminal domain. Mol Cell Biol. 1998;18:3735–43. doi: 10.1128/mcb.18.7.3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta E, Pai SK, Zhan R, et al. Fatty acid synthase gene is upregulated by hypoxia via activation of Akt and sterol regulatory element binding protein-1. Cancer Res. 2008;68:1003–11. doi: 10.1158/0008-5472.CAN-07-2489. [DOI] [PubMed] [Google Scholar]
- Gao P, Tchernyshyov I, Chang TC, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458:762–5. doi: 10.1038/nature07823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geering B, Cutillas PR, Nock G, Gharbi SI, Vanhaesebroeck B. Class IA phosphoinositide 3-kinases are obligate p85-p110 heterodimers. Proc Natl Acad Sci U S A. 2007a;104:7809–14. doi: 10.1073/pnas.0700373104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geering B, Cutillas PR, Vanhaesebroeck B. Regulation of class IA PI3Ks:is there a role for monomeric PI3K subunits? Biochem Soc Trans. 2007b;3:199–203. doi: 10.1042/BST0350199. [DOI] [PubMed] [Google Scholar]
- Gingras AC, Raught B, Sonenberg N. Regulation of translation initiation by FRAP/mTOR. Genes Dev. 2001;15:807–26. doi: 10.1101/gad.887201. [DOI] [PubMed] [Google Scholar]
- Gordan JD, Bertout JA, Hu CJ, Diehl JA, Simon MC. HIF2αpromotes hypoxic cell proliferation by enhancing c-Myc transcriptional activity. Cancer Cell. 2007;11:335–47. doi: 10.1016/j.ccr.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordan JD, Thompson CB, Simon MC. HIF and c-Myc:sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell. 2007;12:108–213. doi: 10.1016/j.ccr.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb E, Tomlinson IP. Mitochondrial tumor suppressors:a genetic and biochemical update. Nat Rev Cancer. 2005;5:857–66. doi: 10.1038/nrc1737. [DOI] [PubMed] [Google Scholar]
- Gottlob K, Majewski N, Kennedy S, et al. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 2001;15:1406–18. doi: 10.1101/gad.889901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo G, Cui Y. New perspective on targeting the tumor suppressor p53 pathway in the tumor microenvironment to enhance the efficacy of immunotherapy. J Immunother Cancer. 2015;3:9. doi: 10.1186/s40425-015-0053-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzy RD, Hoyos B, Robin E. Mitochondrial complex III is required for hypoxia induced ROS production and cellular oxygen sensing. Cell Metab. 2005;1:401–8. doi: 10.1016/j.cmet.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Guzy RD, Sharma B, Bell E, Chandel NS, Schumacker PT. Loss of the SdhB, but Not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Mol Cell Biol. 2008;28:718–31. doi: 10.1128/MCB.01338-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwinn DM, Shackelford DB, Egan DF, et al. AMPK phosphorylationof raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–26. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer:the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hardie DG. AMP-activated/SNF1 protein kinases:conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774–85. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- Hawley SA, Boudeau J, Reid JL, et al. Complexes between the LKB1 tumor suppressor, STRADα/βand MO25α/βare upstream kinases in the AMP-activated protein kinase cascade. J Biol. 2003;2:28. doi: 10.1186/1475-4924-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley SA, Pan DA, Mustard KJ, et al. Calmodulin-dependent protein kinase kinase-βis an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2:9–19. doi: 10.1016/j.cmet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Hay N. Reprogramming glucose metabolism in cancer:can it be exploited for cancer therapy? Nat Rev Cancer. 2016;16:635–49. doi: 10.1038/nrc.2016.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey MM, Simon MC. Regulation of angiogenesis by hypoxia and hypoxia-inducible factors. Curr Top Dev Biol. 2006;76:217–57. doi: 10.1016/S0070-2153(06)76007-0. [DOI] [PubMed] [Google Scholar]
