

Abstract
Phosphorylation modulates many cellular processes during cell cycle progression. The yeast centrosome (called the spindle pole body, SPB) is regulated by the protein kinases Mps1 and Cdc28/Cdk1 as it nucleates microtubules to separate chromosomes during mitosis. Previously we completed an SPB phosphoproteome, identifying 297 sites on 17 of the 18 SPB components. Here we describe mutagenic analysis of phosphorylation events on Spc29 and Spc42, two SPB core components that were shown in the phosphoproteome to be heavily phosphorylated. Mutagenesis at multiple sites in Spc29 and Spc42 suggests that much of the phosphorylation on these two proteins is not essential but enhances several steps of mitosis. Of the 65 sites examined on both proteins, phosphorylation of the Mps1 sites Spc29-T18 and Spc29-T240 was shown to be critical for function. Interestingly, these two sites primarily influence distinct successive steps; Spc29-T240 is important for the interaction of Spc29 with Spc42, likely during satellite formation, and Spc29-T18 facilitates insertion of the new SPB into the nuclear envelope and promotes anaphase spindle elongation. Phosphorylation sites within Cdk1 motifs affect function to varying degrees, but mutations only have significant effects in the presence of an MPS1 mutation, supporting a theme of coregulation by these two kinases.
INTRODUCTION
Phosphorylation is a widespread form of posttranslational protein modification; the most comprehensive study to date estimates that over 75% of human proteins are phosphorylated (Sharma et al., 2014). Cells from a wide variety of biological states have been found to share many phosphorylation events, but 20% are found exclusively in mitotic cells, which also contain some of the highest stoichiometry of sites (Sharma et al., 2014).
In yeast cells, there are a single cyclin-dependent kinase (Cdk1/Cdc28) and nine cyclins that regulate its activity toward particular targets, thereby advancing the cell cycle through G1-S-G2-M (Mendenhall and Hodge, 1998). In addition, Mps1 kinase and Cdc5 polo kinase have been shown to be required for cell cycle progression (Winey et al., 1991; Segal, 2011; Elserafy, Šaric´, Neuner, et al., 2014; Botchkarev and Haber, 2018). Like DNA duplication, duplication of the SPB is a highly regulated cell cycle event necessary for chromosome segregation in mitosis. Although SPBs are structurally distinct from centrosomes, study of SPB duplication and spindle formation has led to insights into mechanisms of spindle assembly, microtubule nucleation, asymmetric cell division, and spindle positioning relevant to higher organisms (Cavanaugh and Jaspersen, 2017).
In budding yeast, the nuclear envelope stays intact throughout the cell cycle and the SPB remains embedded in the nuclear envelope, nucleating both cytoplasmic and nuclear microtubules. All of the components of the SPB have been identified, and much has been established about the interaction and function of the various proteins through a number of techniques, including two-hybrid analysis, Förster resonance energy transfer (FRET), and genetics. Recently, a model of the molecular architecture of the SPB core was determined using integrative structure modeling incorporating the large volume of data known about the SPB (Viswanath, Bonomi, et al., 2017). The steps of SPB duplication also have been well described (reviewed in Rüthnick and Schiebel, 2016; Cavanaugh and Jaspersen, 2017) using both electron microscopy (Byers and Goetsch, 1974, 1975; Adams and Kilmartin, 1999) and high-resolution fluorescence techniques revealing the order of addition of particular components (Burns et al., 2015; Seybold, Elserafy, et al., 2015; Rüthnick et al., 2017). SPB duplication entails elongation of the “half-bridge” (see pathway in Figure 9 later in this paper), deposition of a “satellite,” starting with Spc42 quickly followed by Spc29, and maturation into a duplicate plaque, which becomes the new SPB. A Bbp1–Mps2 complex is thought to aid insertion of the duplicate plaque into the nuclear envelope (Schramm et al., 2000). Mutations that block this step (Winey et al., 1991) result in a detached pole (sometimes referred to as a “dead pole”; Araki, Lau, et al., 2006) that can separate from the mother SPB and remains associated with, but does not insert into, the nuclear envelope. Interestingly, duplication plaque insertion also involves clustering of nuclear pore complexes and additional membrane-bound proteins, as well as interaction of the new SPB core with nuclear Spc110 (Rüthnick et al., 2017). Next the core SPB-Spc110 connects the nuclear gamma tubulin complex, initiating nucleation of microtubules (Burns et al., 2015; Rüthnick et al., 2017), and the duplicated SPBs separate to opposite sides of the nucleus, leading to the formation of a bipolar spindle. Errors at any step of SPB duplication result in activation of the spindle checkpoint, which transiently delays the cell cycle in an attempt to correct the spindle assembly defect (as reviewed in Amon, 1999).
FIGURE 9:

Model showing role of phosphorylation of Spc29-T18A and Spc29-T240A in sequential steps of SPB duplication. Relevant substructures on the SPB are indicated. Small yellow spheres containing P = phosphorylation events at noted residue. MPS1 mutations mps1-8, mps1-412, and mps1-737 are different mutant alleles with distinct SPB duplication blocks described in the text.
The restriction of duplication to once per cell cycle is accomplished by an elegant mechanism involving, in part, phosphorylation/dephosphorylation of the conserved Sfi1 protein, which is a structural component of the half-bridge (Avena et al., 2014; Elserafy, Šaric´, Neuner, et al., 2014). Phosphorylation of two core central plaque components of the SPB, Spc42 and Spc29, has also been investigated. Proper Spc42 assembly at the SPB and normal spindle function require two verified Cdk1 sites (Spc42-S4T6) as well as Mps1 activity (Castillo et al., 2002; Jaspersen et al., 2004). Spc29 also has been shown to be phosphorylated by both Cdk1 and Mps1, and several Mps1 sites have been mapped in Spc29 (Holinger et al., 2009; Araki et al., 2010). The mutation spc29-T240A (as well as a mutation changing all of the mapped Mps1 sites to alanines) is lethal in single copy (Holinger et al., 2009; Araki et al., 2010), and two copies of T240A in tandem lead to a monopolar spindle phenotype at the nonpermissive temperature (Holinger et al., 2009). Spc29 has been shown to bind Bbp1 (Schramm et al., 2000), and Spc29 phosphorylation increases binding (Araki et al., 2010). Furthermore, biochemical studies have provided reconstitution of an Spc29-Bbp1-Mps2 complex (Kupke et al., 2017).
Despite analysis of Spc29 phosphorylation, studies investigating effects on specific protein–protein interactions have involved alleles with multiple mutations; thus, the mechanism of action of single essential phosphorylation events remains to be elucidated. Also, many of the sites in both Spc29 and Spc42 identified in the phosphoproteomic analysis of the SPB (Keck, Jones, et al., 2011) have not been investigated. To determine how modification of both known sites and these additional residues influences SPB duplication and spindle assembly, we describe a comprehensive analysis of the role of phosphorylation in the SPB core components Spc29 and Spc42, examining effects on cell growth, cell cycle progression, SPB duplication, mitotic spindle morphology, protein localization, and protein–protein interactions. Our work provides insight into the mechanism by which particular phosphorylation events influence distinct steps in this complex pathway.
RESULTS
Phosphosite analysis
We previously performed a phosphoproteome analysis of the Saccharomyces cerevisiae centrosome (Keck, Jones, et al., 2011). This work identified 297 (227 novel) phosphorylated residues on the 18 known SPB components in enriched SPB preparations analyzed by mass spectrometry. Twenty-one percent of these (65 sites) were found on the core SPB binding partners, Spc29 and Spc42. In the current study, we performed a comprehensive mutagenic analysis in the W303 yeast strain of all serine/threonine residues in Spc29 and Spc42 found in our previous work (or another, Jaspersen et al., 2004) to be phosphorylated in vivo. See Table 2 later in the paper for a summary of their relative growth rates.
TABLE 2:
Estimated growth (%) for SPC29 and SPC42 alleles (W303 strain).
| Mutant strain | 24°C | 24°C mad2∆ | 37°C | 37°C mad2∆ | Reference |
|---|---|---|---|---|---|
| SPC29-wildtype (integ) | 100 | 100 | 100 | 100 | This study |
| spc29-T240A | — | nd | — | nd | Holinger et al., 2009, Araki et al., 2010 |
| spc29-T240A (2 tandem copies) | 100 | nd | — | nd | Holinger et al., 2009 |
| spc29-T18A | 20 | 0.2 | — | — | This studya |
| spc29-T18D | 100 | nd | 100 | nd | This studya |
| spc29-30A | 100 | 100 | 100 | 100 | This study |
| spc29-4A | 100 | nd | 100 | nd | This study |
| SPC42-wildtype (integ) | 100 | 100 | 100 | 100 | This study |
| spc42-24A | 100 | 100 | 4 | — | This study |
| Spc29-wildtype; Spc42-wildtype | 100 | 100 | 100 | 100 | This study |
| spc29-30A; spc42-24A | 100 | 100 | 0.2 | — | This study |
| mps1-8 | 100 | nd | — | nd | Castillo et al., 2002 |
| spc42-Cdk-8A | 20 | 20 | 4 | 0.8 | This studyb |
| spc42-Cdk-8A; mps1-8 | 4 | nd | — | nd | This study |
| spc29-Cdk-4A; spc42-Cdk-8A | 100 | nd | 20 | nd | This study |
nd = not determined; — = dead.
aThese effects are more severe in strain S288C (Araki et al., 2010).
bMutation spc42-Cdk-8A was remade; in Keck, Jones, et al. (2011), an additional mutation was present.
