

Abstract
Background
Tumor mutational burden (TMB) is an increasingly important biomarker for immune checkpoint inhibitors. Recent publications have described strong association between high TMB and objective response to mono- and combination immunotherapies in several cancer types. Existing methods to estimate TMB require large amount of input DNA, which may not always be available.
Methods
In this study, we develop a method to estimate TMB using the Oncomine Tumor Mutation Load (TML) Assay with 20 ng of DNA, and we characterize the performance of this method on various formalin-fixed, paraffin-embedded (FFPE) research samples of several cancer types. We measure the analytical performance of TML workflow through comparison with control samples with known truth, and we compare performance with an orthogonal method which uses matched normal sample to remove germline variants. We perform whole exome sequencing (WES) on a batch of FFPE samples and compare the WES TMB values with TMB estimates by the TML assay.
Results
In-silico analyses demonstrated the Oncomine TML panel has sufficient genomic coverage to estimate somatic mutations with a strong correlation (r2=0.986) to WES. Further, in silico prediction using WES data from three separate cohorts and comparing with a subset of the WES overlapping with the TML panel, confirmed the ability to stratify responders and non-responders to immune checkpoint inhibitors with high statistical significance. We found the rate of somatic mutations with the TML assay on cell lines and control samples were similar to the known truth. We verified the performance of germline filtering using only a tumor sample in comparison to a matched tumor-normal experimental design to remove germline variants. We compared TMB estimates by the TML assay with that from WES on a batch of FFPE research samples and found high correlation (r2=0.83). We found biologically interesting tumorigenesis signatures on FFPE research samples of colorectal cancer (CRC), lung, and melanoma origin. Further, we assessed TMB on a cohort of FFPE research samples including lung, colon, and melanoma tumors to discover the biologically relevant range of TMB values.
Conclusions
These results show that the TML assay targeting a 1.7-Mb genomic footprint can accurately predict TMB values that are comparable to the WES. The TML assay workflow incorporates a simple workflow using the Ion GeneStudio S5 System. Further, the AmpliSeq chemistry allows the use of low input DNA to estimate mutational burden from FFPE samples. This TMB assay enables scalable, robust research into immuno-oncology biomarkers with scarce samples.
Keywords: Cancer genomics, checkpoint inhibitors, tumor mutational burden (TMB), Oncomine Tumor Mutation Load (TML) Assay, immuno-oncology
Introduction
Therapeutic antibodies targeting cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) and programmed cell death protein 1 (PD-1) immune checkpoints result in increased activation of the immune response and have shown remarkable clinical benefit in patients with diverse cancer types (1-7). However, although these approaches have transformed treatment strategies in certain cancer types, most cancer patients are not yet effectively treated with these approaches and a minority experience serious adverse events due to sustained immune activation (8,9).
For PD-1 pathway inhibitors, the expression level of programmed cell death 1 ligand 1 (PD-L1) as measured by immunohistochemistry is associated with a higher clinical response rate relative to patients with low or absent expression although not all patients are effectively stratified with this approach (10-12). In contrast, the protein expression level of CTLA-4 or its ligands has not proven to predict response to anti-CTLA-4 immunotherapies. In addition, new immunotherapy agents targeting other T-cell co-inhibitory receptor proteins have entered clinical development (13,14). Thus, there remains an unmet need to develop effective predictive biomarkers for diverse immunotherapies.
Early clinical trials of PD-1 pathway inhibitors employed whole exome sequencing (WES) to explore mutational predictors of response. From these studies, it was observed that tumor mutational burden (TMB) was associated with clinical response (15-17). In parallel, microsatellite instability was associated with response to PD-1 checkpoint inhibitors, first in advanced colorectal cancers (CRC) and then in diverse cancer types with mismatch repair (MMR) deficiency (18,19). In patients with MMR deficiency, high somatic mutation loads were associated with positive clinical response (19). More recently, an association between the median TMB and objective response rate across multiple cancer types was established (20).
TMB is an emerging predictive biomarker for immune checkpoint inhibitors, but questions remain whether this biomarker can be effectively translated from WES to a targeted sequencing approach. We describe the development of a targeted sequencing panel based on Ion AmpliSeq technology that provides a cost-effective and robust TMB estimate using minimal sample input from formalin fixed paraffin embedded tumor samples.
Methods
Oncomine Tumor Mutation Load (TML) Assay
The OncomineTM Tumor Mutation Load Assay is a PCR-based next-generation sequencing assay. The panel covers 1.7 megabases (Mb) of 409 genes with known cancer associations. The panel consists of 15,513 PCR targets that are evenly distributed in two pools. The covered genomic space divides into 1.2 Mb exonic and 0.45 Mb intronic region.
