

Abstract
Background
Coccidiosis is a major contributor to losses in poultry production. With emerging constraints on the use of in-feed prophylactic anticoccidial drugs and the relatively high costs of effective vaccines, there are commercial incentives to breed chickens with greater resistance to this important production disease. To identify phenotypic biomarkers that are associated with the production impacts of coccidiosis, and to assess their covariance and heritability, 942 Cobb500 commercial broilers were subjected to a defined challenge with Eimeria tenella (Houghton). Three traits were measured: weight gain (WG) during the period of infection, caecal lesion score (CLS) post mortem, and the level of a serum biomarker of intestinal inflammation, i.e. circulating interleukin 10 (IL-10), measured at the height of the infection.
Results
Phenotypic analysis of the challenged chicken cohort revealed a significant positive correlation between CLS and IL-10, with significant negative correlations of both these traits with WG. Eigenanalysis of phenotypic covariances between measured traits revealed three distinct eigenvectors. Trait weightings of the first eigenvector, (EV1, eigenvalue = 59%), were biologically interpreted as representing a response of birds that were susceptible to infection, with low WG, high CLS and high IL-10. Similarly, the second eigenvector represented infection resilience/resistance (EV2, 22%; high WG, low CLS and high IL-10), and the third eigenvector tolerance (EV3, 19%; high WG, high CLS and low IL-10), respectively. Genome-wide association studies (GWAS) identified two SNPs that were associated with WG at the suggestive level.
Conclusions
Eigenanalysis separated the phenotypic impact of a defined challenge with E. tenella on WG, caecal inflammation/pathology, and production of IL-10 into three major eigenvectors, indicating that the susceptibility-resistance axis is not a single continuous quantitative trait. The SNPs identified by the GWAS for body weight were located in close proximity to two genes that are involved in innate immunity (FAM96B and RRAD).
Electronic supplementary material
The online version of this article (10.1186/s12711-018-0433-7) contains supplementary material, which is available to authorized users.
Background
In chickens, seven species of Eimeria (Apicomplexa, Coccidia) are responsible for the debilitating and sometimes fatal disease coccidiosis that is estimated to cost the international poultry industry around US$3 billion per year, mainly due to reduced productivity and the cost of preventive measures [1, 2]. Current control of coccidiosis relies on the prophylactic use of synthetic or fermented ionophore anticoccidial drugs and on vaccination with formulations of live wild-type or attenuated parasites [3–5]. Use of some anticoccidial drugs has been curtailed by legislation in many countries, while the costs and limited production capacity of live attenuated vaccines compromise their utility in broiler flocks [6]. Thus, identification of traits that may contribute towards selective breeding of chickens to control the consequences of coccidiosis is of great importance to industry.
In principle, the impact of Eimeria infection could be mitigated by selecting chickens that limit their infectious load by being refractory to parasite infection (resistance), by tolerating the consequences of infection (tolerance), or by recovering from pathological consequences of infection sufficiently quickly to maintain their growth and body condition (resilience). Resistance, resilience and tolerance to disease are distinct, individual-specific traits that are likely to have a different genetic basis [7, 8]. However, phenotypic distinction between these three traits is difficult without a longitudinal measurement of pathogen load [9–11]. Nevertheless, there is evidence for genetic differences among chicken populations in their response to Eimeria parasitism. For example, some native chicken breeds, such as the Egyptian Fayoumi, appear to tolerate the pathological impacts of Eimeria infection [12, 13], while distinct inbred lines of White Leghorn chickens support variable levels of parasite replication by different Eimeria species [14]. Biological pathways that may be indicative of resistance to Eimeria maxima infection have been noted in modern commercial broilers [15, 16]. One genome-wide association study (GWAS) [16] alluded to the mechanisms of resistance to E. maxima including primary innate immune response, tissue repair, and proliferation. Definitions of the immune categories used in this study are presented in Fig. 1 [4, 7, 11, 17, 18].
Fig. 1.

Definitions of immune categories used
Eimeria species that infect chickens have life cycles of 4 to 14 days in vivo that include self-limiting phases of asexual (schizogony) and sexual (gametogony) reproduction. E. tenella causes haemorrhagic coccidiosis in chickens, with morbidity and mortality being highest during maturation and rupture of the second-generation schizonts in the caeca, starting from − 108 h (4.5 days) post-infection [19]. The caecal lesions caused are sometimes exacerbated by bacterial co-infection and E. tenella is therefore associated with local induction of a wide range of pro- and anti-inflammatory cytokines [20]. Selection of chickens with intrinsically high or low levels of pro-inflammatory cytokines and chemokines in response to E. tenella (i.e. IL-6, CXCLi2, and CCLi2) has revealed an association of innate immune response with resistance to the pathology that is associated with coccidial infections [21].
Induction of the anti-inflammatory and immunosuppressive cytokine, interleukin-10 (IL-10), was first characterised in the spleen and intestine of chickens of inbred White Leghorn lines infected with E. maxima [22]. Both constitutive and inducible levels of IL-10 mRNA were higher in “disease-susceptible” than in “disease-resistant” lines. Later, enhanced expression of IL-10 mRNA was shown in the caeca of broilers infected with E. tenella [23]. Monoclonal antibodies and an ELISA (enzyme-linked immunosorbent assay) test to detect IL-10 in chicken blood were recently developed and, as in mice, IL-10 was shown to be an inhibitor of both innate and acquired immune responses, with a marked elevation in chickens challenged with E. tenella [24]. This finding agrees with the proposed specific function of IL-10 in immune regulation in the intestine, based upon spontaneous colitis that develops in IL-10 knockout mice [25].
Mechanisms of innate immunity have been alluded to in many previous studies that investigated genetic resistance to coccidiosis [10, 13, 16, 19, 22, 26, 27]. However, those exploring response to E. tenella were performed using either microsatellite or low-density single nucleotide polymorphism (SNP) panels to map quantitative trait loci (QTL) in inbred chicken lines [13, 27]. A previous large-scale GWAS using the Cobb500 investigated a different spp. (E. maxima) with a commercially available SNP panel [17]. In this study, our aim was to investigate the relationship between circulating IL-10 (IL-10) as a quantitative trait and two standard phenotypic measures of E. tenella infection: weight gain (WG) and caecal lesion score (CLS), to assess the heritability of each trait and to identify candidate genetic markers that impact the outcome of coccidial challenge. We hypothesized a positive relationship of circulating IL-10 with disease pathology (CLS), and negative relationships of both these traits with WG, and the identification of significant SNPs that correlate with these traits.