Mutation of phosphorylated residues in Cdk1 sites is exacerbated in the presence of mps1 mutations
Spc29 has been shown to be phosphorylated in vivo on four serines lying within Cdk1 (S/T(P)) motifs (Holinger, 2007; Keck, Jones, et al., 2011). Mutation of all four residues to alanine (spc29-Cdk-4A, S59A, S65A, S77A, S121A; Table 1) does not result in a growth defect at any temperature checked (Figure 1, A and C). Because SPB components have been shown previously to be regulated by both Cdk1 and Mps1, we examined the effect of this mutation in the presence of a unique MPS1 mutation, mps1-8, that is defective in its SPB duplication function but not in its checkpoint function (Castillo et al., 2002). If the cells also contain mps1-8, there is an additive cost to fitness, decreasing growth at 30°C ∼25-fold compared with that for mps1-8 alone. Mutation of the same residues to aspartic acid also led to reduced growth in the presence of mps1-8. Using the combined defects of spc29-Cdk-4A and mps1-8, we were able to determine the relative contribution of each site and show that spc29-S121A is the most significant mutation of the 4A allele (Figure 1, B and C). This may be due to the fact that S121 is the only one of the four that contains a full Cdk1 motif (S/T-P-X-K/R), which has been shown to be phosphorylated more efficiently than the minimal (S/T-P) sites (Koivomagi et al., 2011).
TABLE 1:
Changed amino acid residues in mutant alleles.
| Allele | Mutations | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Spc29-30A | S9A | S11A | S39A | S40A | T42A | S43A | S58A | S59A | S65A | T67A |
| S70A | S77A | S100A | T107A | T119A | S120A | T125A | S130A | S167A | S187A | |
| S189A | S191A | T195A | S216A | S217A | S230A | S231A | S239A | S249A | S250A | |
| Spc42-24A | S12A | S29A | S43A | T59A | S130A | T131A | S151A | T168A | T193A | S195A |
| S204A | T221A | S246A | S255A | S284A | S294A | S308A | S319A | S321A | S322A | |
| S328A | S329A | S352A | T354A | |||||||
| Spc29-Cdk-4A | S59A | S65A | S77A | S121A | ||||||
| Spc29-Cdk-4D | S59D | S65D | S77D | S121D | ||||||
| Spc42-Cdk-8A | S4A | S6A | T32A | S213A | S217A | S326A | T357A | T359A | ||
FIGURE 1:
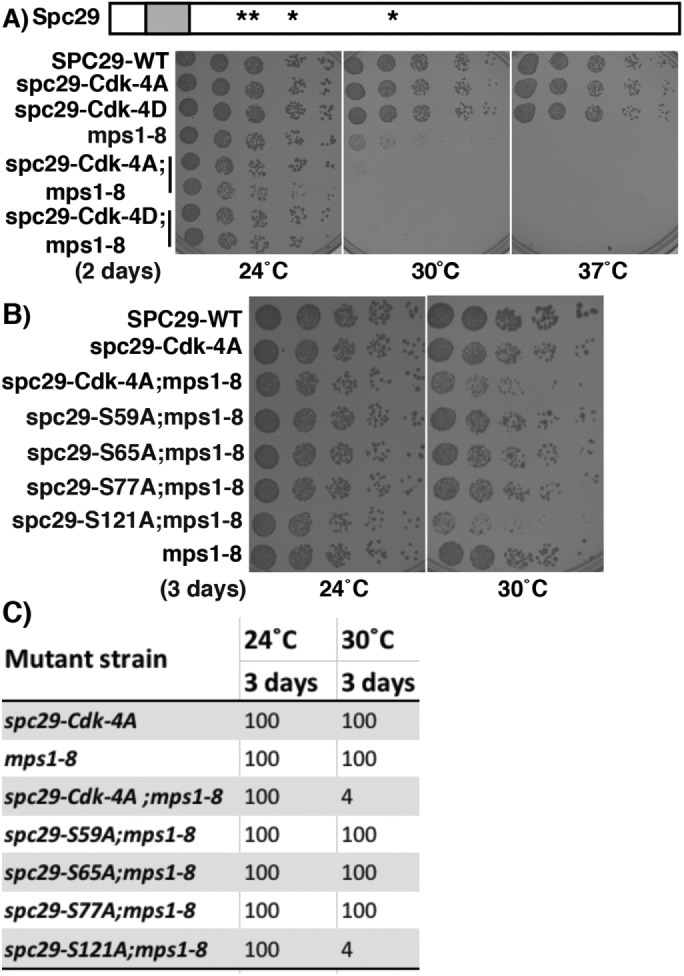
The mutation in a Cdk1 motif, spc29-S121A, confers a growth defect in the presence of mps1-8. (A) Diagram of Spc29 showing position of “4A” and “4D” phosphorylation site mutations (asterisks) and the predicted coiled–coil domain from residues 16–36 (shaded box). spc29-Cdk-4A = spc29-S59A, S65A, S77A, S121A; spc29-Cdk-4D = spc29-S59D, S65D, S77D, S121D. Growth of cells with listed genotypes at 24, 30, and 37°C for 2 d. There are two rows each for spc29-4A;mps1-8 and spc29-4D;mps1-8. (B) Growth of cells with listed genotypes at 24 and 30°C for 3 d. (C) Summary of growth of strains. Numbers indicate estimated percent growth compared with wild-type strain, based on a fivefold dilution assay.
We previously reported that a mutation to alanine in all eight potential Cdk1 sites in Spc42 is lethal and has reduced SPB localization (“8A”; Keck, Jones, et al., 2011). Further examination of this allele revealed that it also contained a single nucleotide deletion at position 37 in the coding sequence. Therefore, the spc42-Cdk-8A allele (residues S4, T6, T32, S213, S217, S326, T357, T359; Table 1) was remade and tested in our dilution assay and also fused to GFP (green fluorescent protein) to monitor localization. Spc42-Cdk-8A leads to a decrease in growth compared with the wild type (Figure 2A and Table 2) and an ∼25-fold decrease at 37°C. Residues S4, T6, T357, and T359 are the S/T(P) sites conserved among budding yeasts in Spc42 (Supplemental Figure 1A; note location of black stars above the sequence to mark sites). Spc42-Cdk-8A cells are more defective in the presence of the checkpoint mutation mad2∆ (37°C, see Figure 4B, later in the paper, for growth of SPC42;mad2∆) or with mps1-8 (24°C). Also, spc42-Cdk-8A;spc29-Cdk-4A growth is slightly more reduced at 37°C than for either alone, and the spc42-8A;spc29-4A;mps1-8 allele is dead (see Figure 1A for growth of spc29-Cdk-4A). Both Spc42-GFP and Spc42-Cdk-8A-GFP localize as one or two foci (depending on cell cycle stage), consistent with normal SPB localization (Figure 2B), and showing that the mutation does not dramatically affect Spc42 levels. Taken together, these data suggest that phosphorylation due to the Mps1 and Cdk1-Cdc28 protein kinases is required directly or indirectly for full function of both Spc29 and Spc42. Because Spc42 is known to interact with both Spc29 and the C-terminus of Spc110 (Adams and Kilmartin, 1999; Elliott et al., 1999), we tested whether the spc42-Cdk-8A mutation affected interactions with these binding partners in a yeast two-hybrid assay. Wildtype Spc42 interacts with itself, Spc29, and the C-terminus of Spc110, as previously reported (Adams and Kilmartin, 1999; Elliott et al., 1999). We demonstrate that Spc42-Cdk-8A is able to interact with Spc42 and with Spc29 but not with the C-terminus of Spc110 (Figure 2C), suggesting that phosphorylation is important for the Spc42-Spc110 interaction.