Utilizing Ion AmpliSeqTM library preparation technology, library preparation requires only 20 ng of input DNA (10 ng per primer pool) extracted from formalin-fixed, paraffin-embedded (FFPE) cancer specimens. Sequencing is performed on high throughput semiconductor sequencing platform, the Ion GeneStudioTM S5 System, on 540 chip to achieve high median coverage (>500×) and uniformity (>90%). Unless otherwise stated, all 8 samples were run on a 540 chip. Reads are aligned to hg19 using Torrent Suite 5.6 and BAM files are transferred to Ion Reporter 5.6 for variant calling and secondary analysis including TMB calculation. The assay is research use only, not for diagnostic procedures.
WES analyses on FFPE samples
WES was performed on 12 FFPE tumors and their matched normal from adjacent tissue.
The Agilent SureSelect Human All Exon V5 kit (Agilent Technologies; with ~50 Mb panel) was used for target capture from 200 ng input DNA followed by sequencing on HiSeqX (Illumina). The paired-end reads were aligned to the UCSC reference human genome (GRCh37/hg19; Feb. 2009 release download) using BWA-MEM aligner (version bwa-0.7.12). The aligned reads were first sorted and then duplicates were removed using Picard (picard-tools-1.115). After removing duplicates, the reads were realigned around the known indels using GenomeAnalysisTK-3.6 toolkit (GATK). After performing realignment, the base re-calibration step was performed using GATK. Somatic variant calling of variants down to 10% allelic frequency was performed using Strelka (v2.0.17). Low-frequency variants to 5% frequency were called using LoFreq (v2.1.2). Variants found in the matched normal sample were removed from the tumor variant calls for clearing putative germline variants in the sample. Mutations were then filtered to remove population single-nucleotide polymorphisms (SNPs) found in dbSNP (v150), Exome Aggregation Consortium (ExAC) and 1000 genome to remove any residual non-somatic variants remaining after matched normal filtering. Sequencing and analyses was performed by MedGenome (https://www.medgenome.com/). Nonsynonymous point mutations at ≥5% allelic frequency from the somatic mutations were used for the WES comparison with TMB by TML assay.
WES data for in-silico analysis
Total 21,056 exomes were downloaded from COSMIC (COSMIC v80; http://cancer.sanger.ac.uk/cosmic). Download comprised specimens from more than 22 cancer types. The somatic mutation as determined by COSMIC were used as the raw somatic mutation count from whole exome. The TML panel estimate for each specimen was computed by only counting mutations overlapping with TML panel.
TMB analysis
First, all single base substitutions at ≥5% allelic frequency were called from TML panel. Initially, we count the raw total somatic mutations from noncoding region and synonymous and non-synonymous mutations from coding region. We use the raw somatic mutation set for maximizing the number of bases used to capture the tumorigenesis signature, described below.
We optimized mapping and variant calling parameters and implemented an analysis workflow with these optimizations in Ion Reporter software (21).
TMB is defined as nonsynonymous somatic mutations per Mb, including missense and nonsense point mutations, in the exonic regions of the genome examined. To count the appropriate number of Mb in the denominator, we include only those bases with sufficient coverage in the sequencing run to sensitively detect variants at 5%. After variant calling, variants were filtered to eliminate germline variants and select for the highest quality somatic variants to further reduce noise for TMB estimation. Germline variants were removed using a germline filter-chain based on population databases: variant alleles present from the 1000 Genome Project, NHLBI GO Exome Sequencing Project (ESP), and ExAC were filtered out. To calculate the TMB, a normalized and calibrated predictor including the nonsynonymous somatic variants is divided by the number of covered Mb.
Control samples for accuracy of somatic mutations
For analyses on human DNA cell line controls, NA12878 (normal) and HCC1143 (breast positive) from NIGMS Human Genetic Cell Repository at the Coriell Institute for Medical Research and ATCC, respectively, were used. HCC1143 has 10 somatic mutations from COSMIC Cell line database (https://cancer.sanger.ac.uk/; expected ~6 raw somatic mutations/Mb) that overlaps TML panel.
Additionally, the limits of somatic variant detection were tested in controlled dilution series using a blend of synthetic DNA and genomic DNA from the Genome in a Bottle cell line GM24385. We prepared a custom AcroMetrix Hotspot Frequency Ladder that contained 555 mutations at 5%, 10%, 12.5%, 15%, 25%, and 50% frequency levels. Each custom AcroMetrix sample contained 339 common tumor mutations (expected TMB ~207 mutations/Mb) interrogated by our assay after germline filtering. Germline filters were expected to remove all variants of GM24385. This count was used to measure the specificity of the raw somatic mutations calls and raw somatic mutation rate.
