

Abstract
When untransformed human cells spend >1.5 h in prometaphase under standard culture conditions, all daughters arrest in G1 despite normal division of their mothers. We investigate what happens during prolonged prometaphase that leads to daughter cell arrest in the absence of DNA damage. We find that progressive loss of anti-apoptotic MCL-1 activity and oxidative stress act in concert to partially activate the apoptosis pathway, resulting in the delayed death of some daughters and senescence for the rest. At physiological oxygen levels, longer prometaphase durations are needed for all daughters to arrest. Partial activation of apoptosis during prolonged prometaphase leads to persistent caspase activity, which activates the kinase cascade mediating the post–mitotic activation of p38. This in turn activates p53, and the consequent expression of p21stops the cell cycle. This mechanism can prevent cells suffering intractable mitotic defects, which modestly prolong mitosis but allow its completion without DNA damage, from producing future cell generations that are susceptible to the evolution of a transformed phenotype.
INTRODUCTION
The mitotic checkpoint maintains genomic integrity by blocking the metaphase–anaphase transition until all sister chromatids attach to opposite spindle poles (reviewed in Musacchio and Salmon [2007] and Lara-Gonzalez et al. [2012]). However, some problems in spindle assembly or mitotic progression eventually allow checkpoint satisfaction and could result in a completed but defective mitosis. For example, in response to low concentrations of microtubule targeting agents the checkpoint becomes satisfied after many hours even though the spindle is short and/or multipolar (Brito et al., 2008; Yang et al., 2009). For cells held in G2 and then released into mitosis, prolonging prometaphase with the Eg5 inhibitor monastrol can lead to centrosome fragmentation and mother–daughter centriole separation (Karki et al., 2017). Reduced spindle size can compromise cleavage completion, and spindle multipolarity can lead to unequal chromosome distribution. In real life, environmental toxins, radiation, or chemotherapeutic agents could produce spindle defects that result in completed but inaccurate mitoses. Also, environmental stresses that prolong prometaphase without diminishing spindle assembly/function can lead to cohesion fatigue that compromises genomic stability (reviewed in Gorbsky [2013]). Whether or not the cell has a way to protect against such mitotic defects that evade the mitotic and DNA damage checkpoints has not been clear.
Once the mitotic checkpoint is satisfied and the cell completes mitosis, it would be logical for the daughter cells to proliferate regardless of prometaphase duration. However, when untransformed cells are continuously treated with microtubule targeting agents, they remain in prometaphase for up to 40 h and after slipping into interphase arrest in G1 (Cross et al., 1995; Minn et al., 1996; Lanni and Jacks, 1998; Rieder and Maiato, 2004; Brito and Rieder, 2006; Demidenko et al., 2008). Similarly, transient (∼16–20 h) prolongations of prometaphase allow completion of mitosis but the daughter cells nevertheless arrest (Ciciarello et al., 2001; Tritarelli et al., 2004). For such grossly prolonged mitoses, daughter cell arrest can be attributed to p53 activation following DNA damage starting after ∼16 h in prometaphase (Orth et al. [2012] also see Dalton et al. [2007], Quignon et al. [2007], and Hayashi et al. [2012]). This DNA damage results from the sublethal activation of the apoptosis pathway during prometaphase and consequent caspase-activated DNase activity (Orth et al., 2012). For transformed cells held continuously in mitosis with microtubule agents such as taxol, robust activation of the apoptosis pathway during prometaphase kills them during mitosis if they do not slip into interphase too soon (reviewed in Topham and Taylor [2013]).
For untransformed human cells, we investigated the relationship between prometaphase duration of mother cells and the proliferative capacity of daughter cells under standard culture conditions (Uetake and Sluder, 2010). Daughter cells proliferated if prometaphase lasted <1.5 h. However, when cells spent >1.5 h in prometaphase, the daughters remained viable but arrested in G1 despite the normal divisions of their mothers. This daughter cell arrest occurred after prometaphase durations less than those needed for apoptotic death in mitosis for cells constitutively arrested in mitosis (Gascoigne and Taylor, 2008; Topham and Taylor, 2013; Sloss et al., 2016). The G1 arrest we observed was p38 and p53 dependent and enforced by p21 expression. The basis for the activation of a p38–p53 response after modest prolongation of prometaphase was a mystery, because we ruled out DNA damage, a high incidence of lagging chromosomes, prolonged activity of the mitotic checkpoint, and p53 accumulation during mitosis for prometaphase durations lasting up to 6 h.
When we prolonged prometaphase for >1.5 h and transiently inhibited p38 activity for at least 3 h just after mitosis, the daughter cells progressed through interphase and divided again. Remarkably, all granddaughter cells arrested in G1, even though they were born from mitoses of normal duration (Uetake and Sluder, 2010). Thus, after more than ∼1.5 h in prometaphase, there is an irreversible change in the properties of the daughter cells that cannot be “erased” by progression through interphase and mitosis.
The purpose of the present study is to gain insight into the basis for the DNA damage and mitotic defect independent G1 arrest of daughter cells after moderate prolongation of prometaphase. We investigate what happens during modestly prolonged prometaphase that leads to a durable change in the properties of daughter cells and the nature of that change.
RESULTS
We used hTERT RPE1 cells, untransformed human cells that show a qualitatively identical response to prolonged prometaphase as primary human fibroblasts (Uetake and Sluder, 2010). We used 0.08 μM nocodazole to diminish spindle size and interfere with chromosome attachment, thereby prolonging mitotic checkpoint activity. No cells slipped out of mitosis during any of the nocodazole treatments. The G1 arrest of daughter cells after prolonged prometaphase is not nocodazole specific; monastrol, taxol, and MG132 produced qualitatively similar results (Uetake and Sluder, 2010).
Death and senescence after prolonged prometaphase
Since grossly prolonged prometaphase leads to the partial activation of the apoptosis pathway (Orth et al., 2012), we tested whether this also holds true for moderate prolongations of prometaphase (up to 6 h) that do not lead to DNA damage under standard culture conditions (Uetake and Sluder, 2010). We started by following the long-term fate of individual daughter cells born of mothers spending up to 6 h in prometaphase. Asynchronous cultures were treated with nocodazole for 4 h and mitotic shake-off cells were kept in nocodazole for another 2 h. Thus, all cells spent 2–6 h in prometaphase, times sufficient to give a G1 arrest of all daughters (Uetake and Sluder, 2010). For 136 daughters continuously filmed for 303 h (almost 13 d), 69/136 (51%) eventually died (dots above the time line, Figure 1A), almost all before the culture became confluent at ∼240 h (asterisk above time line) due to the proliferation of 1% of the cells starting at 72 h (Uetake and Sluder, 2010). This resumption of proliferation in a few cells could be due to reversal of the apoptotic program (Tang et al., 2012). Figure 1B shows two examples of pronounced blebbing activity followed by cell death. To control for handling and observation conditions we treated asynchronous cells with nocodazole for 30 min, shook off mitotic cells, and then continuously followed them after drug washout. All 40 daughters proliferated, and after the culture became confluent at ∼120 h (asterisk under the time line), only 15/1776 cells (0.8%) present at the end of the film runs had died (Figure 1A, dots under the time line).
FIGURE 1:

(A) Times of individual daughter cell death after prolonged prometaphase. Points above the time line: for daughters born of 136 mothers spending 2–6 h in prometaphase, 69/136 (51%) died, and all but one died before the preparation became confluent (asterisk) due to the proliferation of a few cells starting at 72 h. Points below the time line: for daughters born of 40 mothers treated with nocodazole for 30 min, all daughters proliferated and by the end of the film runs 15/1776 (0.8%) of the progeny died, all after the culture became confluent (asterisk). Time line is hours after removal of nocodazole. (B) Two examples of the death of daughters born of mothers spending 2–6 h in prometaphase. First frames show blebbing activity and the second frames show terminal compaction of the same cells (arrows). Times are hh:mm after nocodazole removal. Phase contrast images, bar = 20 μm. (C) For daughters born of mothers held 2–6 h in prometaphase, evidence for activation of the apoptotic pathway at 18 h after nocodazole removal as detected with FITC-tagged annexin V (green contrast) for surface phosphatidylserine. Of the annexin V–positive cells, 14/68 (21%) show propidium iodide PI-positive nuclei (red contrast), indicating surface membrane permeability during later stage apoptosis (right-hand cell). Phase contrast/fluorescence image, bar = 20 μm. (D) Senescence in some G1-arrested daughter cells born of mothers held 2–6 h in prometaphase. First and second frames show examples β-galactosidase positive cells still alive at 72 and 96 h after nocodazole removal. Third frame, none of the daughter cells born of mothers treated with nocodazole for 30 min are β-galactosidase positive at 72 h. Phase contrast images, bar = 20 μm.