FIGURE 2:

Mutations in Spc42 sites lying in Cdk1 motifs impair interaction with Spc110 and cause growth defects that are exacerbated by an SPB-defective mutant in MPS1, but do not impair localization. (A) Diagram of Spc42 showing position of spc42-Cdk-8A phosphorylation site mutations (asterisks) and the predicted coiled–coil domains from residues 62–131 and 251–278 (shaded boxes). spc42-Cdk-8A = S4A, T6A, T32A, S213A, S217A, S326A, T357A, T359A. Growth of cells with listed genotypes at 24 and 37°C for 1 d. (B) Localization of Spc42-GFP and Spc42-Cdk8A-GFP at 24°C. Cells are imaged with DIC, and green is GFP autofluorescence. (C) Two-hybrid analysis on -His plates of interactions between Spc42, Spc42-Cdk-8A, and Spc42-24A with Spc42, Spc29, and the carboxy terminus of Spc110 (from 741-944, “Spc110-C”).
FIGURE 4:
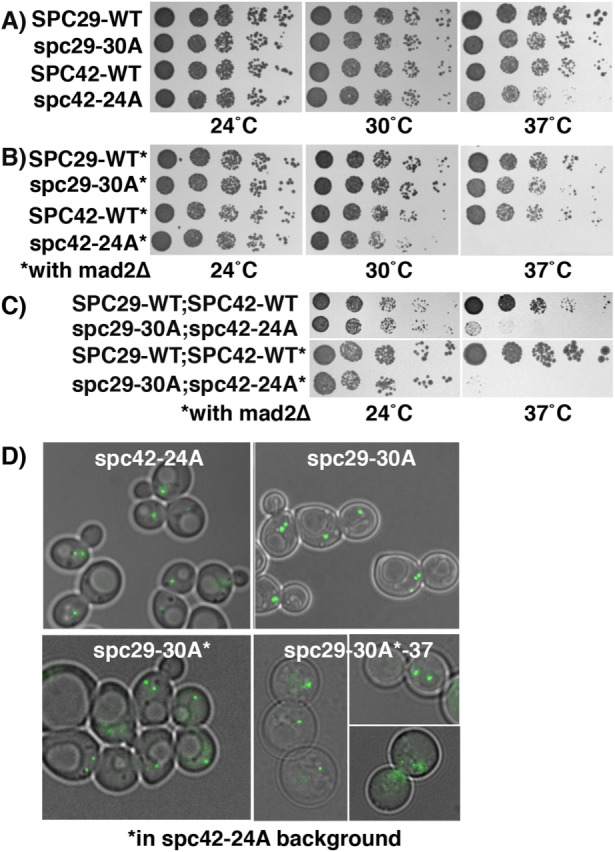
A double mutant of SPC29 and SPC42 is lethal at 37°C but does not impair SPB localization. (A) Growth of cells with listed genotypes and otherwise wildtype backgrounds at 24, 30, and 37°C for 3 d. (B) Growth of cells with listed genotypes in a mad2∆ background (*) for 3 d. (C) Growth of double integrated wild-type or mutant cells at 24 and 37°C with otherwise wild-type or mad2∆ (*) background for 3 d. (D) Localization of Spc42-24A-GFP and localization of Spc29-30A-GFP both in an otherwise wild-type background and in cells also containing spc42-24A (*). Growth in bottom right panels was at 37°C; all others at 24°C. An additional aberrant signal of Spc29-30A was observed in 38% of spc42-24A cells at 37°C (inset, bottom right), n = 50. Cells are imaged with DIC and green is GFP fluorescence.
Additional phosphorylation events are required for robust spindle formation
Next, we focused on mutations of the non-Cdk1 sites in both Spc42 and Spc29. Except for spc29-T18A and spc29-T240A, other single residue alanine mutations did not result in an apparent fitness defect (see Materials and Methods for comparison with Araki et al., 2010). We found that in comparison with the wild type, cells containing the spc29-T18A mutation showed impaired growth (24°C and 30°C), were both cold (16°C)- and heat (37°C)-sensitive, and were more compromised in the presence of mad2∆ (Figure 3A and Table 2). In addition, we show that haploid spc29-T18A cells quickly become apparent diploids (Supplemental Figure 2A), a phenotype observed for mutations in a subset of SPB genes: CDC31, MPS2, KAR1, NDC1, and MPS3, although the mechanism is not fully elucidated (Schild et al., 1981; Winey et al., 1991; Vallen et al., 1994; Chial et al., 1999; Jaspersen et al., 2002). Spc29-T18A-GFP localizes as one or two foci (Figure 3B, 24 and 37°C), suggesting that the lethality caused by spc29-T18A at 37°C is not due to mislocalization. Interestingly, Spc29 functions like the wild type when T18 is mutated to a phosphomimic aspartic acid residue, bolstering the importance of its role as a phosphorylated residue (Figure 3A and Table 2). Both T18 and the other important Mps1 site, T240, lie in the most conserved regions of Spc29 among budding yeasts (Supplemental Figure 1B, Spc29; note location of black stars above the sequence to mark sites).
FIGURE 3:
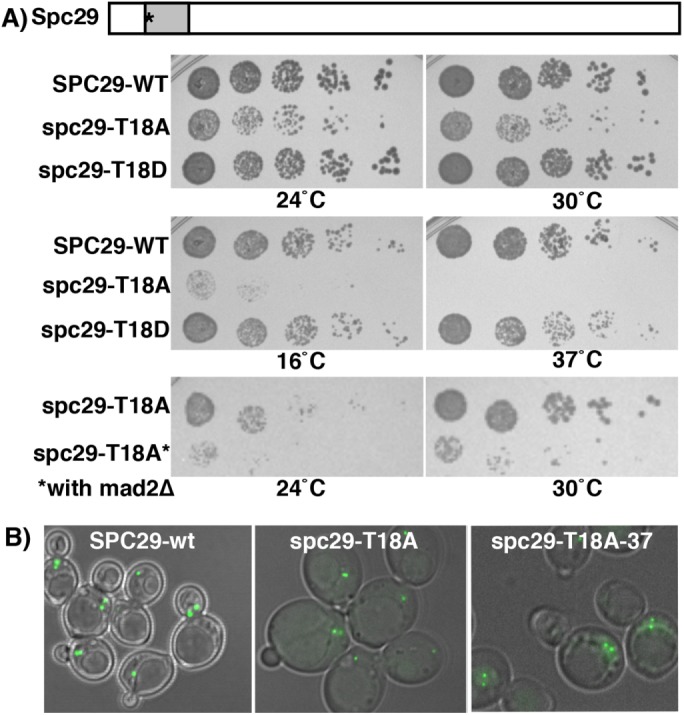
spc29-T18A confers a growth defect but does not impair SPB localization. (A) spc29-T18A cells show growth defects and are both cold- and heat-sensitive. Diagram of Spc29 showing the position of T18A phosphorylation site mutation (asterisk) and the predicted coiled–coil domain from residues 16–36 (shaded box). Growth of cells with listed genotypes (top four panels, 24, 30, 16, and 37°C): 2 d, bottom two panels (24 and 30°C): 3 d in mad2∆ background (*). (B) Localization of Spc29-GFP-WT and Spc29-T18A-GFP at 24 and 37°C Far right panel growth was at 37°C; left and middle panels were at 24°C. Cells are imaged with DIC, and green is GFP autofluorescence.
The lack of an observable growth defect in other single-residue substitution mutants could be due to the highly redundant nature of many posttranslational modifications. Therefore, we mutated the remaining in vivo sites as a group to create spc29-30A and spc42-24A, in which 30 and 24 positions in the respective proteins were mutated to alanine (Table 1). We included mutations in the minimal Cdk motifs in the SPC29 allele because they did not show any growth defects either on their own, or with mps1-8. Surprisingly, this extensive mutagenesis (12% of Spc29; 7% of Spc42) of either gene alone or combined in the same cell (spc29-30A;spc42-24A) results in functional proteins that support growth (Figure 4, A and C, 24°C, and Table 2) and localize to the SPB (Figure 4D). In addition, the levels of these altered Spc29 and Spc42 proteins are not dramatically different from those in the wild-type versions (see Figures 3 and 2, respectively). Consistent with this fitness level, analysis of the propensity to form coiled coils reveals that neither Spc29 nor Spc42 shows dramatic differences in these heavily altered proteins (Supplemental Figure 3; Lupas et al., 1991). However, at elevated temperatures, spc42-24A cells grow less than the wild type and are dead with mad2∆ (Figure 4B, 37°C, and Table 2); additionally, in a yeast two-hybrid system, Spc42-24A shows reduced interaction with the C-terminus of Spc110 (Figure 2C). The double mutant (spc29-30A; spc42-24A) is severely impaired and dead in the presence of mad2∆ at 37°C (Figure 4C and Table 2). Interestingly, under lethal conditions, at 37°C in the presence of spc42-24A, the majority of Spc29-30A-GFP is still able to localize to the SPB (Figure 4D), although there is some additional diffuse signal not at the SPB in about a third of the cells (see the inset).