Methods
Animals and management
In this study, 1200 1-day old infectious bronchitis-vaccinated (Nobilis IB H120, MSD Animal Health, Milton Keynes, UK) commercial four-way crossbred Cobb500 broiler chickens (Gallus gallus domesticus) were acquired from a major UK supplier. These comprised three replicate intakes of 375 (intake1, September 2016), 376 (intake2, October 2016) and 391 (intake3, November 2016) individuals at the time of infection. At the conclusion of the trial, 1142 animals were available for sampling. For each intake, chicks were initially housed together in a coccidia-free environment at a DEFRA (Department for Environment Food & Rural Affairs) approved stocking density of 34 kg/m2 (anticipated end weight) under a UK Home Office project licence. Up to day 10, chickens received a starter feed that contained the anticoccidial drug Maxiban® (Elanco; Greenfield, Indiana, USA; 31.25 g/kg feed). On day 11, an anticoccidial-free grower diet was introduced, and dust-extracted pinewood shaving bedding (StableBed, Essex, UK) was replaced to diminish the risk that anticoccidial residues in the bedding affected future experimental procedures. Consistent with standard commercial broiler rearing management, the grower diet was replaced with finisher pellets on day 25 (Target Feeds Ltd, Shropshire, UK). The light:dark ratio and temperature were changed according to commercial broiler rearing practice.
Experimental design
The design of the study is summarised in Fig. 2. To facilitate animal management, at day 19 each chick intake was randomly divided into two separately housed equally-sized groups, referred to as replicates ‘a’ and ‘b’, that were handled on consecutive days (day1 and day2). At d-2 post-infection (pi), birds were wing tagged for individual identification and weighed (WT1, WT for body weight) to the nearest 0.01 g using digital scales (Kern CXB 3K0.2, KERN & SOHN GmbH, Balingen, Germany). Following WT1, birds were randomly assigned to control (intake1, n = 100; intake2, n = 50; and intake3, n = 50) or infection (intake1, n = 275; intake2, n = 326; and intake3, n = 341) within-replicate groups. On d0pi, control birds received a unique 1.0 mL inoculum of DNase/RNase free H2O. The remaining birds were infected via oral gavage with a unique 1.0 mL inoculation of E. tenella (Houghton strain) sporulated oocysts. Individuals in intakes1, 3 received 42,500 2-month old oocysts, while those in intake2 received 40,000 1-month old oocysts. Oocyst sporulation was verified by light microscopy. Birds assigned to replicate ‘a’ received inoculations on day 21, while those assigned to replicate ‘b’ were inoculated on day 22, i.e. replicate ‘b’ gained an additional day of infection-free growth. Infected and control birds were housed in separate rooms, thus allowing both sufficient space to meet commercial environmental conditions and minimising the risk of accidental infection of the control birds with Eimeria parasites.
Fig. 2.

Experimental design
At d4.5pi (108 h), all birds were weighed (WT2) and a 0.5-mL blood sample was collected from the brachial vein. Blood was immediately transferred to a 1.5 mL microcentrifuge tube (Sigma-Aldrich, Dorset, UK), allowed to clot overnight at 4 °C, and centrifuged at 2000 × g/10 min. Then, the serum was aspirated into a sterile microcentrifuge tube and stored at − 20 °C, for IL-10 assay at a later date. Circulating IL-10 was measured by Capture ELISA according to [24], using ROS-AV164 and biotinylated ROS-AV163, as capture and detection antibodies, respectively.
At d6.5pi (156 h), birds were culled by cervical dislocation and weighed post-mortem (WT3). Both caeca were removed and scored individually for lesion damage [19] by the same experienced operator throughout the entire study. For DNA isolation, a 1-cm section of the base of each caecal pair was transferred to a 7 mL Sterilin™ bijou tube (ThermoFisher Scientific, Waltham, MA, USA) that contained 5–10 volumes of RNAlater® at room temperature (Life Technologies, Carlsbad, CA, USA), according to the manufacturer’s instructions. The DNeasy® Blood and Tissue Kit (QIAGEN, Hilden, Germany) was used to isolate total genomic DNA (gDNA) from each caecal tissue sample, following the manufacturer’s instructions. DNA extracts were stored at − 20 °C. A total of 942 DNA samples were available from infected chickens and were genotyped by GeneSeek, a Neogen Company [28] using a proprietary 62K SNP array (Cobb-Vantress, Arkansas, USA).
Phenotypic statistical analyses
Three phenotypic traits were measured: body weight (WT, g) at time-points d2pi (WT1), d4.5pi (WT2) and d6.5pi (WT3); circulating IL-10 (pg/mL) at d4.5pi, and caecal lesion score (CLS, on a scale of 0-4) post-mortem (d6.5pi). The derived trait, weight gain (WG = WT3–WT1) was also considered. Since the pathology of the two caeca from a bird may differ, CLS were allocated per bird according to the higher score for each caecal pair. A natural log-transformation was applied to IL-10 for it to conform to a Gaussian distribution. The same transformation did not improve the negative distribution skew of CLS, thus these data were not adjusted. Formal testing of differences in weight traits and in the three phenotypic traits of interest (WG, CLS and IL-10) between control and infected birds was carried out in ASReml 4.0 [29] using a simple linear univariate model Eq. (1) to generate predicted means:
| 1 |
where is the vector of observations, is the design matrix relating fixed effects to observations, is the vector of fixed effects, and ε is the vector of residual effects. Fixed effects included intake (a three-level factor), trial replicate (a two-level management within intake factor, accounting for the 1-day handling age difference: ‘a’ and ‘b’), sex (male or female), and all significant interaction terms. Weight at d-2pi (WT1) was fitted as a covariate in all analyses of weight traits except WT1. To ascertain the most parsimonious model for each phenotypic trait, fixed effects were formally tested and removed from the individual trait model if their significance was above the 5% threshold based on a conditional Wald F-test.
All three traits of interest were subsequently rescaled (mean-centred/standard deviation) to adjust for differences in units of measurements. Using ASReml 4.0 [29] and referring to the univariate models for the assignment of significant fixed effects to each trait, a multivariate linear model was used to estimate the phenotypic variance–covariance matrix () among traits, i.e. no genetic effect was fitted at this stage. Likelihood ratio tests (LRT) were used to determine the significance of trait (co)variances by comparing the full multiple trait model with a model that constrained trait covariances to zero. This approach avoided differences in environmental variance (or measurement error) that can generate spurious support for correlation significance.
An eigen decomposition was applied to P and the percentage of variance and trait loadings for each eigenvector (EV) was derived. Trait loadings for each EV were plotted on a histogram to facilitate interpretation of the between-trait correlations, following [30].
