

Abstract
Intramolecular hydrogen-atom transfer is an established approach for the site-specific functionalization of unactivated, aliphatic C–H bonds. Transformations using this strategy typically require unstable intermediates formed using strong oxidants and have mainly targeted C–H halogenations or intramolecular aminations. Herein, we report a site-specific C–H functionalization that significantly increases the synthetic scope and convergency of reactions proceeding via intramolecular hydrogen atom transfer. Stable, isolable N-dithiocarbamates are used as precursors to amidyl radicals formed via either light or radical initiation to efficiently deliver highly versatile alkyl dithiocarbamates across a wide range of complex structures.
Keywords: radical reactions, hydrogen atom transfer, C–H functionalization, synthetic methods, regioselectivity
Graphical Abstract
We report a site-specific C–H functionalization that significantly increases the synthetic scope and convergency of reactions proceeding via intramolecular hydrogen atom transfer. Stable, isolable dithiocarbamates are used as precursors to amidyl radicals formed via either light or radical initiation to efficiently deliver highly versatile alkyl dithiocarbamates across a wide range of complex structures.
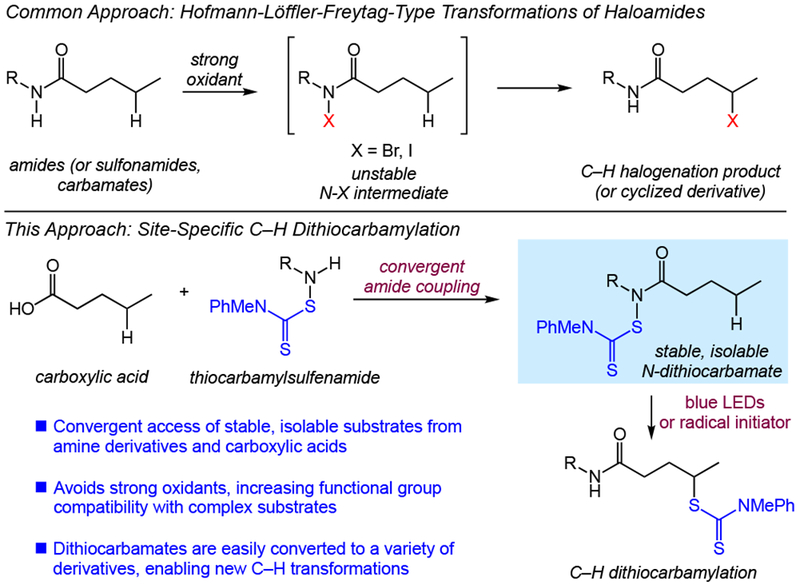
Site-specific C–H functionalization using intramolecular strategies holds significant promise in the synthesis and late-stage derivatization of complex structures.[1] The venerable Hofmann-Löffler-Freytag (HLF) reaction and related variants are prototypical examples of transformations of this type that proceed via intramolecular hydrogen atom transfer of reactive nitrogen-centered radicals.[2,3] With few exceptions,[3b,4] a major limitation of this approach in complex synthesis is the use of strong oxidants to generate unstable halogenated N–X derivatives.[5] Moreover, the marked reliance on C–H halogenation (or subsequent intramolecular aminations) in these processes ultimately has limited the synthetic scope and value of these reactions.[2,3,5]
Prior efforts from our laboratory have demonstrated the utility of functionalized amides to achieve site-selective, intermolecular C–H functionalizations.[6,7] In particular, we recently reported a C–H xanthylation as a synthetic platform unlocking a diverse array of aliphatic C–H transformations.[7] We hypothesized that a site-specific, intramolecular variant of this transformation would offer significant advantages over standard HLF-type reactions as the impressive synthetic versatility of dithiocarbonyl products would allow access to a diverse range of derivatives via both polar and radical processes. As such, we considered a convergent approach involving coupling of a suitable functionalized amine and a carboxylic acid (Figure 1). In this way, stable, isolable N-dithiocarbonyl substrates would be prepared by standard amide coupling in contrast to highly reactive N-halo derivatives that would require exposing complex substrates to strong oxidants. We anticipated this aspect to be a major advantage in applications involving highly functionalized, bioactive small molecules.