For daughters of mothers held 2–6 h in prometaphase, 68/109 cells (62%) showed surface exposure of phosphatidylserine by 18 h after nocodazole removal, indicative of early-stage apoptosis as detected with fluorescein isothiocyanate (FITC)-tagged annexin V (reviewed in Krysko et al. [2008]) (Figure 1C, all cells). By 96 h all of the annexin V–positive cells had propidium iodide positive nuclei indicating surface membrane permeability in later stage apoptosis. For daughters still living at 72 h, 228/242 (94%) exhibited β-galactosidase staining, indicative of senescence (Figure 1D, first panel, and middle panel for 96 h), unlike any of 312 cells in the control culture treated for 30 min with nocodazole (Figure 1D, right-hand panel). Together these results reveal that prometaphase durations of 6 h or less lead to the activation of the apoptosis pathway but not in an immediately lethal manner.
MCL-1 activity loss during prolonged prometaphase
We next investigated the basis for the partial activation of the apoptosis pathway during prometaphase. The experimental platform used is described in Uetake and Sluder (2010) and here under Materials and Methods. Briefly, we prolonged prometaphase to varying extents in mother cells and followed the proliferative capacity of their daughters. Asynchronous cultures were treated for 6 h with 0.08 μM nocodazole to diminish spindle assembly and maintain mitotic checkpoint activation. Fields of cells were continuously followed so that the times when individual cells entered mitosis were known. Those entering mitosis early spent more time in prometaphase than those entering mitosis near the end of the 6-h treatment. After drug washout, the same cells were continuously followed to determine the proliferative capacity of their daughters. By following individual cells, we precisely knew their prometaphase durations and the behavior of their daughters. All experiments each involved multiple preparations and time-lapse runs conducted on separate days.
The consequences of prolonging prometaphase without additional intervention are shown in Figure 2A. This we call the “basic” experiment. Each vertical bar represents a daughter cell and the height of the bar indicates the duration of prometaphase for its mother cell. Daughters that proliferated are shown as light-colored bars and those that arrested in G1 are shown as dark bars. Some bars are single because one of the two daughter cells crawled off the field of view. The vertical dashed line indicates the prometaphase duration of the mother cells (1.5 h) beyond which all daughters arrest in G1. Thus, the temporal tolerance of RPE1 cells for prolonged prometaphase is 1.5 h under standard culture conditions. Prometaphase in control cells averages 18 min (range 9–30 min, n = 117).
FIGURE 2:

(A) Relationship between prometaphase duration and daughter cell proliferation under standard culture conditions—the “basic” experiment (redrawn from Figure 1B of Uetake and Sluder [2010]). Asynchronous cultures were treated with nocodazole for 6 h and entry of individual cells into mitosis followed. After drug washout, daughters of previously followed mothers were continuously followed. Each vertical bar represents a daughter cell remaining in the field of view and the height of the bar indicates the prometaphase duration for its mother cell. The bars are ordered by the duration of prometaphase for the mother cells. Daughters that proliferated are shown as light-colored bars, and those that arrested in G1 are shown as dark colored bars. The vertical dashed line indicates the prometaphase duration of the mother cells (90 min) beyond which all daughter cells arrested in G1. (B) Partial inhibition of MCL1 activity during prometaphase reduces the temporal tolerance for prolonged prometaphase. Asynchronous cultures were treated with nocodazole plus MIM1 for 6 h and both drugs washed out. Significantly fewer daughters born of mothers spending <1.5 h in prometaphase proliferated relative to the basic experiment (A): p = 0.0019. For the daughters born of mothers spending >1.5 h in prometaphase, there was no significant increase in the proportion of cells that proliferated (p = 1.0). (C) Knockdown of the F-box protein FBW7 allows some daughter cells to proliferate even though their mothers spent up to 4.6 h in prometaphase. Forty-eight hours after siRNA transfection, asynchronous cultures were treated with nocodazole for 6 h and the progeny of individual mother cell were continuously followed. For three pairs of daughter cells born of mothers spending >1.5 h in prometaphase, one daughter proliferated while its sister arrested. Significantly more daughters born of mothers spending >1.5 h in prometaphase proliferated relative to the basic experiment (A): p = 0.00012. For the daughters born of mothers spending <1.5 h in prometaphase, there was no significant decrease in the proportion of cells that proliferated (p = 1.0).
The expression and hence activity levels of the antiapoptotic protein MCL-1 gradually decline during prometaphase in cells constitutively held in mitosis with nocodazole or Taxol (Harley et al., 2010; Wertz et al., 2011; Sloss et al., 2016). We tested whether reduction of MCL-1 activity influences how long mother cells must spend in prometaphase before the future daughter cells become committed to arrest in G1. We partially diminished MCL-1 activity with the specific small molecule inhibitor MIM1 at a concentration that did not kill control cells or change their cell-cycle timing. Our rationale was that partial diminishment of MCL-1 activity should reveal its influence on determining the range of prometaphase durations beyond which daughter cells arrested in G1. At 10 μM continuous exposure (IC50 4.8 μM; Cohen et al. [2012]), cells repeatedly cycled with normal morphology and timing (average 19 h, range 18–23 h, n = 41 vs. average of 20 h, range 16–26 h, n = 40 for untreated cells). This equivalency indicates that the drug per se, at the concentration used, does not lead to a G1 arrest after normal mitosis or have obvious pleiotropic activity.
We applied 10 μM MIM1 and 0.08 μM nocodazole for 6 h and then removed both. For prometaphase durations up to 48 min, none of the daughters arrested. However, for prometaphase durations 48–90 min, 24/32 (75%) of the daughters arrested (Figure 2B). For the progeny of mother cells that spent >90 min in prometaphase, all arrested. These results reveal that reduction in MCL-1 activity reduces the temporal tolerance for prolonged prometaphase.
This reduction in the temporal tolerance for prometaphase duration should not be due to residual MIM1, if any, in the daughter cells. First, continuous application of this concentration of MIM1 to control cells does not alter the cell cycle or produce a G1 arrest in any daughter cells. Second, continuous treatment with MIM1 starting at nocodazole washout does not alter the temporal tolerance for prolonged prometaphase. All daughters of mothers spending <1.5 h in prometaphase proliferated (n = 32) while all those born of mothers spending >1.5 h in prometaphase arrested (n = 21).
To back up our observations with MIM1 we knocked down MCL-1 and obtained similar results. Forty-eight hours after small interfering RNA (siRNA) transfection (knockdown efficacy shown in Supplemental Figure S1A), asynchronous cultures were treated with nocodazole for 6 h. Thirty-three of 74 (45%) daughters arrested though their mothers spent <1.5 h in prometaphase (Supplemental Figure S1C) a clear departure from the basic experiment (Figure 2A). Cells transfected with a control siRNA showed a similar response to prolonged prometaphase as untransfected cells (Figure 2A vs. Supplemental Figure S1D). Thus, reduced MCL-1 expression leads to a reduced temporal tolerance for prolonged prometaphase.
The gradual loss of MCL-1 during prolonged mitosis involves SCFFbw7-mediated degradation in some but not all cell types (Inuzuka et al., 2011; Wertz et al., 2011) (reviewed in Millman and Pagano [2011] and Sloss et al. [2016]). We next sought to slow the loss of MCL-1 during prolonged prometaphase by knocking down Fbw7. To test the practical effect of Fbw7 knockdown for prometaphase MCL1 levels in our RPE1 cells, asynchronous cultures were transfected with Fbw7 siRNA and 48 h later treated with 0.08 μM nocodazole for 3 h. Mitotic shake-off cells were then cultured for another 3 h in nocodazole so that all cells assayed spent 3–6 h in prometaphase. We also ran parallel cultures of untransfected cells for comparison. In two experiments, MCL1 levels in Fbw7 knockdown cells were 3.1 and 3.7 times higher than those of the untransfected cells (Supplemental Figure S1B), consistent with the results of Wertz et al. (2011) for the same siRNA sequence (Wei et al., 2005).
We tested the effects of Fbw7 knockdown on the temporal tolerance for prolonged prometaphase. Forty-eight hours after siRNA transfection, asynchronous cultures were treated with nocodazole for 6 h. After drug washout, 15/58 (26%) of the daughters proliferated though their mothers spent 1.5–4.5 h in prometaphase (Figure 2C). The observation that not all daughters born of mothers spending >1.5 h in prometaphase proliferated may be due to cell-to-cell variability in Fbw7 knockdown in conjunction with both continued MCL1 synthesis during mitosis, balanced against its ubiquitin-independent proteolytic degradation (Sloss et al., 2016). Although Fbw7 knockdown could alter the stability of other proteins, we did not observe any morphological or cell-cycle defects in daughter cells born of mothers spending <1.5 h in prometaphase. Additionally, FBW7 knockdown cells, not treated with nocodazole, repeatedly cycled with normal morphology and timing, indicating the lack of obvious pleiotropic effects (average 20 h, range 15–23 h, n = 12, vs. average of 20 h, range 16–26 h, n = 40, for untreated cells). Together, these results indicate that partial stabilization of MCL1 levels prolongs the temporal tolerance for prolonged prometaphase.