Genetic statistical analyses
Genotyping the 942 infected individuals resulted in 62,732 SNPs located on 28 autosomes, the W and Z sex chromosomes, and four unplaced scaffolds. GenABEL [31] was used to construct a genetic relationship matrix (GRM) based on the SNP genotypes and used in a univariate linear mixed model for each trait (using ASReml 4.0 [29]) to estimate variance components and genetic parameters Eq. (2):
| 2 |
where , , and ε are defined as in Eq. (1), is the vector of additive genetic effects, and is a design matrix relating random effects to observations. Weight at d-2pi was included as a covariate for WG trait. Heritability of each trait was calculated as the ratio of additive genetic and phenotypic variance. Genetic correlations between traits were estimated by bivariate analyses using Model (2).
For GWAS, quality control removed individuals and SNPs that failed according to the following criteria: minor allele frequency lower than 0.05 to distinguish common polymorphisms from rare variants); call rate lower than 90%; cut off individual call-rate lower than 90%; identity by state threshold less than 1; Hardy–Weinberg equilibrium P 10−6. Thus, 921 individuals and 46,836 SNPs remained for further analyses. Classical multidimensional scaling of the SNP genotypes was conducted to confirm the genetic homogeneity of the sample before analysis. Since no obvious substructure was detected (see Additional file 1: Figure S1), we did not consider it in further analyses.
A separate GWAS was performed for each trait in GenABEL [32], fitting sex, intake, and replicate as fixed effects and the GRM as a polygenic effect to correct for genetic relationships. The −log10 P value from the Wald test was corrected for possible inflation of lambda [32]. Thresholds for genome-wide (P ≤ 0.05) and suggestive (i.e. accounting for one false discovery per genome-scan; significance levels were calculated using the Bonferroni correction for multiple-testing, i.e. −Log10 (0.05/total number of SNPs) and −Log10 (1.0/total number of SNPs), respectively. To determine the effects associated with significant SNPs identified in the GWAS, single-marker association analyses were carried out using ASReml v 4.1, as in Eq. (2). Predicted trait values for each SNP genotype, represented by , and , and SNP allele frequencies and were used to estimate the additive (a = (AA − BB)/2) and dominance (d = AB − [(AA + BB)/2] effects and the variance at the SNP (], assuming Hardy–Weinberg equilibrium genotype frequencies. The heritability of each SNP was then calculated as divided by the of the trait.
To identify candidate genes, the significant SNPs identified by GWAS were converted from the Gallus-gallus-4.0 to the Gallus-gallus-5.0 assembly using the LiftOver tool [33] Then, genes located 250 kb up- and down-stream were annotated using the data mining tool BioMart [34].
Results
Data from the six replicates (2 replicates for 3 intakes) were pooled according to the assigned infection status. The numbers of birds falling into each category are summarised in Table S1 (see Additional file 2: Table S1).
Phenotypic analyses
Body weight and weight gain
Significant differences were found between the unadjusted weights of control and infected birds data (Fig. 3). Univariate analysis of mean-adjusted phenotypic data revealed significant differences (P < 0.001) between control and infected chickens for body weight at d-2pi (WT1), d4.5pi (WT2), and d6.5pi (WT3) and for weight gain (Fig. 4a; see Additional file 3: Table S2). Mean weights and mean weight gains were also significantly (P < 0.001) different between the three experimental intakes (see Additional file 3: Table S2). Males were significantly heavier than females for all measures (P < 0.001; Fig. 4b; see Additional file 3: Table S2).
Fig. 3.
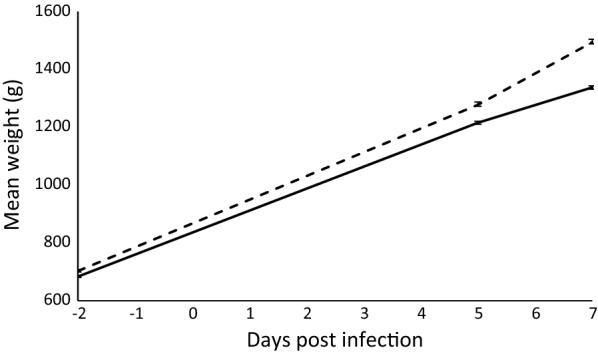
Mean weight (g) of control (n = 200, dashed-line) and infected (n = 942, solid-line) birds throughout the trial period, with standard error bars. See also (Additional file 2: Table S2) for full details
Fig. 4.

Box plot of body weight for a control and infected birds and b the two sexes at d6.5pi
Relationships between WG, CLS, and serum IL-10
No caecal pathology was detected in the control chickens (CLS score = 0). Phenotypic variance in CLS was observed in infected chickens, with the full spectrum of scores (0–4) observed across both sexes (Fig. 5a). Both intake and replicate explained some of the variance in CLS (P < 0.001; see Additional file 3: Table S2).
Fig. 5.
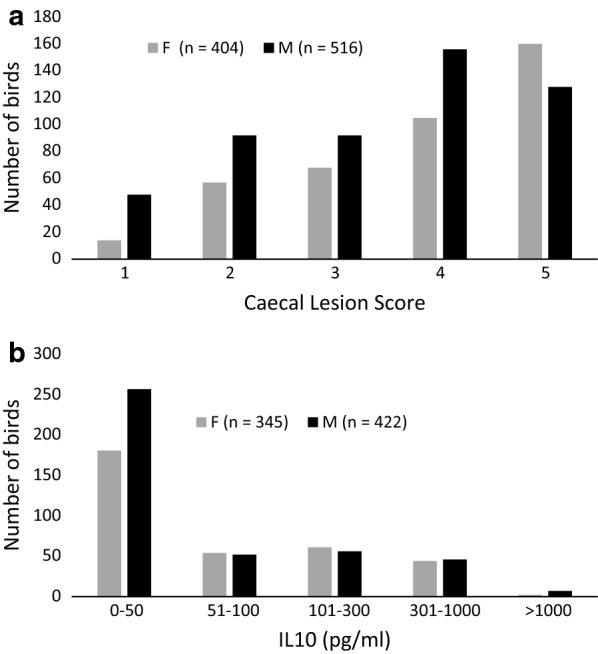
Distribution of a caecal lesion score and b IL-10 for male (M) and female (F) infected birds
Serum IL-10 concentrations in the control chickens were below the assay limit of detection but were elevated to varying extents in birds challenged with Eimeria, as previously described [22]. The difference between male and female infected chickens in the production of IL-10 was not significant (P = 0.54; Fig. 5b; and (see Additional file 3: Table S2).
A linear model multivariate analysis was used to estimate phenotypic correlations () between the three measured traits (P < 0.001, Table 1). WG was negatively correlated with both CLS (− 0.37 ± 0.03) and IL-10 (− 0.35 ± 0.03), while the correlation between CLS and IL-10 was positive (0.39 ± 0.03; Fig. 6). Overall, birds with high CLS tended to have high IL-10 and low WG.
Table 1.