To measure the accuracy of the TML assay in calling the engineered variants in custom AcroMetrix sample, the specificity was measured by calculating positive predictive value (PPV, i.e., percent true positives w.r.t. true positive and false positives). The synthetic DNA of AcroMetrix has a high number of engineered variants by design. One effect of this high number of linked variants is that some variants within the amplicon inserts will be in a haplotype with a variant under primer site, and variants under primer sites will interfere with amplification. Hence variants in a haplotype with a variant under a primer will not always be expected to be amplified and were not included in measures of sensitivity. A revised truth set of 234 that met this criterion was used to measure sensitivity of somatic variant detection.
FFPE research samples
Twenty-three FFPE research samples (20 CRC, 2 Melanoma, and 1 lung tumors) were used for multiple analyses, to an average depth of 500×. Duplicates were generated for replication studies. Matched normal sample adjacent to tumor was used in comparison with matched normal analyses.
A cohort of 33 research samples (13 lung, 12 colon, 3 melanoma, 2 gastrointestinal stromal, and 3 breast/ovarian tumors) was separately sequenced. For this group, deeper sequencing (4 samples per 540 chip to an average depth of 1,000×) was performed. In addition, one sample failed QC due to insufficient sequencing depth (<100) and two failed QC due to too many (>60) and high proportion of variants consistent with de-amination artifacts.
Results
In-silico evaluation of panel design to measure TMB
We first sought to determine the suitability of the size and targets of TML panel to predict the TMB as measured by WES. For each sample, we used 21,056 exomes from COSMIC, to determine the number of somatic mutations by WES and compared that to the number of somatic mutations covered by the TML panel. We found the results were highly correlated (r2=0.986; Figure 1A). We also performed similar analyses on samples of one of the four common cancer types (colorectal, melanoma, lung, or endometrial) to obtain high correlation in each of these tumor types (r2=0.975, r2=0.976, r2=0.935, or r2=0.995, respectively) in each set (Figure 1B). This confirms that a panel of this size 1.7 Mb is theoretically sufficient to measure TMB.
Figure 1.

Suitability of TML panel’s size and targets in assessing TMB. (A) Comparison of somatic mutations by WES with somatic mutations covered by TML panel. This pan-cancer in-silico analysis was performed on 21,056 exomes from COSMIC v80 covering more than 22 cancer types; (B) in-silico comparison of somatic mutations by TML with that from WES for four common cancer types: colorectal, melanoma, lung, and endometrial cancer. Next, exomes from three separate cohorts were downloaded, and for each exome, TML panel covered somatic mutations were used to stratify responders and non-responders to obtain high statistical significance in, (C) NSCLC (ref. Rizvi; P=0.00568); (D) melanoma (ref. Snyder; P=0.000348), and (E) melanoma (ref. Van; P=0.00498). TML, tumor mutation load; TMB, tumor mutational burden; WES, whole exome sequencing; NSCLC, non-small cell lung cancer.
We assessed the ability of our panel in stratifying responders and non-responders to immune checkpoint inhibitors through in-silico analyses on exomes from three cohorts. First, we downloaded clinical trial WES data for (I) 31 NSCLC subjects treated with pembrolizumab (anti-PD1) from Rizvi et al. study (15), (II) 64 melanoma subjects treated with ipilimumab (anti-CTLA4) and tremelimumab (anti-CTLA4) from Snyder et al. study (17), and (III) 100 melanoma subjects treated with ipilimumab (anti-CTLA4) from Van Allen et al. (16). Each sample was annotated with response or non-response to the respective immune checkpoint inhibitor. We computed predicted somatic mutation counts using only the genomic region covered by the TML panel and measured the ability of this measurement to separate the responders vs. non-responders for the three experiments. We performed Mann-Whitney Exact test to examine the significance in difference of means between two groups. We observed high statistical significance between mutation counts of responders and non-responders for each cohort [P=0.00568 for NSCLC (15); Figure 1C, P=0.000348 for melanoma (17); Figure 1D, P=0.00498 for melanoma (16); Figure 1E]. This confirms that a panel of this size can, in principle, measure significant differences in the TMB of responders from non-responders based.
Performance of filter chain in removing germline variants
The workflow of TML assay requires tumor samples only. There is no need for a matched normal sample. The workflow removes the germ-line variants, along with common artifacts present in population databases, through the application of filters based on variants prevalent in population databases. The allele frequency distribution of all variants called on TML panel effectively shows the median of homozygous and heterozygous germline variants at 100% and 50% allelic frequency, respectively (Figure 2A). The median and distribution of allele frequencies of somatic variants depends on the tumor content of the sample, the heterogeneity of the sample and the existence of tumor clonal evolution. Population databases effectively eliminate the germ-line variants as evaluated by allele frequency distribution yardstick (Figure 2A). Normalization approaches may model the residual germline variants that may be left after germline database filtering, which is expected to be less than 3% for most global populations, though populations with African ancestry have somewhat more genetic diversity and a smaller proportion of these continental alleles represented in population databases (22). However, allele frequency cannot be solely used for detecting somatic mutations due to the possibility of high tumor content and loss of heterozygosity (LOH; Figure 2B). Samples with high tumor content may recapitulate germline allele ratios, as may loci in regions with LOH.