To understand the defect conferred by the double mutation at 37°C, we analyzed DNA content in wild-type and spc29-30A; spc42-24A cells before and after the shift to 37°C, and cell morphology at 37°C. Compared with wild-type cells, the double mutant cells display a mitotic delay after the shift, indicated by an increase of cells with high G2-M DNA content and large buds (Supplemental Figure 4, A and B). Immunofluorescence shows that after the shift to 37°C, the large-budded spc29-30A; spc42-24A cells contained mostly short spindles (∼60%) but also single microtubule asters (with an unduplicated or two closely spaced SPBs, 25%) and elongated spindles (15%), while cells containing the integrated wild-type alleles contained a mix of short and long spindles (Figure 5). The pleiotropic effects of this double mutant allele suggests that these sites are not required for a specific step in SPB duplication or spindle formation. Further, our results imply that there is considerable plasticity or redundancy within the core SPB to accommodate large changes in amino acid sequence, as in Spc29-30A and Spc42-24A.
FIGURE 5:
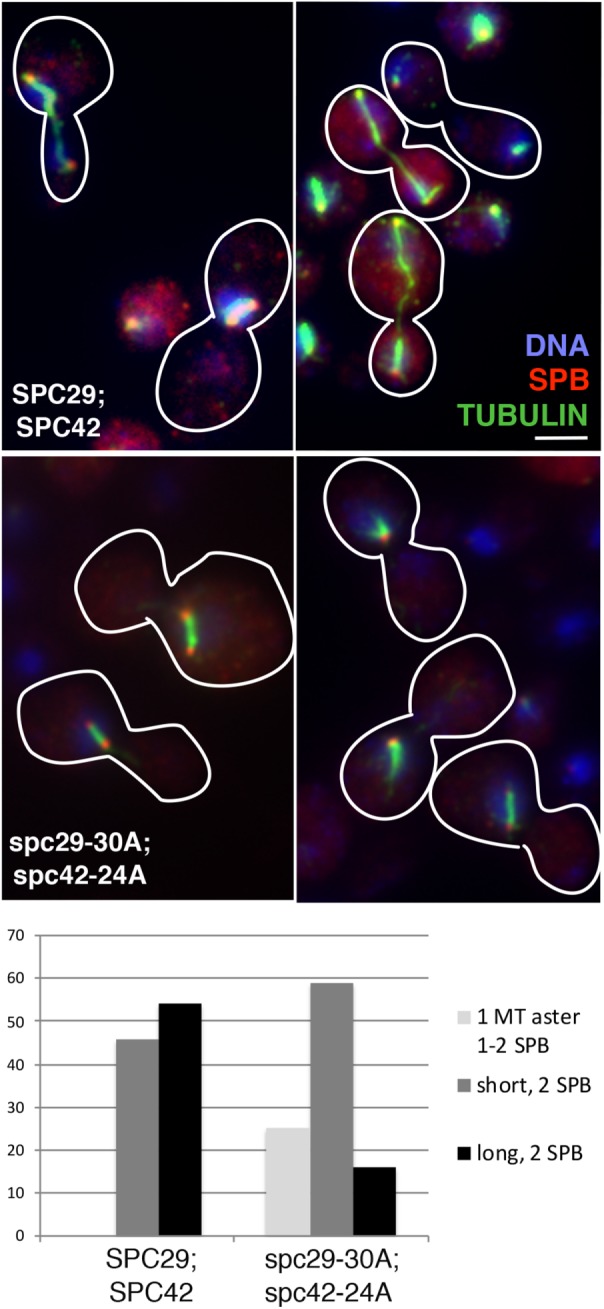
Mutation of multiple phosphorylation sites in Spc29 and Spc42 leads to SPB/spindle defects at several stages in mitosis. Immunofluorescence of cells containing either integrated wild-type alleles of SPC29 and SPC42 (SPC29;SPC42) or spc29-30A and spc42-24A alleles (spc29-30A;spc42-24A) grown for 6 h at 37°C. Bar = 2 μm. Blue = DNA (DAPI); red = SPB (Tub4); green = tubulin (Tub1). Below, different spindle phenotypes are quantified for SPC29;SPC42 (n = 100) or spc29-30A;spc42-24A (n = 134). MT = microtubule.
Phosphorylation of Spc29-T18 and of Spc29-T240 have distinct roles in SPB function
Because of the severe growth defect resulting from the point mutation spc29-T18A, we were interested in examining its effect on the SPB cycle. spc29-T18A cells were synchronized in G1 and released to 37°C, and then analyzed for DNA content and cell morphology to check cell cycle progression and subjected to immunofluorescence to examine the spindle and SPBs. Unlike wild-type cells, which are mostly small-budded and have approximately equal amounts of G1 and G2/M DNA content after a shift to 37°C, most spc29-T18A cells are large-budded and have G2/M DNA content, indicating a mitotic delay (Supplemental Figure 2, B and C, 37°C). Immunofluorescence analysis demonstrates that the majority of wild-type large-budded cells contain duplicated SPBs, an extended anaphase spindle, and separated DNA masses (Figure 6). In contrast, while all spc29-T18A large-budded cells appear to have duplicated SPBs, most have short spindles with a single DNA mass, and a subset (14%) contain a detached SPB that is not associated with microtubules or the DNA staining inside the nucleus (Figure 6, white arrowheads). The observed diploidization in spc29-T18A cells likely results from large-budded cells that contain short spindles and DNA only in one bud (inappropriate for this late stage) that undergo cytokinesis, resulting in a diploid and an aploid cell.
FIGURE 6:
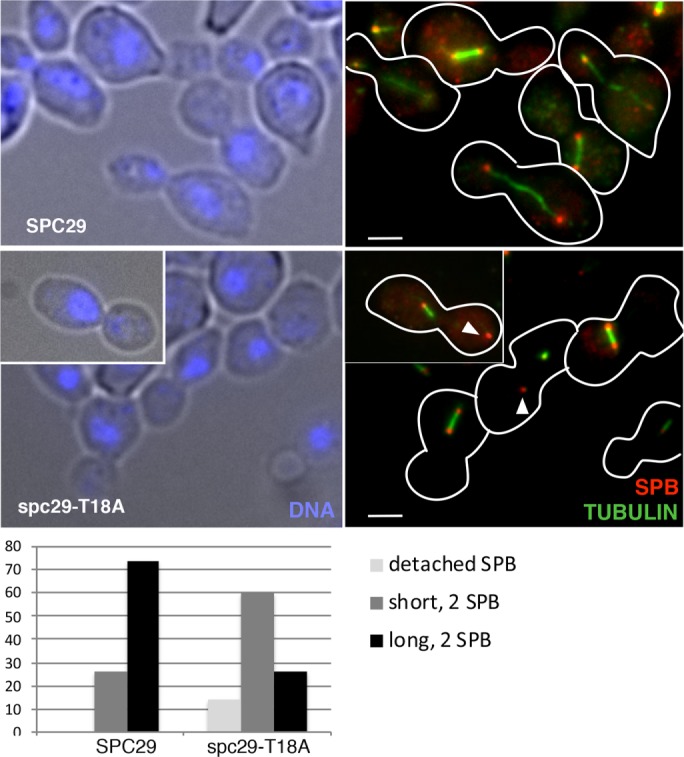
The spc29-T18A mutation causes defects in spindle elongation and in SPB insertion in a subset of cells. Immunofluorescence of SPC29 and spc29-T18A cells shifted from 24°C α-factor arrest to 37°C for 3 h: left panels, DIC/DAP1 (blue); right panels, tubulin (green), Tub4/SPB (red). Arrowheads in right bottom panel and inset show detached poles. Bar = 2 μm. Below, spindle phenotypes are quantified: SPC29 (n = 100) and spc29-T18A (n = 190).