Estimates of the phenotypic variances, covariances, and correlations produced by multivariate mixed model analysis
| WG | CLS | IL-10 | |
|---|---|---|---|
| WG | 0.679 (0.032) | − 0.366 (0.029) | − 0.350 (0.032) |
| CLS | − 0.291 (0.028) | 0.928 (0.043) | 0.393 (0.030) |
| IL-10 | − 0.275 (0.030) | 0.361 (0.035) | 0.903 (0.046) |
Between-trait covariances are below the diagonal (italics), phenotypic trait variances are on the diagonal (bold), with between-trait correlations above the diagonal. Measured traits are weight gain (WG), caecal lesion score (CLS) and serum interleukin 10 (IL-10). Standard errors (± SE) are presented in parentheses
Fig. 6.
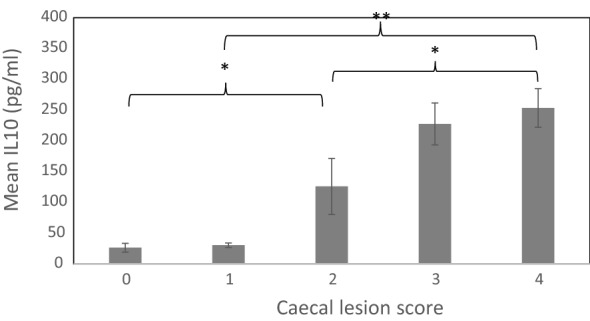
Visualisation of the relationship between caecal lesion score and IL-10 (pg/ml) in infected birds. *P < 0.05; **P < 0.001
Using the univariate linear mixed model (Eq. 2), genetic variance and heritability were evident for WG and CLS but not for IL-10. Only the heritability estimate for WG was significantly different from zero (0.19 ± 0.09; Table 2). The multivariate linear mixed model and the bivariate models that included IL-10 as a response variable did converge, likely due to the absence of detectable genetic variance for IL-10. The estimate of the genetic correlation between WG and CLS was negative and significant (− 0.94 ± 0.32).
Table 2.
Suggestively significant SNPs with their GalGal 5.0 location (chromosome and position in base pairs) and P values for each of the measured phenotypic traits
| SNP | Location Chr: bp | P value | trait (SE) | trait (SE) | Minor allele frequency | SNP | (SE) | P () | (SE) | P () |
|---|---|---|---|---|---|---|---|---|---|---|
| WG_1 | 11:4,244,594 | 1.34 × 10−5 | 0.191 (0.093) | 0.683 (0.033) | 0.43 | 0.026 | 21.05 (4.99) | < 0.001 | − 6.37 (6.37) | 0.241 |
| WG_2 | 11:11,383,252 | 1.49 × 10−5 | 0.42 | 0.026 | 22.42 (5.00) | < 0.001 | 3.46 (6.46) | 0.346 | ||
| CLS_1 | 12:17,993,212 | 4.62 × 10−5 | 0.071 (0.083) | 0.930 (0.044) | 0.33 | 0.001 | 0.19 (0.08) | 0.019 | 0.054 (0.094) | 0.337 |
| IL-10_1 | 4:7,575,042 | 2.763 × 10−5 | 0 (0) | 0.921 (0.047) | 0.28 | – | – | – | – | – |
Univariate linear mixed models performed using Eq. 2 (ASReml 4.0), provided the trait phenotypic variance (; means-adjusted) and trait heritability () with standard errors (SE). The non-means-centered additive () and dominance () effects of the SNPs are presented with their significance values (P). SNP heritabilities ( SNP) were calculated as additive variance of the SNP divided by of the trait. Major () and minor () SNP allele frequencies are also presented. Non-estimable values for the IL-10 SNP are indicated (−)
Eigen analysis
The modest phenotypic correlations between traits were investigated further using an eigen decomposition of the phenotypic covariance (Fig. 7). Trait weightings of the first eigenvector, (EV1, eigenvalue = 59%), were low WG, high CLS and high IL-10, and likely represent a susceptible spectrum, as defined in Fig. 1. In these birds, it is assumed that the low WG is a consequence of the parasite-induced caecal pathology. The second and third eigenvectors (EV2, 22%; high WG, low CLS and high IL-10, and EV3, 19%, high WG, high CLS and low IL-10) separate birds that may be considered as resistant/resilient or tolerant, respectively. The birds with high values for EV2 may produce an effective immune response, including production of IL-10, that either prevents pathogen invasion/replication or mitigates the pathology, i.e. they are either resistant or resilient; these alternatives cannot be distinguished. The birds with high values for EV3 gain weight despite caecal pathology and the lack of an innate immune response (low IL-10), i.e. they are phenotypically tolerant. Plotting the trait loadings for each EV, as in Fig. 7, enabled a visualisation of the multivariate distribution of partly overlapping immune categories which include susceptible, resilient/resistant and tolerant.
Fig. 7.

Eigenvector decomposition of the phenotypic covariance matrix. Vector loadings for the three measured traits [weight gain (WG), caecal lesion score (CLS) and serum interleukin-10 (IL-10)] are presented in standard deviation units on the y-axis. The percentage of variance explained by each eigen vector (EV) is in brackets on the x-axis
Genotypic analysis
Association analysis
Following quality control of the data, 921 of the 942 infected birds were available to detect significant associations between SNPs and the analysed traits. After Bonferroni correction for multiple testing and adjustment for the test inflation factor, two suggestive SNPs were identified for the derived trait, WG, during the period of infection. Both were located on chromosome 11 (Table 2; Fig. 8a). These SNPs were 7.76 Mb apart (i.e. at opposite ends of the microchromosome) and not in linkage disequilibrium (LD). The strongest association with CLS was observed for a SNP on chromosome 12 (Table 2; Fig. 8b). However, after correction, this association was not significant. Lastly, one SNP was associated with IL-10 level at the height of infection, on chromosome 4 (Table 2; Fig. 8c). Given the lack of detectable genetic variance for this trait, this is likely a false positive. Corresponding quantile–quantile (q–q) plots for the three GWAS analyses are in Fig. 6a, b and c. Small additive effects (P < 0.003) were estimated for the significant SNPs, (Table 2). Estimates of dominance effects were not significant. The effect of the significant SNPs were also found to be significant (P < 0.001) in the univariate linear mixed models.
Fig. 8.