Other antiapoptotic Bcl-2 family members
Although the expression levels of the antiapoptotic proteins Bcl-xL, Bcl-2, and Bcl-w do not change during prometaphase, their activity diminishes during prolonged prometaphase due to phosphorylation by Cdk1 (Terrano et al. [2010] and references therein). We next tested whether activity loss for these antiapoptotic Bcl family members during prolonged prometaphase participates in the G1 arrest of daughter cells. We did this because Bcl-xL is reported to be a primary negative regulator of apoptosis during mitotic arrest of some cancer cells (Shi et al., 2011); its inhibition sensitizes cells to mitotic blockers (Bennett et al., 2016); and Bcl-2/Bcl-xL inhibition potentiates apoptosis after prolonged mitosis in transformed cells (Colin et al., 2015).
We treated asynchronous cultures with 1 μM ABT-263 (Navitoclax) plus nocodazole for 6 h and then washed both out. ABT-263 inhibits Bcl-xL, Bcl-2, and Bcl-w (IC50s: <0.5 nM, <1 nM, and <1 nM, respectively; Vogler et al. [2009]). We found that all 41 daughters born of mothers spending <1.5 h in prometaphase proliferated (Supplemental Figure S2, top). Thus, inhibition of these other antiapoptotic Bcl-2 family members does not additionally predispose daughter cells to arrest for prometaphase durations <1.5 h, unlike the case for partial MCL-1 inhibition (Figure 2B). Thirty of 67 (45%) of the daughters of mothers spending >1.5 h in prometaphase arrested but remained alive (Supplemental Figure S2, top). Another three (asterisks) died at times ranging from 63–104 h after nocodazole removal. We also observed that 22 mother cells died during prometaphase (Supplemental Figure S2, bottom), consistent with the report that continuous application of 1 μM ABT-263 promotes cancer cell killing during or just after slippage out of mitosis (Shi et al., 2011). Surprisingly, 31 daughters still in the fields of view proliferated even though their mothers spent between 1.75 and 2.75 h in prometaphase. This is consistent with reports that MCL-1 can be translated during mitosis (Sloss et al., 2016) and ABT-263 inhibition of Bcl-xL, Bcl-2, and Bcl-w can lead to increased expression and activity of MCL-1 (Williams et al., 2017).
Effectors of apoptosis
To further test whether partial activation of the apoptotic pathway leads to daughter cell arrest, we tested whether diminishing proapoptotic elements of this pathway increases the temporal tolerance of cells for prolonged prometaphase. We treated asynchronous cultures with 2.5 μM Bax channel blocker iMAC1 (IC50 520 nM) and nocodazole for 6 h and washed out both. Sixteen of 46 (35%) daughters proliferated, even though their mothers spent between 1.5 and 5 h in prometaphase (Figure 3A). The fact that not all daughters proliferated may reflect cell-to-cell variability in the loss of MCL-1, Bak activity, and incomplete inhibition of Bax channels.
FIGURE 3:

(A) Application of the Bax channel blocker iMAC1 just during prolonged prometaphase allows some daughter cells to proliferate even though their mothers spent between 1.5 and 5 h in prometaphase. Asynchronous cultures were treated with nocodazole plus 2.5 μM iMAC1 for 6 h and both drugs washed out. For two pairs of daughter cells born of mothers spending >1.5 h in prometaphase, one daughter proliferated while its sister arrested. Significantly more daughters born of mothers spending >1.5 h in prometaphase proliferated relative to the basic experiment (Figure 2A): p = 3.1 × 10–6. For the daughters born of mothers spending <1.5 h in prometaphase, there was no significant decrease in the proportion of cells that proliferated (p = 1.0). (B) Pancaspase inhibitor allows some daughter cells to proliferate even though their mothers spent between 1.5 and 5.5 h in prometaphase. Asynchronous cultures were treated with 20 μM z-VAD (OMe)-fmk for 12 h to allow drug entry/cleavage. After this agent was washed out, the cultures were treated with nocodazole for 6 h and daughter cells were continuously followed. Significantly more daughters born of mothers spending >1.5 h in prometaphase proliferated relative to the basic experiment (Figure 2A): p = 3.2 × 10–12. For the daughters born of mothers spending <1.5 h in prometaphase, there was no significant decrease in the proportion of cells that proliferated (p = 1.0).
To back up these observations we knocked down Bak and Bax with a mixture of validated siRNA constructs (Supplemental Figure S3A) and obtained similar results. Forty-eight hours after siRNA transfection, asynchronous cultures were treated with nocodazole for 6 h. We found that 9/42 (21%) daughters born from mothers spending 2–4 h in prometaphase proliferated. Together these results indicate that diminishing the apoptotic response increases the temporal tolerance for prolonged prometaphase.
To test the involvement of caspase activities in the G1 arrest of daughter cells after sufficiently prolonged prometaphase, we partially inhibited caspase activities during and after prolonged prometaphase with cell permeable z-VAD (OMe)-fmk, which is converted to the pan-caspase inhibitor z-VAD-fmk by intracellular esterase activity. We treated asynchronous cultures with 20 μM z-VAD (OMe)-fmk for 12 h to allow drug entry/cleavage and after washout treated with nocodazole for 6 h. After nocodazole removal, 46/77 (60%) of the daughters remaining in the fields of view proliferated, even though their mothers spent 1.5–5.5 h in prometaphase (Figure 3B). The arrest of other daughters may be due to insufficient caspase inhibition in those particular cells. These results point to the functional importance of caspase activity in effecting the arrest of daughter cells after prolonged prometaphase.
To back up this functional approach, we assayed for the presence of activated caspase-3, an effector caspase, during prolonged prometaphase. This caspase is expressed as an inactive procaspase that is activated by proteolytic removal of the pro-domain and cleavage of the intersubunit linker (Riedl and Shi, 2004). We treated asynchronous cultures with nocodazole for 6 h and immediately fixed them for immunofluorescence labeling with a monoclonal antibody that recognizes only the large fragment of cleaved/activated caspase-3, not full-length caspase-3 (Supplemental Figure S3C). For comparison, we did the same for parallel cultures treated for 30 min with nocodazole. We then measured the fluorescence intensity in individual mitotic cells in both populations. The distributions of individual mitotic cell gray-scale values for the two populations are shown in Supplemental Figure S3B. The mean values for the two populations are significantly different (p < 0.00001). These results reveal the presence of activated caspase-3 in mitotic cells and an increase in its levels during prolonged prometaphase.
Oxidative stress during prolonged prometaphase
Since oxidative stress can trigger apoptosis (reviewed in Wu et al. [2015] and Hampton and O’Connor [2016]) and lead to MCL-1 inactivation (Inoshita et al., 2002), we tested whether oxidative stress participates during prolonged prometaphase in arresting daughter cells. We did this because oxygen levels under standard culture conditions (∼20% or a pO2 of 140 mm Hg at sea level) is higher than physiological conditions. In vivo mammalian levels range from 3% to 14% depending on the tissue (Sen and Roy, 2010).
To increase oxidative stress, asynchronous cultures were exposed to nocodazole and the oxidant KBrO3 (1 mM) for 6 h and both washed out. Twenty-one of 30 (70%) of the daughters born of mothers spending <1.5 h in prometaphase arrested as did all daughters born of longer prometaphase durations; none later died (Figure 4A). This is not due to persistent oxidative damage because same preparation cells that were in interphase during the nocodazole/oxidant treatment cycled after KBrO3 removal with the same timing (average 20.5 h, range 14–29 h, n = 11) as cells of separate control cultures (average 21 h, range 15–28 h, n = 20). Thus, increased oxidative stress reduced prometaphase durations that lead to daughter cell arrest.
FIGURE 4:
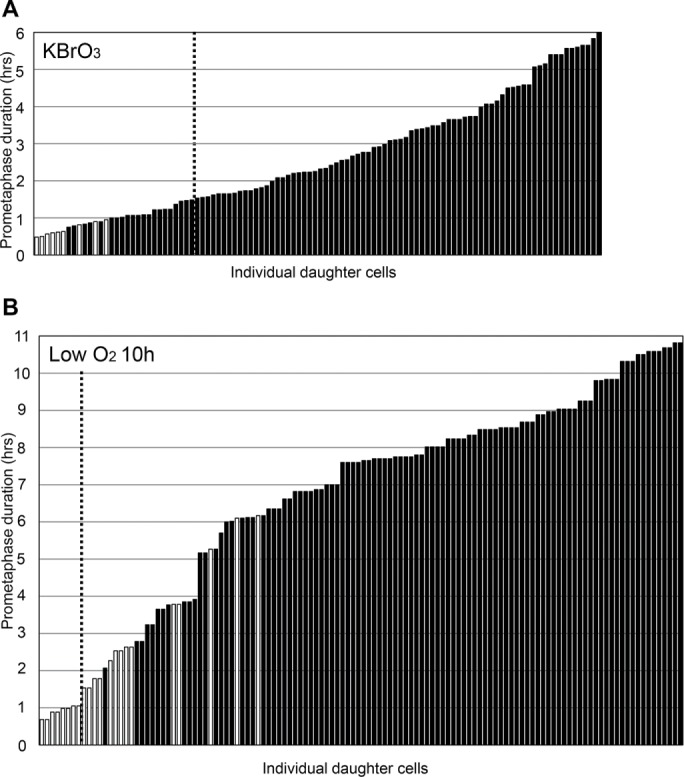
(A) Increased oxidative stress just during prometaphase reduces the temporal tolerance for prolonged prometaphase. Asynchronous cultures were treated with nocodazole plus KBrO3 for 6 h and then both were washed out. Significantly fewer daughters born of mothers spending <1.5 h in prometaphase proliferated relative to the basic experiment (Figure 2A): p = 4.8 × 10–5. For the daughters born of mothers spending >1.5 h in prometaphase, there was no significant increase in the proportion of cells that proliferated (p = 1.0). (B) Reduced oxygen level increases the temporal tolerance for prolonged prometaphase up to 6 h but does not abrogate the G1 arrest of daughter cells born of longer prometaphase durations. Asynchronous populations were treated with nocodazole for 10 h in medium equilibrated with 3% oxygen. After nocodazole washout, the daughter cells were followed at ambient oxygen levels.