To examine whether the structure and position of the new SPB were altered in spc29-T18A cells, we examined wild-type and spc29-T18A cells using serial thin-section electron microscopy. Wild-type (SPC29) cells displayed normal bipolar spindle assembly and SPB morphology, with both SPBs embedded in the nuclear envelope, facing toward each other in proper orientation to nucleate interdigitated microtubules (Figure 7A, black arrows and insets, n = 8). In contrast, over half of the spc29-T18A cells (n = 20) displayed dramatic defects with respect to bipolar spindle assembly and positioning of SPBs. The defects included abnormal positioning of SPBs in anaphase (Figure 7B, class I, n = 3), an early block to insertion of the duplication plaque (Figure 7B, class II, n = 4), and invagination of SPBs with large monopolar spindles (Figure 7B, class III, n = 5). In the first class of spc29-T18A cells, the aberrant pole in spc29-T18A cells (Figure 7B, class I, bottom inset) does not appear to be correctly inserted into the nuclear envelope. Also, the mother SPB and the daughter SPB are not facing each other; the lower SPB is in longitudinal view and the upper SPB is en face so that only the microtubules in cross-section are visible (Figure 7B, class I, top inset). The upper “active” SPB in the spc29-T18A class I cell appears to be nucleating more microtubules than is normal for a wild-type SPB in late anaphase (wild-type cells have 16 kinetochore and four polar microtubules for ∼20 total), consistent with a delay in mitosis (O’Toole et al., 1997). We used electron tomography to image the details of spindle structure of five large-budded spc29-T18A cells. Two cells showed large monopolar spindles with 63 (Figure 7C, left panel) and 46 microtubules, respectively. Two cells had other spindle defects such as improper bipolar spindle formation and anaphase elongation (Figure 7C, middle and right panels), and one cell appeared to have a normal bipolar spindle. In summary, spc29-T18A cells arrest at a nonpermissive temperature with a few long, but mostly short spindles with a variety of defects and a detached pole in a subset of the cells that does not nucleate nuclear microtubules.
FIGURE 7:

Electron microscopy and electron tomography of spc29-T18A cells reveals abnormal SPBs and spindle defects. Line drawings for each panel show the SPB and nuclear envelope. (A) Two sections from the same large-budded wild-type (SPC29) cell with a typical anaphase SPB orientation (arrows; insets are higher magnifications of SPBs) with microtubules forming a central spindle (n = 8). Bar = 500 nm; insets = 100 nm. (B) Electron microscopy of spc29-T18A cells (n = 20): Twelve cells were defective and a representative from each of the three classes observed is shown. Class I cells (n = 3) display mispositioning of SPBs in anaphase cells. Arrows indicate position of the SPBs; insets show a higher-magnification view. Bar = 500 nm. Class II cells (n = 4) display duplication plaque abnormalities such as delayed insertion in large budded cells (arrows; inset is higher magnification of SPB indicated by the arrow). Class III cells (n = 5) display invagination of SPBs with monopolar spindles. (C) Electron tomography of spc29-T18A cells (n = 5). Left: example of a monopolar spindle containing 63 microtubules (green = microtubules; light blue spheres = microtubules plus ends). Middle: bipolar spindle with abnormal SPB positioning. SPBs nucleate 28 and 33 microtubules (green, pink) that do not interdigitate properly. Right: late anaphase spindle contained mostly in the mother cell, resulting in a severely bowed spindle. Bar = 250 nm.
We have shown previously that spc29-T240A cells have a notably different phenotype than that just described for spc29-T18A cells. At a nonpermissive temperature, spc29-T240A cells have a complete block in SPB duplication, which results in a single SPB shown by both immunofluorescence and electron microscopy (Holinger et al., 2009). The spc29-T18A defect is also distinct from a quadruple mutant spc29-T18A,S159A,S187A,T240A. In the latter case essentially all the cells showed either a single SPB or duplicated SPBs with one detached, resulting in no bipolar spindles (Araki et al., 2010).
The observation that mutation of T18 and T240 in Spc29 does not result in the same SPB defect suggests that different steps of duplication, and thus different Spc29 interactions, may be regulated through phosphorylation of each residue. We wondered whether the early defect in SPB duplication in spc29-T240A mutant cells could be due to an inability of the mutant to interact either with itself to form multimers, or with other core factors such as Spc42. To test this idea, we examined the interactions of Spc29, Spc29-T18A/D, and Spc29-T240A with Spc29, Spc42, Bbp1, and Spc110, employing the yeast two-hybrid system, which has been used previously to observe interactions between these proteins (Elliott et al., 1999; Schramm et al., 2000). We show that none of the altered Spc29 proteins examined, including Spc29-T240A, cause a defect in Spc29–Spc29 interactions (Figure 8A). However, Spc29-T240A, but not Spc29-T18A or D, blocks the interaction between Spc29 and Spc42 (Figure 8B). This is consistent with a failure of spc29-T240A cells early in SPB duplication when Spc29 binds Spc42 at the satellite (Burns et al., 2015). We next tested interactions with Bbp1 and Spc110, which have both been shown to affect insertion (Schramm et al., 2000; Rüthnick et al., 2017). While wild-type Spc29, Spc29-T18D, and Spc29-T240A are able to interact with Bbp1, Spc29-T18A is defective (Figure 8C). Interaction with Spc110 does not appear to be disrupted by an alanine at position Spc29-T18 (T18A, Figure 8B); however, a phosphomimic aspartate residue (T18D) increases binding. Thus, our two-hybrid analysis reveals that residues Spc29-T18 and Spc29-T240 have disparate effects on binding partners of Spc29. Additional evidence suggesting that spc29-T18A may impact interactions with SPC110 comes from genetic synergism between spc29-T18A and two different C-terminal SPC110 mutations (spc110-220 and spc110-226, Figure 8D; see Figure 3A, 24°C, for spc29-T18A alone; Sundberg et al., 1996; Sundberg and Davis, 1997).
FIGURE 8:

Spc29-T18A and Spc29-T240A show different defects in interaction with Spc29 binding partners and can complement each other in a diploid. (A–C) Two-hybrid analyses on -His plates. (A) Spc29 and all mutant versions of Spc29 interact with each other robustly (with 30 mM AT). (B) Spc29-T240A disrupts interaction with Spc42 and Spc29-T18D enhances interaction with the carboxy terminus of Spc110 (from 741 to 944, “Spc110-C”). (C) Spc29-T18A disrupts interaction with Bbp1 (with 20 mM AT). (D) Spc29-T18A shows genetic interaction with spc110-220 and spc110-226 at 24°C. (E) spc29-18A and spc29-T240A (2 tandem copies) partially complement each other in a diploid at 37°C.
A prediction that stems from the idea that Spc29-T18 and Spc29-T240 have roles in different functions is that the alleles would show genetic complementation. We demonstrate using a functional in vivo test that the temperature-sensitive lethal alleles spc29-T18A and spc29-T240A:(2 tandem copies) partially complement each other when both are present in a diploid cell (Figure 8E). Collectively, the fact that mutant alleles show diverging SPB duplication defects, different protein interaction preferences, and complementation strongly suggests that these two key Mps1 sites in Spc29 have distinct roles in SPB duplication, with phosphorylation of Spc29-T240 acting earlier and affecting interaction with Spc42, presumably during satellite assembly, and phosphorylation of Spc29-T18 primarily influencing insertion of the duplicated SPB into the nuclear envelope and subsequent spindle elongation. Overall, we have shown that phosphorylation of the SPB core central plaque proteins, Spc29 and Spc42, affects a variety of steps in the pathway from a single SPB to duplicated SPBs that nucleate a bipolar spindle during mitosis.
DISCUSSION
We have completed an extensive mutational analysis of phosphorylation events on the SPB core proteins, Spc29 and Spc42. Three types of mutations were analyzed in Spc29 and Spc42: those in Cdk1 consensus sites (spc29-121A, spc42-Cdk-8A), those in known Mps1 sites in Spc29 (spc29-T18A and spc29-T240A), and those in the remaining sites that were identified in the SPB phosphoproteome study (spc42-24A, spc29-30A; Keck, Jones, et al., 2011). Interestingly, all of the mutations studied that showed growth defects were more deficient at higher temperatures. Temperature-sensitive dysfunction of altered proteins can result from a variety of causes; for example, mutation of buried residues often leads to protein instability due to disruption of protein structure and mutation of surface residues can impair protein–protein (or protein-nucleic acid) interaction (Mondal, Dastidar, et al., 2007; Bajaj et al., 2008). The fact that all our mutant GFP-fusion proteins appear to be sufficiently stable and functional to localize to the SPB suggests that binding, not stability, is the more likely mechanism of temperature sensitivity. Indeed, specific protein–protein interactions were shown to be affected both in the two mutations studied in Spc42 and in the Mps1 site mutations in Spc29. Also, it has been demonstrated previously that phosphorylation can modulate protein binding (Yu et al., 2003; Nishi et al., 2011). However, it is possible that small differences in stability between wild-type and mutant proteins may also contribute to phenotypes. The relatively less severe viability defects observed for mutations than for their defects in the two-hybrid assay (between pairs of proteins) is likely due to the additional stabilizing protein–protein interactions that exist within the context of the entire SPB.