Figure 1.
Approaches to intramolecular, site-specific C–H functionalizations using nitrogen-centered radicals.
From the outset, we anticipated that this strategy would require a different dithiocarbonyl derivative from the xanthate used in the intermolecular C–H functionalization,[7] as amine and xanthate functionality are generally incompatible with each other owing to the electrophilicity of the thiocarbonyl group resulting in rapid degradation of the xanthate.[8,9] We thus considered a less electrophilic dithiocarbonyl derivative–a dithiocarbamate–as the transfer group for our studies. Notably, the dithiocarbamate group is also a useful pharmacophore with roles in enzyme catalysis, protein folding, and redox signaling and regulation.[10] The strong affinity of the dithiocarbamate for metal ions also confers antitumor and enzyme inhibitory properties.[11] Overall, these properties make C–H dithiocarbamylation itself a highly desirable reaction in the context of drug discovery and development.
The functionalized amine coupling partners in these studies were easily accessed from primary amines by a reaction with dilute NaOCl followed by addition of a dithiocarbamate salt.[12] While this approach dictates that the functionality present on the amine coupling partner is stable to NaOCl, this is a milder oxidant with better functional group compatibility[13] than reagents typically used to generate N-halo intermediates for HLF-type reactions (e.g., in situ generated AcOI).[2] Standard amide couplings of the thiocarbamylsulfenamides with carboxylic acids then enabled a convergent synthesis of the N-dithiocarbamate substrates under mild conditions.[14] In contrast to the N-halo derivatives commonly used in classical HLF reactions, these substrates are both light and air stable and may be isolated and purified by silica gel chromatography and stored for future use, as desired. Following a survey of different N-dithiocarbamates, we elected to use methylaniline dithiocarbamate derivatives owing to the higher rate of group transfer as compared to other dithiocarbamates.[15]
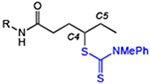
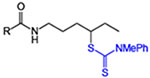
We began with simple aliphatic substrates to identify suitable conditions for the C–H functionalization. Both irradiation with blue LEDs under ambient conditions as well as standard radical initiation using 10 mol % dicumyl peroxide at 120 °C under an inert atmosphere provided the desired products efficiently (Table 1). A range of different N-alkyl hexanamides worked well in the reaction (entries 1–4), with the nPr amide substrate for 3 providing the 1,6-functionalization isomer as a minor product.[16] With suitable methylene C–H sites on both the N- and C-termini of the substrate (entry 4), functionalization on the carboxyl side chain was slightly favored. Site-specific functionalization of the amine coupling partner was also facile under these conditions (entries 5–6), which is a unique feature of our approach as compared to standard HLF-type transformations that often require specific N-substitution (e.g., Ts).[2–5] The C–H dithiocarbamylation is not limited to methylene functionalization, as primary C–H functionalization was successful (entries 7–8) in contrast to related reactions that require an electron-withdrawing group on nitrogen to achieve primary functionalization.[3c,17] Cyclic substrates also reacted efficiently in a site-specific manner to deliver products bearing 1,2- or 1,3-substitution in good yields (entries 9–10). In most cases, comparable yields were obtained with either mode of initiation.
Table 1.