Figure 2.

Germ-line filtering and distribution of variants called by TML assay. (A) Allele frequency distribution of all variants before applying germ-line filtering and only somatic variants after applying germ-line filtering from an FFPE tumor; (B) allele frequency distribution of a possible LOH FFPE tumor and matched normal before applying germ-line filtering; (C) allele frequency distribution of variants in individual chromosomes of LOH tumor from B. TML, tumor mutation load; LOH, loss of heterozygosity; FFPE, formalin-fixed, paraffin-embedded.
Most FFPE samples exhibit characteristic peaks at allele ratios of 1 and 0.5, consistent with expected germline homozygote and heterozygote variants, along with a peak of somatic mutations; this somatic peak varies in amplitude and allele ratio depending on the characteristic of the tumor. (Figure 2A). However, occasionally samples will exhibit different characteristics, with the histogram of allele ratios being broadly distributed across the spectrum. Such samples may have high levels of aneuploidy and other chromosomal aberrations (Figure 2B). In one such sample, we examined the allele ratio distribution by chromosome to see whether this broad distribution of allele ratios was similar in every chromosome (Figure 2C). Interestingly, we discovered it was not. In some chromosomes (chromosome 8), the normal pattern of three peaks at putative homozygote, heterozygote, and somatic variant frequencies was present. In another (chromosome 7), a trimodal distribution was present, with two equal sized peaks, and the mean allele ratio of each peak corresponding approximately to that which would be expected if the chromosome was triploid. In another (chromosome 1), there was a wide distribution of allele ratios, suggesting aneuploidy in the chromosome.
Comparison with tumor-normal analyses
A robust method for evaluating the ability of a tumor-only method to predict the number of somatic mutations is to compare the tumor-only mutations with the equivalent metric computed on matched tumor-normal samples. Using a matched normal sample enables the removal of all germline variants, including private mutations as well as population variants. It also enables the removal of any systematic noise. We evaluated the performance of germ-line filtering by comparing the raw somatic mutation estimates of 12 FFPE tumors with mutation estimates from tumor-normal analyses using matched normal samples. We observed strong correlation between two approaches (r2=0.94; Figure 3).
Figure 3.

Comparison with matched normal analysis workflow. Correlation of raw somatic mutations using tumor only TML assay workflow with that from tumor-normal workflow, which relies on matched normal to clear germ-line mutations and noise, on FFPE tumor samples. TML, tumor mutation load; FFPE, formalin-fixed, paraffin-embedded.
Reproducibility
We also assessed the reproducibility of the rate of somatic mutations as measured by the TML assay. We compared raw somatic mutations per megabase between two replicates of 21 FFPE samples and two replicates of two cell lines. We observed a high reproducibility between replicates, with a correlation of 0.97 (Figure 4). The mean and median of absolute differences between replicates for all 21 samples was 2.49 and 1.85, respectively.
Figure 4.
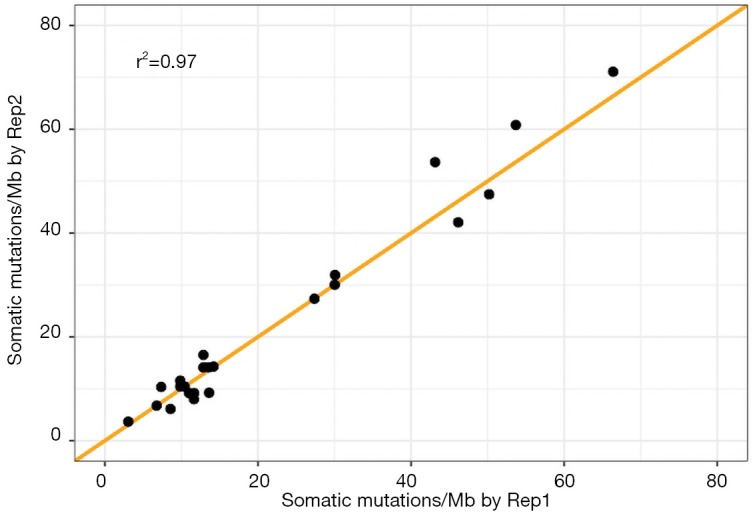
Assay reproducibility. Comparison of raw somatic mutations/Mb estimates by TML assay in two replicates of 21 FFPE and 2 cell line samples. The line y = x is plotted in orange. TML, tumor mutation load; FFPE, formalin-fixed, paraffin-embedded.