- Hirsila M, Koivunen P, Gunzler V, Kivirikko KI, Myllyharju J. Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. J Biol Chem. 2003;278:30772–80. doi: 10.1074/jbc.M304982200. [DOI] [PubMed] [Google Scholar]
- Hitosugi T, Zhou L, Elf S, et al. Phosphoglycerate mutase 1 coordinates glycolysis and biosynthesis to promote tumor growth. Cancer Cell. 2012;22:585–600. doi: 10.1016/j.ccr.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollander MC, Blumenthal GM, Dennis PA. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat Rev Cancer. 2011;11:289–301. doi: 10.1038/nrc3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Locasale JW, Bielas JH, et al. Heterogeneity of tumor-induced gene expression changes in the human metabolic network. Nat Biotechnol. 2013;31:522–9. doi: 10.1038/nbt.2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Zhang C, Wu R. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci U S A. 2010;107:7455–60. doi: 10.1073/pnas.1001006107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley RL, Anderson KA, Franzone JM. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J Biol Chem. 2005;280:29060–66. doi: 10.1074/jbc.M503824200. [DOI] [PubMed] [Google Scholar]
- Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–90. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- Jason R, Cantor JR, David M, Sabatini DM. Cancer cell metabolism:One Hallmark, Many Faces. Cancer Discov. 2012;2:1–18. doi: 10.1158/2159-8290.CD-12-0345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenne DE. Peutz Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nature Genet. 1998;18:38–43. doi: 10.1038/ng0198-38. [DOI] [PubMed] [Google Scholar]
- Ji H. LKB1 modulates lung cancer differentiation and metastasis. Nature. 2007;448:807–10. doi: 10.1038/nature06030. [DOI] [PubMed] [Google Scholar]
- Jiang BH, Jiang G, Zheng JZ, et al. Phosphatidylinositol3-kinase signaling controls levels of hypoxia-inducible factor 1. Cell Growth Differ. 2001;12:36–9. [PubMed] [Google Scholar]
- Jiang P, Du W, Wang X, et al. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat Cell Biol. 2011;13:310–16. doi: 10.1038/ncb2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RF, Perkins ND. Nuclear factor-kappa B, p53, and mitochondria:regulation of cellular metabolism and the Warburg affect. Trends Biochem Sci. 2012;37:317–24. doi: 10.1016/j.tibs.2012.04.002. [DOI] [PubMed] [Google Scholar]
- Jonas SK, Benedetto C, Flatman A, et al. Increased activity of 6-phosphogluconate dehydrogenase and glucose-6-phosphate dehydrogenase in purified cell suspensions and single cells from the uterine cervix in cervical intraepithelial neoplasia. Br J Cancer. 1992;66:185–91. doi: 10.1038/bjc.1992.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RG, Plas DR, et al. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005;18:283–93. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- Kawauchi K, Araki K, Tobiume K, Tanaka N. p53 regulates glucose metabolism through an IKK-NF-kappa B pathway and inhibits cell transformation. Nat Cell Biol. 2008;10:611–8. doi: 10.1038/ncb1724. [DOI] [PubMed] [Google Scholar]
- Kim E, Giese A, Deppert W. Wild-type p53 in cancer cells:When a guardian turns into a blackguard. Biochem. Pharmacol. 2009;77:11–20. doi: 10.1016/j.bcp.2008.08.030. [DOI] [PubMed] [Google Scholar]
- Kim JW, Dang CV. Multifaceted roles of glycolytic enzymes. Trends Biochem Sci. 2005;30:142–50. doi: 10.1016/j.tibs.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase:A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–85. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Klimova T, Chandel NS. Mitochondrial complex III regulates hypoxic activation of HIF. Cell Death Differ. 2008;15:660–6. doi: 10.1038/sj.cdd.4402307. [DOI] [PubMed] [Google Scholar]