Site-specific C–H dithiocarbamylation of simple substrates.[a]
| entry | product | blue LEDs yield (%)[b] | radical initiator yield (%)[c] |
|---|---|---|---|
 |
|||
| 1 | 1: R = iPr | 91 | 73 |
| 2 | 2: R = tBu | 61 | 56 |
| 3 | 3: R = nPr | 79 (3:1 C4:C5) | 85 (3:1 C4:C5) |
| 4 | 4: R = nHex | 87 (2.5:1 rr)[d] | 87 (1.6:1 rr)[d] |
 |
|||
| 5 | 5: R = Ph | 82 | 82 |
| 6 | 6: R = CF3 | 78 | 86 |
| 7 | 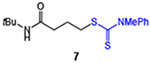 |
65 | 76 |
| 8 | 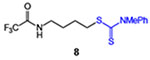 |
21 (35% brsm) | 52 |
| 9 | 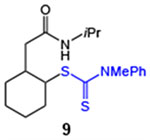 |
89 (2:1 dr) | 79 (2:1 dr) |
| 10 | 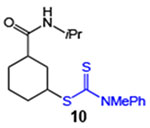 |
81 (2:1 dr) | 87 (2:1 dr) |
Isolated yields.
Reactions were performed with blue LEDs in PhCF3 under ambient conditions for 14 h under Ar
Reactions were performed in PhCl at 120 °C with 10 mol % dicumyl peroxide for 14 h under Ar
Regioisomeric mixture resulting from 1,5-functionalization of the amide sidechain.
The site specificity of this intramolecular C–H functionalization offers important benefits over our prior work involving intermolecular C–H xanthylation (Figure 2).[7] For example, while the intermolecular C–H xanthylation of linear substrates favors functionalization at the most distal methylene site, a mixture of regioisomers is still observed. In cases where complete site specificity is desired, the present approach provides a useful alternative. In addition, this intramolecular protocol may be used to provide a completely different regioisomer as compared to the intermolecular C–H xanthylation. The intermolecular functionalization of a norleucine derivative, for instance, favors the distal methylene site; using the N-dithiocarbamate derivative instead provides the ω−2 product shown as the sole isomer. We anticipate that the complementary nature of these two strategies will constitute general, broadly applicable C–H functionalizations in synthesis.
Figure 2.
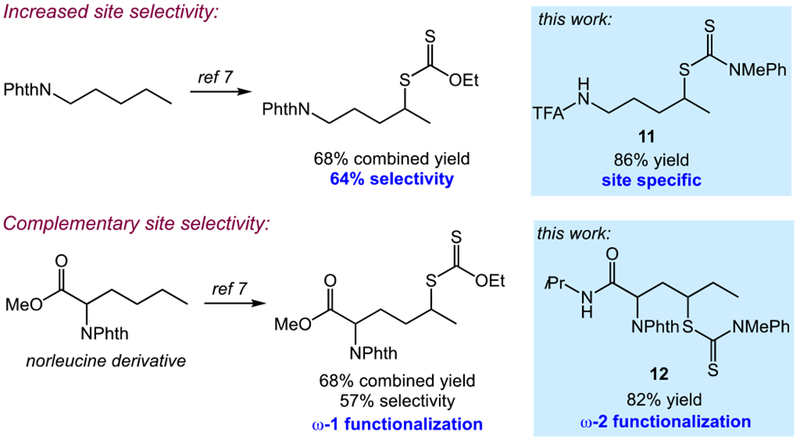
Comparing the C–H dithiocarbamylation to previous work involving intermolecular C–H xanthylation. Reactions were performed with blue LEDs in PhCF3 under ambient conditions for 14 h.
In light of the mild reaction conditions and synthetic convergency of our approach, we anticipate powerful applications in the C–H functionalization of complex molecules. As such, we applied the C–H dithiocarbamylation to a diverse set of complex natural product and drug derivatives to test the limits of the transformation (Figure 3). The reaction of the N-dithiocarbamate derived from rimantadine delivered 13 as the sole product in good yield, with no functionalization of the adamantyl core that is the major product of intermolecular C–H functionalization of related substrates.[6a,6b,7,18] A gemfibrozil derived substrate afforded 14, demonstrating successful C–H dithiocarbamylation in the presence of an electron-rich arene prone to react with the strong oxidants common to HLF-type reactions. The C–H functionalization of substrates derived from the amino acids norleucine and valine provided products 12 and 15, respectively, with functionalizations at either secondary or primary C–H sites, providing avenues for unnatural amino acid synthesis.[19] Reaction of the N-dithiocarbamate derived from the clotting agent tranexamic acid proceeded in high yield to deliver functionalization at the C–H site adjacent to the amino functionality of the cyclohexane core.