Correlation with WES
We sought to compare TMB estimated by TML assay with TMB measured from WES with a matched normal sample. We counted the number of nonsynonymous point mutations from 12 FFPE tumors using WES and compared that to TMB estimated by the TML assay. We observed strong correlation between the two approaches (r2=0.83; Figure 5). This result confirms that the TML panel can accurately estimate TMB even though the panel is only 3.6% of the size of the whole exome, uses 10% of total input amount of tumor DNA required by WES, and without a matched normal sample.
Figure 5.

Comparison of somatic mutations by TML panel with that from WES. WES analyses performed on tumor-normal samples, while TML assay ran on only tumor samples. TML, tumor mutation load; WES, whole exome sequencing.
Accuracy on control samples
We assessed that average raw somatic mutations per Mb estimates by our assay on normal and positive cell lines are 3.37 and 6.76 mutations/Mb (Figure 6). This estimate is close to expected (~6 raw somatic mutations/Mb) on positive cell line. Except for the 5% custom AcroMetrix sample, the observed raw somatic mutations/Mb estimates by TML assay are within 16% (mean 13.62%) from expected rate of raw somatic mutations.
Figure 6.

Estimation of raw somatic mutations/Mb on control samples. (A) Raw somatic mutations/Mb on replicates of NA12878 (normal cell line), HCC1143 (breast cancer positive cell line), and custom AcroMetrix Hotspot Frequency Ladder containing engineered variants at 5%, 10%, 12.5%, 15%, 25%, and 50% frequency levels; (B) sensitivity and (C) specificity in detecting true, post-filter variants at different frequency levels.
At a threshold of 5%, mutations at 5% frequency in the reads will be detected. However, in the 5% custom AcroMetrix sample, it is expected that half of the genomic positions of interest will have reads containing just under 5% of the alternative allele, so it is expected that a 5% LOD workflow will start to have reduced sensitivity on the 5% custom AcroMetrix sample.
We finally sought to assess the accuracy of the TML assay in calling the engineered variants of AcroMetrix. We assessed specificity by calculating PPV (i.e., percent true positives w.r.t. true positive and false positives). We obtained high PPV (mean 96.98%; min 97.17% and max 97.66%) for all custom frequencies (Figure 6). In the 5% custom AcroMetrix sample, sensitivity was ≥93%. In all other dilutions, we obtained high sensitivity (mean 97.18%; min 96.58% and max 97.86%; Figure 6).
Separation of mutation high and low samples
High TMB is associated with MMR deficiency which results from mutations in genes coding DNA MMR pathways (MSH2, MSH6, MLH1, PMS2). MMR deficiency, also called microsatellite instability, is often typed though polymerase chain reaction or immunohistochemistry assays. We tested four definitions of TMB using TML assay based on the power of separating microsatellite instable (MSI) and microsatellite stable (MSS) CRC samples. Our four definitions included, (I) rate of all somatic mutations at ≥5% allelic frequency in the coding and noncoding region of TML panel, (II) rate of nonsynonymous somatic mutations in the coding region of TML panel, (III) rate of all somatic mutations in the coding region of TML panel, and (IV) rate of all somatic mutations at ≥10% allelic frequency in the coding and noncoding region of TML panel. We observed statistical significance (P=0.0019 in I; P=0.00069 in II; P=0.00092 in III; P=0.012 in IV) in separating two groups through all four approaches (Figure 7). However, TMB as defined by rate of nonsynonymous mutations in the coding region provides the strongest separation (Figure 7).
Figure 7.

Comparison of four approaches for defining TMB through stratifying eight MSI and twelve MSS CRC tumors. In all cases, allele ratio is set at 5% except for D. The denominator is every base in the panel for case A and D or every exonic base for B and C; additional scaling factors in the denominator to normalize for the number of bases considered are not applied. (A) High statistical significance in grouping when TMB is defined as per megabase rate of all somatic mutations from the full TML panel (including coding and non-coding regions); (B) high statistical significance in grouping when TMB is defined with high statistical significance when TMB is defined as per megabase rate of nonsynonymous somatic mutations in the exonic region on TML panel; (C) high statistical significance in grouping when TMB is defined as per megabase rate of all somatic mutations in the exonic region on TML panel; (D) moderate statistical significance in grouping when TMB is defined as per megabase rate of all somatic mutations at and above 10% allelic frequency from the full TML panel (including coding and non-coding regions). TMB, tumor mutational burden; MSI, microsatellite instable; MSS, microsatellite stable; TML, tumor mutation load.