Manhattan and corresponding QQ plots from the GWAS for: a weight gain; b caecal lesion score; c serum IL-10. The log10 P value is plotted for each SNP on the relevant chromosome (x-axis). Bonferroni corrected thresholds were set as P ≤ 1.1×10−6 and P ≤ 2.1×10−5 for genome-wide (P ≤ 0.05) and suggestive (i.e. one false discovery per genome-scan) levels, corresponding to −log10 P values of 5.97 (blue) and 4.67 (red) lines
Discussion
The objective of this study was to estimate the variance and investigate the genomic architecture of chicken response to E. tenella infection, using the Cobb500 commercial broiler. GWAS of the three analysed phenotypic traits revealed the presence of suggestive significant SNPs for body weight gain (WG) but not for caecal lesions or IL-10. Significant genetic variance and heritability was detected for WG only. For CLS both these estimates had large standard errors. We could not detect genetic variance for IL-10, and the reasons for this may be complex. In addition, it was clear that IL-10 production was not strongly correlated with caecal pathology. A future study will focus on the relationship between quantified pathogen load and circulating IL-10 to determine whether IL-10 production is an independently heritable trait. Based on estimates of variance and the extensive heterozygosity of the four-way crossbred population, it is now clear that our study was underpowered to identify the genetic control of the response to Eimeria challenge. The Cobb500 genotype was selected deliberately to establish whether substantial phenotypic variation exists in response of commercial broiler chickens following a defined infection with E. tenella, the cause of caecal coccidiosis. The phenotypic results indicate that this is clearly the case. We were, however, unable to estimate heritabilities that were significantly different from zero, but it is unclear if this was a result of the four-way cross or that variance in CLS and IL-10 has no underlying genetic basis. Further investigation is required to determine if the assays used here could be applied to pedigree lines to select for the desired phenotype(s).
Innate immune response is highly conserved across chordates and IL-10 is known to be critical for resistance to gastrointestinal parasite infections in mice [35]. As noted above, selection for enhanced production of pro-inflammatory cytokines has been reported to increase resistance to E. tenella [21] and increased expression of these cytokines in the intestinal mucosa of birds infected with E. acervulina has been documented e.g. [36]. However, the published studies have assessed neither the utility of these responses as biomarkers, nor their variance on a population scale in commercial chicken lines. Here, we focused on the anti-inflammatory cytokine IL-10 that was previously reported to be significantly elevated in the serum of E. tenella challenged birds [24]. Although the function of IL-10 in host defence in Eimeria infection is unclear, there is evidence that it contributes to the pathology in some manner, as the use of oral antibodies against IL-10 has been reported to mitigate the growth rate suppression that occurs in broiler chickens infected with Eimeria [37, 38]. Another reason for our focus on IL-10 was to identify a quantifiable phenotype in the early stage of disease challenge that could provide a biomarker for genetic selection.
Quantification of individual disease severity was assessed post-mortem by scoring caecal lesions, with males exhibiting, on average, less damage compared to females. Interestingly, a previous study [39] found that significantly more male embryos survived infection with E. tenella, suggesting that innate coccidiosis resistance differs between sexes. In line with our hypothesis that birds with high WG have, on average, lower CLS and IL-10, the estimated phenotypic correlations of WG with CLS and IL-10 were negative (− 0.37 ± 0.03 and − 0.35 ± 0.03, respectively), indicating that birds failed to thrive due to, at least in part, the direct effects of E. tenella infection. These phenotypic correlation estimates are consistent with other reports e.g. [40] (− 0.14), [41] (only a negative direction is indicated) and [42]. The positive phenotypic correlation between CLS and IL-10 (0.39 ± 0.03) was consistent with the relationship between susceptibility and high IL-10 mRNA production in mucosa among inbred chicken lines [22]. A weakness of our study is that circulating IL-10 was measured at a single time-point only. In that sense, the 0.39 phenotypic correlation with caecal lesion score is remarkable.
We concentrated on the use of eigenvalue decomposition of the phenotypic variance–covariance matrix among traits, rather than principal components because the eigenvector trait loadings provide population evidence for variance in the immune categories described in Fig. 1 [43]. If phenotypic immune response was a simple susceptible/resistant axis, then all the variance should be explained by the first eigenvector. However, this was not the case, with 41% of the variance shared between eigenvectors 2 and 3.
Trait loadings for EV1 reflected the estimated between-trait correlation directions that are indicative of susceptibility: i.e. the ability/inability of a host to control pathogen invasion or replication, resulting in a lack of weight gain, a high level of pathology and a strong response from the innate immune system. In the most affected birds, a high level of circulating IL-10 is likely to be the consequence of pathology and local immune responses, and is consistent with the variation observed among inbred birds that differ in resistance [22]. Certainly, E. tenella infection impairs normal caecal function such as fermentation of complex carbohydrates, nitrogen absorption, and water retention. In field situations, haemorrhagic disease induced by E. tenella causes diarrhoea, leaves birds weak and dehydrated with reduced appetites and the damaged intestine susceptible to secondary infection by opportunistic intestinal bacteria. EV2 displayed no loading for WG, and a negative loading on CLS, thus indicating that part of the variation in CLS is independent of WG. Nevertheless, the high IL-10 loading for EV2 indicates that there was a strong response to the pathogen, and we interpret this vector to be indicative of resistance/resilience. Finally, the EV3 trait loadings displayed severe pathology while simultaneously retaining growth performance although the loading on IL-10 was very low, thus our biological interpretation is that this eigenvector represents a tolerant phenotype.
We speculate that elevation of IL-10 could induce systemic immunosuppression that may underlie the observed negative correlation of serum antibody titres against bacterial and viral pathogens with parasite load in field studies of native birds in Africa [42]. In mammalian species, there is strong evidence that polymorphisms in the IL-10 and IL-10 receptor genes are responsible for susceptibility to gastrointestinal inflammatory disease [44].
As noted above, the population used in this study may not be ideal for a genetic analysis due to its four-way cross construction. Other studies that reported significant SNP associations with WG and Eimeria infection used larger populations of the same broiler [16] and/or different population structures [13, 15, 45–47]. Two suggestive SNPs on chromosome 11 were associated with body weight. Putative candidate genes FAM96B (family with sequence similarity 96 member B) and RRAD (Ras related glycolysis inhibitor and calcium channel regulator) for these SNPs are associated with gastrointestinal and metabolic diseases, and with the development of the digestive system and disorder networks, [48–50].
A separate study that used Cobb500 broilers infected with the small intestine parasite E. maxima, found several significant QTL for disease resistance but none of these were identified in our study [16]. This could be due to one or more of the many differences between these two studies, including those relating to the pathology and immune responses induced by E maxima versus E. tenella, the phenotypes measured, sample size, sex ratio, age of birds at infection, and density of SNP information available. Keeping the birds for a further 7 days may have elucidated the difference between resistance and resilience by enabling measurement of the extent of recovery, but it would have precluded collection of CLS that must be scored between 5 and 7 days post-infection [19] since pathological lesions resolve after this time. Two previous QTL mapping studies on resistance to E. tenella infection used a Fayoumi White Leghorn intercross rather than a commercial line, measured phenotypes that, apart from WG, could not be compared to our study, [13, 27]. Only one of these studies [27] located QTL on chromosomes 4 and 11 but none of these were located in similar regions or were linked to phenotypes of the suggestive SNPs found our study.