- Ko YH, Smith BL, Wang Y, et al. Advanced cancers:eradicationin all cases using 3-bromopyruvate therapy to deplete ATP. Biochem Biophys Res Commun. 2004;324:269–75. doi: 10.1016/j.bbrc.2004.09.047. [DOI] [PubMed] [Google Scholar]
- Kondoh H, Lleonart ME, Gil J, et al. Glycolytic enzymes can modulate cellular life span. Cancer Res. 2005;65:177–85. [PubMed] [Google Scholar]
- Koppenol WH, Bounds PL, Dang CV. Otto Warburg's contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11:325–7. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- Kruse JP, Gu W. Modes of p53 regulation. Cell. 2009;137:609–22. doi: 10.1016/j.cell.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by mdm2. Nat. 1997;387:299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- Kuhajda FP. AMP activated protein kinase and human cancer:cancer metabolism revisited. Int J Obest. 2008;32:36–41. doi: 10.1038/ijo.2008.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EJ, Kim TJ, Kim DS, et al. p53 alteration independently predicts poor outcomes in patients with endometrial cancer:a clinicopathologic study of 131 cases and literature review. Gynecoloncol. 2010;116:533–8. doi: 10.1016/j.ygyno.2009.11.018. [DOI] [PubMed] [Google Scholar]
- Levine AJ, Puzio-Kuter AM. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science. 2010;330:1340–4. doi: 10.1126/science.1193494. [DOI] [PubMed] [Google Scholar]
- Li W, Saud SM, Young MR, Chen G, Hua B. Targeting AMPK for cancer prevention and treatment. Oncotarget. 2015;6:7365–72. doi: 10.18632/oncotarget.3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Wang Y, Zeller KI, et al. Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol Cell Biol. 2005;25:6225–34. doi: 10.1128/MCB.25.14.6225-6234.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodish H, Berk A, Zipursky SL, et al. Molecular Cell Biology. 4th edition. Vol. 2000. New York: W. H. Freeman; 2000. Section 24.2, Proto-Oncogenes and Tumor-Suppressor. [Google Scholar]
- Long YC, Zierath JR. AMP-activated protein kinase signaling in metabolic regulation. J Clin Investig. 2006;116:1776–81. doi: 10.1172/JCI29044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Tan M, Cai Q. The Warburg effect in tumor progression:Mitochondrial oxidative metabolism as an anti-metastasis mechanism. Cancer Lett. 2015;356:156–64. doi: 10.1016/j.canlet.2014.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W, Hu H, Chang R, et al. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell. 2011;145:732–44. doi: 10.1016/j.cell.2011.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Z, Zang M, Guo W. AMPK as a metabolic tumor suppressor:control of metabolism and cell growth. Future Oncol. 2010;6:457–70. doi: 10.2217/fon.09.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv X, Li J, Zhang C, et al. The role of hypoxia-inducible factors in tumor angiogenesis and cell metabolism. Genes Dis. 2017;4:19–24. doi: 10.1016/j.gendis.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marine JC, Francoz S, Maetens M, et al. Keeping p53 in check:Essential and synergistic functions of mdm2 and mdm4. Cell Death Differ. 2006;13:927–34. doi: 10.1038/sj.cdd.4401912. [DOI] [PubMed] [Google Scholar]
- Masoud GN, Lin W. HIF-1αpathway:role, regulation and intervention for cancer therapy. Acta Pharmaceutica Sinica B. 2015;5:378–89. doi: 10.1016/j.apsb.2015.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathupala SP, Heese C, Pedersen PL. Glucose catabolism in cancer cells. The type II hexokinase promoter contains functionally active response elements for the tumor suppressor p53. J Biol Chem. 1997;272:22776–80. doi: 10.1074/jbc.272.36.22776. [DOI] [PubMed] [Google Scholar]