Signatures of genomic instability
Biological process causing mutations in somatic cells leaves a mutational signature (23). These signatures can be examined through substitution type and context of somatic mutations using the TML assay. Single base substitutions are often classified into six subtypes; C:G > A:T, C:G > G:C, C:G > T:A, T:A > A:T, T:A > C:G, and T:A > G:C. This classification can be further refined by including the sequence context of each mutated base by including adjacent 5' and 3' bases. For example, a C:G > T:A mutation can be characterized as TpCpG > TpTpG (where mutated base is underlined, and preceded and followed by thymine and guanine, respectively). The inclusion of 5' and 3' bases generates 96 possible substitution type and context classes (6 types of substitutions × 4 types of 5' base × 4 types of 3' base). We computed metrics and graphs characterizing this tumorigenesis signature.
We hypothesized that the tumorigenesis signature would be consistent with the expected mutation source or published signatures in tumors of different types, and this in fact is what we observed. An elevated rate of spontaneous deamination of 5-methylcytosine through C:G > T:A transitions at cytosine-guanine (CpG) dinucleotide sites was observed in CRC tumors, as published in the literature (23) (Figure 8A). We found high prevalence of C:G > A:T substitutions in the lung tumor sample consistent with characteristic tobacco carcinogen DNA damage (24) (Figure 8B). In a melanoma sample, we found two novel signatures: high C:G > T:A at TpC, high C:G > T:A at CpC and CpC sites, consistent with DNA damage signatures of ultraviolet radiation exposure in melanoma (25) (Figure 8C).
Figure 8.

Signature of tumorigenesis and FFPE fixation error through substitution type and context of somatic mutations. (A) Spontaneous deamination of 5-methylcytosine as observed in CRC tumor; (B) signature pattern of tobacco damage as observed in lung tumor; (C) signature pattern of UV damage as observed in melanoma tumor; (D) signature pattern of FFPE sample fixation error; (E) pie charts quantifying the proportion of respective signature. From left to right, pie charts represent elevated rate of (I) spontaneous deamination of 5-methylcytosine footprint (red and blue segments) in CRC tumor, (II) tobacco damage footprint (orange segment) in lung tumor, (III) UV damage footprint (blue, green, and yellow segments) in melanoma tumor, (IV) fixation error footprint (green and purple segments). FFPE, formalin-fixed, paraffin-embedded; CRC, colorectal cancer.
Clinical research specimens from molecular pathology laboratories are typically fixed in formalin for detailed morphological assessment (26). However, formalin fixation can cause DNA damage such as fragmentation and de-amination, which can in turn create loss of coverage and sequencing artefacts. These fixation artifacts can artificially inflate the somatic mutations and impact the TMB estimate. In samples with fixation error, we observed disproportionate levels of C:G > T:A transitions in the 1–10% allele frequency range in all substitution contexts, except that we see a notably lower occurrence of deamination at CpG sites in many samples (Figure 8D). While both CRC tumors and FFPE damage exhibit high C:G > T:A transitions, there is a noticeable difference in the sequence context in which these transitions occur. In CRC samples, they occur preferentially at the CpG sites: in samples with deamination damage, they occur preferentially away from CpG sites. This pattern can be dramatically observed in the visual representation of the signatures (Figure 8A,D,E).
The lack of reproducibility of variants consistent with deamination of replicates of samples with FFPE damage further suggest that these variants are likely to be artifacts rather than somatic variants present in the original sample. We developed a deamination metric based on the frequency of C:G > T:A transitions in the low allelic frequency variants and consistently monitored this metric in all FFPE libraries. In samples with low tumor content, the deamination metric will count true somatic mutations as well as variants whose source is deamination. Nonetheless, in practice the deamination metric very accurately accounted for true deamination in compromised samples and was also accompanied by low coverage depth in many instances. Samples with high numbers of variants consistent with deamination damage may exhibit high TMB, but the source is the deamination damage rather than somatic mutations resulting in neo-antigen production.
TMB analysis on cohort of FFPE samples
We assessed TMB on a cohort of 30 FFPE research samples (12 lung, 10 colon, 3 melanoma, 2 gastrointestinal stromal, and 3 breast/ovarian tumors) to obtain results consistent with the literature for individual tumor types (23,27) (Figure 9). Encouragingly, we did not see evidence of many samples with deamination damage artificially increasing the TMB estimate: only two samples had such evidence of deamination, and this was clearly detectable with our reported QC metrics. On lung samples, we obtained mean TMB 8.73 (min 1.67; max 20.31) with 7 out of 12 samples having TMB values lower than 10. The mean TMB on colon samples was 8.75 (min 2.51; max 21.95) with 7 out of 10 samples having TMB values lower than 10. We discovered a broad range of TMB values in melanoma samples with two samples at 0 or 1.68 and the third at 36.84. Similar broad range of TMB was observed in gastrointestinal stromal tumors with one sample at 1.7 and another at 53.4. All three breast or ovarian tumors had TMB values under 7.
