

Abstract
Idiopathic pulmonary fibrosis (IPF) is an incurable disease of the lung that is characterized by excessive deposition of extracellular matrix (ECM), resulting in disruption of normal lung function. The signals regulating fibrosis include both transforming growth factor beta (TGF-β) and tissue rigidity and a major signaling pathway implicated in fibrosis involves activation of the GTPase RhoA. During studies exploring how elevated RhoA activity is sustained in IPF, we discovered that not only is RhoA activated by profibrotic stimuli but also that the expression of Rnd3, a major antagonist of RhoA activity, and the activity of p190RhoGAP (p190), a Rnd3 effector, are both suppressed in IPF fibroblasts. Restoration of Rnd3 levels in IPF fibroblasts results in an increase in p190 activity, a decrease in RhoA activity and a decrease in the overall fibrotic phenotype. We also find that treatment with IPF drugs nintedanib and pirfenidone decreases the fibrotic phenotype and RhoA activity through up-regulation of Rnd3 expression and p190 activity. These data provide evidence for a pathway in IPF where fibroblasts down-regulate Rnd3 levels and p190 activity to enhance RhoA activity and drive the fibrotic phenotype.
INTRODUCTION
Idiopathic pulmonary fibrosis (IPF) is a progressive lethal lung disease of unknown cause. In the United States, IPF affects 150,000–200,000 people and causes 40,000 deaths per year (Raghu et al., 2014). The disease is characterized by patchy fibrotic areas with persistent fibroblast and myofibroblast proliferation and excess deposition of extracellular matrix (ECM) that results in a permanent distortion of lung architecture and a progressive irreversible loss of lung function (King, Jr. et al., 2001).
IPF is categorized as a fibroproliferative disorder characterized by an aberrant wound healing response (Raghu et al., 2011). Myofibroblasts function transiently during wound healing where they deposit and remodel ECM and exert mechanical tension to restore tissue integrity (Gabbiani et al., 1971; Tomasek et al., 2002). Myofibroblast function is temporally and spatially regulated in normal acute wounds but not in pathogenic fibrotic conditions. The persistence of myofibroblasts, long past when they are needed, is a well-established feature of fibrosis. These cells are key effectors in IPF, and targeting myofibroblasts is an important therapeutic option (Eickelberg and Laurent, 2010). Although the precise mechanism through which the persistent myofibroblast phenotype is acquired remains unclear, transforming growth factor beta (TGF-β) and mechanical stiffness have been identified as key factors (Vaughan et al., 2000; Hinz, 2010).
The mechanical properties of the fibrotic lung play a pivotal role in regulating the fibrotic phenotype. The excess ECM deposition observed during IPF results in the production of stiff scar tissue that obstructs normal lung function (Hinz, 2015). Substrates with the enhanced stiffness of fibrotic tissues have been shown to drive healthy cells toward a fibrogenic phenotype (Hinz, 2010; Saums et al., 2014). Conversely, softer substrates that mimic normal tissue stiffness can suppress the fibrotic phenotype in a number of cell types, including lung fibroblasts (Balestrini et al., 2012).
TGF-β is the most potent profibrotic cytokine known and the main chemical factor in myofibroblast differentiation (Hinz et al., 2007). Interestingly, a mechanical mechanism of TGF-β activation has been described whereby integrin contractile forces on the matrix release active TGF-β (Wipff et al., 2007; Wipff and Hinz, 2008). These findings suggest a model where the combination of TGF-β and matrix stiffness could engage in a feed forward signaling loop (Marinkovic et al., 2012). The enhanced ECM deposition by the myofibroblasts leads to the production of a stiffer matrix with a higher mechanical load when compared with healthy ECM produced by fibroblasts (Klingberg et al., 2014). This mechanically stiffer ECM, in turn, leads to more efficient activation of TGF-β, and the increased levels of TGF-β further enhance myofibroblast differentiation leading to positive feedback that sustains the profibrotic environment of the lung. Conversely, healthy fibroblasts produce softer matrices which fail to promote TGF-β activation, prevent the feed-forward loop, and maintain the compliant lung matrix (Marinkovic et al., 2012). It seems then that the combination of both the chemical and mechanical signals provide linked cues to drive cells into a disease state (Shimbori et al., 2013).
While there is strong evidence to suggest that the intersection of TGF-β and mechanical signaling pathways are key regulators of the IPF phenotype, there is still a major gap in understanding the signaling events that occur downstream of these pathways. Interestingly, the TGF-β and mechanical signals seem to converge on RhoA, a key regulator of cell contractility and the actin cytoskeleton (Amano et al., 1996; Chrzanowska-Wodnicka and Burridge, 1996; Kimura et al., 1996). Fibroblasts are known to activate RhoA in response to a number of mechanical stimuli (Lessey et al., 2012) and in response to TGF-β stimulation (Vardouli et al., 2005; Johnson et al., 2014). Rho GTPases act as molecular switches, active when GTP-bound and inactive when GDP-bound. Guanine nucleotide exchange factors (GEFs) activate Rho proteins by catalyzing the exchange of GDP for GTP, whereas GTPase activating proteins (GAPs) stimulate the intrinsic GTPase activity of Rho proteins, rendering them inactive (Schmidt and Hall, 2002; Jaffe and Hall, 2005). A recent study demonstrated that inhibition of Rho-associated protein kinase/Rho-associated, coiled-coil–containing protein kinase (ROCK) signaling (a known downstream effector of RhoA) decreases fibrosis in a mouse model of IPF (Jiang et al., 2012; Bei et al., 2013; Zhou et al., 2013), further suggesting that the RhoA pathway may regulate IPF.
In this study, we seek to determine how RhoA is regulated downstream of mechanical and TGF-β signaling. Determining the pathways that regulate Rho proteins in IPF should provide enhanced understanding of the molecular mechanisms underlying this disease and may identify potential therapeutic targets.
RESULTS
RhoA activity drives the IPF phenotype
Previous work has demonstrated that RhoA/ROCK signaling is up-regulated in IPF fibroblasts (Jiang et al., 2012; Bei et al., 2013; Zhou et al., 2013). However, little is known about the upstream regulators of RhoA activity in IPF. Because upstream regulators may provide potential therapeutic targets, we investigated the molecular mechanisms that regulate RhoA signaling in IPF cell lines. Comparing two IPF fibroblast lines (LL29 and LL97a) with normal pulmonary fibroblasts (MRC5) revealed elevated expression of FN and collagen I, as well as expression of smooth muscle actin (SMA), a marker for myofibroblast differentiation (Darby et al., 1990) (Figure 1A). These data recapitulate the IPF fibroblast phenotype observed in patients (Kuhn and McDonald, 1991). RhoA activity was enhanced in the two IPF cell lines compared with the normal fibroblasts (Figure 1, B and C). To determine whether the fibrosis phenotype of elevated FN, collagen, and SMA is dependent on RhoA activity, we used an adenovirally delivered RhoA microRNA (miRNA) to knock down RhoA expression in the LL29 and LL97a IPF cell lines and a myc-tagged RhoA construct (RhoA-NT) with five silent mutations within the RNA interference (RNAi) targeting sequence, thus allowing reexpression. These constructs were validated in an earlier publication (Aghajanian et al., 2009). We found that knocking down RhoA expression decreased FN, collagen, and SMA levels while reexpression of RhoA with the RhoA-NT restored with IPF phenotype (Figure 1D), consistent with the idea that RhoA activity is critical in driving the fibrosis phenotype.
FIGURE 1:
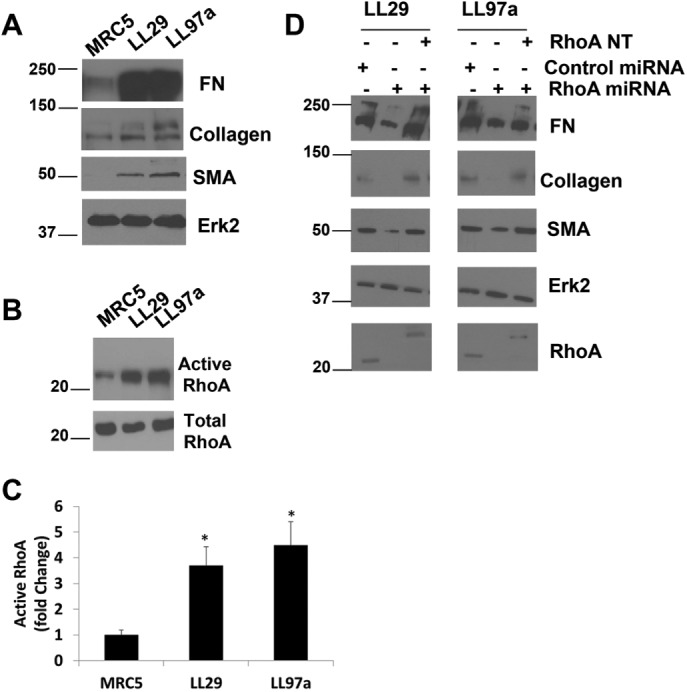
RhoA is required for the IPF phenotype. (A) MRC5, LL29, and LL97a cell lysates were analyzed by Western blotting for FN, collagen I, SMA, and Erk2 (as a loading control). (B) MRC5, LL29, and LL97a cells were lysed, and active RhoA was precipitated from lysates using GST-RBD and immunoblotted with RhoA antibodies. (C) Quantification of RhoA activity in three independent assays. p < 0.05 vs. MRC5 as determined by a t test. (D) LL29 and LL97a cells were infected with an adenoviral miRNA against RhoA for 48 h to knock down RhoA expression. Cell lysates were analyzed by Western blot for expression of RhoA, FN, collagen I, SMA, and Erk2. (D) LL29 and LL97a cells were infected with RhoA miRNA-encoding adenovirus or a control adenovirus for 48 h. After 48 h, cells were transfected with a myc-RhoA NT construct for 24 h, where indicated. After a total of 72 h, total cell lysates were analyzed by Western blot for FN, collagen, SMA, Erk2, and RhoA expression. Note that the position of the myc-RhoA NT construct was detected higher in the blot than the endogenous RhoA.