Figure 3.
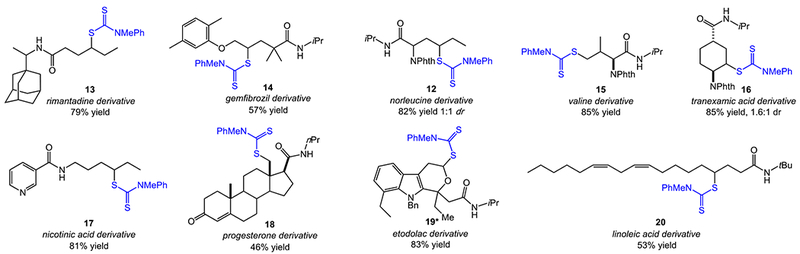
Site-specific C–H dithiocarbamylations of complex natural product and pharmaceutical derivatives. Reactions were performed with blue LEDs in PhCF3 under ambient conditions for 12-14 h under Ar. *Reaction was performed in PhCl at 120 °C with 10 mol % dicumyl peroxide for 14 h under Ar.
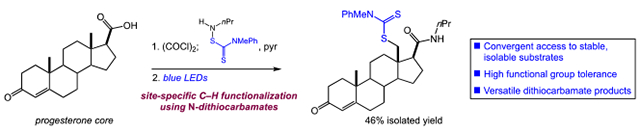
The C–H functionalization of complex molecules containing Lewis-basic heterocycles or substrate unsaturation using HLF chemistry–or other intermolecular C–H functionalizations–remains a major challenge owing to the strongly oxidizing reagents or intermediates involved.[20] The advantages of the current approach in the functionalization of such substrates are clear from the synthesis of products 17-20. The substrate derived from nicotinic acid delivered 17 site-specifically in good yield. The sensitive enone functionality of progesterone was also tolerated, permitting the primary C–H functionalization of the axial methyl group in proximity to the N-dithiocarbamate functionality (18). The N-dithiocarbamate derivative of NSAID etodolac provided 19 with 1,6-functionalization adjacent to the oxygen atom of the tetrahydropyran ring instead of 1,5-functionalization of the pendant ethyl group. Finally, we viewed linoleic acid as a highly challenging substrate for C–H functionalization with its sensitive skipped diene. In the event, site-specific functionalization of the unsaturated sidechain was successful, albeit in moderate yield (20). This example clearly highlights the unique functional group compatibility of our approach, as the addition of electrophilic oxidants would surely lead to undesired side products.
We found the versatility of the dithiocarbamate group to be similar to that of the xanthate group, with a wide variety of C–H transformations easily achieved via subsequent radical or polar processes (Figure 4).[21] For example, radical mediated allylation, deuteration,[21b] and azide transfer provide valuable C–H diversification products in good yields. Chugaev-type elimination of the alkyl dithiocarbamate provides formal dehydrogenation product 24 in nearly quantitative yield.[21c] Facile cleavage of the dithiocarbamate also enables site-specific C–H thiolation in good yield.[21d] Additional functional group transformations are easily envisioned.[7]
Figure 4.