Figure 9.

TMB estimates by TML assay on a batch of 27 FFPE research samples (12 lung, 10 colon, 3 melanoma, and 2 breast/ovarian tumors). TMB, tumor mutational burden; TML, tumor mutation load; FFPE, formalin-fixed, paraffin-embedded.
This confirms that the TML assay predicts TMB values from a range of tumor types that are consistent with the range reported in the literature.
Discussion and conclusions
Since the first exploration of mutational burden as a biomarker to predict clinical benefit from ipilimumab in advanced melanoma, evidence for the predictive value of TMB as a standalone biomarker has expanded with recent clinical trial readouts (17,28). For example, TMB was predictive of response to nivolumab plus ipilimumab combination therapy in a phase 3 clinical trial for NSCLC (28), and TMB above the mean was associated with enhanced response to nivolumab + ipilimumab in NSCLC in the CheckMate 568 study (CheckMate 568 ClinicalTrials.gov number, NCT02659059).
WES is a comprehensive method for computing TMB and can identify all predicted neo-antigens. However, the high cost, large input DNA amount, and workflow and analysis complexity limits its applicability in practice. To overcome these limitations, a number of methods are being developed to estimate TMB using next-generation DNA sequencing gene panels (29,30). The ability of targeted sequencing panels to predict TMB is limited by statistical sampling effects if the panel size is not large enough. Typically, larger panels are less subject to sampling effects and can more precisely estimate TMB levels, and the question of how large a panel must be is an important one. Multiple studies have independently assessed the size of the targeted panel that is sufficient in estimating WES TMB, and many recommend a panel larger than 1 Mb in size (30). Through in-silico experiments using thousands of exomes from COSMIC, we showed that the 1.7 Mb TML panel is large enough to estimate WES TMB accurately. Indeed, the Oncomine TML assay results agreed well with that from WES on a batch of FFPE research samples.
We have demonstrated the analytical capabilities of the TML assay in accurately estimating TMB from FFPE research samples. The paucity of available matched normal germline samples in many situations motivated us to design a tumor only workflow which incorporates population databases to filter out germline alterations. On cell lines and synthetic controls, we found the estimates of raw somatic mutations to be close to expected. Further, the high correlation of raw somatic mutations by TML assay with that from tumor-normal analyses, a workflow using matched normal DNA for filtering out germline variants, verified the effectiveness of germline elimination. Comparing replicates of FFPE samples, the raw somatic mutation estimates of TML assay showed high reproducibility. These findings indicate that Oncomine TML is an accurate, reproducible, and scalable assay for TMB calculation.
Pediatric malignancies tend to have the lowest mutational burden, while cancers associated with environmental DNA damage such as ultraviolet radiation or smoking history are more likely to be highly mutated (15). Further, the specific signature of the mutagen is often characteristic. We found that a 1.7 Mb gene panel can effectively show the tumorigenesis signatures in various cancers. For example, we often observed high C:G > A:T substitutions consistent with tobacco carcinogen damage in lung research samples. While efficacy to pembrolizumab has been correlated with molecular smoking signature (15), which also associates with TMB, tumorigenesis signatures may serve as supplemental evidence and help in understanding genomic landscape of lung and other epithelial cancers associated with environmental DNA damage.
Since this panel uses Ion AmpliSeq chemistry, low amounts of input DNA are sufficient; the TML assay has been successfully tested with 20 ng input DNA for both control and FFPE samples. An assay with low input DNA requirement empowers researchers to perform multiple experiments and explore a rich variety of scientific questions on a single FFPE sample using multiple assays. The quick turnaround time and the performance of the assay across a range of common cancer types confirm this TMB solution has ability to impact the space of immuno-oncology research.
Acknowledgements
We acknowledge the support of Rosella Petraroli and Chris Allen.
Footnotes
Conflicts of Interest: Employees of Thermo Fisher Scientific: R Chaudhary, D Cyanam, V Mittal, W Tom, J Au-Young, S Sadis, F Hyland. The other authors have no conflicts of interest to declare.