In previous studies, either an MPS1 mutation or an spc42-S4AT6A mutation (Castillo et al., 2002; Jaspersen et al., 2004) leads to aberration in Spc42 assembly using a “super-plaque” assay, in which overexpressed Spc42 has been shown by electron microscopy to lead to an outward expansion of the central plaque beyond the SPB in a layer on the nuclear envelope (Donaldson and Kilmartin, 1996). We found, however, that mutating more of the Spc42 S/T residues that lie in Cdk1 motifs (Spc42-Cdk-8A) results in growth defects, indicating increased aberrancy either in Spc42 assembly or in another Spc42 function. It should be noted that Spc42-S4 and T6 account for the majority of phosphorylation (>95%) by Cdk1 in vitro, revealed by comparing a wild-type substrate with one containing these residues changed to alanine (Jaspersen et al., 2004). If this is an accurate reflection of in vivo Cdk1 phosphorylation, it implies that the remaining S/T-P sites are either phosphorylated at a much lower stoichiometry or potentially by another proline-directed protein kinase (Mok et al., 2010). Further study will be required to clarify the contribution of different proline-directed protein kinases to Spc42 phosphorylation.
Mutation of multiple residues not present in Cdk1 motifs in both Spc29 (spc29-30A) and Spc42 (spc42-24A) showed surprisingly mild phenotypes at lower temperatures even when combined (spc29-30A; spc42-24A) and produced GFP fusion proteins that appeared by autofluorescence to be correctly localized to the SPB. It is possible that during mitosis at permissive temperature, these phosphorylation events provide no advantage to Spc29 and Spc42. However, even when phosphorylation sites are not essential, there is evidence that they may facilitate a switchlike response of a protein (Wang et al., 2010), which, in this case, could promote progression through mitosis, particularly noticeable at higher temperatures. Finally, we observed that diploid cells containing spc42-24A were unable to produce four-spored asci, pointing to a role for these phosphorylated residues in meiosis (unpublished data), and suggesting that phosphorylation sites may also be conserved for functions other than SPB duplication during vegetative growth (Neiman, 2011).
By far the greatest effect of single alanine substitution mutations (nonphosphorylatable) in SPC29 and SPC42 is spc29-T18A and spc29-T240A, which have been shown previously to affect growth (Holinger et al., 2009; Araki et al., 2010). Importantly, we extend this work by showing that these two mutations lead to different defects both in bipolar spindle formation and in binding interactions with Spc42, Bbp1, and Spc110. In addition, the interallelic complementation of the mutations is consistent with each providing the function the other lacks as part of an oligomeric complex, most likely a trimer (Zizlsperger et al., 2008; Viswanath, Bonomi, et al., 2017). Thus our data are consistent with a model where two Mps1 sites on Spc29 play roles in two distinct successive steps in the spindle pole body assembly pathway (Figure 9). First, Mps1 phosphorylates Spc29 at T240, promoting its association with satellite-localized Spc42. Subsequently, Mps1 phosphorylates Spc29 at T18, initiating interaction with the soluble Mps2-binding protein Bbp1and thereby linking the nascent SPB with the nuclear envelope through Mps2 to begin membrane insertion. Next, phosphorylated Spc29 facilitates interaction of the Spc42-Spc29 core with nuclear Spc110 to complete insertion. Interaction of Spc29-T18-P with Spc110 is suggested by both the elevated two-hybrid signal between Spc29-T18D and Spc110 and the genetic synergism combining spc29-T18A with either spc110-220 or spc110-226. The more severe genetic combination is with spc110-220, which contains a C911R mutation in the Cmd1 binding site; a mutant allele with this same mutation (spc110-124; Stirling et al., 1996) has been shown to reduce binding of Spc110 to Spc29 (Elliott et al., 1999). FRET analysis of spc110-226, on the other hand, shows a decreased signal between Spc110 and Spc42 (Yoder et al., 2005). Either of these defects could act in combination with spc29-T18A mutation to further destabilize interaction of the central plaque with Spc110.
Mutations in the protein kinase gene MPS1 have been shown to result in a variety of defects in the SPB duplication pathway (Winey et al., 1991; Schutz and Winey, 1998; Castillo et al., 2002). The mutation mps1-8 blocks duplication at a very early step before half-bridge elongation (Figure 9). Defects caused by two other mps1 mutations, both lying in the catalytic domain of Mps1, were found to impede progression on the pathway at different steps, both from each other and from mps1-8 (Schutz and Winey, 1998). While mps1-412 resulted in a block after half-bridge elongation, mps1-737 allowed progression through the first step to production of a duplicate plaque that was not inserted into the nuclear envelope. Furthermore, mps1-737 and mps1-412 were able to complement each other in a diploid. The fact that Mps1-737 protein has the same or lower in vitro protein kinase activity as Mps1-412 argues against the earlier step simply requiring a lower level of overall kinase activity. A more likely scenario is that mps1-412 and mps1-737 negatively affect interactions with substrates that promote satellite formation and duplicate plaque insertion, respectively; while other targets are certainly possible, the C-terminus of Spc29 (at T240) and the N-terminus (at T18) would be excellent candidate substrates for such a mechanism. This mode would be similar to that of Plk4 sequentially phosphorylating Ana2/STIL, first to localize it to the centriole and subsequently to recruit Sas6 (Dzhindzhev et al., 2017).
A recent new study of SPB phosphorylation was conducted (Fong et al., 2018), primarily to increase coverage of the gamma-tubulin complex proteins by focusing on a new phosphoproteomic arrest state (G1/S), but it also included SPBs from asynchronous and mitotic cells, as in Keck, Jones, et al. (2011). While, with the use of current analysis methods and more stringent statistical cutoffs, not all of the sites described in (Keck, Jones, et al., 2011) were recovered, the most critical residues described here, Spc29-T18 and T240, as well as all the Cdk sites in both Spc29 and Spc42 (which were covered in their analysis) were shared by the two studies. Furthermore, because only a single new phosphorylated residue in these two proteins (Spc29-Y3 or S4) was identified, the contribution of the vast majority of phosphorylation in Spc29 and Spc42 has been evaluated.
The SPB core architecture model determined by Bayesian integrative structure modeling (Viswanath, Bonomi, et al., 2017) contains an interface between a region on Spc110 near its C-terminus, N-terminal Spc42, and Spc29, all three lying just beneath a layer of Cmd1. In the two major clusters of models, Spc29 is elongated and flexible, with the C-terminus likely on the cytoplasmic side. The model was derived in part by FRET and two-hybrid analysis in vivo, which were presumably influenced by the phosphorylation state of the proteins under study. Therefore, it will be interesting to learn in more detail the placement and consequences of individual phosphorylated residues in the multiple interactions between these proteins. Our work is a powerful example of the multilayered control of SPB duplication and serves as a model for analysis of centrosome phosphorylation sites.
MATERIALS AND METHODS
Strains and plasmids
Standard Escherichia coli and yeast genetic techniques were used. All yeast strains (Supplemental Table 1) were derived from W303. Null strains were made using a kanMX-marked plasmid (Longtine et al., 1998) in a PCR for transformation of a diploid wildtype strain. Mutations were made in a modified pRS304-kanMX-(TRP1) plasmid (Longtine et al., 1998) containing either Spc42 or Spc29, either using PCR-directed mutagenesis or synthesized by GenScript (genscript.com). “Wild-type” SPC29 contained two silent mutations, Bglll at position +325 and Nde1 at position +594 relative to the start of the coding sequence, and Sma1 and Sal1 restriction sites at positions –9 and +13 relative to the coding sequence, which were used for swapping domains. “Wild-type” SPC42 contains a silent Sph1 site at nucleotide position +993 relative to the start of the coding sequence and a Sma1 site added at position -43 relative to the coding sequence. These “wild-type” plasmids or mutant allele plasmids were integrated into the kanMX locus before sporulation and dissection to obtain the desired haploid mutant strains. All mutant alleles were validated as single-copy in the yeast genome unless otherwise stated. C-terminal tagging of alleles with GFP was done, again using PCR transformation with the haploid mutant strains and selecting for the introduced nutritional marker. Strains were verified by PCR and subsequent sequencing using oligonucleotide primers within the GPF gene and the gene of interest.
Mutant analysis, culture, and cytological techniques
Mutations were made at two types of sites, those present in potential Cdk1 motifs [S/T(P)] and those not in Cdk1 motifs. Most of the changes were to alanine (A), an amino acid that cannot be phosphorylated, and some were to aspartic acid (D), which can act as a phosphomimic in some cases, because of its negative charge. If a growth defect was not detected resulting from the change, mutations were combined with other mutations, either in the same gene or in genes encoding proteins known to interact with or regulate the first protein, such as a mutation in MPS1. Because spc29-T18A and spc29-T240A were already known to cause growth defects (Holinger et al., 2009; Araki et al., 2010), these alleles were not combined with mutations in other residues, but were examined individually. In many cases, mutations were also tested in the presence of a MAD2 deletion to assess the ability of the strain to grow in the absence of the spindle checkpoint. Growth of strains was measured using fivefold dilution series with cells at an initial OD/ml 0.1 (5 μl on plate) in the leftmost column and fivefold diluted for each successive column. They were grown at the indicated temperatures for the indicated numbers of days. Approximate quantification relative to a wild-type control was as follows: control strains were considered 100% and growth in each successive column to the left was considered fivefold reduced, taking into account the size of the colonies if notably different from the control. Growth experiments were often done in duplicate or with distinct strains containing the identical genotype, and were repeated at least three times for each strain, with highly similar results. Mutations spc29-T18D and spc29-S187A, which were reported as lethal mutations in previous work (Araki et al., 2010), did not show defects in our growth assays. In general, mutations studied in our strain background (W303) cause lesser growth defects than in the strain background (S288C) used in Araki et al. (2010).