Rnd3/p190 regulate RhoA activity in IPF
As we continued our analysis comparing the IPF fibroblasts with normal lung fibroblasts, we evaluated the expression levels of the Rnd family of Rho proteins (Figure 2). Rnd1 was expressed at equal levels in the IPF and normal lung fibroblasts, and no detectable levels of Rnd2 were observed in any of the cell lines. However, examination of lysates prepared from testis, a tissue known to express Rnd2 (Nobes et al., 1998), did show successful antibody detection of Rnd2 (Supplemental Figure S1). Rnd3, however, showed decreased expression in the IPF cells when compared with their normal counterpart (Figure 2, A and B). Interestingly, Rnd3 is known to be antagonistic to RhoA signaling through activation of p190RhoGAP (p190) (Wennerberg et al., 2003). To determine whether p190 activity was altered in the IPF cells compared with the normal lung fibroblasts, we used a constitutively active RhoA pull-down assay (Garcia-Mata et al., 2006). Western blot analysis of active GAPs pulled down from MRC5, LL29, and LL97a cell lysates revealed that p190 activity was depressed in the IPF fibroblasts, LL29 and LL97a, when compared with the normal lung fibroblast line, MRC5 (Figure 2, C and D). To examine the interaction between p190 and Rnd3, coimmunoprecipitation analysis was performed. P190 was precipitated from MRC5, LL29, and LL97a cells and the precipitates were examined for the presence of Rnd3. MRC5 cells showed a greater interaction between p190 and Rnd3 (Figure 2E), consistent with the enhanced activity of p190 observed in the MRC5 cells.
FIGURE 2:
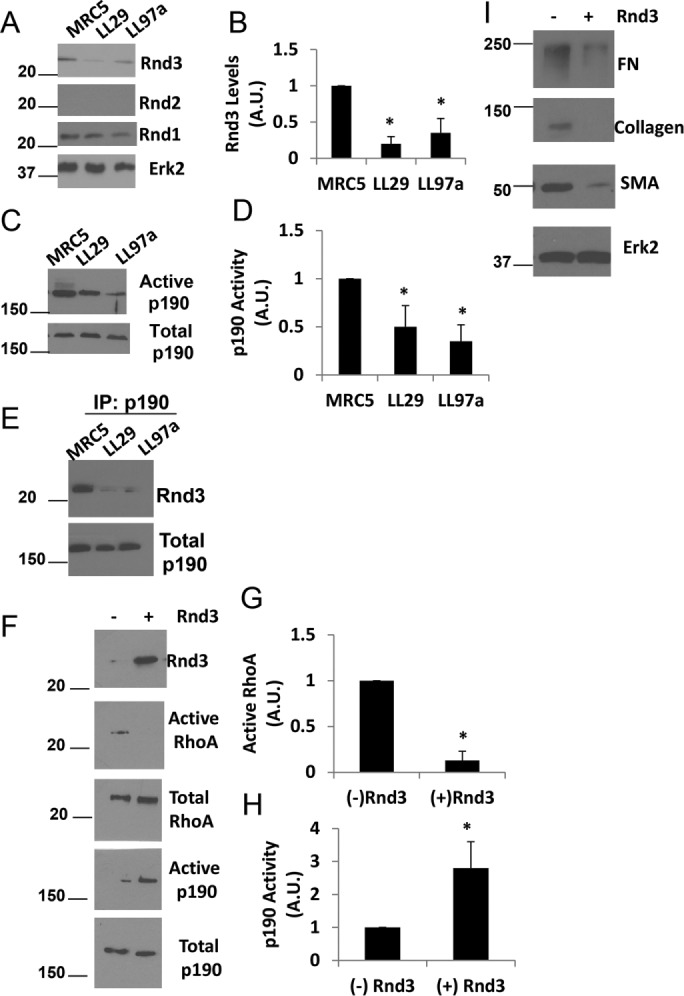
Rnd3 and p190 regulate RhoA activity in IPF cells. (A) MRC5, LL29, and LL97a cell lysates were analyzed by Western blotting for Rnd1, Rnd2, Rnd3, and Erk2 (loading control). (B) Quantification of Rnd3 expression from three independent assays. p < 0.05 vs. MRC5 as determined by a t test. (C) MRC5, LL29, and LL97a cells were lysed and activation of p190 was determined using the GST-RhoAQ63L pull-down assay and immunoblotting with p190 antibodies. (D) Quantification of p190 activity from three independent assays. p < 0.05 vs. MRC5 as determined by a t test. (E) MRC5, LL29, and LL97a cells were lysed in immunoprecipitation buffer and p190 was immunoprecipitated from the cell lysates. Immunoprecipitates were then blotted for the presence of Rnd3. (F) LL29 cells were transfected with Rnd3 cDNA. Cell lysates were then analyzed for RhoA activity through a GST-RBD pull-down assay and p190 activity through a GST-RhoAQ63L pull-down assay. Western blot analysis of pull downs and total cell lysates were analyzed for levels of Rnd3, RhoA, and p190. (G, H) Quantification of RhoA activity (G) and p190 activity (H) from three independent assays. *p < 0.05 vs. (–) Rnd3 as determined by a t test. (I) LL29 cells were transfected with Rnd3 cDNA. Cell lysates were subjected to Western blot analysis for FN, collagen I, and SMA, as well as Erk2 (loading control).
The reciprocal relationship between RhoA activity and Rnd3 expression/p190 activity is interesting, but we wanted to determine whether Rnd3 was regulating RhoA activity via its activation of p190. To address this relationship, Rnd3 was exogenously expressed in LL29 IPF cells. Rnd3 overexpression in IPF cells increased p190 activity (Figure 2, F and H) and decreased RhoA activity (Figure 2, F and G). Additionally, enhanced expression of Rnd3 in the LL29 cells decreased the expression of FN, collagen, and SMA (Figure 2I). To explore the morphological consequences of Rnd3 overexpression in IPF cells we examined stress fiber formation, as it is a well-characterized readout of RhoA activity (Ridley and Hall, 1992). LL29 IPF cells and LL29 cells transfected with Rnd3 were plated onto fibronectin-coated coverslipts for 24 h. The cells were then fixed and F-actin visualized with a Texas-red-labeled phalloidin (Supplemental Figure S2). The LL29 cells transfected with Rnd3 showed less prominent stress fibers and an overall cell rounding. This is in agreement with earlier studies in Cos7 cells, demonstrating that Rnd3 overexpression results in stress fiber collapse and cell rounding (Wennerberg et al., 2003). Taken together, these data suggest that the decreased levels of Rnd3 in the IPF cells result in decreased p190 activity, causing increased RhoA activity. Furthermore, these results indicate that Rnd3 control of RhoA signaling is a key regulator of the IPF phenotype.
Loss of Rnd3 drives the fibrotic phenotype
We asked whether decreasing Rnd3 levels in normal pulmonary fibroblasts would drive a more fibrotic phenotype in these cells. Using two different small interfering RNAs (siRNAs) against Rnd3 (Rnd3 #1 and Rnd3 #2) to knock down Rnd3 expression in MRC5 cells (Figure 3A), we observed that knockdown of Rnd3 caused a decrease in p190 activity (Figure 3, B and C), an increase in RhoA activity (Figure 3, D and E), and an increase in the expression of FN, collagen, and SMA (Figure 3F). These data demonstrate that depression of Rnd3 drives a more fibrotic phenotype in normal lung fibroblasts.
FIGURE 3:
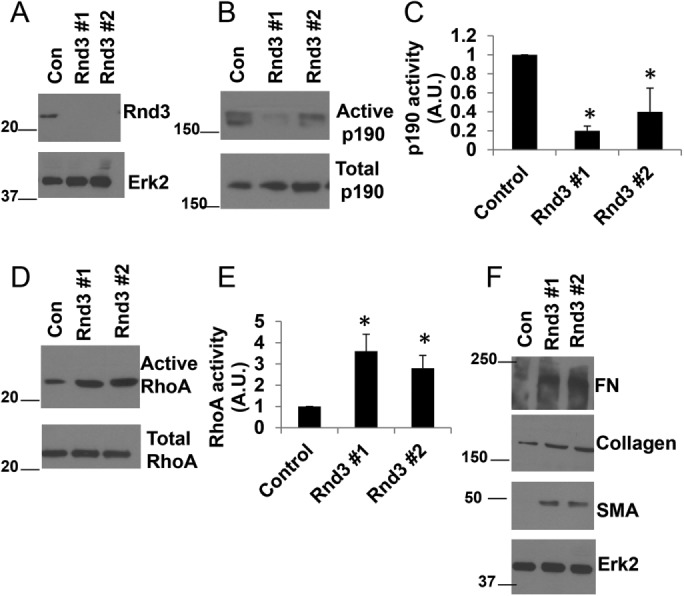
Rnd3 knockdown enhances the fibrotic phenotype in normal lung fibroblasts. MRC5 cells were transfected with control, Rnd3 #1, or Rnd3 #2 siRNA for 72 h. (A) Cell lysates were analyzed by Western blotting for Rnd3 and Erk2 (loading control). (B) Cell lysates were analyzed for p190 activity through a GST-RhoAQ63L pull-down assay. (C) Quantification of p190 activity from three independent assays. (D) Cell lysates were analyzed for RhoA activity through a GST-RBD pull-down assay. (E) Quantification of RhoA activity from three independent assays. (F) Total cell lysates were analyzed by Western blot for expression of FN, collagen I, SMA, and Erk2 (loading control). *p < 0.05 vs control as determined by a t test.