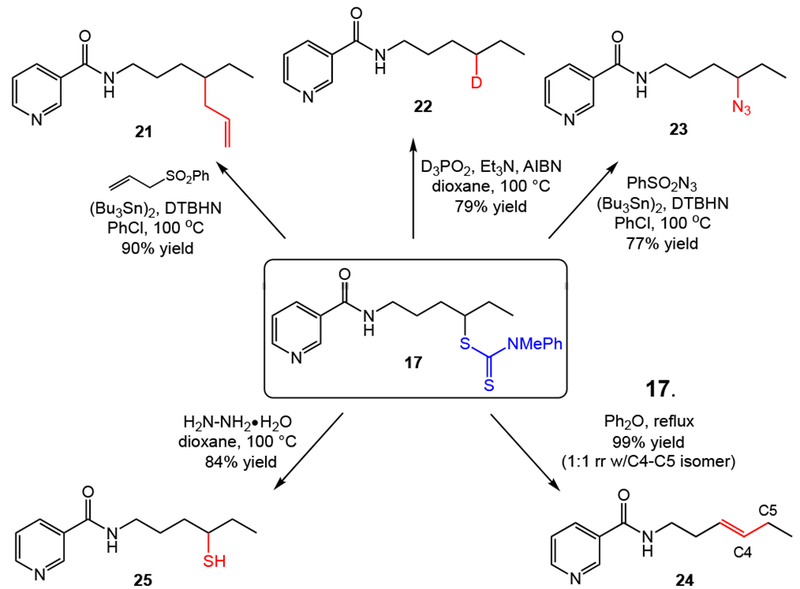
C–H diversification via polar and radical functional group interconversions of the nicotinic acid dithiocarbamate product 17.
In summary, we have developed a general, convergent approach to site-specific C–H functionalization via hydrogen atom transfer using N-dithiocarbamates. This general process is applicable to the diversification of both primary and secondary C–H sites of highly functionalized complex natural products and drug derivatives. We expect these studies to significantly expand the synthetic breadth of C–H functionalizations involving intramolecular hydrogen atom transfer owing to the versatility of the alkyl dithiocarbamate products for accessing a range of C–H transformations. We anticipate this strategy will find widespread use in the synthesis and late-stage modification of complex molecules.
Supplementary Material
Acknowledgements
Financial support was provided by Award R01 GM 120163 from the National Institute of General Medical Sciences. C.G.N. thanks the NSF for a Graduate Research Fellowhip and the Graduate School for a Royster Fellowship.
References
- [1].a) W. R. Gutekunst, P. S. Baran, Chem. Soc. Rev 2011, 40, 1976;21298176 [Google Scholar]; b) J. Yamaguchi, A. D. Yamaguchi, K. Itami, Angew. Chem. Int. Ed 2012,51,8960 [Google Scholar]; c) J. C. K. Chu, T. Rovis, Angew. Chem. Int. Ed 2018, 57, 62. [Google Scholar]
- [2].a) S. Chiba, H. Chen, Org. Biomol. Chem 2014, 12, 4051. [DOI] [PubMed] [Google Scholar]; b) L. M. Stateman, K. M. Nakafuku, D. A. Nagib Synthesis 2018, 50, 156929755145 [Google Scholar]; c) D. Šakić, H. Zipse, Adv. Synth. Catal 2016, 358, 3983. [Google Scholar]
- [3].For recent examples of HLF-type reactions, see [Google Scholar]; a) C. Q. O’Broin, P. Fernández, C. Martínez, K. Muñiz, Org. Lett 2016,18,436;26782641 [Google Scholar]; b) E. A. Wappes, S. C. Fosu, T. C. Chopko, D. A. Nagib, Angew. Chem.Int.Ed 2016,55,9974 [Google Scholar]; c) J. Richers, M. Heilmann, M. Drees, K. Tiefenbacher, Org. Lett 2016,18,6472. [DOI] [PubMed] [Google Scholar]; d) T. Liu, M. C. Myers, J.-Q. Yu, Angew. Chem. Int. Ed 2017, 56, 306 [Google Scholar]; e) P. Becker, T. Duhamel, J. C. Stein, M. Reiher, K. Muñiz, Angew. Chem. Int. Ed 2017, 56, 8004 [Google Scholar]; f) W. Shu, C. Nevado, Angew. Chem. Int. Ed 2017, 56, 1881 [Google Scholar]; g) S. Sathyamoorthi, S. Banerjee, J. Du Bois, N. Z. Burns, R. N. Zare, Chem. Sci 2018, 9, 10029629078 [Google Scholar]; h) M. A. Short, J. M. Blackburn, J. L. Roizen, Angew. Chem. Int. Ed 2018,57,296 [Google Scholar]; i) E. A. Wappes, A. Vanitcha, D. A. Nagib, Chem. Sci 2018, 9, 450029896392 [Google Scholar]; j) D. Chang, R. Zhao, C. Wei, Y. Yao, Y. Liu, L. Shi, J. Org. Chem 2018,83,330529484885 [Google Scholar]; k) D. Zhang, H. Wang, H. Cheng, J. G. Hernández, C. Bolm, Adv. Synth. Catal 2017, 359, 4274. [Google Scholar]