We next used two means to determine whether reduced oxidative stress during mitosis would increase a cell’s tolerance for prolonged prometaphase. First, we used external application of reduced glutathione, a tripeptide antioxidant normally present at 0.5–10 mM inside cells (Franco and Cidlowski, 2009). Asynchronous cultures were exposed to nocodazole and 50 μM reduced glutathione for 6 h and both washed out. We observed the proliferation of 25/71 (35%) of the daughters born of mothers spending >1.5 h in prometaphase (Supplemental Figure S4A), a significant difference from the basic experiment (Figure 2A). Generally, cells do not import glutathione, but uptake has been reported in primary cultures of human RPE cells (Kannan et al., 2001). In contrast, external application of 50 μM oxidized glutathione did not promote the proliferation of daughters born of prometaphase durations >1.5 h (Supplemental Figure S4B). These results indicate that reduced oxidative stress can increase the temporal tolerance for prolonged prometaphase.
To further test whether reduced oxidative stress can influence the temporal tolerance for prolonged prometaphase, we worked with cells held at reduced oxygen levels. For control cells cultured at 3% oxygen, cell-cycle timing was normal (mitosis average 35 min. range 19–48 min, n = 20, and interphase 21 h, range 15–28 h, n = 20, values closely similar to those observed at atmospheric oxygen levels—mitosis average 35-min range, 18–57 min, n = 20, and interphase 20-h range, 16–26 h, n = 20).
Asynchronous cultures were treated with nocodazole for 10 h in medium equilibrated with 3% oxygen and then followed under atmospheric oxygen levels after drug washout. For daughters born of mothers spending 1.5–6.2 h in prometaphase, 14/35 (40%) proliferated while none proliferated for longer prometaphase durations (Figure 4B).
The arrest of daughter cells at low oxygen levels can occur without DNA damage. Even though 6-h prometaphase durations do not lead to DNA damage in our RPE1 cells (Uetake and Sluder, 2010), we tested for DNA damage in cells spending 6–10 h in prometaphase. Immunofluorescence localization for H2AX foci in individual daughter cells previously followed in vivo revealed that 24/50 (48%) of the daughter cells born of mothers spending 6.8–10.7 h in prometaphase showed low levels of DNA damage that should contribute to their arrest (Supplemental Figure S4C). However, the other 26/50 (52%) of this group showed no DNA damage, yet they arrested. Together, these results indicate that reduced oxygen levels can increase the temporal tolerance for prolonged prometaphase up to ∼6 h for some cells but does not prevent the arrest of cells held longer in prometaphase, even those that do not show DNA damage.
In separate experiments with up to 6-h prometaphase durations, we found that reduction of MCL1 activity at 3% oxygen shortened the temporal tolerance for prolonged prometaphase. As a baseline, we treated asynchronous cultures with nocodazole for 6 h at 3% oxygen and then followed daughter cells under atmospheric oxygen levels after drug removal. Consistent with the previous experiment, 34/52 (65%) of the daughter cells born of mothers held 1.5–6 h in prometaphase proliferated (Figure 5A). When the same experiment was conducted with 10 μM MIM1 present just during the nocodazole treatment, all daughters of mothers spending >1.5 h in prometaphase arrested (Figure 5B). For mother cells that spent 1.2–1.5 h in prometaphase, 9/11 (82%) of the daughters arrested. Together these results support the importance of MCL-1 activity loss in triggering a G1 arrest of daughter cells at physiological oxygen levels.
FIGURE 5:
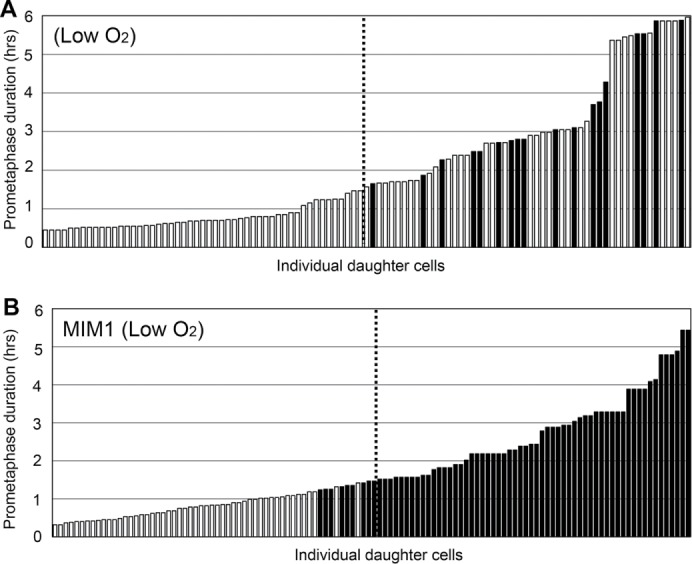
(A) Reduced oxygen increases the temporal tolerance for prometaphase prolonged up to 6 h. Asynchronous populations were treated with nocodazole for 6 h in medium equilibrated with 3% oxygen. After nocodazole removal, the daughter cells were followed at ambient oxygen levels. Significantly more daughters born of mothers spending >1.5 h in prometaphase proliferated relative to the basic experiment (Figure 2A): p = 1.5 × 10–14. For the daughters born of mothers spending <1.5 h in prometaphase, there was no significant decrease in the proportion of cells that proliferated (p = 1.0). (B) In contrast, partial inhibition of MCL1 activity at reduced oxygen level diminishes the temporal tolerance for prolonged prometaphase. Cells were treated with MIM1 plus nocodazole at ∼3% oxygen for 6 h, and after washout of both drugs, daughter cells were continuously followed at ambient oxygen levels.
Post–mitotic activation of p38
The G1 arrest of daughters after prolonged prometaphase requires p38 activity after the mother cells complete mitosis. Inhibition of p38 activity just during prolonged prometaphase does not abrogate the G1 arrest of daughters while its continuous inhibition after nocodazole washout allows all daughters to proliferate regardless of the prometaphase duration for their mothers (Uetake and Sluder, 2010). To gain insight into these observations we determined when p38 is activated. We treated asynchronous cultures with nocodazole for 4 h and cultured shake-off mitotic cells for another 2 h in nocodazole. After drug removal, aliquots of shake-off cells were processed for western blotting at 0 min (prometaphase), 40 min (anaphase/telophase), 1.5 h (early G1), and 5 h (mid to late G1). Shake-off cells from control populations showed activating phosphorylations on Thr180 and Tyr182 of p38 (Figure 6, first lane), as previously reported for cells early in mitosis (Takenaka et al., 1998; Campos et al., 2002; Fan et al., 2005; Tang et al., 2008; Lee et al., 2010). In contrast, after 2–6 h in prometaphase, activating phosphorylation levels in mitosis were low (0 min lane) and remained so through the end of mitosis (40 min lane). In early G1, activating phosphorylation levels markedly increased (1.5 h lane) and were low again by 5 h after drug removal (5 h lane). Such transient p38 activation is consistent with the report that continuous exposure to anisomycin leads to a ∼1-h activation of p38 (Lu et al., 2007). These results reveal that p38 activation occurs downstream of prolonged mitosis.
FIGURE 6:
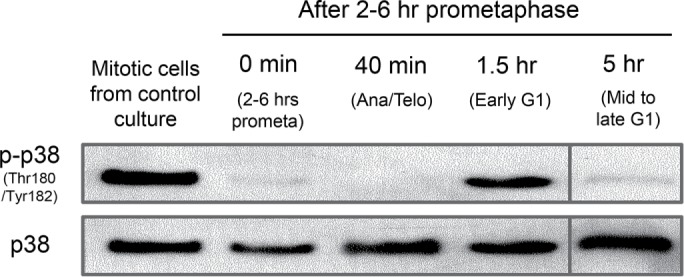
Transient post–mitotic activation of p38 in daughter cells born of mothers that spent 2–6 h in prometaphase. Western blots for activated p38, top row (phospho Thr180/Tyr182), and total p38, bottom row. First lane: mitotic shake-off cells from control culture. 0 min–5 h lanes: asynchronous cultures were treated with nocodazole for 4 h; mitotic cells were shaken off and cultured another 2 h in nocodazole before drug removal. Populations were prepared for blotting at the indicated times after nocodazole removal. 0 min lane: prometaphase cells collected just after drug removal. 40 min lane: late anaphase/telophase cells harvested 40 min after nocodazole removal. 1.5 h lane: G1 cells collected 1.5 h after nocodazole removal. 5 h lane: G1 cells harvested 5 h after nocodazole removal (from a different blot). The blots shown are representative of five experiments that showed identical results.