Resilient birds are attractive to industry since they are able to overcome disease, acquire immunity, and return to their original healthy condition. However, pathogens can mutate rapidly to overcome host resistance that can lead to a selective advantage of mutants in resistant individuals. It is unclear whether tolerance would be a desirable production phenotype without information on other commercially important traits such as feed conversion efficiency. Genetic change of the pathogen may be smaller if it can complete its life cycle in tolerant birds [51], but birds that can tolerate disease could also be “super-shedders” that drive environmental oocyst numbers upward. IL-10 production may be beneficial or detrimental to the host. Other studies have suggested that IL-10 is directly involved in the reduced body weight gain observed in Eimeria-infected birds [37, 38]. The existence of birds that produce high levels of IL-10 but gain weight normally argues against such a direct role.
We anticipate extending our studies to pedigree lines where a simpler genetic relationship matrix can be established, and to natural infection, over a longer time course, where we can also measure the trajectory of circulating IL-10, pathogen load, shedding, and feed conversion efficiency. There is also a need to test whether circulating IL-10 is specific to the parasite infection or could provide a more general indication of intestinal health.
Conclusions
We have confirmed the existence of substantial variance for weight gain in birds infected with E. tenella, a trait that is used traditionally as a measure of resilience/resistance to coccidiosis caused by this pathogen. Our data indicate that breeding for increased weight gain under challenge alone would not distinguish between resilience/resistance and tolerance. Eigen analysis of the phenotypic variance–covariance structure among weight gain, caecal inflammation/pathology and production of IL-10 separated the phenotypic impact of the defined challenge with E. tenella into three major eigenvectors, indicating that the susceptibility-resistance axis in this population is complex. We were unable to demonstrate significant genetic basis for the variation in CLS and IL-10. However, suggestive SNPs identified by the GWAS for body weight were located in close proximity with two genes known to be involved in innate immunity (FAM96B and RRAD).
Additional files
Additional file 1: Figure S1. Plot of population substructure, as explored by classical multidimensional scaling using GenABEL.
Additional file 2: Table S1. Number of birds used in this 3 × 2 × 2 × 2 design by category:-Intake (1, 2, 3), disease status (control or infected), replicate (a, b) and sex.
Additional file 3: Table S2. Univariate linear model analyses performed in ASReml 4.0 were used to predict the means (PM) of the fixed effects on each of the measured traits, with standard errors (± SE) and significance (P value).
Authors’ contributions
PK, SB, DB and FT conceived, designed and secured funding for this study; MN, DB and KB designed the experimental trials; MN, KH, DB, and KH undertook the experimental work; ZW performed IL-10 assays; KB performed statistical analyses with input from KW, GWAS with input from VR and AP, and prepared the manuscript with input from MN, VR, AP, DB, DH, and FT. VR, DH, DB, AP, MN, and FT assisted with interpretation of results. RH and MA supplied advice on the study, birds for the trials and genotyping. All authors read and approved the final manuscript.
Acknowledgements
We are grateful to Cobb-Vantress Inc. for their continuing involvement and enthusiasm in our work. We also thank the staff of the Biological Services Unit at RVC for their support during our studies. Thanks also to Alice Vismarra, Iván Pastor Fernández, Tatiana Küster, Sarah Macdonald, Virginia Marugán-Hernández, James Pritchard, Lucy Freem, Jenny Geddes, and Rakhi Harne, who all assisted with sample collection; Enrique Sanchez-Molano and Oswald Matika for help and advice with the GWAS; Ricardo Pong-Wong for help with quantitative genetics; Andrea Doeschl-Wilson for lively discussions on immune categories and eigen analysis. Finally, we would like to thank two anonymous reviewers for their comments that have helped in shaping this manuscript.
Stephen C. Bishop, Pete Kaiser: Deceased.
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The authors declare that they have no competing interests.
Availability of data and materials
Raw genotypes and SNP details from the proprietary 62K SNP array (Cobb-Vantress) are not available for public release, with the exception of the suggestive SNPs uncovered in this study. The dataset used for mixed model analysis is available from the public repository DRYAD (https://datadryad.org/).
Ethics approval
These trials were conducted under the Home Office Project Licence in accordance with Home Office regulations under the Animals (Scientific Procedures) Act 1986 and the guidelines set down by the RVC Animal Welfare and Ethics Review Body. Throughout the study, birds were checked twice daily for welfare issues. In addition, the frequency of observations during the 96- to 128-h post-infection period, corresponding to the most pathogenic part of the parasite’s life cycle, was increased to every hour.
Funding
This study was funded by BBSRC via Animal Research Club (ARC) Grants (BB/L004046 and BB/L004003). Commercial birds and genotyping were provided by Cobb-Vantress Inc. The funding bodies did not contribute to the design of the study, sample collection, analysis, interpretation of data, or in writing the manuscript.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Kay Boulton, Email: kay.boulton@roslin.ed.ac.uk.
Matthew J. Nolan, Email: mnolan@rvc.ac.uk
Zhiguang Wu, Email: Zhiguang.wu@roslin.ed.ac.uk.
Androniki Psifidi, Email: apfisidi@rvc.ac.uk.
Valentina Riggio, Email: valentina.riggio@roslin.ed.ac.uk.
Kimberley Harman, Email: kharman@rvc.ac.uk.
Mitchell S. Abrahamsen, Email: Mitchell.Abrahamsen@cobb-vantress.com
Rachel Hawken, Email: Rachel.Hawken@cobb-vantress.com.