TGF-β signals through the Rnd3/p190/RhoA pathway
TGF-β has been implicated in IPF, and TGF-β signaling is known to activate RhoA in a number of cell types (Bhowmick et al., 2001; Shen et al., 2001; Edlund et al., 2002; Chen et al., 2006). We sought to determine whether TGF-β regulated the Rnd3/p190/RhoA pathway in IPF. As patients with IPF show enhanced levels of TGF-β, we compared levels of active TGF-β present in cell culture supernatants between the IPF fibroblasts and normal lung fibroblasts. Supernatants from MRC5, LL29, and LL97a cultures were assayed for active TGF-β by enzyme-linked immunosorbent assay (ELISA). Active TGF-β levels were approximately fourfold higher in the conditioned media of the IPF cell lines LL29 and LL97a compared with the conditioned media from the MRC5 normal lung fibroblasts (Figure 4A). To test whether TGF-β applied to normal lung fibroblasts promotes a more IPF-like phenotype, MRC5 cells were treated with TGF-β and Western blot analysis was performed to determine the expression of the IPF phenotype markers. MRC5 cells treated with TGF-β elevated the levels of FN, collagen, and SMA (Figure 4B). TGF-β treatment enhanced RhoA activity in the MRC5 cells (Figure 4, C and D). Further analysis revealed that TGF-β treatment resulted in decreased p190 activity (Figure 4, E and F) and decreased Rnd3 levels (Figure 4, G and H), while levels of Rnd1 remain unaffected and Rnd2 remains unexpressed (Figure 4G).
FIGURE 4:

TGF-β induces the IPF phenotype. (A) Conditioned medium was collected from MRC5, LL29, and LL97a cells and assayed for active TGF-β present in the supernatant by ELISA. (B) MRC5 cells were treated with 10 ng/ml TGF-β for 24 h. Cells were lysed, and lysates were evaluated by Western blot for FN, collagen I, SMA, and Erk2. (C) MRC5 cells were treated with 10 ng/ml TGF-β for 24 h. Cells were lysed, and active RhoA was precipitated from lysates using GST-RBD and immunoblotted with RhoA antibodies. (D) Quantification of RhoA activity from three independent assays. (E) MRC5 cells were treated with 10 ng/ml TGF-β for 24 h. Cells were lysed, and p190 GAP activity was assessed through a GST-RhoAQ63L pull-down assay. (F) Quantification of p190 GAP activity from three independent assays. (G) MRC5 cells were treated with 10 ng/ml TGF-β for 24 h. Cells were lysed, and lysates were evaluated by Western blot with antibodies against Rnd3, Rnd2, and Rnd1. (H) Quantification of Rnd3 expression in three independent assays. *p < 0.05 vs. (–)TGF-β as determined by a t test.
As TGF-β promoted the IPF phenotype, we hypothesized that treatment of MRC5 cells with IPF-conditioned media would also induce the Rho-dependent IPF phenotype. To test this hypothesis, we incubated MRC5 cells with conditioned medium from IPF cells LL29 and LL97a or from normal MRC5 cells. We then examined the Rnd3/p190/RhoA signaling pathway in the MRC5 cells treated with these conditioned media. MRC5 cells incubated with IPF-conditioned media (from the LL29 and LL97a cells) displayed a decrease in both Rnd3 levels (Supplemental Figure S3, A and B) and p190 activity (Supplemental Figure S3, C and D), as well as a concomitant increase in RhoA activity (Supplemental Figure S3, E and F). To further analyze the effects of IPF cell–conditioned media on normal MRC5 lung cells, the IPF phenotype markers were examined by Western blot. Treatment of normal lung cells with IPF cell–conditioned media enhanced the expression of the IPF markers FN, collagen, and SMA (Supplemental Figure S3G).
The role for TGF-β signaling in regulating the IPF phenotype through the Rnd3/p190/RhoA pathway was further investigated using SB431542 to inhibit TGF-β receptor signaling (Laping et al., 2002). IPF cells LL29 and LL97a were treated with SB431542, and expression of the IPF phenotype markers was assessed. Inhibition of TGF-β signaling with SB431542 caused a decrease in the expression of FN, collagen, and SMA in the IPF cell lines (Figure 5A). To determine whether the inhibition of TGF-β signaling was affecting the IPF phenotype through alterations in the Rnd3/p190/RhoA pathway, we examined the level of Rnd3 expression and the activities of p190 and RhoA. SB431542 increased the levels of Rnd3 in IPF cells, while levels of Rnd1 remained unchanged and levels of Rnd2 remained undetectable (Figure 5, B and C). Along with the increased Rnd3 expression, we observed an increase in p190 activity (Figure 5, D and E) and a decrease in RhoA activity in IPF cells treated with SB431542 (Figure 5, F and G). TGF-β signaling occurs through both Smad-dependent and Smad-independent pathways (Zhang, 2009). To determine whether the changes observed in Rnd3 expression were Smad dependent, we treated the LL29 IPF cells with the Smad3 inhibitor, SIS3, and measured Rnd3 expression by Western blot. We found no change in Rnd3 levels on SIS3 treatment, indicating that the TGF-β signaling regulating Rnd3 expression in IPF cells is Smad independent (Figure 5H).
FIGURE 5:
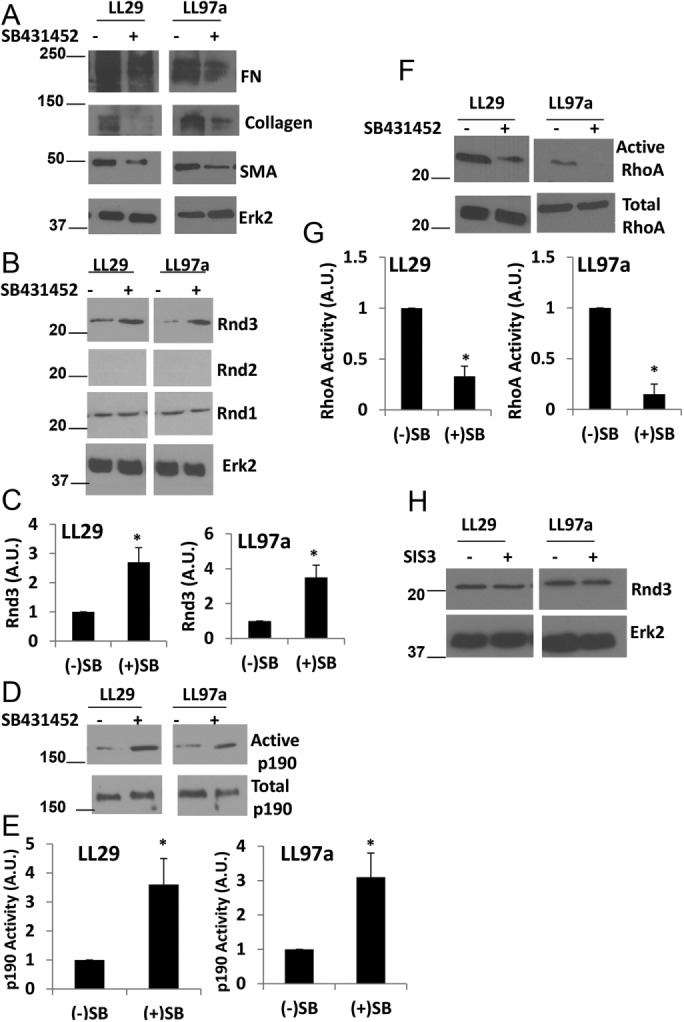
TGF-β signaling regulates the RhoA signaling pathway in IPF. Cells were treated, with or without 10 µM of the TGF-β receptor inhibitor SB431542 for 24 h. (A) LL29 and LL97a cells were lysed and levels of FN, collagen I, SMA, and Erk2 were assessed by Western blot. (B) LL29 and LL97a lysates were analyzed for expression of Rnd3, Rnd2, Rnd1, and Erk2 (loading control). (C) Quantification of Rnd3 expression from three independent experiments. (D) LL29 and LL97a cells were then analyzed for p190 activity using a GST-RhoAQ63L pull-down assay. (E) Quantification of p190 activity from three independent assays. (F) LL29 and LL97a lysates were then analyzed for the activation of RhoA measured through a GST-RBD pull-down assay as described under Materials and Methods. (G) Quantification of RhoA activity from three independent assays. (H) Cells were treated with or without the SMAD3 inhibitor SIS3 at a concentration of 10 µM for 24 h. Cells were then lysed and Rnd3 levels were evaluated by Western blot. *p < 0.05 vs. control (–) SB431542 cells as determined by a t test.
We reasoned that inhibition of TGF-β signaling may prevent the IPF cell–conditioned media from promoting an IPF-like phenotype in the normal lung cells. To test this, we incubated MRC5 cells with IPF cell–conditioned media (from the LL29 and LL97a cells) in the presence or absence of the TGF-β receptor inhibitor, SB431542. MRC5 cells treated with the SB inhibitor greatly abrogated the effects of the IPF cell–conditioned media. MRC5 cells in the presence of SB431542 showed higher levels of Rnd3 than control cells when treated with IPF cell–conditioned media (Supplemental Figure S4, A and B). Likewise, the cells treated with SB431542 showed higher levels of p190 activity than control cells in the presence of IPF-conditioned media (Supplemental Figure S4, C and D). Finally, RhoA activity levels were decreased by SB431542 relative to control treated cells (Supplemental Figure S4, E and F). We also wanted to determine whether TGF-β signaling was necessary to drive the IPF-like phenotype in normal lung cells treated with IPF cell–conditioned media. Indeed, we found that in response to IPF-conditioned media, SB431542-treated cells showed reduced levels of FN, collagen, and SMA when compared with control cells treated with the same IPF-conditioned media (Supplemental Figure S4G). Collectively, these results suggest that TGF-β is a strong upstream regulator of the Rnd3/p190/RhoA pathway in IPF fibroblasts and confirm the role of TGF-β as a major cytokine driving the IPF phenotype.