P38 activation after caspase inhibition
In this study, we earlier provided functional evidence that caspase activity participates in the G1 arrest of daughter cells after prolonged prometaphase. Given that p38 activity is required for daughter cell arrest after prolonged prometaphase, we tested whether caspase inhibition would influence the levels of activated p38 after prolonged mitosis. In three experiments, we treated asynchronous cultures with 20 μM z-VAD (OMe)-fmk for 12 h to allow drug entry/cleavage. After inhibitor washout, the cells were treated with nocodazole for 4 h, and mitotic shake-off cells were kept in nocodazole for another 2 h. One and a half hours after nocodazole washout, the daughter cells were processed for Western blotting to quantify phospho-p38 levels. As a control, asynchronous cultures were treated same way with nocodazole but without 20 μM z-VAD (OMe)-fmk pretreatment. For cultures treated with the caspase inhibitor, the levels of activated p38 were 42%, 64%, and 48% those of the parallel control cultures (example shown in Supplemental Figure S3D). These results reveal that caspase activity participates in the post–mitotic activation of p38. The fact that the levels of activated p38 were not zero in the caspase-inhibited cultures may be due to incomplete caspase inhibition in some cells, especially after the stress inherent in the shake off of their mothers.
Persistence of caspase activity
We previously found that when we prolonged prometaphase for >1.5 h and transiently inhibited p38 activity for at least 3 h just after mitosis to allow the daughters to get through G1, they progressed through interphase and divided again. Remarkably, all granddaughters arrested in G1, even though they were born from mitoses of normal duration (Uetake and Sluder, 2010). Thus, prolonged prometaphase can lead to an irreversible change in the properties of the daughter cells that cannot be “erased” by progression through interphase and normal mitosis.
To test whether persistent caspase activity participates in the granddaughter cell arrest, we first repeated the experiment. We treated asynchronous cultures with nocodazole for 6 h and, just after its removal, treated with the p38 inhibitor SB 203580 (10 μM) for 8 h and then washed it out. All daughter cells born of mothers spending >1.5 h in prometaphase progressed through interphase and divided again. All granddaughters arrested, confirming our previous report (Uetake and Sluder 2010) (Figure 7A). We then did the same experiment but this time included the pan-caspase inhibitor z-VAD(Ome)-fmk (20 μM) along with the SB 203580 for 8 h. For mothers spending >1.5 h in prometaphase 100/115 (87%) of their granddaughters proliferated (Figure 7B). For the same preparation mothers spending <1.5 h in prometaphase, all 60 granddaughters proliferated (Figure 7C). These results provide functional evidence that sufficiently prolonged prometaphase in the mother cell leads to persistent caspase activity that later participates in granddaughter arrest.
FIGURE 7:

Persistent caspase activity leads to granddaughter cell arrest. (A) Asynchronous cultures were treated with nocodazole for 6 h and immediately after drug washout treated with the p38 inhibitor SB 203580 for 8 h. All daughters born of mothers spending >1.5 h in prometaphase progressed through interphase and divided again. However, all granddaughters still in the fields of view arrested. For the same preparation daughters born of mothers spending <1.5 h in prometaphase all granddaughters proliferated (not diagrammed). (B) The same experiment except that the pan-caspase inhibitor z-VAD(Ome)-fmk was included during the 8 h SB 203580 treatment. All daughter cells born of mothers spending >1.5 h in prometaphase progressed through interphase and divided again, and 87% of the granddaughters proliferated. (C) For the same preparation, daughters born of mothers spending <1.5 h in prometaphase all granddaughters proliferated.
DISCUSSION
Response to prolonged prometaphase
Moderate prolongation of prometaphase leads to the activation of the apoptosis pathway in the absence of DNA damage but not in an immediately lethal manner. This apoptotic response is slow for half of the daughter cells in which it runs to completion (death 33–288 h after mother cell mitosis), and for the other half it leads to senescence. Cells can commit to either apoptotic death or senescence—with apoptosis favored by stronger stresses (Childs et al., 2014). Our observations are consistent with the report of sublethal activation of the apoptosis pathway during greatly prolonged prometaphase (Orth et al., 2012) and the finding that cells can show a graded apoptotic response in which a small fraction of the mitochondria execute MOMP leading to sublethal levels of caspase activity (Ichim et al., 2015).
Activation of the apoptotic pathway and daughter cell arrest involve the progressive loss of MCL-1 activity during prometaphase acting in concert with oxidative stress, which can promote MCL-1 inactivation (Inoshita et al., 2002). Over time, these tip the balance of pro- and anti-apoptotic activities toward a consequential, albeit partial, apoptotic response. Though oxidative stress can participate, it is not needed. At 3% oxygen levels the temporal tolerance for prolonged prometaphase is increased up to 6 h for many cells but all daughters arrest after longer prometaphase durations. At such lowered oxygen levels, the extent of MCL-1 activity loss is still important, because when we diminish its activity during prometaphase, the temporal tolerance for prolonged prometaphase is reduced from 6 to ∼1.2 h.
For cells in the living mammal, those exposed to atmospheric oxygen, such as those of a healing surface wound, ∼90 min in prometaphase may be the tipping point for a block to daughter cell proliferation. For cells inside the body that experience ∼3% oxygen levels the tolerance for prometaphase duration would be ∼6 h. Cells of tissues, such as those in lung alveoli and the pulmonary aorta, which normally experience up to 14% oxygen, may have an intermediate tolerance for prolonged prometaphase.
Although the expression levels of the antiapoptotic proteins Bcl-xL, Bcl-2, and Bcl-w do not change during prometaphase, their activity diminishes due to phosphorylation by Cdk1 (Terrano et al., 2010). Unlike partial MCL-1 inhibition, diminishment of their activities did not promote the arrest of daughters born of mothers spending <1.5 h in prometaphase. Thus, for RPE1 cells MCL1 activity loss during prolonged prometaphase more directly promotes daughter cell arrest than activity loss for these other antiapoptotic Bcl-2 family members.
In our initial study (Uetake and Sluder, 2010), we prolonged prometaphase with the proteasome inhibitor MG132 at atmospheric oxygen levels and found that all daughter cells arrested if born of mothers spending >1.5 h in prometaphase. This seems paradoxical because MG132, while present, should protect MCL-1 from proteolytic degradation, thereby allowing daughter cell proliferation regardless of prometaphase duration. An explanation for this daughter cell arrest is that mother cells spending longer in prometaphase suffer more accumulated oxidative stress. MG132 is said to induce apoptosis through the formation of reactive oxygen species (Wu et al., 2002; Han and Park, 2010; Guo and Peng, 2013). Also, Inoshita et al. (2002) report that oxidative stress induces c-Jun N-terminal kinase (JNK) activation that leads to the phosphorylation and inactivation of MCL-1. This in turn would promote daughter cell arrest despite inhibition of the proteolytic loss of MCL-1.
When prometaphase is prolonged with nocodazole or monastrol at atmospheric oxygen levels, there is a sharp break between prometaphase durations that do and do not allow daughter cell proliferation (Uetake and Sluder, 2010). For all other experimental conditions, there is a range of prometaphase durations over which some daughters arrest and some proliferate. During prolonged prometaphase, should there be any cell-to-cell variation in the changing balance of pro- and anti- apoptotic activities when cells are at the cusp of activating a consequential apoptotic response (see Flusberg and Sorger [2015]), the lack of a sharp break between daughter cell proliferation and arrest is not surprising. Under less than extreme circumstances apoptosis is not a digital live or die phenomenon; it can be partial (Orth et al., 2012; Ichim et al., 2015) and even be reversible (Tang et al., 2012). This may explain why partially stabilizing MCL-1 levels or partially inhibiting proapoptotic factors such as BAX or caspases allows some but not all of the daughters to proliferate after their mothers spent >1.5 h in prometaphase. In this regard, we noted in some experiments a few cases in which one daughter arrested while its sister proliferated after prolonged prometaphase. Since Mcl-1 activity participates in the intrinsic (mitochondrion based) apoptotic pathway, this phenomenon could be due to unequal inheritance of the small percentage of mitochondria undergoing MOMP in response to sublethal stress (Ichim et al., 2015).