Kellie A. Watson, Email: kellie.watson@ed.ac.uk
Fiona M. Tomley, Email: ftomley@rvc.ac.uk
Damer P. Blake, Email: dblake@rvc.ac.uk
David A. Hume, Email: david.hume@uq.edu.au
References
- 1.Shirley M, Smith A, Tomley F. The biology of avian Eimeria with an emphasis on their control by vaccination. Adv Parasitol. 2005;60:285–330. doi: 10.1016/S0065-308X(05)60005-X. [DOI] [PubMed] [Google Scholar]
- 2.Williams RB. A compartmentalised model for the estimation of the cost of coccidiosis to the world’s chicken production industry. Int J Parasitol. 1999;29:1209–1229. doi: 10.1016/S0020-7519(99)00086-7. [DOI] [PubMed] [Google Scholar]
- 3.Crouch CF, Andrews SJ, Ward RG, Francis MJ. Protective efficacy of a live attenuated anti-coccidial vaccine administered to 1-day-old chickens. Avian Pathol. 2003;32:297–304. doi: 10.1080/10307945031000097912. [DOI] [PubMed] [Google Scholar]
- 4.Doeschl-Wilson AB, Villanueva B, Kyriazakis I. The first step toward genetic selection for host tolerance to infectious pathogens: obtaining the tolerance phenotype through group estimates. Front Genet. 2012;3:265. doi: 10.3389/fgene.2012.00265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McDonald V, Shirley MW. Past and future: vaccination against Eimeria. Parasitology. 2009;136:1477–1489. doi: 10.1017/S0031182009006349. [DOI] [PubMed] [Google Scholar]
- 6.Shivaramaiah C, Barta JR, Hernandez-Velasco X, Téllez G, Hargis B. Coccidiosis: recent advancements in the immunobiology of Eimeria species, preventive measures, and the importance of vaccination as a control tool against these Apicomplexan parasites. Vet Med. 2014;5:23–34. doi: 10.2147/VMRR.S57839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bishop SC, Woolliams JA. Genomics and disease resistance studies in livestock. Livest Sci. 2014;166:190–198. doi: 10.1016/j.livsci.2014.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Warren HS, Fitting C, Hoff E, Adib-Conquy M, Beasley-Topliffe L, Tesini B, et al. Resilience to bacterial infection: difference between species could be due to proteins in serum. J Infect Dis. 2010;201:223–232. doi: 10.1086/649557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kause A. Genetic analysis of tolerance to infections using random regressions: a simulation study. Genet Res. 2011;93:291–302. doi: 10.1017/S0016672311000176. [DOI] [PubMed] [Google Scholar]
- 10.Kause A, van Dalen S, Bovenhuis H. Genetics of ascites resistance and tolerance in chicken: a random regression approach. G3 (Bethesda) 2012;2:527–535. doi: 10.1534/g3.112.002311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mulder HA, Rashidi H. Selection on resilience improves disease resistance and tolerance to infections. J Anim Sci. 2017;95:3346–3358. doi: 10.2527/jas.2017.1479. [DOI] [PubMed] [Google Scholar]
- 12.Borrmann E, Berndt A, Hänel I, Köhler H. Campylobacter-induced interleukin-8 responses in human intestinal epithelial cells and primary intestinal chick cells. Vet Microbiol. 2007;124:115–124. doi: 10.1016/j.vetmic.2007.04.041. [DOI] [PubMed] [Google Scholar]
- 13.Pinard-van der Laan MH, Bed’hom B, Coville JL, Pitel F, Feve K, Leroux S, et al. Microsatellite mapping of QTLs affecting resistance to coccidiosis (Eimeria tenella) in a Fayoumi × White Leghorn cross. BMC Genomics. 2009;10:31. doi: 10.1186/1471-2164-10-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bumstead N, Millard B. Genetics of resistance to coccidiosis: Response of inbred chicken lines to infection by Eimeria tenella and Eimeria maxima. Br Poult Sci. 1987;28:705–715. doi: 10.1080/00071668708417006. [DOI] [PubMed] [Google Scholar]
- 15.Hamzic E, Bed’Hom B, Juin H, Hawken R, Abrahamsen MS, Elsen JM, et al. Large-scale investigation of the parameters in response to Eimeria maxima challenge in broilers. J Anim Sci. 2015;93:1830–1840. doi: 10.2527/jas.2014-8592. [DOI] [PubMed] [Google Scholar]
- 16.Hamzic E, Buitenhuis B, Herault F, Hawken R, Abrahamsen MS, Servin B, et al. Genome-wide association study and biological pathway analysis of the Eimeria maxima response in broilers. Genet Sel Evol. 2015;47:91. doi: 10.1186/s12711-015-0170-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bishop SC. A consideration of resistance and tolerance for ruminant nematode infections. Front Genet. 2012;3:168. doi: 10.3389/fgene.2012.00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lough G, Rashidi H, Kyriazakis I, Dekkers JCM, Hess A, Hess M, et al. Use of multi-trait and random regression models to identify genetic variation in tolerance to porcine reproductive and respiratory syndrome virus. Genet Sel Evol. 2017;49:37. doi: 10.1186/s12711-017-0312-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson J, Reid WM. Anticoccidial drugs: lesion scoring techniques in battery and floor-pen experiments with chickens. Exp Parasitol. 1970;28:30–36. doi: 10.1016/0014-4894(70)90063-9. [DOI] [PubMed] [Google Scholar]
- 20.Park SS, Lillehoj HS, Allen PC, Park DW, FitzCoy S, Bautista DA, et al. Immunopathology and cytokine responses in broiler chickens coinfected with Eimeria maxima and Clostridium perfringens with the use of an animal model of necrotic enteritis. Avian Dis. 2008;52:14–22. doi: 10.1637/7997-041707-Reg. [DOI] [PubMed] [Google Scholar]
- 21.Swaggerty CL, Pevzner IY, Kogut MH. Selection for pro-inflammatory mediators produces chickens more resistant to Eimeria tenella. Poult Sci. 2015;94:37–42. doi: 10.3382/ps/peu053. [DOI] [PubMed] [Google Scholar]
- 22.Rothwell L, Young JR, Zoorob R, Whittaker CA, Hesketh P, Archer A, et al. Cloning and characterization of chicken IL-10 and its role in the immune response to Eimeria maxima. J Immunol. 2004;173:2675–2682. doi: 10.4049/jimmunol.173.4.2675. [DOI] [PubMed] [Google Scholar]
- 23.Haritova AM, Stanilova SA. Enhanced expression of IL-10 in contrast to IL-12B mRNA in poultry with experimental coccidiosis. Exp Parasitol. 2012;132:378–382. doi: 10.1016/j.exppara.2012.08.017. [DOI] [PubMed] [Google Scholar]
- 24.Wu Z, Hu T, Rothwell L, Vervelde L, Kaiser P, Boulton K, et al. Analysis of the function of IL-10 in chickens using specific neutralising antibodies and a sensitive capture ELISA. Dev Comp Immunol. 2016;63:206–212. doi: 10.1016/j.dci.2016.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scheinin T, Butler DM, Salway F, Scallon B, Feldmann M. Validation of the interleukin-10 knockout mouse model of colitis: antitumour necrosis factor-antibodies suppress the progression of colitis. Clin Exp Immunol. 2003;133:38–43. doi: 10.1046/j.1365-2249.2003.02193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park H, Jacobsson L, Wahlberg P, Siegel P, Andersson L. QTL analysis of body composition and metabolic traits in an intercross between chicken lines divergently selected for growth. Physiol Genomics. 2006;25:216–223. doi: 10.1152/physiolgenomics.00113.2005. [DOI] [PubMed] [Google Scholar]
- 27.Bacciu N, Bed’Hom B, Filangi O, Rome H, Gourichon D, Reperant J, et al. QTL detection for coccidiosis (Eimeria tenella) resistance in a Fayoumi × Leghorn F-2 cross, using a medium-density SNP panel. Genet Sel Evol. 2014;46:14. doi: 10.1186/1297-9686-46-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu JJ, Lillehoj HS, Allen P, Van Tassell CP, Sonstegard TS, Cheng HH, et al. Mapping quantitative trait loci associated with resistance to coccidiosis and growth. Poult Sci. 2003;82:9–16. doi: 10.1093/ps/82.1.9. [DOI] [PubMed] [Google Scholar]
- 29.Gilmour AR, Gogel BJ, Cullis BR, Welham SJ, Thompson R. ASReml user guide release 4.1 structural specification. Hemel Hempstead: VSN International Ltd; 2015. [Google Scholar]
- 30.Boulton K, Couto E, Grimmer AJ, Earley RL, Canario AVM, Wilson AJ, et al. How integrated are behavioral and endocrine stress response traits? a repeated measures approach to testing the stress-coping style model. Ecol Evol. 2015;5:618–633. doi: 10.1002/ece3.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aulchenko YS, Ripke S, Isaacs A, Duijn CM. GenABEL: an R library for genome-wide association analysis. Bioinformatics. 2007;23:1294–1296. doi: 10.1093/bioinformatics/btm108. [DOI] [PubMed] [Google Scholar]
- 32.Amin N, van Duijn CM, Aulchenko YS. A genomic background based method for association analysis in related individuals. PLoS One. 2007;2:e1274. doi: 10.1371/journal.pone.0001274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Batch Coordinate Conversion (liftOver). http://genome.ucsc.edu/. Accessed 1 Sept 2018.