Our data demonstrate that TGF-β signaling can affect Rnd3 levels. We have also shown that alterations in Rnd3 expression regulate the IPF phenotype. To determine whether TGF-β signaling mediates the IPF phenotype through modulation of Rnd3 levels, Rnd3 was overexpressed in LL29 cells, and then these cells were treated with TGF-β or with SB431542 to stimulate or inhibit the TGF-β pathway, respectively. Overexpression of Rnd3 rendered the IPF phenotype unresponsive to changes in TGF-β signaling (Figure 6A). In earlier work (Figure 3), we showed that knockdown of Rnd3 expression induced the IPF phenotype. Just as driving Rnd3 expression in MRC5 cells inhibited this response to TGF-β, we asked whether removal of Rnd3 would similarly block the response of MRC5 cells to TGF-β. To examine this possibility, Rnd3 expression was knocked down in the MRC5 cells using siRNA, and the IPF phenotype measured in response to TGF-β and SB431542. Rnd3 knockdown rendered the cells unable to regulate the IPF phenotype in response to alterations in TGF-β signaling (Figure 6B). Collectively, these data suggests that TGF-β regulation of the IPF phenotype requires the modulation of Rnd3 levels.
FIGURE 6:

TGF-β regulation of the IPF phenotype requires modulation of Rnd3 levels. (A) LL29 cells were transfected with GST-Rnd3 cDNA to yield LL29 Rnd3 overexpressing cells. Cells were then treated with 10 ng/ml TGF-β or 10 μM SB431542. Cells were lysed, and lysates evaluated for the expression of FN, collagen I, SMA, Erk2 (loading control), and Rnd3. (B) MRC5 cells were transfected with Rnd3 #1 siRNA for 72 h and then treated with 10 ng/ml TGF-β or 10 uM SB431542. Cells were lysed, and lysates evaluated for the expression of FN, collagen I, SMA, Erk2 (loading control), and Rnd3.
Mechanical stiffness modulates Rnd3/p190/RhoA signaling
One of the hallmarks of IPF is the increased stiffness of the lung that occurs as the disease progresses. Normal lung tissue has a stiffness of approximately 2 kPa, while fibrotic lung tissue can show an increase in stiffness to approximately 20 kPa (Liu et al., 2010; Booth et al., 2012). To mimic the progression of stiffness observed during IPF, cells were cultured on substrates of 2, 8, and 25 kPa. Plating these cells on substrata of increasing stiffness increased the levels of TGF-β produced by the cells (Figure 7A), consistent with previous findings (Wipff et al., 2007). As we earlier determined that TGF-β regulates RhoA GTPase signaling in IPF cells, we sought to determine whether changes in substrate stiffness also regulate RhoA activity. The IPF fibroblasts showed higher levels of RhoA activity and concomitant lower levels of Rnd3 and p190 activity on stiffer substrates when compared with less stiff substrates (Figure 7, B–G). Analysis of the IPF phenotype across enhanced substrate rigidities revealed a stiffness-dependent increase in FN, collagen, and SMA (Figure 7H). These data are consistent with earlier studies demonstrating that enhanced substrate stiffness exacerbates the signaling pathways that regulate the IPF phenotype (Hinz, 2012).
FIGURE 7:
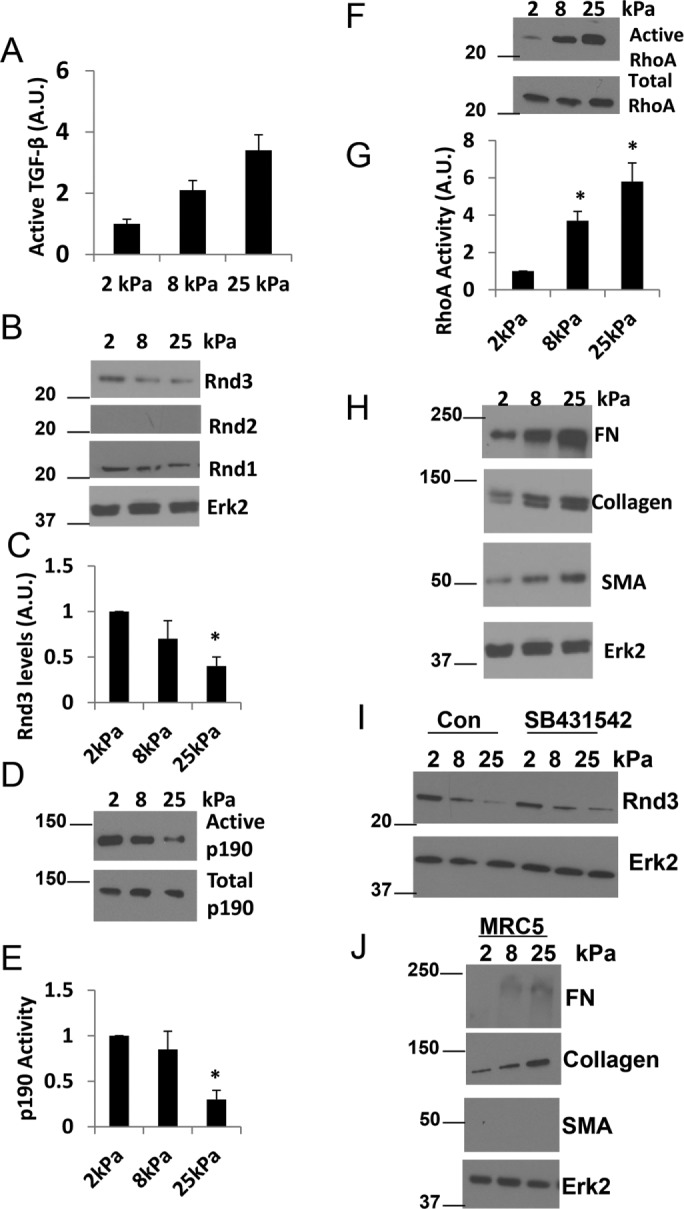
Increasing substrate rigidity enhances the RhoA signaling pathway and IPF phenotype. LL29 cells were cultured on substrates of different rigidities: 2, 8, and 25kPa. (A) Conditioned medium was collected from LL29 cells and assayed for active TGF-β by ELISA. (B) LL29 cells were then lysed and Rnd1, Rnd2, andRnd3 levels analyzed by Western blot. (C) Quantification of Rnd3 expression from three independent assays. (D) LL29 cells were then analyzed for p190 activity using a GST-RhoAQ63L pull-down assay. (E) Quantification of p190 activity from three independent assays. (F) LL29 lysates were then analyzed for the activation of RhoA measured through a GST-RBD pull-down assay as described under Materials and Methods. (G) Quantification of RhoA activity from three independent assays. (H) LL29 cells were lysed, and lysates evaluated by Western blot for FN, collagen I, SMA, and Erk2 levels. Note these samples were from the same experiment as in B and the Erk2 loading control is the same blot. (I) Cells were treated with 10 µM SB431542 for 24 h where indicated. Cells were lysed and Rnd3 and Erk2 (loading control) levels were assessed by Western blot. *p < 0.05 vs. 2 kPa as determined by a t test. (J) MRC5 cells were cultured on increasing substrate rigidities (2, 8, and 25kPa). Cells were lysed, and lysates evaluated by Western blot for FN, collagen I, SMA, and Erk2 levels (loading control).
Is the effect of substratum stiffness mediated by effects on TGF-β?
Having demonstrated that Rnd3 expression is a critical regulator of the IPF phenotype, we wanted to examine the relationship between Rnd3 expression, TGF-β signaling, and substrate stiffness in more detail. To determine whether the change in substrate stiffness was critical in regulating Rnd3 expression levels or whether the effects of stiffness were mediated by enhanced TGF-β signaling, we plated LL29 cells on substrates of increasing stiffness and inhibited TGF-β signaling with SB431542. Analysis of Rnd3 levels showed that the IPF cells decreased their expression of Rnd3 both in the presence of TGF-β signaling, as well as in the absence of TGF-β signaling (after SB431542 treatment) (Figure 7I). These data indicate that alterations in substrate stiffness are sufficient to drive changes in Rnd3 levels in IPF cells, greater stiffness leading to lower levels of Rnd3 expression regardless of the presence or absence of TGF-β signaling.
To examine whether increasing substrate stiffness would be sufficient to initiate the fibrotic phenotype in the MRC5 normal lung fibroblasts, MRC5 cells were plated on increasing substrate rigidities (2, 8, and 25 kPa) and the IPF phenotype markers measured (Figure 7J). Increasing substrate stiffness increased the deposition of FN and collagen but was insufficient to induce expression of SMA. This result is consistent with our earlier results demonstrating that MRC5 cells require TGF-β treatment even when plated on an extremely stiff substrate (plastic) to induce SMA expression (Figure 4B).