For liquid culture growth, wild-type and mutant cells were grown to log phase at 24°C and either shifted to 37°C or synchronized with alpha factor before release to 37°C for the times indicated. Cells containing spc29-T18A increase their ploidy over successive cell cycles; therefore, the strain was covered with a wild-type SPC29 plasmid, which was selected against just before the experiment. Flow cytometry and budding index analysis were performed to monitor DNA content and cell morphology. For immunofluorescence, samples were fixed with 4% formaldehyde for 1 h, washed with phosphate-buffered saline, treated with zymolyase, and subjected on slides to treatment with YOL1/34 rat anti-tubulin antibody (1/200), FITC anti-rat secondary antibody (1/3000), rabbit anti-Tub4 antibody (1/500), Texas Red anti-rabbit secondary antibody (1/200), and 2-(4-amidinophenyl)-6-indolecarbamidine (DAPI) to detect DNA. In a microscope field, all large-budded cells were counted and at least 100 cells were counted for each condition. Experiments with a particular strain were repeated at least three times with highly similar results. Live cell fluorescence of cells containing GFP-fusion protein was performed on cells from 0.1 to 1 OD/ml, using differential interference contrast (DIC) and FITC channels. Cells appeared uniformly, expressing the GFP fusion, and images were taken to include budded cells. Cells were visualized on an Eclipse Ti inverted microscope (Nikon, Japan) fitted with a CFI PlanApo VC 60× H numerical aperture 1.4 objective (Nikon) and a CoolSNAP hq2 charge coupled device camera (Photometrics) was used. Images were analyzed using MetaMorph Imaging software (Molecular Devices).
Electron microscopy and electron tomography
Cells were prepared for electron microscopy using high-pressure freezing and freeze substitution as previously described (Giddings et al., 2001). Briefly, cells grown in liquid culture were collected onto a 0.45-μm Millipore filter using vacuum filtration and then high-pressure frozen using a Wohlwend Compact 02 high-pressure freezer (Technotrade International). The frozen cells were freeze-substituted in acetone containing 0.25% glutaraldehyde and 0.1% uranyl acetate and embedded in Lowicryl HM20. Serial thin sections (80 nm) were cut using a Leica UCT microtome and the grids poststained with 2% aqueous uranyl acetate and Reynold’s lead citrate and imaged using a Tecnai T12 microscope operating at 100 kV. SPBs were identified and ∼3–8 serial sections were imaged to capture both SPBs and the complete spindle. A total of eight SPC29 cells and 20 spc29-T18A cells were imaged. For electron tomography, serial semithick (250-nm) sections were collected onto slot grids and post stained in 2% aqueous uranyl acetate and Reynold’s lead citrate. Tilt series were collected using a Tecnai F30 microscope operating at 300 kV. Serial section tomograms were computed, joined, and modeled using the IMOD software package (Kremer et al., 1996; Mastronarde, 1997). A total of five spc29-T18A cells were reconstructed.
Yeast two-hybrid analysis
DNA-binding domain (BD) and activating domain (AD) constructs were made by amplifying genes of interest from the W303 strain genomic DNA or from plasmids containing mutant alleles using BamHI and SalI and ligating into pGBDU-C1(URA3) or pGAD-C1(LEU2) cleaved with the same enzymes (James et al., 1996). Plasmids were all sequenced to confirm correct sequence and cotransfected into PJ69-4A, MATa trp1-901 leu2-3 leu2-112 ura3-52 his3-200 gal4Δ gal80Δ LYS2::GAL1-HIS3 GAL2-ADE2 met2::GAL7-lacZ (James et al., 1996). Colonies were selected on -Ura-Leu plates, and growth was tested on -His plates. 3-AT (3-amino-1,2,4-triazole; Sigma-Aldrich) at 5–20 mM was added to plates to reduce background growth in the Spc29AD-containing strains (in the absence of the BD plasmid to be tested).
For diagrams in Figures 1–3, predicted coiled-coil domains were those residues with a propensity above 0.5 to be coiled–coil in 21 amino acid stretches surrounding the residue (Lupas et al., 1991).
Supplementary Material
Acknowledgments
We are grateful to Alex Stemm-Wolf for discussion and critical reading of the manuscript, Shyla Shirk for technical assistance, members of the Winey laboratory and the PO1 group for advice, and the reviewers for helpful suggestions. We acknowledge Kevin Jones and the University of Colorado, Boulder, Department of Molecular, Cellular and Developmental Biology for providing laboratory space for Michele Jones to finish this work. Electron microscopy was performed in the Boulder Electron Microscopy Facility in the Department of Molecular, Cellular and Developmental Biology, University of Colorado, Boulder. This work was supported in part by National Institutes of Health Grants PO1 GM105537 (to M.W.) and R01 GM121443 (to S.L.J.).
Abbreviations used:
- DAPI
2-(4-amidinophenyl)-6-indolecarbamidine
- DIC
differential interference contrast
- FRET
Förster resonance energy transfer
- GFP
green fluorescent protein
- SPB
spindle pole body
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E18-05-0296) on July 25, 2018.
REFERENCES
Boldface names denote co–first authors.
- Adams IR, Kilmartin JV. (1999). Localization of core spindle pole body (SPB) components during SPB duplication in Saccharomyces cerevisiae. J Cell Biol , 809–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amon A. (1999). The spindle checkpoint. Curr Opin Genet Dev , 69–75. [DOI] [PubMed] [Google Scholar]
- Araki Y, Gombos L, Migueleti SPS, Sivashanmugam L, Antony C, Schiebel E. (2010). N-terminal regions of Mps1 kinase determine functional bifurcation. J Cell Biol , 41–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki Y, Lau CK, Maekawa H, Jaspersen SL, Giddings THJ, Schiebel E, Winey M. (2006). The Saccharomyces cerevisiae spindle pole body (SPB) component Nbp1p is required for SPB membrane insertion and interacts with the integral membrane proteins Ndc1p and Mps2p. Mol Biol Cell , 1959–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avena JS, Burns S, Yu Z, Ebmeier CC, Old WM, Jaspersen SL, Winey M. (2014). Licensing of yeast centrosome duplication requires phosphoregulation of sfi1. PLoS Genet , e1004666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajaj K, Dewan PC, Chakrabarti P, Goswami D, Barua B, Baliga C, Varadarajan R. (2008). Structural correlates of the temperature sensitive phenotype derived from saturation mutagenesis studies of CcdB. Biochemistry (Mosc) , 12964–12973. [DOI] [PubMed] [Google Scholar]
- Botchkarev VVJ, Haber JE. (2018). Functions and regulation of the Polo-like kinase Cdc5 in the absence and presence of DNA damage. Curr Genet , 87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns S, Avena JS, Unruh JR, Yu Z, Smith SE, Slaughter BD, Winey M, Jaspersen SL. (2015). Structured illumination with particle averaging reveals novel roles for yeast centrosome components during duplication. ELife , e08586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byers B, Goetsch L. (1974). Duplication of spindle plaques and integration of the yeast cell cycle. Cold Spring Harb Symp Quant Biol , 123–131. [DOI] [PubMed] [Google Scholar]
- Byers B, Goetsch L. (1975). Behavior of spindles and spindle plaques in the cell cycle and conjugation of Saccharomyces cerevisiae. J Bacteriol , 511–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo AR, Meehl JB, Morgan G, Schutz-Geschwender A, Winey M. (2002). The yeast protein kinase Mps1p is required for assembly of the integral spindle pole body component Spc42p. J Cell Biol , 453–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanaugh AM, Jaspersen SL. (2017). Big lessons from little yeast: Budding and fission yeast centrosome structure, duplication, and function. Annu Rev Genet , 361–383. [DOI] [PubMed] [Google Scholar]
- Chial HJ, Giddings THJ, Siewert EA, Hoyt MA, Winey M. (1999). Altered dosage of the Saccharomyces cerevisiae spindle pole body duplication gene, NDC1, leads to aneuploidy and polyploidy. Proc Natl Acad Sci USA , 10200–10205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson AD, Kilmartin JV. (1996). Spc42p: a phosphorylated component of the S. cerevisiae spindle pole body (SPD) with an essential function during SPB duplication. J Cell Biol , 887–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzhindzhev NS, Tzolovsky G, Lipinszki Z, Abdelaziz M, Debski J, Dadlez M, Glover DM. (2017). Two-step phosphorylation of Ana2 by Plk4 is required for the sequential loading of Ana2 and Sas6 to initiate procentriole formation. Open Biol , 170247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott S, Knop M, Schlenstedt G, Schiebel E. (1999). Spc29p is a component of the Spc110p subcomplex and is essential for spindle pole body duplication. Proc Natl Acad Sci USA , 6205–6210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elserafy M, Šaric´ M, Neuner A, Lin T, Zhang W, Seybold C, Sivashanmugam L, Schiebel E. (2014). Molecular mechanisms that restrict yeast centrosome duplication to one event per cell cycle. Curr Biol , 1456–1466. [DOI] [PubMed] [Google Scholar]
- Fong KK, Zelter A, Graczyk B, Hoyt JM, Riffle M, Johnson R, MacCoss MJ, Davis TN. (2018). Novel phosphorylation states of the yeast spindle pole body. Biol Open. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giddings THJ, O’Toole ET, Morphew M, Mastronarde DN, McIntosh JR, Winey M. (2001). Using rapid freeze and freeze-substitution for the preparation of yeast cells for electron microscopy and three-dimensional analysis. Methods Cell Biol , 27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holinger EP. (2007). The Spindle Pole Body Phosphoproteome and the Importance of Phosphorylation on the S. cerevisiae Spindle Pole Body. PhD Thesis. University of Colorado, Boulder.