The p53 stabilizing activities of 53BP1 and USP28 are required for daughter cell arrest downstream from prolonged prometaphase (Fong et al., 2016; Lambrus et al., 2016; Meitinger et al., 2016). How the p53-53BP1-USP28 complex is assembled and initiates p53 stabilization after prolonged mitosis, in the absence of DNA damage, is an intriguing issue that has yet to be investigated (reviewed in Lambrus and Holland [2017]). P53 stabilization does not appear to be due to activity of this complex during prometaphase, because we did not find an increase in p53 levels for cells held in prometaphase for almost 4 h (Uetake and Sluder, 2010). Moreover, at low oxygen levels, prometaphase durations up to 6 h allowed many daughter cells proliferate, suggesting no significant prometaphase accumulation of p53.
The prometaphase loss of 53BP1 from kinetochores (in ∼88% of the cells by 60 min into mitosis) raised the issue that this delocalization could be how cells sense prometaphase duration and trigger a block of daughter cell proliferation (Fong et al., 2016). However, under low-oxygen conditions we find that prometaphase durations up to 6 h allow many daughter cells to proliferate. Thus, daughter cells can proliferate when born from mitoses in which 53BP1 should be gone from all kinetochores well before anaphase.
For cells synchronized in G2 and then released into mitosis, prolonging prometaphase with the Eg5 inhibitor monastrol can lead to centrosome fragmentation and mother–daughter centriole separation (Karki et al., 2017). We also observed centrosome expansion during prolonged prometaphase in the presence of monastrol. However, we did not observe centriole separation or spindle multipolarity when prometaphase was prolonged up to 6 h with nocodazole.
Apoptosis and daughter cell arrest
An important question is how activation of the apoptosis pathway during prolonged prometaphase leads to the G1 arrest of the daughter cells. This arrest is dependent on p38 activity after mitosis and the subsequent activity of the p53 (Uetake and Sluder, 2010; Fong et al., 2016; Lambrus et al., 2016; Meitinger et al., 2016). Since p38 activates p53 (Cuenda and Rousseau, 2007), the question is how the apoptotic response during prometaphase activates p38. This occurs by caspase cleavage of MEKK-1, which produces a kinase active C-terminal fragment that activates MKK3 and MKK6 that in turn phosphorylate the p38 proteins at activating sites (Cardone et al., 1997; Nakagami et al., 2001; Zarubin and Han, 2005; Cuenda and Rousseau, 2007). P38 activates p53 after mitosis and 53BP1/USP28 activities stabilize p53 to allow its accumulation (Fong et al., 2016; Lambrus et al., 2016; Meitinger et al., 2016). P53 activation leads to p21 expression that arrests the cell cycle in G1.
Activation of p38 downstream of the apoptotic pathway (Nakagami et al., 2001) is supported by our finding that p38 does not show activating phosphorylations at the end of prolonged prometaphase; rather it is transiently phosphorylated ∼1.5 h after nocodazole removal. Also, inhibition of p38 activity just during prolonged prometaphase does not abrogate the G1 arrest of daughter cells while its continuous inhibition after prolonged mitosis allows all daughters to proliferate regardless of prometaphase duration for their mothers (Uetake and Sluder, 2010).
The importance of caspase activity for the G1 arrest of daughter cells after prolonged prometaphase is evident from our finding that caspase inhibition during and after prolonged prometaphase allows over half of the daughters to proliferate even though their mothers spent between 1.5 and 5.5 h in prometaphase. This is in contrast to no daughter cell proliferation in the absence of caspase inhibition after >1.5 h prometaphase for the mothers. We also found that caspase inhibition reduced the levels of p38 activation after prolonged prometaphase.
For the daughters of mother cells that spent >1.5 h in prometaphase, transient inhibition of p38 activity just after mother-cell mitosis allows them to progress through interphase and a subsequent mitosis of normal duration. Remarkably, all granddaughters arrest suggesting an irreversible change in the properties of the progeny of such mother cells (Uetake and Sluder [2010] and present study). Here we provide functional evidence that this change involves persistent caspase activity in the daughter cells. When we inhibited caspase activity starting at the transient block to p38 activity after prolonged mitosis of the mother, the great majority of the granddaughters proliferated without additional inhibition of p38.
Backing up the mitotic checkpoint
The human cell appears to have layers of protection against mitotic defects that can lead to genomic instability and the consequent risk of transformation. The mitotic checkpoint will hold cells in mitosis for hours until defects in bipolar chromosome attachment are resolved. If prometaphase is grossly prolonged, the sustained activation of the apoptotic pathway leads to DNA damage and its consequent p53 response that prevents daughter-cell proliferation. However, environmental perturbations can lead to unresolved mitotic defects that prolong prometaphase but eventually allow for mitotic checkpoint satisfaction and completion of an error prone mitosis (Brito et al., 2008). In real life, such defects could arise from environmental toxins, heat, radiation, or oxidative stress in tissues that experience higher oxygen levels. Our present results reveal that partial activation of the apoptotic pathway protects the cell from mitotic defects that modestly prolong prometaphase yet allow the satisfaction of the mitotic checkpoint before DNA damage occurs.
MATERIALS AND METHODS
Cell culture and reagents
hTERT-RPE1 cells were cultured as described previously (Uetake and Sluder, 2004). Nocodazole (#M1404), l-glutathione reduced (#G4251), l-glutathione oxidized (#G4376), and KBrO3 (#309087) were purchased from Sigma-Aldrich. MCL-1 inhibitor MIM1 (#444130), Bax channel blocker (iMAC1, #196805), SB 203580 (#559389), and pan-caspase inhibitor (z-VAD(OMe)-fmk, #627609) were purchased from EMD Millipore. ABT-263 (Navitoclax) was purchased from ApexBio. Annexin V-FITC early apoptosis detection kit (#6592) was purchased from Cell Signaling and used according to the manufacturer’s instructions. Senescence-specific β-galactosidase staining was performed according to Dimri et al. (1995) with reagents purchased from Sigma-Aldrich. Anti-p38 (#9218), anti-phospho-p38 (#9211; Thr180/Tyr182), anti-MCL-1 (#4572), anti-cleaved caspase-3 (#9664; Asp175), and anti-α-tubulin (#3873) antibodies were obtained from Cell Signaling. Western blotting was performed as described previously (Uetake and Sluder, 2010). Cells stained for β-galactosidase were observed with phase contrast optics and color images recorded with a Canon Rebel XS DSLR camera and EOS Utility software (Canon). The images were processed with Adobe Photoshop to enhance the blue color of the β-galactosidase reaction product; all images were processed identically and full frame. Immunostaining of cleaved caspase-3 was performed according to the protocol provided by the manufacturer. The cells were observed on a Leica DMI 6000B using a 20×/0.40 objective, and images were taken with a Leica DFC365 FX camera. Fluorescence intensity of Alexa fluor 488 was measured using Leica Rebel XS software.
RNA interference
Certified siRNAs against MCL-1(SignalSilence Mcl-1 siRNA I, #6315), Bak (SignalSilence Bak siRNA I, #6486), and Bax (SignalSilence Bax siRNA I, #6321) were purchased from Cell Signaling. All SignalSilence siRNA products from Cell Signaling Technology were tested by the manufacturer and shown by the company’s Western blot analysis to efficiently knock down the target proteins. Annealed siRNA against FBW7 (same sequence as in Wertz et al. [2011] and Wei et al. [2005]), and control siRNA (siGENOME Non-targeting siRNA#1) were obtained from GE Dharmacon. All siRNAs were diluted to 20 μM with nuclease-free water as a stock solution. Two microliters of stock solution was mixed with 5 μl of Lipofectamine (Invitrogen) and then introduced onto the cells for 2 h; the cells were then washed with culture medium and cultured for 48 h (Uetake and Sluder, 2010).
Prolonging prometaphase
Asynchronous cultures were mounted in observation chambers (Uetake and Sluder, 2012) for 6 h in the presence of 0.08 μM nocodazole. Fields were marked with a diamond scribe and continuously followed by video time-lapse microscopy to record when individuals entered mitosis. Cells entering mitosis assembled short bipolar spindles or monopolar spindles, both with monooriented chromosomes that prevented mitotic checkpoint satisfaction. None slipped into interphase in 6 h. Cells entering mitosis early spent several hours in prometaphase before drug washout while cells entering mitosis shortly before drug removal spent little extra time in prometaphase. After 6 h, the bottom of the observation chamber was removed, the drug washed out with fresh medium several times, and the chamber reassembled with fresh medium for further observation of the same fields for up to 100 h. In this way, we knew precisely the prometaphase duration for each mother cell—here defined as mitosis entry to anaphase onset—and the fate of her daughters. All experiments involved multiple preparations and time-lapse runs conducted on separate days. During the film runs, the medium was changed every 2 d. Live-cell imaging was conducted with Leica DMRXE microscopes equipped with phase-contrast optics (10×, 0.3 NA objectives). Images were taken with Orca ER, Orca 100 (Hamamatsu), and RetigaEXi (QImaging Corp.) cameras, acquired with HCImage software (Hamamatsu), and exported as AVI movies.