Exogenous expression of Rnd3 inhibits stiffness-enhanced expression of fibronectin, collagen, and SMA
Our data suggest that decreased expression of Rnd3 drives enhanced RhoA signaling and leads to a more fibrotic phenotype in IPF. These data also suggest that Rnd3 expression is responsive to substrate stiffness, with stiffer substrates showing lower levels of Rnd3. To determine whether the enhanced fibrotic phenotype seen on stiffer substrates requires modulation of Rnd3 levels, Rnd3 was exogenously expressed in the LL29 IPF cells. The cells were plated on substrates of increasing rigidity (Figure 8A). Cells expressing Rnd3 exogenously were no longer able to decrease p190 activity in response to increased substrate stiffness (Figure 8B). In addition, cells expressing Rnd3 no longer showed increasing RhoA activity with increased substrate stiffness (Figure 8C). The exogenous expression of Rnd3 in the IPF cells abrogated the expression of FN, collagen, and SMA in response to enhanced substrate stiffness (Figure 8D).
FIGURE 8:

Rnd3 overexpression abolishes IPF phenotype in response to substrate stiffness. LL29 cells were transfected with Rnd3 (where indicated). Cells were then plated on substrates with stiffnesses of 2, 8, and 25 kPa. (A) Cells were lysed and levels of GST-Rnd3 and Erk2 (loading control) were evaluated by Western blot. (B) Cells were then analyzed for p190 activity using a GST-RhoAQ63L pull-down assay. (C) Cell lysates were then analyzed for the activation of RhoA measured through a GST-RBD pull-down assay. (D) LL29 cells were lysed, and lysates evaluated by Western blot for FN, collagen I, SMA, and Erk2 levels.
Nintedanib and pirfenidone impact the Rnd3/p190/RhoA pathway
In 2014, pirfenidone and nintedanib were approved in the United States to treat IPF. Nintedanib is a tyrosine kinase inhibitor (Hilberg et al., 2008), whereas pirfenidone is a drug of unknown mechanism with anti-fibrotic properties in multiple animal models (Schaefer et al., 2011). However, much remains to be discovered about the anti-fibrotic actions of these drugs. We treated LL29 IPF cells with pirfenidone or nintedanib and examined the Rho signaling pathway. Pirfenidone elevates Rnd3 expression (Figure 9, A and B), enhances p190 activity (Figure 9, C and D), and decreases RhoA activity (Figure 9, E and F). Similar results were obtained when IPF cells were treated with nintedanib (Figure 9, A–F). Interestingly, when we examined the IPF phenotype after pirfenidone or nintedanib treatment, we found a reduction in the levels of FN and collagen but SMA levels remained high (Figure 9G). This result is consistent with the observation that while both drugs are beneficial to a percentage of patients with IPF, either drug only slows the progression of disease (Myllarniemi and Kaarteenaho, 2015; Troy and Corte, 2016). To examine the morphological consequences of nintedanib and pirfenidone treatment on IPF cells, we stained for stress fibers. Control LL29 IPF cells showed prominent stress fibers, whereas LL29 cells treated with nintedanib or pirfenidone showed decreased cell spreading and decreased stress fiber formation (Figure 9H), consistent with the phenotype observed on Rnd3 overexpression.
FIGURE 9:

Treatment with pirfenidone and nintedanib impacts the RhoA signaling pathway in IPF cells. LL29 cells were treated with pirfenidone (1 mM) and nintedanib (1 μM) for 24 h. (A) The LL29 cells were then lysed, and Rnd3 levels analyzed by Western blot. (B) Quantification of Rnd3 expression from three independent assays. (C) The LL29 cells were then analyzed for p190 activity using a GST-RhoAQ63L pull-down assay. (D) Quantification of p190 activity from three independent assays. (E) LL29 lysates were then analyzed for the activation of RhoA measured through a GST-RBD pull-down assay as described under Materials and Methods. (F) Quantification of RhoA activity from three independent assays. (G) LL29 cells were lysed, and lysates evaluated by Western blot for FN, collagen I, SMA, and Erk2 levels. * p < 0.05 vs. (–) pirfenidone or (–) nintedanib as determined by a t test. (H) LL29 cells were plated onto FN-coated coverslips for 8 h and subsequently either untreated or treated with nintedanib (1 μM) or pirfenidone (1 mM) for an additional 16 h. F-actin staining was detected with Texas-red-labeled phalloidin.
To test whether the elevation of Rnd3 levels induced by pirfenidone and nintedanib is responsible for the ability of these drugs to decrease the IPF phenotype, we used siRNA to deplete the expression of Rnd3 in LL29 IPF cells and examined their response following treatment with nintedanib and pirfenidone (Figure 10A). Whereas in control cells, treatment with either drug raised p190 activity and lowered RhoA activity, cells treated with siRNA were no longer able to alter p190 or Rho activity (Figure 10, B and C). Furthermore, nintedanib and pirfenidone were no longer able to impact the IPF phenotype (FN, collagen levels) in the Rnd3 siRNA-treated cells (Figure 10D). These data suggest that modulation of Rnd3 levels is critical to the anti-fibrotic action of nintedanib and pirfenidone.
FIGURE 10:
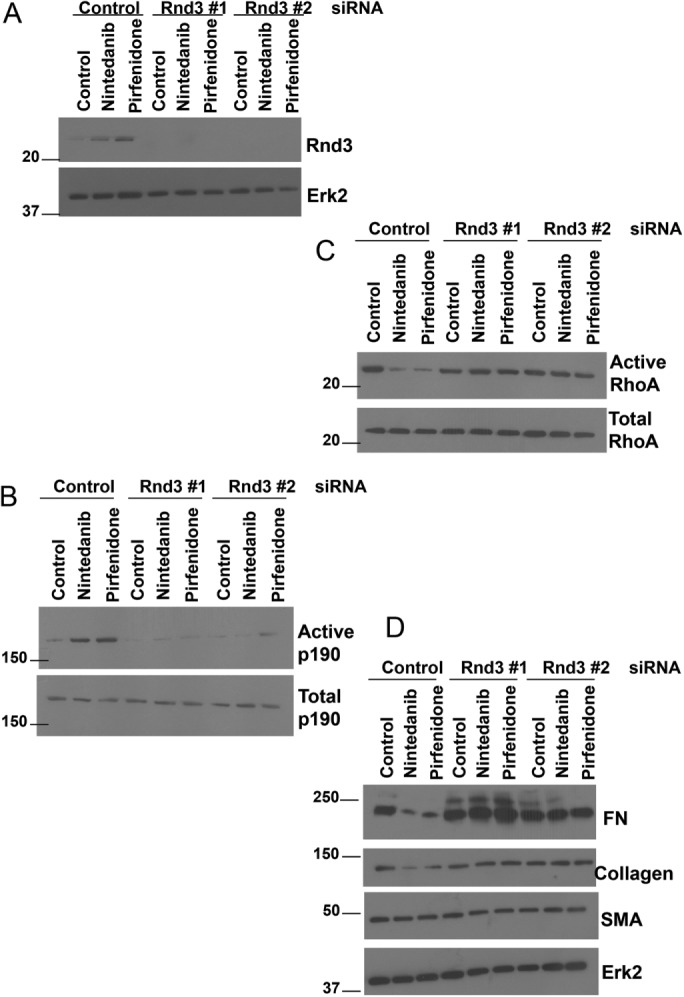
Knockdown of Rnd3 decreases IPF fibroblast sensitivity to nintedanib and pirfenidone. LL29 cells were transfected with control, Rnd3 #1, or Rnd3 #2 siRNA for 72 h and treated with nintedanib (1 μM) or pirfenidone (1 mM) for the final 24 h where indicated. (A) The LL29 cells were then lysed and Rnd3 levels analyzed by Western blot. (B) The LL29 cells were then analyzed for p190 activity using a GST-RhoAQ63L pull-down assay. (C) LL29 lysates were then analyzed for the activation of RhoA measured through a GST-RBD pull-down assay. (D) LL29 cells were lysed, and lysates evaluated by Western blot for FN, collagen I, SMA, and Erk2 levels.
DISCUSSION
Profibrotic stimuli elevate RhoA activity by depressing Rnd3 expression and p190RhoGAP activity
The patchy fibrotic areas that are the pathological hallmark of IPF are characterized by the presence of highly active fibroblasts and myofibroblasts. The origin of these myofibroblasts in IPF tissues has been controversial. It has been argued that they derive from endogenous pulmonary fibroblasts, from the bone marrow, from circulating fibrocytes, from pericytes, and from alveolar epithelial cells that have undergone epithelial to mesenchymal transition (Rock et al., 2011; Noble et al., 2012; Barkauskas and Noble, 2014). Regardless of the origin of the myofibroblasts, there is broad agreement that these cells are key effectors in IPF and that targeting these cells is an important therapeutic option (Eickelberg and Laurent, 2010). Although the precise mechanism through which the persistent myofibroblast phenotype is acquired remains unclear, TGF-β (Vaughan et al., 2000) and mechanical stiffness (Hinz, 2010) have been identified as key factors. Here we report that in IPF, TGF-β and mechanical stiffness signal through a RhoA pathway to regulate the IPF phenotype. Moreover, we find that RhoA activity in IPF is regulated via a Rnd3 and p190RhoGAP pathway.