- Mathupala SP, Ko YH, Pedersen PL. Hexokinase II:cancer's double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene. 2006;25:4777–86. doi: 10.1038/sj.onc.1209603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathupala SP, Ko YH, Pedersen PL. Hexokinase-2 bound to mitochondria:cancer's stygian link to the “Warburg Effect”and a pivotal target for effective therapy. Semin Cancer Biol. 2009;19:17–24. doi: 10.1016/j.semcancer.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matoba S, Kang JG, Patino WD, et al. p53 regulates mitochondrial respiration. Science. 2006;312:1650–3. doi: 10.1126/science.1126863. [DOI] [PubMed] [Google Scholar]
- Maxwell PH, Pugh CW, Ratcliffe PJ. Activation of the HIF pathway in cancer. Curr Opin Genet Dev. 2001;11:29–39. doi: 10.1016/s0959-437x(00)00193-3. [DOI] [PubMed] [Google Scholar]
- Maynard MA, Evans AJ, Hosomi T, et al. Human HIF-3alpha4 is a dominant-negative regulator of HIF-1 and is down-regulated in renal cell carcinoma. FASEB J. 2015;19:1396–1406. doi: 10.1096/fj.05-3788com. [DOI] [PubMed] [Google Scholar]
- McCubrey JA, Steelman LS, Chappell WH, et al. Mutations and deregulation of Ras/Raf/MEK/ERK and PI3K/ PTEN/Akt/mTOR cascades which alter therapy response. Oncol Target. 2012;3:954–87. doi: 10.18632/oncotarget.652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina RA, Owen GI. Glucose transporters:expression, regulation and cancer. Biol Res. 2002;35:9–26. doi: 10.4067/s0716-97602002000100004. [DOI] [PubMed] [Google Scholar]
- Meichle A, Philipp A, Eilers M. The functions of Mycproteins. Biochim Biophys Acta. 1992;1114:129–46. doi: 10.1016/0304-419x(92)90011-m. [DOI] [PubMed] [Google Scholar]
- Minchenko O, Opentanova I, Caro J. Hypoxic regulation of the 6-phosphofructo-2- kinase/fructose-2,6-bisphosphatase gene family (PFKFB-1-4) expression in vivo. FEBS J. 2003;554:264–70. doi: 10.1016/s0014-5793(03)01179-7. [DOI] [PubMed] [Google Scholar]
- Miyamoto S, Murphy AN, Brown JH. Akt mediates mitochondrial protection in cardiomyocytes through phosphorylation of mitochondrial hexokinase-II. Cell Death Differ. 2008;15:521–529. doi: 10.1038/sj.cdd.4402285. [DOI] [PubMed] [Google Scholar]
- Momcilovic M, Hong SP, Carlson M. Mammalian TAK1 activates Snf1 protein kinase in yeast and phosphorylates AMP-activated protein kinase in vitro. J Biol Chem. 2006;281:25336–43. doi: 10.1074/jbc.M604399200. [DOI] [PubMed] [Google Scholar]
- Muller PA, Vousden KH. P53 mutations in cancer. Nat Cell Biol. 2013;15:2–8. doi: 10.1038/ncb2641. [DOI] [PubMed] [Google Scholar]
- Mutter GL. PTEN, a protein tumor suppressor. Am J Pathol. 2001;158:1895–1980. doi: 10.1016/S0002-9440(10)64656-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogueira V, Patra KC, Hay N. Selective eradication of cancer displaying hyperactive Akt by exploiting the metabolic consequences of Akt activation. eLife. 2018;7:e32213. doi: 10.7554/eLife.32213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Normanno N, Tejpar S, Morgillo F, et al. Implications for KRAS status and EGFR-targetedtherapies in metastatic CRC. Nat Rev Clin Oncol. 2009;6:519–27. doi: 10.1038/nrclinonc.2009.111. [DOI] [PubMed] [Google Scholar]
- Okar DA, Lange AJ. Fructose-2,6-bisphosphate and control of carbohydrate metabolism in eukaryotes. Biofactors. 1999;10:1–14. doi: 10.1002/biof.5520100101. [DOI] [PubMed] [Google Scholar]
- Okar DA, Manzano A, Navarro- Sabate A, et al. PFK- 2/FBPase-2:maker and breaker of the essential bio factor fructose-2, 6-bisphosphate. Trends Biochem Sci. 2001;26:30–5. doi: 10.1016/s0968-0004(00)01699-6. [DOI] [PubMed] [Google Scholar]
- Osthus RC, Shim H, Kim S. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J Biol Chem. 2000;275:2179–7800. doi: 10.1074/jbc.C000023200. [DOI] [PubMed] [Google Scholar]
- Palaskas N, Larson SM, Schultz N, et al. 18F-fluorodeoxy-glucose positron emission tomography marks MYC overexpressing human basal-like breast cancers. Cancer Res. 2011;71:5164–74. doi: 10.1158/0008-5472.CAN-10-4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively down regulating mitochondrial oxygen consumption. Cell Metab. 2006;3:187–97. doi: 10.1016/j.cmet.2006.01.012. [DOI] [PubMed] [Google Scholar]