- 34.Ensemble. https://www.ensembl.org/biomart/martview/. Accessed 1 Sept 2018.
- 35.Schopf LR, Hoffmann KF, Cheever AW, Urban JF, Jr, Wynn TA. IL-10 is critical for host resistance and survival during gastrointestinal helminth infection. J Immunol. 2002;168:2383–2392. doi: 10.4049/jimmunol.168.5.2383. [DOI] [PubMed] [Google Scholar]
- 36.Yun CH, Lillehoj HS, Lillehoj EP. Intestinal immune responses to coccidiosis. Dev Comp Immunol. 2000;24:303–324. doi: 10.1016/S0145-305X(99)00080-4. [DOI] [PubMed] [Google Scholar]
- 37.Arendt MK, Sand JM, Marcone TM, Cook ME. Interleukin-10 neutralizing antibody for detection of intestinal luminal levels and as a dietary additive in Eimeria challenged broiler chicks. Poult Sci. 2016;95:430–438. doi: 10.3382/ps/pev365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sand JM, Arendt MK, Repasy A, Deniz G, Cook ME. Oral antibody to interleukin-10 reduces growth rate depression due to Eimeria spp. infection in broiler chickens. Poult Sci. 2016;95:439–446. doi: 10.3382/ps/pev352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jeffers TK, Wagenbach GE. Sex differences in embryonic response to Eimeria tenella infection. J Parasitol. 1969;55:949–951. doi: 10.2307/3277156. [DOI] [PubMed] [Google Scholar]
- 40.Mathis GF, Washburn KW, McDougald LR. Genetic variability of resistance to Eimeria acervulina and E. tenella in chickens. Theor Appl Genet. 1984;68:385–389. doi: 10.1007/BF00254803. [DOI] [PubMed] [Google Scholar]
- 41.Conway DP, McKenzie ME, Dayton AD. Relationship of coccidial lesion scores and weight gain in infections of Eimeria acervulina, E. maxima and E. tenella in broilers. Avian Pathol. 1990;19:489–496. doi: 10.1080/03079459008418702. [DOI] [PubMed] [Google Scholar]
- 42.Psifidi A, Banos G, Matika O, Desta T, Bettridge J, Hume D, et al. Genome-wide association studies of immune, disease and production traits in indigenous chicken ecotypes. Genet Sel Evol. 2016;48:74. doi: 10.1186/s12711-016-0252-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2:e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Engelhardt KR, Grimbacher B. IL-10 in humans: lessons from the gut, IL-10/IL-10 receptor deficiencies, and IL-10 polymorphisms. Curr Top Microbiol Immunol. 2014;380:1–18. doi: 10.1007/978-3-662-43492-5_1. [DOI] [PubMed] [Google Scholar]
- 45.Demeure O, Hamzic E, Juin H, Naciri M, Juul-Madsen HR, Okimoto R, et al. Investigation of immune response to Eimeria maxima in broilers. Scand J Immunol. 2013;77:276–277. [Google Scholar]
- 46.Hamzic E, Bed’Hom B, Juin H, Hawken R, Abrahamsen M, Elsen J, et al. Plasma components as traits for resistance to coccidiosis in chicken. In 10th world congress on genetics applied to livestock production. 17–22 August 2014, Vancouver; 2014.
- 47.Pinard MH, Hamet N, Thomas M, Monvoisin JL, Pery P. Genetic resistance to coccidiosis (Eimeria tenella) in various lines of outbred chickens. Anim Genet. 1994;25:5. doi: 10.1111/j.1365-2052.1994.tb00397.x. [DOI] [PubMed] [Google Scholar]
- 48.Stehling O, Mascarenhas J, Vashisht AA, Sheftel AD, Niggemeyer B, Rösser R, et al. Human CIA2A (FAM96A) and CIA2B (FAM96B) integrate maturation of different subsets of cytosolic-nuclear iron-sulfur proteins and iron homeostasis. Cell Metab. 2013;18:187–198. doi: 10.1016/j.cmet.2013.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weerapana E, Wang C, Simon GM, Richter F, Khare S, Dillon MBD, et al. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature. 2010;468:790–795. doi: 10.1038/nature09472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Genatlas. http://genatlas.medecine.univ-paris5.fr/fiche.php?onglet=3&n=30808. Accessed 25 Oct 2017.
- 51.Bishop SC, Stear MJ. Modeling of host genetics and resistance to infectious diseases: understanding and controlling nematode infections. Vet Parasitol. 2003;115:147–166. doi: 10.1016/S0304-4017(03)00204-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure S1. Plot of population substructure, as explored by classical multidimensional scaling using GenABEL.
Additional file 2: Table S1. Number of birds used in this 3 × 2 × 2 × 2 design by category:-Intake (1, 2, 3), disease status (control or infected), replicate (a, b) and sex.
Additional file 3: Table S2. Univariate linear model analyses performed in ASReml 4.0 were used to predict the means (PM) of the fixed effects on each of the measured traits, with standard errors (± SE) and significance (P value).
Data Availability Statement
Raw genotypes and SNP details from the proprietary 62K SNP array (Cobb-Vantress) are not available for public release, with the exception of the suggestive SNPs uncovered in this study. The dataset used for mixed model analysis is available from the public repository DRYAD (https://datadryad.org/).