At the outset of this work we anticipated that fibroblasts from IPF patients would reveal elevated levels of active RhoA. Similarly, on the basis of the literature, we expected that fibroblasts responding to TGF-β would have higher RhoA activity (Bhowmick et al., 2001; Shen et al., 2001; Edlund et al., 2002; Chen et al., 2006). This is indeed what we have found. Additionally, we have shown that the fibrotic phenotype (increased expression of SMA, collagen, and fibronectin) is decreased by depleting RhoA expression. Our results support the idea that elevated RhoA activity is a major factor in IPF and are consistent with studies showing that inhibition of RhoA signaling using ROCK inhibitors is beneficial in animal models of IPF (Jiang et al., 2012; Bei et al., 2013; Zhou et al., 2013). Our goal has been to identify the upstream signaling pathways involved in RhoA activation in IPF cells, and we expected to identify the RhoA GEFs responsible for this activation. However, our preliminary analysis did not reveal major changes in the activity of several prominent GEFs that are expressed in these cells. Instead, we discovered that IPF fibroblasts express lower levels of Rnd3, a close RhoA family member that is antagonistic to RhoA. Rnd3 (also known as RhoE) is an unusual member of the Rho protein family because it lacks GTPase activity, and no GAPs or GEFs have been found that act on Rnd3 (Foster et al., 1996; Nobes et al., 1998). In contrast to most Rho proteins, Rnd3 appears to bind GTP at all times, and, consequently, to be constitutively active. Rnd3 was originally discovered as a protein, which, when expressed exogenously in fibroblasts, promotes disassembly of stress fibers and induces a round (circular) phenotype (Guasch et al., 1998; Nobes et al., 1998). It was subsequently shown to oppose RhoA signaling both upstream and downstream. It binds to and inhibits ROCK (Rho kinase) (Riento et al., 2005), a major RhoA downstream effector. However, it also acts upstream of RhoA by binding and activating p190RhoGAP (Wennerberg et al., 2003) leading to the inactive GDP-bound form of RhoA. Additionally, Rnd3 binds to and inhibits the activation of the RhoA GEF Syx (Goh and Manser, 2010). Investigating p190RhoGAP, we found that it was expressed equally in normal pulmonary and IPF fibroblasts, but its activity was significantly lower in the IPF lines, consistent with these cells having higher RhoA activity. Overexpressing Rnd3 in IPF cells decreased RhoA activity and decreased the IPF phenotype (SMA, collagen I, and fibronectin expression).
IPF patients show higher levels of TGF-β in their plasma (Yong et al., 2001) and bronchoalveolar lavage fluid (Hiwatari et al., 1997). Additionally, TGF-β is overexpressed in the lung tissue of IPF patients (Coker et al., 2001; Bergeron et al., 2003). Not only have we confirmed that the IPF cells generate higher levels of active TGF-β, but also that inhibiting the TGF-β receptor diminishes the IPF phenotype. Multiple studies have shown that TGF-β treatment elevates RhoA activity (Bhowmick et al., 2001; Shen et al., 2001; Edlund et al., 2002; Chen et al., 2006). Notably, we have found that TGF-β decreases Rnd3 expression and p190RhoGAP activity, whereas inhibiting the TGF-β receptor elevates Rnd3 expression and p190RhoGAP activity and decreases RhoA activity. The mechanical properties of fibrotic tissues have been known for some time to contribute to the pathology of fibrosis. Fibroblasts differentiate into myofibroblasts when plated on substrates with an elastic modulus of pathologically stiff fibrotic tissue (Balestrini et al., 2012). Furthermore, decellularized ECM derived from the fibrotic lung is sufficient to induce fibrotic traits in normal lung fibroblasts, including the expression of SMA and enhanced levels of collagen deposition (Booth et al., 2012; Parker et al., 2014). Exploring whether increasing matrix rigidity may activate RhoA by depressing Rnd3 levels and p190 activity, we examined the expression and activity of these in IPF cells plated on substrata of different rigidity. Our results revealed that cells plated on high rigidity substrata have low Rnd3 expression, low p190RhoGAP activity, and correspondingly elevated RhoA activity. On the more rigid substrata, the IPF cells also displayed increased SMA, collagen, and fibronectin expression. All of these results point toward the Rnd3/p190 pathway being a critical negative regulator of RhoA activity and being a pathway that is inhibited under profibrotic conditions.
Both TGF-β and a rigid matrix decrease Rnd3 expression, resulting in decreased p190RhoGAP activity and increased RhoA activity. However, a stiff matrix also contributes to the activation of TGF-β (Wipff et al., 2007), leading to the question of whether these stimuli of increased rigidity and TGF-β depress Rnd3 levels independently or whether the decrease in Rnd3 expression by stiff matrix is downstream of TGF-β. To get at this question, we cultured the IPF fibroblasts on substrata of different rigidities in the presence and absence of a TGF-β receptor inhibitor. Even in the presence of the inhibitor, the stiff matrix decreased Rnd3 levels. We conclude that a stiff matrix and TGF-β activate RhoA independently. However, in the context of a fibrotic tissue, they will synergize and contribute to increased fibrosis in a positive feed forward loop because they both contribute to increased matrix assembly and consequent increased tissue stiffness, with elevated RhoA activity playing a part in this cycle. Our results also raise the interesting possibility of Rnd3 transcription being regulated by mechanoresponsive elements.
IPF drugs pirfenidone and nintedanib elevate Rnd3 expression to depress RhoA activity
Currently, there are no effective therapies for IPF, which is typically fatal within 2–3 yr of diagnosis. However, in 2014 two drugs, pirfenidone and nintedanib, were approved for IPF patients after being found to be beneficial although not curative (Myllarniemi and Kaarteenaho, 2015; Troy and Corte, 2016). Nintedanib is a tyrosine kinase inhibitor that acts on several receptor tyrosine kinases (PDGFR, FGFR, and VEGFR) (Hilberg et al., 2008). Pirfenidone is a drug of unknown mechanism with anti-fibrotic properties in multiple animal models, including pulmonary, cardiac, renal, and hepatic fibrosis (Schaefer et al., 2011). Curious as to their mode of action, we investigated their effects on the Rnd3/p190/RhoA pathway. Strikingly, both drugs elevate Rnd3 expression and p190RhoGAP activity and lead to decreased RhoA activity. Interestingly, while both drugs decreased the levels of FN and collagen I associated with the IPF cells, neither drug induced a significant decrease in SMA levels. The centrality of RhoA signaling to the IPF phenotype makes targeting the regulation of RhoA activity through the Rnd3/p190 pathway an appealing therapeutic strategy that merits further investigation.
Since the discovery that RhoA activity is modulated via Rnd3’s activation of p190RhoGAP, this has been an intriguing pathway but one in search of both initiating upstream stimuli and physiological relevance. Our results identify this pathway as potentially important downstream from TGF-β and one that contributes to TGF-β’s pathological role in fibrosis. Our finding that nintedanib, a tyrosine kinase inhibitor, elevates Rnd3 expression and p190RhoGAP activity raises the possibility that this pathway may also be inhibited by other tyrosine kinases that elevate RhoA activity. We plan to investigate these potential signaling pathways in future work. RhoA activity is negatively regulated by Rnd3 expression levels, but changing the level of a protein’s expression is often a relatively slow response that depends on rates of transcription/translation as well as on its rate of degradation. Consequently, we suspect that the Rnd3/p190 pathway is important in regulating the baseline level of RhoA activity and in mediating slow changes in this baseline. In contrast, GEF activity is typically modulated rapidly in response to external signals and signaling pathways, resulting in more immediate changes in RhoA activity. It will be interesting to investigate further the interplay between these different modes of regulation and to determine in what other situations RhoA activity is controlled by the Rnd3/p190 pathway.
MATERIALS AND METHODS
Antibodies and reagents
The following antibodies were used: anti-FN (Chen et al., 1978); anti-collagen I, anti-Erk2 (Santa Cruz Biotechnology); anti-SMA (Abcam); anti-RhoA (Cell Signaling), anti-p190RhoGAP (Cell Signaling); anti-Rnd3 (Millipore); anti-Rnd1 (abCam); anti-Rnd2 (abCam); anti-myc (Santa Cruz) and horseradish peroxidase (HRP)-conjugated anti-mouse and anti-rabbit secondary antibodies (Jackson ImmunoResearch). IPF drugs pirfenidone (1 mM) (Santa Cruz Biotechnology) and nintedanib (1 µM) (Boehringer) were used. All other reagents were from Sigma-Aldrich unless otherwise noted.
Cell culture
MRC5, LL29, and LL97a cells were obtained from the American Type Culture Collection and grown in high-glucose DMEM supplemented with 10% fetal bovine serum (FBS) and antibiotic–antimycotic solution (all from Life Technologies/ThermoFisher Scientific). In some experiments cells were treated for 24 h with 10 µM SB431452 (Sigma) or 10 ng/ml TGF-β. For substrate rigidity experiments cells were cultured on hydrogels of 2, 8, or 25 kPa (Matrigen) for 24 h prior to harvesting.
Knockdown of RhoA using miRNA adenovirus
Adenoviral miRNA constructs against RhoA and RhoA NT were designed as previously described (Aghajanian et al., 2009). Briefly, miRNA adenoviral constructs were designed and engineered using the BLOCK-iT Pol II miR RNAi expression vector system (Invitrogen) according to the manufacturer’s protocol. Double-stranded oligonucleotides were designed to form an engineered pre-miRNA sequence structure that targets a conserved region in human, mouse, and rat RhoA: 5′TGCTGAAGACTATGAGCAAGCATGTCGTTTTGGCCACTGACTGACGACATGCTCTCATAGTCTT3′ (21–base pair antisense target sequence underlined). Synthesized oligonucleotides were annealed and ligated into pcDNA 6.2-GW/EmGFP-miR. The EmGFP-miRNA cassette was subsequently shuttled through pDONR221(Invitrogen) by Gateway BP recombination and then into pAd-CMV-Dest Gateway vector by LR recombination. Each construct was sequence verified and virus was produced in 293A packaging cell line with the ViraPower Adenoviral Expression System (Invitrogen) using the manufacturer’s recommended protocol. For experiments, cells were infected by 48 h incubation with adenovirus-containing media.