Reduced oxygen levels
Culture medium was equilibrated with an atmosphere of 3% oxygen, 5% CO2, 92% nitrogen overnight in a sealed enclosure (Modular Incubator Chamber from Billups-Rothenberg) in the incubator. Observation chambers were filled with the equilibrated media and then sealed with a cell bearing coverslip within 30 s under atmospheric conditions. At the end of 6 or 10 h, depending on the experiment, the drugs were washed out with medium that was equilibrated with atmospheric oxygen level.
Statistical analysis
Statistical tests were performed using “R” Core Team (RC Team, 2016). The significance of differences in the proliferative capacity of daughter cells between the basic experiment (Figure 2A) and other experimental conditions (Figures 2, B and C, 3, A and B, 4A, and 5A and Supplemental Figures S1, C and D, S3A, and S4, A and B) was assessed using pairwise Fisher’s exact tests on the 2 × 2 contingency tables of counts of proliferating versus nonproliferating daughter cells, separately in two bins based on prometaphase duration: a one-tailed test in the “≤90 min” bin was used to test for a decreased proportion of proliferating cells compared with the basic experiment (in which 52 of 57 daughter cells were proliferating), and a one-tailed test in the “>90 min” bin was used to test for an increased proportion of proliferating cells compared with the basic experiment (in which 0 of 54 daughter cells were proliferating). Even within these bins, if the probability of nonproliferation increases as the prometaphase duration increases, then differences in the distributions of durations between conditions can bias these statistical tests. Thus, we also performed analogous Fisher’s exact tests on subsets of cells selected to counter this bias, as detailed below; all differences that were significant at p < 0.01 remained so when using the more conservative tests, and it is these more conservative p values.
Because requiring distributions of durations to be nearly the same between conditions can result in many cells being excluded, we allowed the distributions to differ, but only in a direction that biases the one-tailed tests to be more conservative. For the “≤90 minute” bin, in which we tested for a decreased proportion of proliferating cells compared with the basic experiment, we allowed the distribution to be skewed toward shorter durations than for the basic experiment. In this case, we used the Hungarian method (function solve_LSAP in the R package clue; Hornik [2005]) to find a maximum-sized pairing of cells between experiments subject to the constraint that each cell in the basic experiment can only be paired with a cell having shorter-or-equal prometaphase duration. Viewing the pairing as a matching in a bipartite graph with cells as vertices, we assigned weight 0 to illegal edges (those that violate the constraint) and weight 1 + αe–|δ| to other edges, where δ is the difference in prometaphase durations (which gives a preference to pairings between cells with more-similar durations) and α = 1/(N + 1), where N is the lesser of the cell counts for the two experiments being compared; this α is small enough to ensure that the maximum-weight matching also has the maximum cardinality of legal edges, so that as few cells as possible are excluded. The maximum-weight matching need not be unique, so to avoid bias the algorithm was run without regard to proliferation status and with cell order randomized. For the “>90 minute” bin, in which we tested for an increased proportion of proliferating cells compared with the basic experiment, we allowed the distribution to be skewed toward longer durations than for the basic experiment, using an analogous procedure.
Supplementary Material
ACKNOWLEDGMENTS
We greatly thank Oliver King of the Wellstone Foundation at UMass Medical School for designing and conducting the statistical analysis of the results of selected experiments. This work was supported by National Institutes of Health grant GM30758 to G.S.
Abbreviations used:
- FITC
fluorescein isothiocyanate
- JNK
c-Jun N-terminal kinase
- Mcl-1
myeloid cell leukemia factor 1
- RPE
retinal pigment epithelium.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E18-01-0026) on August 22, 2018.
REFERENCES
- Bennett A, Sloss O, Topham C, Nelson L, Tighe A, Taylor SS. (2016). Inhibition of Bcl-xL sensitizes cells to mitotic blockers, but not mitotic drivers. Open Biol , 160134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brito DA, Rieder CL. (2006). Mitotic checkpoint slippage in humans occurs via cyclin B destruction in the presence of an active checkpoint. Curr Biol , 1194–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brito DA, Yang Z, Rieder CL. (2008). Microtubules do not promote mitotic slippage when the spindle assembly checkpoint cannot be satisfied. J Cell Biol , 623–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos CB, Bedard PA, Linden R. (2002). Activation of p38 mitogen-activated protein kinase during normal mitosis in the developing retina. Neuroscience , 583–591. [DOI] [PubMed] [Google Scholar]
- Cardone MH, Salvesen GS, Widmann C, Johnson G, Frisch SM. (1997). The regulation of anoikis: MEKK-1 activation requires cleavage by caspases. Cell , 315–323. [DOI] [PubMed] [Google Scholar]
- Childs BG, Baker DJ, Kirkland JL, Campisi J, van Deursen JM. (2014). Senescence and apoptosis: dueling or complementary cell fates? EMBO Rep , 1139–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciciarello M, Mangiacasale R, Casenghi M, Zaira Limongi M, D’Angelo M, Soddu S, Lavia P, Cundari E. (2001). p53 displacement from centrosomes and p53-mediated G1 arrest following transient inhibition of the mitotic spindle. J Biol Chem , 19205–19213. [DOI] [PubMed] [Google Scholar]
- Cohen NA, Stewart ML, Gavathiotis E, Tepper JL, Bruekner SR, Koss B, Opferman JT, Walensky LD. (2012). A competitive stapled peptide screen identifies a selective small molecule that overcomes MCL-1-dependent leukemia cell survival. Chem Biol , 1175–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colin DJ, Hain KO, Allan LA, Clarke PR. (2015). Cellular responses to a prolonged delay in mitosis are determined by a DNA damage response controlled by Bcl-2 family proteins. Open Biol , 140156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross SM, Sanchez CA, Morgan CA, Schimke MK, Ramel S, Idzerda RL, Raskind WH, Reid BJ. (1995). A p53-dependent mouse spindle checkpoint. Science , 1353–1356. [DOI] [PubMed] [Google Scholar]
- Cuenda A, Rousseau S. (2007). p38 MAP-kinases pathway regulation, function and role in human diseases. Biochim Biophys Acta , 1358–1375. [DOI] [PubMed] [Google Scholar]
- Dalton WB, Nandan MO, Moore RT, Yang VW. (2007). Human cancer cells commonly acquire DNA damage during mitotic arrest. Cancer Res , 11487–11492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demidenko ZN, Kalurupalle S, Hanko C, Lim CU, Broude E, Blagosklonny MV. (2008). Mechanism of G1-like arrest by low concentrations of paclitaxel: next cell cycle p53-dependent arrest with sub G1 DNA content mediated by prolonged mitosis. Oncogene , 4402–4410. [DOI] [PubMed] [Google Scholar]
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, et al. (1995). A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA , 9363–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan L, Yang X, Du J, Marshall M, Blanchard K, Ye X. (2005). A novel role of p38 alpha MAPK in mitotic progression independent of its kinase activity. Cell Cycle , 1616–1624. [DOI] [PubMed] [Google Scholar]
- Flusberg DA, Sorger PK. (2015). Surviving apoptosis: life-death signaling in single cells. Trends Cell Biol , 446–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong CS, Mazo G, Das T, Goodman J, Kim M, O’Rourke BP, Izquierdo D, Tsou MF. (2016). 53BP1 and USP28 mediate p53-dependent cell cycle arrest in response to centrosome loss and prolonged mitosis. Elife . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco R, Cidlowski JA. (2009). Apoptosis and glutathione: beyond an antioxidant. Cell Death Differ , 1303–1314. [DOI] [PubMed] [Google Scholar]
- Gascoigne KE, Taylor SS. (2008). Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell , 111–122. [DOI] [PubMed] [Google Scholar]
- Gorbsky GJ. (2013). Cohesion fatigue. Curr Biol , R986–988. [DOI] [PubMed] [Google Scholar]
- Guo N, Peng Z. (2013). MG132, a proteasome inhibitor, induces apoptosis in tumor cells. Asia Pac J Clin Oncol , 6–11. [DOI] [PubMed] [Google Scholar]
- Hampton MB, O’Connor KM. (2016). Peroxiredoxins and the regulation of cell death. Mol Cells , 72–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han YH, Park WH. (2010). The changes of reactive oxygen species and glutathione by MG132, a proteasome inhibitor affect As4.1 juxtaglomerular cell growth and death. Chem Biol Interact , 319–327. [DOI] [PubMed] [Google Scholar]
- Harley ME, Allan LA, Sanderson HS, Clarke PR. (2010). Phosphorylation of Mcl-1 by CDK1-cyclin B1 initiates its Cdc20-dependent destruction during mitotic arrest. EMBO J , 2407–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi MT, Cesare AJ, Fitzpatrick JA, Lazzerini-Denchi E, Karlseder J. (2012). A telomere-dependent DNA damage checkpoint induced by prolonged mitotic arrest. Nat Struct Mol Biol , 387–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornik K. (2005). A CLUE for CLUster Ensembles. J Stat Softw . [Google Scholar]