- Holinger EP, Old WM, Giddings THJ, Wong C, Yates JR, 3rd, Winey M. (2009). Budding yeast centrosome duplication requires stabilization of Spc29 via Mps1-mediated phosphorylation. J Biol Chem , 12949–12955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James P, Halladay J, Craig EA. (1996). Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics , 1425–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaspersen SL, Giddings TH, Winey M. (2002). Mps3p is a novel component of the yeast spindle pole body that interacts with the yeast centrin homologue Cdc31p. J Cell Biol , 945–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaspersen SL, Huneycutt BJ, Giddings THJ, Resing KA, Ahn NG, Winey M. (2004). Cdc28/Cdk1 regulates spindle pole body duplication through phosphorylation of Spc42 and Mps1. Dev Cell , 263–274. [DOI] [PubMed] [Google Scholar]
- Keck JM, Jones MH, Wong CL, Binkley J, Chen D, Jaspersen SL, Holinger EP, Xu T, Niepel M, Rout MP, et al. (2011). A cell cycle phosphoproteome of the yeast centrosome. Science , 1557–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koivomagi M, Valk E, Venta R, Iofik A, Lepiku M, Morgan DO, Loog M. (2011). Dynamics of Cdk1 substrate specificity during the cell cycle. Mol Cell , 610–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer JR, Mastronarde DN, McIntosh JR. (1996). Computer visualization of three-dimensional image data using IMOD. J Struct Biol , 71–76. [DOI] [PubMed] [Google Scholar]
- Kupke T, Malsam J, Schiebel E. (2017). A ternary membrane protein complex anchors the spindle pole body in the nuclear envelope in budding yeast. J Biol Chem , 8447–8458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A, 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. (1998). Additional modules for versatile and PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast , 953–961. [DOI] [PubMed] [Google Scholar]
- Lupas A, Van Dyke M, Stock J. (1991). Predicting coiled coils from protein sequences. Science , 1162–1164. [DOI] [PubMed] [Google Scholar]
- Mastronarde DN. (1997). Dual-axis tomography: an approach with alignment methods that preserve resolution. J Struct Biol , 343–352. [DOI] [PubMed] [Google Scholar]
- Mendenhall MD, Hodge AE. (1998). Regulation of Cdc28 cyclin-dependent protein kinase activity during the cell cycle of the yeast Saccharomyces cerevisiae. Microbiol Mol Biol Rev , 1191–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok J, Kim PM, Lam HY, Piccirillo S, Zhou X, Jeschke GR, Sheridan DL, Parker SA, Desai V, Jwa M, et al. (2010). Deciphering protein kinase specificity through large-scale analysis of yeast phosphorylation site motifs. Sci Signal , ra12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal K, Dastidar AG, Singh G, Madhusudhanan S, Gande SL, VijayRaghavan K, Varadarajan R. (2007). Design and isolation of temperature-sensitive mutants of Gal4 in yeast and Drosophila. J Mol Biol , 939–950. [DOI] [PubMed] [Google Scholar]
- Neiman AM. (2011). Sporulation in the budding yeast Saccharomyces cerevisiae. Genetics , 737–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi H, Hashimoto K, Panchenko AR. (2011). Phosphorylation in protein-protein binding: effect on stability and function. Struct Lond Engl 1993 , 1807–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Toole ET, Mastronarde DN, Giddings THJ, Winey M, Burke DJ, McIntosh JR. (1997). Three-dimensional analysis and ultrastructural design of mitotic spindles from the cdc20 mutant of Saccharomyces cerevisiae. Mol Biol Cell , 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rüthnick D, Neuner A, Dietrich F, Kirrmaier D, Engel U, Knop M, Schiebel E. (2017). Characterization of spindle pole body duplication reveals a regulatory role for nuclear pore complexes. J Cell Biol , 2425–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rüthnick D, Schiebel E. (2016). Duplication of the yeast spindle pole body once per cell cycle. Mol Cell Biol , 1324–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schild D, Ananthaswamy HN, Mortimer RK. (1981). An endomitotic effect of a cell cycle mutation of Saccharomyces cerevisiae. Genetics , 551–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramm C, Elliott S, Shevchenko A, Schiebel E. (2000). The Bbp1p-Mps2p complex connects the SPB to the nuclear envelope and is essential for SPB duplication. EMBO J , 421–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutz AR, Winey M. (1998). New alleles of the yeast MPS1 gene reveal multiple requirements in spindle pole body duplication. Mol Biol Cell , 759–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal M. (2011). Mitotic exit control: a space and time odyssey. Curr Biol , R857–R859. [DOI] [PubMed] [Google Scholar]
- Seybold C, Elserafy M, Rüthnick D, Ozboyaci M, Neuner A, Flottmann B, Heilemann M, Wade RC, Schiebel E. (2015). Kar1 binding to Sfi1 C-terminal regions anchors the SPB bridge to the nuclear envelope. J Cell Biol , 843–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma K, D’Souza RCJ, Tyanova S, Schaab C, Wis´niewski JR, Cox J, Mann M. (2014). Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Rep , 1583–1594. [DOI] [PubMed] [Google Scholar]
- Stirling DA, Rayner TF, Prescott AR, Stark MJ. (1996). Mutations which block the binding of calmodulin to Spc110p cause multiple mitotic defects. J Cell Sci (Pt 6), 1297–1310. [DOI] [PubMed] [Google Scholar]
- Sundberg HA, Davis TN. (1997). A mutational analysis identifies three functional regions of the spindle pole component Spc110p in Saccharomyces cerevisiae. Mol Biol Cell , 2575–2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundberg HA, Goetsch L, Byers B, Davis TN. (1996). Role of calmodulin and Spc110p interaction in the proper assembly of spindle pole body compenents. J Cell Biol , 111–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallen EA, Ho W, Winey M, Rose MD. (1994). Genetic interactions between CDC31 and KAR1, two genes required for duplication of the microtubule organizing center in Saccharomyces cerevisiae. Genetics , 407–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanath S, Bonomi M, Kim SJ, Klenchin V, Taylor KC, Yabut KC, Umbreit NT, Van Epps HA, Meehl J, Jones MH, et al. (2017). The molecular architecture of the yeast spindle pole body core determined by Bayesian integrative modeling. Mol Biol Cell , 3298–3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Nie Q, Enciso G. (2010). Nonessential sites improve phosphorylation switch. Biophys J , L41–L43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winey M, Goetsch L, Baum P, Byers B. (1991). MPS1 and MPS2: novel yeast genes defining distinct steps of spindle pole body duplication. J Cell Biol , 745–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder TJ, McElwain MA, Francis SE, Bagley J, Muller EGD, Pak B, O’Toole ET, Winey M, Davis TN. (2005). Analysis of a spindle pole body mutant reveals a defect in biorientation and illuminates spindle forces. Mol Biol Cell , 141–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Chini CCS, He M, Mer G, Chen J. (2003). The BRCT domain is a phospho-protein binding domain. Science , 639–642. [DOI] [PubMed] [Google Scholar]
- Zizlsperger N, Malashkevich VN, Pillay S, Keating AE. (2008). Analysis of coiled-coil interactions between core proteins of the spindle pole body. Biochemistry (Mosc) , 11858–11868. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.