Transfection
The generation of the Rnd3 expression construct was described previously (Wennerberg et al., 2003). All cells were transfected using Lipofectamine 2000 (ThermoFisher Scientific) according to the manufacturer’s protocol. Transfected cells were used for experiments 24 h posttransfection.
siRNA
Two different siRNA duplexes targeting Rnd3 were used: Rnd3 #1 (CUACAGUGUUUGAGAAUUAUU) and Rnd3 #2 (GCGGACAGAUGUUAGUACAUU) (Dharmacon). The nontargeting siControl#1 was also used (Dharmacon). The siRNAs were transfected into cells using Oligofectamine (Invitrogen) at a final concentration of 25 nM. After transfection, cells were cultured for an additional 72 h in complete medium, and then they were processed for further analysis.
RhoA activity assay
Cells were lysed for analysis of RhoA (10 mM MgCl2, 500 mM NaCl, 50 mM Tris, pH 7.6, 1% Triton X-100, 0.1% SDS, 0.5% deoxycholate, 1 mM phenylmethylsulfonyl fluoride (PMSF), and 10 μg/ml aprotinin and leupeptin) and cleared at 14,000 × g for 5 min. Lysates were incubated with 50 μg of glutathione-Sepharose–bound glutathione S-transferase (GST)-RBD (Rhotekin-binding domain) for 30 min at 4°C with gentle rocking. Beads were then washed three times in 50 mM Tris, pH 7.6, 10 mM MgCl2, 150 mM NaCl, 1% Triton X-100, 1 mM PMSF, and 10 μg/ml aprotinin and leupeptin. Released proteins and reserved input control were subjected to Western blot analysis as described later.
GAP activity
Active RhoA GAPs were assayed using GST-RhoA Q63L as described previously (Garcia Mata et al., 2006). Cells were lysed in 150 mM NaCl, 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, pH 7.6, 10 mM MgCl2, 1% Triton X-100, 1 mM PMSF, and 10 μg/ml aprotinin and leupeptin and incubated with 50 μg/ml glutathione-Sepharose–bound GST-RhoAQ63L for 30 min at 4°C and washed in the lysis buffer. Samples were then analyzed by Western blotting as described later.
Immunoprecipitation and immunoblotting
For immunoprecipitation, cells were extracted on ice for 30 min in lysis buffer (10 mM Tris, pH 7.4, 150 mM NaCl, 1% Triton X-100, 0.5% Nonidet P-40, protease, and phosphatase inhibitors). After preclearing with protein A/G-agarose beads, lysates were incubated with antibodies to p190 RhoGAP overnight at 4°C with rotation. Protein A/G agarose beads were added for the final hour of incubation. After being washed three times, the beads were resuspended in sample buffer, boiled for 10 min, and analyzed by Western blotting. For whole cell lysate analysis, cells were lysed in 2 × Laemmli sample buffer and boiled for 5 min. For tissue blots, male C57BL/6 J mice were killed by CO2 inhalation and lung and testes were removed. Tissue was lysed in sample buffer by dounce homogenization. Animal experiments were approved by the Institutional Animal Care and Use Committee of the University of North Carolina (IACUC-ID: 16-142). Samples were resolved on polyacrylamide gels in the presence of SDS. Resolved gels were transferred onto nitrocellulose membranes, blocked with 5% BSA in Tris-buffered saline (25 mM Tris, pH 7.6, 150 mM NaCl) plus 0.1% Tween-20 (TBST) and incubated with primary antibody overnight at 4°C with gentle rocking. Blots were washed extensively in TBST before being incubated with species-appropriate HRP-conjugated secondary antibody (Jackson Laboratories) for 1 h at room temperature. Blots were again washed in TBST, and fluorescence was detected using enhanced chemiluminescent reagent (ThermoFisher Scientific) and x-ray film.
Immunofluorescence
LL29 cells were plated onto fibronectin-coated coverslips for 24 h. In some experiments, cells were treated with pirfenidone (1 mM) or nintedanib 1 µM) for the final 16 h of plating before fixing/permeabilizing in 3.7% paraformaldehyde, then 0.02% Triton X-100. Texas Red phalloidin (Molecular Probes) was used to stain F-actin. Immunofluorescence images were taken through a 20× objective (Zeiss plan-Apochromat 20x/0.8) with a Zeiss axiovert 200 M microscope equipped with a Hamamatsu ORCA-ERAG digital camera.
Conditioned medium
Confluent monolayers of cells were incubated in serum-free medium for 48 h to generate conditioned medium. The conditioned medium was then transferred to fresh plates of MRC5 cells for 24 h or analyzed for active TGF-β levels using a commercial ELISA (R&D Quantikine ELISA Assay). In some experiments SB431452 (10 µM) was added to the conditioned medium. After 24 h the MRC5 cells were appropriately lysed for experiments as described.
Statistical analysis
Statistical differences between two groups of data were analyzed with a two-tailed unpaired Student’s t test. All graphical data are represented as a mean with data bars representing SEM.
Supplementary Material
Acknowledgments
We thank Lisa Sharek for technical support and Christophe Guilluy for helpful discussions. We gratefully acknowledge the support of National Institutes of Health grants GM029860 and GM103723 to K.B. and HL114388 to C.D.
Abbreviations used:
- ECM
extracellular matrix
- FN
fibronectin
- IPF
idiopathic pulmonary fibrosis
- ROCK
Rho-associated protein kinase/Rho-associated, coiled-coil–containing protein kinase
- SMA
smooth muscle actin.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E17-11-0642) on July 11, 2018.
REFERENCES
- Aghajanian A, Wittchen ES, Campbell SL, Burridge K. (2009). Direct activation of RhoA by reactive oxygen species requires a redox-sensitive motif. PLoS One , e8045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano M, Ito M, Kimura K, Fukata Y, Chihara K, Nakano T, Matsuura Y, Kaibuchi K. (1996). Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase). J Biol Chem , 20246–20249. [DOI] [PubMed] [Google Scholar]
- Balestrini JL, Chaudhry S, Sarrazy V, Koehler A, Hinz B. (2012). The mechanical memory of lung myofibroblasts. Integr Biol (Camb.) , 410–421. [DOI] [PubMed] [Google Scholar]
- Barkauskas CE, Noble PW. (2014). Cellular mechanisms of tissue fibrosis. 7. New insights into the cellular mechanisms of pulmonary fibrosis. Am J Physiol Cell Physiol , C987–C996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bei Y, Hua-Huy T, Duong-Quy S, Nguyen VH, Chen W, Nicco C, Batteux F, Dinh-Xuan AT. (2013). Long-term treatment with fasudil improves bleomycin-induced pulmonary fibrosis and pulmonary hypertension via inhibition of Smad2/3 phosphorylation. Pulm Pharmacol Ther , 635–643. [DOI] [PubMed] [Google Scholar]
- Bergeron A, Soler P, Kambouchner M, Loiseau P, Milleron B, Valeyre D, Hance AJ, Tazi A. (2003). Cytokine profiles in idiopathic pulmonary fibrosis suggest an important role for TGF-beta and IL-10. Eur Respir J , 69–76. [DOI] [PubMed] [Google Scholar]
- Bhowmick NA, Ghiassi M, Bakin A, Aakre M, Lundquist CA, Engel ME, Arteaga CL, Moses HL. (2001). Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol Biol Cell , 27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth AJ, Hadley R, Cornett AM, Dreffs AA, Matthes SA, Tsui JL, Weiss K, Horowitz JC, Fiore VF, Barker TH, et al. (2012). Acellular normal and fibrotic human lung matrices as a culture system for in vitro investigation. Am J Respir Crit Care Med , 866–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LB, Burridge K, Murray A, Walsh ML, Copple CD, Bushnell A, McDougall JK, Gallimore PH. (1978). Modulation of cell surface glycocalyx: studies on large, external, transformation-sensitive protein. Ann NY Acad Sci , 366–381. [DOI] [PubMed] [Google Scholar]
- Chen S, Crawford M, Day RM, Briones VR, Leader JE, Jose PA, Lechleider RJ. (2006). RhoA modulates Smad signaling during transforming growth factor-beta-induced smooth muscle differentiation. J Biol Chem , 1765–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrzanowska-Wodnicka M, Burridge K. (1996). Rho-stimulated contractility drives the formation of stress fibers and focal adhesions. J Cell Biol , 1403–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coker RK, Laurent GJ, Jeffery PK, du Bois RM, Black CM, McAnulty RJ. (2001). Localisation of transforming growth factor beta1 and beta3 mRNA transcripts in normal and fibrotic human lung. Thorax , 549–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darby I, Skalli O, Gabbiani G. (1990). Alpha-smooth muscle actin is transiently expressed by myofibroblasts during experimental wound healing. Lab Invest , 21–29. [PubMed] [Google Scholar]
- Edlund S, Landstrom M, Heldin CH, Aspenstrom P. (2002). Transforming growth factor-beta-induced mobilization of actin cytoskeleton requires signaling by small GTPases Cdc42 and RhoA. Mol Biol Cell , 902–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eickelberg O, Laurent GJ. (2010). The quest for the initial lesion in idiopathic pulmonary fibrosis: gene expression differences in IPF fibroblasts. Am J Respir Cell Mol Biol , 1–2. [DOI] [PubMed] [Google Scholar]
- Foster R, Hu KQ, Lu Y, Nolan KM, Thissen J, Settleman J. (1996). Identification of a novel human Rho protein with unusual properties: GTPase deficiency and in vivo farnesylation. Mol Cell Biol , 2689–2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabbiani G, Ryan GB, Majne G. (1971). Presence of modified fibroblasts in granulation tissue and their possible role in wound contraction. Experientia , 549–550. [DOI] [PubMed] [Google Scholar]