- Ichim G, Lopez J, Ahmed SU, Muthalagu N, Giampazolias E, Delgado ME, Haller M, Riley JS, Mason SM, Athineos D, et al. (2015). Limited mitochondrial permeabilization causes DNA damage and genomic instability in the absence of cell death. Mol Cell , 860–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoshita S, Takeda K, Hatai T, Terada Y, Sano M, Hata J, Umezawa A, Ichijo H. (2002). Phosphorylation and inactivation of myeloid cell leukemia 1 by JNK in response to oxidative stress. J Biol Chem , 43730–43734. [DOI] [PubMed] [Google Scholar]
- Inuzuka H, Fukushima H, Shaik S, Liu P, Lau AW, Wei W. (2011). Mcl-1 ubiquitination and destruction. Oncotarget , 239–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan R, Tang D, Hu J, Bok D. (2001). Glutathione transport in human retinal pigment epithelial (HRPE) cells: apical localization of sodium-dependent gsh transport. Exp Eye Res , 661–666. [DOI] [PubMed] [Google Scholar]
- Karki M, Keyhaninejad N, Shuster CB. (2017). Precocious centriole disengagement and centrosome fragmentation induced by mitotic delay. Nat Commun , 15803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krysko DV, Vanden Berghe T, Parthoens E, D’Herde K, Vandenabeele P. (2008). Methods for distinguishing apoptotic from necrotic cells and measuring their clearance. Methods Enzymol , 307–341. [DOI] [PubMed] [Google Scholar]
- Lambrus BG, Daggubati V, Uetake Y, Scott PM, Clutario KM, Sluder G, Holland AJ. (2016). A USP28-53BP1-p53-p21 signaling axis arrests growth after centrosome loss or prolonged mitosis. J Cell Biol , 143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrus BG, Holland AJ. (2017). A new mode of mitotic surveillance. Trends Cell Biol , 314–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanni JS, Jacks T. (1998). Characterization of the p53-dependent postmitotic checkpoint following spindle disruption. Mol Cell Biol , 1055–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara-Gonzalez P, Westhorpe FG, Taylor SS. (2012). The spindle assembly checkpoint. Curr Biol , R966–980. [DOI] [PubMed] [Google Scholar]
- Lee K, Kenny AE, Rieder CL. (2010). P38 mitogen-activated protein kinase activity is required during mitosis for timely satisfaction of the mitotic checkpoint but not for the fidelity of chromosome segregation. Mol Biol Cell , 2150–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu D, Chen J, Hai T. (2007). The regulation of ATF3 gene expression by mitogen-activated protein kinases. Biochem J , 559–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meitinger F, Anzola JV, Kaulich M, Richardson A, Stender JD, Benner C, Glass CK, Dowdy SF, Desai A, Shiau AK, Oegema K. (2016). 53BP1 and USP28 mediate p53 activation and G1 arrest after centrosome loss or extended mitotic duration. J Cell Biol , 155–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millman SE, Pagano M. (2011). MCL1 meets its end during mitotic arrest. EMBO Rep , 384–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minn AJ, Boise LH, Thompson CB. (1996). Expression of Bcl-xL and loss of p53 can cooperate to overcome a cell cycle checkpoint induced by mitotic spindle damage. Genes Dev , 2621–2631. [DOI] [PubMed] [Google Scholar]
- Musacchio A, Salmon ED. (2007). The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol , 379–393. [DOI] [PubMed] [Google Scholar]
- Nakagami H, Morishita R, Yamamoto K, Yoshimura SI, Taniyama Y, Aoki M, Matsubara H, Kim S, Kaneda Y, Ogihara T. (2001). Phosphorylation of p38 mitogen-activated protein kinase downstream of bax-caspase-3 pathway leads to cell death induced by high D-glucose in human endothelial cells. Diabetes , 1472–1481. [DOI] [PubMed] [Google Scholar]
- Orth JD, Loewer A, Lahav G, Mitchison TJ. (2012). Prolonged mitotic arrest triggers partial activation of apoptosis, resulting in DNA damage and p53 induction. Mol Biol Cell , 567–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quignon F, Rozier L, Lachages AM, Bieth A, Simili M, Debatisse M. (2007). Sustained mitotic block elicits DNA breaks: one-step alteration of ploidy and chromosome integrity in mammalian cells. Oncogene , 165–172. [DOI] [PubMed] [Google Scholar]
- RC Team. (2016). R: A Language and Environment for Statistical Computing, Vol. , Vienna, Austria: R Foundation for Statistical Computing. [Google Scholar]
- Rieder CL, Maiato H. (2004). Stuck in division or passing through: what happens when cells cannot satisfy the spindle assembly checkpoint. Dev Cell , 637–651. [DOI] [PubMed] [Google Scholar]
- Riedl SJ, Shi Y. (2004). Molecular mechanisms of caspase regulation during apoptosis. Nat Rev Mol Cell Biol , 897–907. [DOI] [PubMed] [Google Scholar]
- Sen CK, Roy S. (2010). Oxygenation state as a driver of myofibroblast differentiation and wound contraction: hypoxia impairs wound closure. J Invest Dermatol , 2701–2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Zhou Y, Huang HC, Mitchison TJ. (2011). Navitoclax (ABT-263) accelerates apoptosis during drug-induced mitotic arrest by antagonizing Bcl-xL. Cancer Res , 4518–4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloss O, Topham C, Diez M, Taylor S. (2016). Mcl-1 dynamics influence mitotic slippage and death in mitosis. Oncotarget , 5176–5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takenaka K, Moriguchi T, Nishida E. (1998). Activation of the protein kinase p38 in the spindle assembly checkpoint and mitotic arrest. Science , 599–602. [DOI] [PubMed] [Google Scholar]
- Tang HL, Tang HM, Mak KH, Hu S, Wang SS, Wong KM, Wong CS, Wu HY, Law HT, Liu K, et al. (2012). Cell survival, DNA damage, and oncogenic transformation after a transient and reversible apoptotic response. Mol Biol Cell , 2240–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, Yang X, Liu X. (2008). Phosphorylation of Plk1 at Ser326 regulates its functions during mitotic progression. Oncogene , 6635–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrano DT, Upreti M, Chambers TC. (2010). Cyclin-dependent kinase 1-mediated Bcl-xL/Bcl-2 phosphorylation acts as a functional link coupling mitotic arrest and apoptosis. Mol Cell Biol , 640–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topham CH, Taylor SS. (2013). Mitosis and apoptosis: how is the balance set? Curr Opin Cell Biol , 780–785. [DOI] [PubMed] [Google Scholar]
- Tritarelli A, Oricchio E, Ciciarello M, Mangiacasale R, Palena A, Lavia P, Soddu S, Cundari E. (2004). p53 localization at centrosomes during mitosis and postmitotic checkpoint are ATM-dependent and require serine 15 phosphorylation. Mol Biol Cell , 3751–3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uetake Y, Sluder G. (2004). Cell cycle progression after cleavage failure: mammalian somatic cells do not possess a “tetraploidy checkpoint.” J Cell Biol , 609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uetake Y, Sluder G. (2010). Prolonged prometaphase blocks daughter cell proliferation despite normal completion of mitosis. Curr Biol , 1666–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uetake Y, Sluder G. (2012). Practical methodology for long-term recordings of live human cells. Biomedical Optical Phase Microscopy and Nanoscopy, ed. Shaked NT, Zalevsky Z, Satterwhite LL, Cambridge, MA: Academic Press; 43–52. [Google Scholar]
- Vogler M, Dinsdale D, Dyer MJ, Cohen GM. (2009). Bcl-2 inhibitors: small molecules with a big impact on cancer therapy. Cell Death Differ , 360–367. [DOI] [PubMed] [Google Scholar]
- Wei W, Jin J, Schlisio S, Harper JW, Kaelin WG., Jr (2005). The v-Jun point mutation allows c-Jun to escape GSK3-dependent recognition and destruction by the Fbw7 ubiquitin ligase. Cancer Cell , 25–33. [DOI] [PubMed] [Google Scholar]
- Wertz IE, Kusam S, Lam C, Okamoto T, Sandoval W, Anderson DJ, Helgason E, Ernst JA, Eby M, Liu J, et al. (2011). Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature , 110–114. [DOI] [PubMed] [Google Scholar]
- Williams MM, Lee L, Hicks DJ, Joly MM, Elion D, Rahman B, McKernan C, Sanchez V, Balko JM, Stricker T, et al. (2017). Key survival factor, Mcl-1, correlates with sensitivity to combined Bcl-2/Bcl-xL blockade. Mol Cancer Res , 259–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F, Tian FJ, Lin Y. (2015). Oxidative stress in placenta: health and diseases. Biomed Res Int , 293271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HM, Chi KH, Lin WW. (2002). Proteasome inhibitors stimulate activator protein-1 pathway via reactive oxygen species production. FEBS Lett , 101–105. [DOI] [PubMed] [Google Scholar]
- Yang Z, Kenny AE, Brito DA, Rieder CL. (2009). Cells satisfy the mitotic checkpoint in Taxol, and do so faster in concentrations that stabilize syntelic attachments. J Cell Biol , 675–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarubin T, Han J. (2005). Activation and signaling of the p38 MAP kinase pathway. Cell Res , 11–18. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.