- [4].a) G. J. Choi, Q. Zhu, D. C. Miller, C. J. Gu, R. R. Knowles, Nature 2016,539,26827732585 [Google Scholar]; b) J. C. K. Chu, T. Rovis, Nature 2016, 539, 27227732580 [Google Scholar]; c) D.-F. Chen, J. C. K. Chu, T. Rovis, J. Am. Chem. Soc 2017, 139, 14897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].For select examples, see [Google Scholar]; a) P. S. Baran, K. Chen, J. M. Richter, J. Am. Chem. Soc 2008, 130, 7247. [DOI] [PubMed] [Google Scholar]; b) R. Fan, D. Pu, F. Wen, J. Wu, J. Org. Chem 2007, 72, 8994; [DOI] [PubMed] [Google Scholar]; c) L. R. Reddy, B. V. S. Reddy, E. J. Corey, Org. Lett 2005, 8, 2819 [Google Scholar]; d) C. G Francisco, A. J. Herrera, E. Suárez, J. Org. Chem 2003, 68, 1012.12558429 [Google Scholar]
- [6].a) V. A. Schmidt, R. K. Quinn, A. T. Brusoe, E. J. Alexanian, J. Am. Chem. Soc 2014, 136, 1438925232995 [Google Scholar]; b) R. K. Quinn, Z. A. Könst, S. E. Michalak, Y. Schmidt, A. R. Szklarski, A. R. Flores, S. Nam, D. A. Horne, C. D. Vanderwal, E. J. Alexanian, J. Am. Chem. Soc 2016, 138, 69626694767 [Google Scholar]; c) A. M. Carestia, D. Ravelli, E. J. Alexanian, Chem. Sci 2018, published online 5/14/18. [Google Scholar]
- [7].Czaplyski WL, Na CG, Alexanian EJ, J. Am. Chem. Soc 2016, 138, 13854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].For a stepwise synthesis of N-(O-ethyl thiocarbonylsulfanyl)amides with limited stability from amides using strong base, see: Gagosz, C. Moutrille, S. Z. Zard, Org. Lett 2002, 4, 2707. [DOI] [PubMed] [Google Scholar]
- [9].Le Neindre M, Magny B, Nicolaÿ R, Polym. Chem 2013, 4, 5577. [Google Scholar]
- [10].For a review on dithiocarbamates in medicinal chemistry, see: N. Lal, J. Chem. Bio. Interfaces 2014, 4, 321. [Google Scholar]
- [11].a) L. Ronconi, C. Marzano, P. Zanello, M. Corsini, G. Miolo, C. Maccà, A. Trevisan, D. Regona, J. Med. Chem 2006, 49, 1648;16509581 [Google Scholar]; b) B. Czek, V. Milacic, J. Taraba, Q. P. Dou, J. Med. Chem 2008, 51, 6256; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) H. Khan, A. Badshah, G. Murtaz, M. Said, Z. Rehman, C. Neuhausen, M. Todorova, B. J. Jean-Claude, I. S. Butler, Eur. J. Med. Chem 2011, 46, 4071;21724304 [Google Scholar]; d) P. Gaspari, T. Banerjee, W. P. Malachowski, A. J. Muller, G. C. Prendergast, J. DuHadaway, S. Bennett, A. M. Donovan, J. Med. Chem 2006, 49, 684.16420054 [Google Scholar]