- Garcia-Mata R, Wennerberg K, Arthur WT, Noren NK, Ellerbroek SM, Burridge K. (2006). Analysis of activated GAPs and GEFs in cell lysates. Methods Enzymol , 425–437. [DOI] [PubMed] [Google Scholar]
- Goh LL, Manser E. (2010). The RhoA GEF Syx is a target of Rnd3 and regulated via a Raf1-like ubiquitin-related domain. PLoS One , e12409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guasch RM, Scambler P, Jones GE, Ridley AJ. (1998). RhoE regulates actin cytoskeleton organization and cell migration. Mol Cell Biol , 4761–4771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilberg F, Roth GJ, Krssak M, Kautschitsch S, Sommergruber W, Tontsch-Grunt U, Garin-Chesa P, Bader G, Zoephel A, Quant J, et al. (2008). BIBF 1120: triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res , 4774–4782. [DOI] [PubMed] [Google Scholar]
- Hinz B. (2010). The myofibroblast: paradigm for a mechanically active cell. J Biomech , 146–155. [DOI] [PubMed] [Google Scholar]
- Hinz B. (2012). Mechanical aspects of lung fibrosis: a spotlight on the myofibroblast. Proc Am Thorac Soc , 137–147. [DOI] [PubMed] [Google Scholar]
- Hinz B. (2015). The extracellular matrix and transforming growth factor-beta1: tale of a strained relationship. Matrix Biol , 54–65. [DOI] [PubMed] [Google Scholar]
- Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G. (2007). The myofibroblast: one function, multiple origins. Am J Pathol , 1807–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiwatari N, Shimura S, Yamauchi K, Nara M, Hida W, Shirato K. (1997). Significance of elevated procollagen-III-peptide and transforming growth factor-beta levels of bronchoalveolar lavage fluids from idiopathic pulmonary fibrosis patients. Tohoku J Exp Med , 285–295. [DOI] [PubMed] [Google Scholar]
- Jaffe AB, Hall A. (2005). Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol , 247–269. [DOI] [PubMed] [Google Scholar]
- Jiang C, Huang H, Liu J, Wang Y, Lu Z, Xu Z. (2012). Fasudil, a Rho-kinase inhibitor, attenuates bleomycin-induced pulmonary fibrosis in mice. Int J Mol Sci , 8293–8307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LA, Rodansky ES, Haak AJ, Larsen SD, Neubig RR, Higgins PD. (2014). Novel Rho/MRTF/SRF inhibitors block matrix-stiffness and TGF-beta-induced fibrogenesis in human colonic myofibroblasts. Inflamm Bowel Dis , 154–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, Yamamori B, Feng J, Nakano T, Okawa K, et al. (1996). Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science , 245–248. [DOI] [PubMed] [Google Scholar]
- King TE, Jr, Schwarz MI, Brown K, Tooze JA, Colby TV, Waldron JA, Jr, Flint A, Thurlbeck W, Cherniack RM. (2001). Idiopathic pulmonary fibrosis: relationship between histopathologic features and mortality. Am J Respir Crit Care Med , 1025–1032. [DOI] [PubMed] [Google Scholar]
- Klingberg F, Chow ML, Koehler A, Boo S, Buscemi L, Quinn TM, Costell M, Alman BA, Genot E, Hinz B. (2014). Prestress in the extracellular matrix sensitizes latent TGF-beta1 for activation. J Cell Biol , 283–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn C, McDonald JA. (1991). The roles of the myofibroblast in idiopathic pulmonary fibrosis. Ultrastructural and immunohistochemical features of sites of active extracellular matrix synthesis. Am J Pathol , 1257–1265. [PMC free article] [PubMed] [Google Scholar]
- Laping NJ, Grygielko E, Mathur A, Butter S, Bomberger J, Tweed C, Martin W, Fornwald J, Lehr R, Harling J, et al. (2002). Inhibition of transforming growth factor (TGF)-beta1-induced extracellular matrix with a novel inhibitor of the TGF-beta type I receptor kinase activity: SB-431542. Mol Pharmacol , 58–64. [DOI] [PubMed] [Google Scholar]
- Lessey EC, Guilluy C, Burridge K. (2012). From mechanical force to RhoA activation. Biochemistry , 7420–7432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Mih JD, Shea BS, Kho AT, Sharif AS, Tager AM, Tschumperlin DJ. (2010). Feedback amplification of fibrosis through matrix stiffening and COX-2 suppression. J Cell Biol , 693–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinkovic A, Mih JD, Park JA, Liu F, Tschumperlin DJ. (2012). Improved throughput traction microscopy reveals pivotal role for matrix stiffness in fibroblast contractility and TGF-beta responsiveness. Am J Physiol Lung Cell Mol Physiol , L169–L180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myllarniemi M, Kaarteenaho R. (2015). Pharmacological treatment of idiopathic pulmonary fibrosis—preclinical and clinical studies of pirfenidone, nintedanib, and N-acetylcysteine. Eur Clin Respir J , 26385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobes CD, Lauritzen I, Mattei MG, Paris S, Hall A, Chardin P. (1998). A new member of the Rho family, Rnd1, promotes disassembly of actin filament structures and loss of cell adhesion. J Cell Biol , 187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble PW, Barkauskas CE, Jiang D. (2012). Pulmonary fibrosis: patterns and perpetrators. J Clin Invest , 2756–2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker MW, Rossi D, Peterson M, Smith K, Sikstrom K, White ES, Connett JE, Henke CA, Larsson O, Bitterman PB. (2014). Fibrotic extracellular matrix activates a profibrotic positive feedback loop. J Clin Invest , 1622–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR, Lasky JA, et al. (2011). An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med , 788–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghu G, Lynch D, Godwin JD, Webb R, Colby TV, Leslie KO, Behr J, Brown KK, Egan JJ, Flaherty KR, et al. (2014). Diagnosis of idiopathic pulmonary fibrosis with high-resolution CT in patients with little or no radiological evidence of honeycombing: secondary analysis of a randomised, controlled trial. Lancet Respir Med , 277–284. [DOI] [PubMed] [Google Scholar]
- Ridley AJ, Hall A. (1992). The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell , 389–399. [DOI] [PubMed] [Google Scholar]
- Riento K, Totty N, Villalonga P, Garg R, Guasch R, Ridley AJ. (2005). RhoE function is regulated by ROCK I-mediated phosphorylation. EMBO J , 1170–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock JR, Barkauskas CE, Cronce MJ, Xue Y, Harris JR, Liang J, Noble PW, Hogan BL. (2011). Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci USA , E1475–E1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saums MK, Wang W, Han B, Madhavan L, Han L, Lee D, Wells RG. (2014). Mechanically and chemically tunable cell culture system for studying the myofibroblast phenotype. Langmuir , 5481–5487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer CJ, Ruhrmund DW, Pan L, Seiwert SD, Kossen K. (2011). Antifibrotic activities of pirfenidone in animal models. Eur Respir Rev , 85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt A, Hall A. (2002). Guanine nucleotide exchange factors for Rho GTPases: turning on the switch. Genes Dev , 1587–1609. [DOI] [PubMed] [Google Scholar]
- Shen X, Li J, Hu PP, Waddell D, Zhang J, Wang XF. (2001). The activity of guanine exchange factor NET1 is essential for transforming growth factor-beta-mediated stress fiber formation. J Biol Chem , 15362–15368. [DOI] [PubMed] [Google Scholar]
- Shimbori C, Gauldie J, Kolb M. (2013). Extracellular matrix microenvironment contributes actively to pulmonary fibrosis. Curr Opin Pulm Med , 446–452. [DOI] [PubMed] [Google Scholar]
- Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. (2002). Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol , 349–363. [DOI] [PubMed] [Google Scholar]
- Troy LK, Corte TJ. (2016). Therapy for idiopathic pulmonary fibrosis: lessons from pooled data analyses. Eur Respir J , 27–30. [DOI] [PubMed] [Google Scholar]
- Vardouli L, Moustakas A, Stournaras C. (2005). LIM-kinase 2 and cofilin phosphorylation mediate actin cytoskeleton reorganization induced by transforming growth factor-beta. J Biol Chem , 11448–11457. [DOI] [PubMed] [Google Scholar]
- Vaughan MB, Howard EW, Tomasek JJ. (2000). Transforming growth factor-beta1 promotes the morphological and functional differentiation of the myofibroblast. Exp Cell Res , 180–189. [DOI] [PubMed] [Google Scholar]
- Wennerberg K, Forget MA, Ellerbroek SM, Arthur WT, Burridge K, Settleman J, Der CJ, Hansen SH. (2003). Rnd proteins function as RhoA antagonists by activating p190 RhoGAP. Curr Biol , 1106–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wipff PJ, Hinz B. (2008). Integrins and the activation of latent transforming growth factor beta1—an intimate relationship. Eur J Cell Biol , 601–615. [DOI] [PubMed] [Google Scholar]
- Wipff PJ, Rifkin DB, Meister JJ, Hinz B. (2007). Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J Cell Biol , 1311–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong SJ, Adlakha A, Limper AH. (2001). Circulating transforming growth factor-beta(1): a potential marker of disease activity during idiopathic pulmonary fibrosis. Chest , 68S–70S. [DOI] [PubMed] [Google Scholar]
- Zhang YE. (2009). Non-Smad pathways in TGF-beta signaling. Cell Res , 129–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Huang X, Hecker L, Kurundkar D, Kurundkar A, Liu H, Jin TH, Desai L, Bernard K, Thannickal VJ. (2013). Inhibition of mechanosensitive signaling in myofibroblasts ameliorates experimental pulmonary fibrosis. J Clin Invest , 1096–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.