- Pasanen A, Heikkila¨ M, Rautavuoma K, et al. Hypoxia-inducible factor (HIF)-3alpha is subject to extensive alternative splicing in human tissues and cancer cells and is regulated by HIF-1 but not HIF-2. Int J Biochem Cell Biol. 2010;42:1189–1200. doi: 10.1016/j.biocel.2010.04.008. [DOI] [PubMed] [Google Scholar]
- Pastorino JG, Shulga N, Hoek JB. Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. J Biol Chem. 2002;277:7610–18. doi: 10.1074/jbc.M109950200. [DOI] [PubMed] [Google Scholar]
- Phan LM, Yeung SJ, Lee MH. Cancer metabolic reprogramming:importance, main features, and potentials for precise targeted anti-cancer therapies. Cancer Biol Med. 2014;11:1–19. doi: 10.7497/j.issn.2095-3941.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard PJ, Ratcliffe PJ. Cancer Puzzling patterns of predisposition. Science. 2009;324:192–204. doi: 10.1126/science.1173362. [DOI] [PubMed] [Google Scholar]
- Porporato PE, Dhup S, Dadhich RK, Copetti T, Sonveaux P. Tumor glycolysis:a therapeutic target. Front Pharmacol. 2011;2:1–12. doi: 10.3389/fphar.2011.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prendergast GC, Ziff EB. A new bind for Myc. Trends Genet. 1992;8:91–96. doi: 10.1016/0168-9525(92)90196-b. [DOI] [PubMed] [Google Scholar]
- Riganti C, Gazzano E, Polimeni M, Aldieri E, Ghigo D. The pentose phosphate pathway:an antioxidant defense and a crossroad in tumor cell fate. Free Radic Biol Med. 2012;53:421–36. doi: 10.1016/j.freeradbiomed.2012.05.006. [DOI] [PubMed] [Google Scholar]
- Robeya RB, Hayd N. Is Akt the “Warburg kinase”?-Akt-energy metabolism interactions and oncogenesis. Semin Cancer Biol. 2009;19:1–9. doi: 10.1016/j.semcancer.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sang N, Stiehl DP, Bohensky J, et al. MAPK signaling up-regulates the activity of hypoxia-inducible factors by its effects on p300. J Biol Chem. 2003;278:14013–19. doi: 10.1074/jbc.M209702200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansal I, Sellers WR. The biology and clinical relevance of the PTEN tumor sup- pressor pathway. J Clin Oncol. 2004;22:295–463. doi: 10.1200/JCO.2004.02.141. [DOI] [PubMed] [Google Scholar]
- Schwartzenberg-Bar-Yoseph F, Armoni M, Karnieli E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 2004;64:262–733. doi: 10.1158/0008-5472.can-03-0846. [DOI] [PubMed] [Google Scholar]
- Semenza G. Signal transduction to hypoxia-inducible factor 1. Biochem Pharmacol. 2002;64:993–8. doi: 10.1016/s0006-2952(02)01168-1. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Hypoxia-inducible factors:mediators of cancer progression and targets for cancer therapy. Trends Pharmacol Sci. 2012;33:207–14. doi: 10.1016/j.tips.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. HIF-1 and human disease:one highly involved factor. Genes Dev. 2000a;14:198–391. [PubMed] [Google Scholar]
- Semenza GL. Hypoxia, clonal selection, and the role of HIF-1 in tumor progression. Crit Rev Biochem Mol Biol. 2000b;35:71–103. doi: 10.1080/10409230091169186. [DOI] [PubMed] [Google Scholar]
- Semenza GL. HIF-1and mechanisms of hypoxia sensing. Curr Opin Cell Biol. 2001;13:167–71. doi: 10.1016/s0955-0674(00)00194-0. [DOI] [PubMed] [Google Scholar]