- [12].Smith GE Jr, Alliger G, Carr EL, Young KC, J. Org. Chem 1949, 14, 935. [Google Scholar]
- [13].Galvin JM, Jacobsen EN, Palucki M, Frederick MO, e-EROS 2013, DOI: 10.1002/047084289X.rs084.pub3, and references cited therein. [Google Scholar]
- [14].If desired, the thiocarbamylsulfenamide may be used in crude form in the substrate synthesis. For example, N-((methyl(phenyl)carbamothioyl)thio)-N-isopropylhexanamide (the substrate used in Table 1, entry 1) was prepared using crude N-(((isopropylamino)thio)carbonothioyl)-N-methylaniline (0.48 g, 2.0 mmol, 2.0 equiv), hexanoyl chloride (0.14 mL, 1.0 mmol, 1.0 equiv), and pyridine (81 μL, 1.0 mmol, 1.0 equiv) in CH2Cl2 (4.0 mL) to give the title compound in good yield (0.29 g, 87% yield). See Supporting Information for detailed experimental procedures.
- [15].See Supporting Information for further details.
- [16].The same ratio of products was obtained at lower concentrations (0.1 M), suggesting that the minor 1,6-product is not the result of background intermolecular functionalization.
- [17].a) Q. Qin, S. Yu, Org. Lett 2015, 17, 1894; [DOI] [PubMed] [Google Scholar]; b) N. R. Paz, D. Rodríguez-Sosa, H. Valdés, R. Marticorena, D. Melián, M. B. Copano, C. C. González, A. J. Herrera, Org. Lett 2015,17,2370;25932748 [Google Scholar]; c) C. Martínez, K. Muñiz, Angew. Chem. Int. Ed 2015, 54, 8287. [Google Scholar]
- [18].a) A. A. Fokin, P. T. Schreiner, Adv. Synth. Catal 2003, 345,1035; [Google Scholar]; b) A. Sharma, J. F. Hartwig, Nature 2015, 517, 600; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) X. Huang, T. M. Bergsten, J. T. Groves, J. Am. Chem. Soc 2015,137,5300; [DOI] [PubMed] [Google Scholar]; d) K. A. Margrey, W. L Czaplyski, D. A. Nicewicz, E. J. Alexanian, J. Am. Chem. Soc 2018, 140, 4213.29522330 [Google Scholar]
- [19].Noisier AFM, Brimble MA, Chem. Rev 2014, 114, 8775. [DOI] [PubMed] [Google Scholar]
- [20].a) T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal, S. W. Krska, Chem. Soc. Rev 2016, 45, 546;26507237 [Google Scholar]; b) J. Wencel-Delord, F. Glorius, Nat. Chem 2013, 5, 369.23609086 [Google Scholar]
- [21].a) R. S. Grainger, E. J. Welsh, Angew. Chem. Int. Ed 2007,46,5377; [Google Scholar]; b) C. McMaster, R. N. Bream, R. S. Grainger, Org. Biomol. Chem 2012,10,4752; [DOI] [PubMed] [Google Scholar]; c) M. Betou, L. Male, J. W. Steed, R. S. Grainger, Chem. Eur. J 2014,20,6505;24711140 [Google Scholar]; d) B. Shakya, P. N. Yadav, J. Ueda, S. Awale, Bioorg. Med. Chem. Lett 2014, 24, 458. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.