References
- 1.Phan GQ, Yang JC, Sherry RM, et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc Natl Acad Sci U S A 2003;100:8372-7. 10.1073/pnas.1533209100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ribas A, Camacho LH, Lopez-Berestein G, et al. Antitumor activity in melanoma and anti-self responses in a phase I trial with the anti-cytotoxic T lymphocyte-associated antigen 4 monoclonal antibody CP-675,206. J Clin Oncol 2005;23:8968-77. 10.1200/JCO.2005.01.109 [DOI] [PubMed] [Google Scholar]
- 3.Ascierto PA, Marincola FM, Ribas A. Anti-CTLA4 monoclonal antibodies: the past and the future in clinical application. J Transl Med 2011;9:196. 10.1186/1479-5876-9-196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brahmer JR, Drake CG, Wollner I, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol 2010;28:3167-75. 10.1200/JCO.2009.26.7609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366:2443-54. 10.1056/NEJMoa1200690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med 2013;369:134-44. 10.1056/NEJMoa1305133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buchbinder EI, Desai A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am J Clin Oncol 2016;39:98-106. 10.1097/COC.0000000000000239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Michot JM, Bigenwald C, Champiat S, et al. Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur J Cancer 2016;54:139-48. 10.1016/j.ejca.2015.11.016 [DOI] [PubMed] [Google Scholar]
- 9.Cousin S, Italiano A. Molecular Pathways: Immune Checkpoint Antibodies and their Toxicities. Clin Cancer Res 2016;22:4550-5. 10.1158/1078-0432.CCR-15-2569 [DOI] [PubMed] [Google Scholar]
- 10.Herbst RS, Baas P, Kim DW, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet 2016;387:1540-50. 10.1016/S0140-6736(15)01281-7 [DOI] [PubMed] [Google Scholar]
- 11.Garon EB, Rizvi NA, Hui R, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med 2015;372:2018-28. 10.1056/NEJMoa1501824 [DOI] [PubMed] [Google Scholar]
- 12.Chatterjee M, Turner DC, Felip E, et al. Systematic evaluation of pembrolizumab dosing in patients with advanced non-small-cell lung cancer. Ann Oncol 2016;27:1291-8. 10.1093/annonc/mdw174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dempke WCM, Fenchel K, Uciechowski P, et al. Second- and third-generation drugs for immuno-oncology treatment-The more the better? Eur J Cancer 2017;74:55-72. 10.1016/j.ejca.2017.01.001 [DOI] [PubMed] [Google Scholar]
- 14.Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016;44:989-1004. 10.1016/j.immuni.2016.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015;348:124-8. 10.1126/science.aaa1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van Allen EM, Miao D, Schilling B, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015;350:207-11. Erratum in: Science 2015;350:aad8366. Science 2016;352. 10.1126/science.aad0095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 2014;371:2189-99. 10.1056/NEJMoa1406498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O'Neil BH, Wallmark JM, Lorente D, et al. Safety and antitumor activity of the anti-PD-1 antibody pembrolizumab in patients with advanced colorectal carcinoma. PLoS One 2017;12:e0189848. 10.1371/journal.pone.0189848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017;357:409-13. 10.1126/science.aan6733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yarchoan M, Hopkins A, Jaffee EM. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N Engl J Med 2017;377:2500-1. 10.1056/NEJMc1713444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Merriman B, Ion Torrent R, Team D, Rothberg JM. Progress in ion torrent semiconductor chip based sequencing. Electrophoresis 2012;33:3397-417. Erratum in: Electrophoresis 2013;34:619. 10.1002/elps.201200424 [DOI] [PubMed] [Google Scholar]
- 22.1000 Genomes Project Consortium, Abecasis GR, Altshuler D, et al. A map of human genome variation from population-scale sequencing. Nature 2010;467:1061-73. Erratum in: Nature. 2011;473:544. [DOI] [PMC free article] [PubMed]
- 23.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature 2013;500:415-21. Erratum in: Nature 2013;502:258. 10.1038/nature12477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alexandrov LB, Ju YS, Haase K, et al. Mutational signatures associated with tobacco smoking in human cancer. Science 2016;354:618-22. 10.1126/science.aag0299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hayward NK, Wilmott JS, Waddell N, et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017;545:175-80. 10.1038/nature22071 [DOI] [PubMed] [Google Scholar]
- 26.Wong SQ, Li J, Tan AY, et al. Sequence artefacts in a prospective series of formalin-fixed tumours tested for mutations in hotspot regions by massively parallel sequencing. BMC Med Genomics 2014;7:23. 10.1186/1755-8794-7-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lawrence MS, Stojanov P, Polak P, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013;499:214-8. 10.1038/nature12213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hellmann MD, Ciuleanu TE, Pluzanski A, et al. Nivolumab plus Ipilimumab in Lung Cancer with a High Tumor Mutational Burden. N Engl J Med 2018;378:2093-104. 10.1056/NEJMoa1801946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zehir A, Benayed R, Shah RH, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med 2017;23:703-13. Erratum in: Nat Med 2017;23:1004. 10.1038/nm.4333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chalmers ZR, Connelly CF, Fabrizio D, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med 2017;9:34. 10.1186/s13073-017-0424-2 [DOI] [PMC free article] [PubMed] [Google Scholar]