- Semenza GL. HIF-1:upstream and downstream of cancer metabolism. Curr Opin Genet Dev. 2010;20:51–6. doi: 10.1016/j.gde.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shackelford DB, Shaw RJ. The LKB1 AMPK pathway:metabolism and growth control in tumour suppression. Nature Rev Cancer. 2009;9:563–75. doi: 10.1038/nrc2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RJ, Kosmatka M, Bardeesy N, et al. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A. 2004;101:3329–35. doi: 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim H, Dolde C, Lewis BC. c-Myc transactivation of LDH-A:implications for tumor metabolism and growth. Proc Natl Acad Sci U S A. 1997;94:665–863. doi: 10.1073/pnas.94.13.6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soga T. Cancer metabolism:Key players in metabolicReprogramming. Cancer Sci. 2013;104:275–81. doi: 10.1111/cas.12085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol. 2012;13:283–96. doi: 10.1038/nrm3330. [DOI] [PubMed] [Google Scholar]
- Sonveaux P, Vegran F, Schroeder T, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest. 2008;118:3930–42. doi: 10.1172/JCI36843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spoden GA, Mazurek S, Morandell D, et al. Isotype specific inhibitors of the glycolytic key regulator pyruvate kinase subtype M2 moderately decelerate tumor cell proliferation. Int J Cancer. 2008;123:312–21. doi: 10.1002/ijc.23512. [DOI] [PubMed] [Google Scholar]
- Susana RM, Maria MB, Moreno A, Heriberto PG, Franciso SG. Lactate contribution to Tumor Microenvironment:Mechanism, Effect on Immune Cells and Theraeutic Relavance. Front Immunol. 2016;7:1–8. doi: 10.3389/fimmu.2016.00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S, Tanaka T, Poyurovsky MV. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci U S A. 2010;107:7461–7560. doi: 10.1073/pnas.1002459107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennant DA. PK-M2 makes cells sweeter on HIF1. Cell. 2011;145:647–9. doi: 10.1016/j.cell.2011.05.009. [DOI] [PubMed] [Google Scholar]
- Tran Q, Lee H, Park J, Kim SH. Targeting cancer metabolism - revisiting the warburg effects. Toxicol Res. 2016;32:177–93. doi: 10.5487/TR.2016.32.3.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the warburg effect:the metabolic requirements of cell proliferation. Science. 2009;324:1029–33. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev LT, Vu BT, Graves B, et al. In vivo activation of the p53 pathway by small-molecule antagonists of mdm2. Science. 2004;303:844–8. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- Villunger A, Michalak EM, Coultas L, et al. P53 and drug-induced apoptotic responses mediated by bh3-only proteins puma and noxa. Science. 2003;302:1036–8. doi: 10.1126/science.1090072. [DOI] [PubMed] [Google Scholar]
- Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- Vousden KH, Ryan KM. p53 and metabolism. Nat Rev Cancer. 2009;9:691–700. doi: 10.1038/nrc2715. [DOI] [PubMed] [Google Scholar]
- Wade M, Wang YV, Wahl GM. The p53 orchestra:Mdm2 and mdmx set the tone. Trends Cell Biol. 2010;20:299–309. doi: 10.1016/j.tcb.2010.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995;92:55–104. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Chen X, Hay N. Akt as a target for cancer therapy:more is not always better (lessons from studies in mice) Br J Cancer. 2017;117:159–63. doi: 10.1038/bjc.2017.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward PS, Thompson CB. Metabolic reprogramming:a cancer hallmark even Warburg did not anticipate. Cancer Cell. 2012;21:297–308. doi: 10.1016/j.ccr.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinhouse S, Warburg O, Burk D, Schade AL. On respiratory impairment in cancer cells. Science. 1956;124:267–72. doi: 10.1126/science.124.3215.267. [DOI] [PubMed] [Google Scholar]
- Wiesener MS, Jurgensen JS, Rosenberger C. Widespread hypoxia inducible expression of HIF-2 alpha in distinct cell populations of different organs. \FASEB J. 2003;17:27–130. doi: 10.1096/fj.02-0445fje. [DOI] [PubMed] [Google Scholar]
- Wingo SN. Somatic LKB1 mutations promote cervical cancer progression. PLoS One. 2009;4:5137. doi: 10.1371/journal.pone.0005137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise DR, DeBerardinis RJ, Mancuso A, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A. 2008;105:18782–7. doi: 10.1073/pnas.0810199105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong KB, DeDecker BS, Freund SM, et al. Hot-spot mutants of p53 core domain evince characteristic local structural changes. Proc Natl Acad Sci U S A. 1999;96:8438–42. doi: 10.1073/pnas.96.15.8438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong KK, Engelman JA, Cantley LC. Targeting the PI3K signaling pathway in cancer. Curr Opin Genet Dev. 2010;20:87–90. doi: 10.1016/j.gde.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong N, De Melo J, Tang D. PKM2, a central point of regulation in cancer metabolism. Int J Cell Biol. 2013;2013:242–513. doi: 10.1155/2013/242513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods A, Dickerson K, Heath R, et al. Ca2+/calmodulin-dependent protein kinase kinase-βacts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005;2:21–33. doi: 10.1016/j.cmet.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Woods A, Johnstone SR, Dickerson K, et al. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol. 2003;13:2004–8. doi: 10.1016/j.cub.2003.10.031. [DOI] [PubMed] [Google Scholar]
- Xie M, Zhang D, Dyck JR, et al. A pivotal role for endogenous TGF-β-activated kinase-1 in the LKB1/AMP-activated protein kinase energy-sensor pathway. Proc Natl Acad Sci U S A. 2006;103:17378–83. doi: 10.1073/pnas.0604708103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing M. Genetic alterations in the phosphatidylinositol-3 kinase/Akt pathway in thyroid cancer. Thyroid. 2010;20:697–706. doi: 10.1089/thy.2010.1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yalcin A, Telang S, Clem B, Chesney J. Regulation of glucose metabolism by6-phosphofructo-2-kinase/fructose-2,6-bisphosphatases in cancer. Exp Mol Pathol. 2009;86:174–9. doi: 10.1016/j.yexmp.2009.01.003. [DOI] [PubMed] [Google Scholar]
- Ye J, Mancuso A, Tong X. Pyruvate kinase m2 promotes de novo serine synthesis to sustain mtorc1 activity and cell proliferation. Proc Natl Acad Sci U S A. 2012;109:6904–9. doi: 10.1073/pnas.1204176109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung SJ, Pan J, Lee MH. Roles of p53, MYC and HIF-1 in regulating glycolysis:the seventh hallmark of cancer. Cell Mol Life Sci. 2008;65:3981–99. doi: 10.1007/s00018-008-8224-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuneva M, Zamboni N, Oefner P, Sachidanandam R, Lazebnik Y. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. Cell Biol. 2007;178:93–105. doi: 10.1083/jcb.200703099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zache N, Lambert JM, Wiman KG, Bykov VJ. Prima-1met inhibits growth of mouse tumors carrying mutant p53. Cell Oncol. 2008;30:411–8. doi: 10.3233/CLO-2008-0440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Juan L, Yiangjian L. Tumour-associated mutant p53 drives the Warburg effect. Nat Comm. 2013;4:1–15. doi: 10.1038/ncomms3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Bosch-Marce M, Shimoda LA, et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283:10892–903. doi: 10.1074/jbc.M800102200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zhang S, Yu D. PI(3)kinase apart PTEN's role in cancer. Clin Cancer Res. 2010;16:4325–30. doi: 10.1158/1078-0432.CCR-09-2990. [DOI] [PubMed] [Google Scholar]
- Zhao L, Vogt PK. Class I PI3K in oncogenic cellular transformation. Oncogene. 2008;27:5486–96. doi: 10.1038/onc.2008.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J. Energy metabolism of cancer:Glycolysis versus oxidative phosphorylation (Review) Oncol Lett. 2012;4:1151–7. doi: 10.3892/ol.2012.928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G, Wang J, Zhao M, et al. Gain-of-function mutant p53 promotes cell growth and cancer cell metabolism via inhibition of AMPK activation. Mol Cell. 2014;54:960–74. doi: 10.1016/j.molcel.2014.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]