

Abstract
Mitochondria are widely recognized as sources of reactive oxygen species in animal cells, with H2O2 being of particular note because it can act not only in oxidative stress but also is important to several signalling pathways. Lesser recognized is that mitochondria can have far greater capacity to consume H2O2 than to produce it; however, the consumption of H2O2 may be kinetically constrained by H2O2 availability especially at the low nanomolar (or lower) concentrations that occur in vivo. The production of H2O2 is a function of many factors, not the least of which are respiratory substrate availability and the protonmotive force (Δp). The Δp, which is predominantly membrane potential (ΔΨ), can be a strong indicator of mitochondrial energy status, particularly if respiratory substrate supply is either not meeting or exceeding demand. The notion that mitochondria may functionally act in regulating H2O2 concentrations may be somewhat implicit but little evidence demonstrating this is available. Here we demonstrate key assumptions that are required for mitochondria to act as regulators of H2O2 by an integrated system of production and concomitant consumption. In particular we show the steady-state level of H2O2 mitochondria approach is a function of both mitochondrial H2O2 consumption and production capacity, the latter of which is strongly influenced by ΔΨ. Our results are consistent with mitochondria being able to manipulate extramitochondrial H2O2 as a means of signalling mitochondrial energetic status, in particular the Δp or ΔΨ. Such a redox-based signal could operate with some independence from other energy sensing mechanisms such as those that transmit information using the cytosolic adenylate pool.
Keywords: Reactive oxygen species (ROS), Peroxiredoxins, Antioxidants, Peroxidase, Energy sensing
Graphical abstract

1. Introduction
Hydrogen peroxide is an important signalling molecule that works via multiple molecular pathways including glutathione (GSH) linked redox switches which can coordinate multiple aspects of cellular function including the interplay between some metabolic fuel supply systems and energy sensing (reviewed in [19], [22], [27], [28]). Although mitochondria have long been recognized as one of the many cellular sites of H2O2 formation [15], [21], [6] mitochondria also have a substantial capacity to eliminate H2O2. Indeed, mitochondria from several tissues, including brain, liver, heart and skeletal muscle appear to have far greater maximal capacity to consume H2O2 than to produce it; however, in all tissues other than liver the rate of H2O2 consumption requires respiratory substrate to be maximized [10], [16], [24], [25], [30], [32], [35]. The respiration-dependency of mitochondrial H2O2 consumption comes largely from the GSH-peroxidase and thioredoxin-dependent peroxiredoxin pathways, both of which require a supply of electrons, via NADPH, to remain active [2]. The importance of mitochondrial H2O2 scavenging has been shown in mammalian cells exposed to an exogenous H2O2 challenge [9]. Knockdown of enzymes central to either mitochondrial H2O2 consumption pathways or the NADPH systems that supply the consumption pathways led to elevated cytosolic and mitochondrial H2O2 levels, indicating that mitochondrial H2O2 scavenging was contributing to cytosolic H2O2 balance [9].
Although the maximal capacity to consume H2O2 can be much higher in mitochondria than the production rates, the consumption of H2O2 becomes limited as the concentration of H2O2 declines [10], [16], [3], [30], [32]. Recently Starkov and colleagues [30] combined this concentration-dependent capacity for mitochondrial H2O2 consumption into an elegantly simple model to describe H2O2 metabolism in mouse brain mitochondria. The model assumes the rate of H2O2 production by mitochondria (VP) is set by the respiratory substrate availability and H2O2 consumption follows simple first order kinetics in regards to the concentration of H2O2, described by a single rate constant (k). Therefore as mitochondria produce H2O2 the rate of consumption should steadily increase until production and consumption are equal and a steady-state concentration of H2O2 ([H2O2]ss) becomes established.
Our group confirmed similar results using rat skeletal muscle mitochondria [32] but also expanded this simple model to account for the fact that although H2O2 traverses membranes, via aquaporins in mitochondria [23], [4], [5], compartmentalization of production and consumption away from the extramitochondrial space where measurements were being made leads to underestimations, making most extramitochondrial based values inaccurate when estimating rates of production and consumption [32].
This simple kinetic model of mitochondria as regulators of the H2O2 concentration in their surroundings, rather than being simple point sources of H2O2, has only been shown with rodent brain and skeletal muscle mitochondria using saturating levels of succinate [30], [32]. Such high levels of succinate induce a very high rate of H2O2 efflux via electron leak as superoxide production at complex I [33] but this likely far exceeds the rates of electron leak that are generally found under physiological conditions [12] or at least under non-pathological conditions [8]. A core problem in testing if mitochondria can act as regulators of H2O2 concentrations is that when rates of H2O2 efflux are low, and likely more physiologically relevant, the consumption appears to overwhelm production such that H2O2 in the medium remains well below detectable limits [30], [32]. This difficulty has prevented further evaluation of the kinetic model under conditions of lower production rates of H2O2.
However, despite analytical challenges, core assumptions of this kinetic model of mitochondrial regulation of H2O2 are still testable. For example, if mitochondria regulate the concentration of H2O2 around them, then the H2O2 concentration that mitochondria regulate towards ([H2O2]ss) should be a function of both production and consumption capacity. We have characterized in rat skeletal muscle mitochondria that production of H2O2 can be manipulated by many means whereas consumption can be pharmacologically manipulated. In the current work we capitalize on our previous characterization of rat skeletal muscle mitochondria by initially evaluating the effect of altered H2O2 consumption on [H2O2]ss using the thioredoxin reductase inhibitor auranofin. Auranofin compromises the peroxiredoxin-dependent H2O2 consumption route in isolated mitochondria [24], [32]. The auranofin inhibited mitochondria provided a system that would then let us manipulate mitochondrial bioenergetics to establish what relationship may occur between production, consumption and [H2O2]ss as we manipulated the mitochondrial energetic state.
One of the quintessential aspects of mitochondrial energy transformation is the protonmotive force (Δp), where chemical potential energy is transformed into an electrochemical gradient across the mitochondrial inner membrane. The bulk of this energy-difference across the inner membrane in mitochondria is made up of the membrane potential (ΔΨ) [14], [18]. The capacity for mitochondria to produce H2O2 from several respiratory substrates, alone or in combinations, is a function of the ΔΨ, or Δp [17], [18], [20], [26], [29], [34]. Therefore, if k for the consumption of H2O2 is relatively constant in energized mitochondria, as argued elsewhere [25], [30], [32], and VP is a function of ΔΨ, then the simple kinetic model of mitochondrial regulation would predict that the [H2O2]ss can reflect the energy gradient across the mitochondrial inner membrane. If so, this could provide a mechanistic link between the intramitochondrial energetics and extramitochondrial signalling by changes in H2O2.
The goals of the current study were two-fold: i) evaluating core assumptions, or requirements of this model of mitochondria as regulators of H2O2 and ii) testing a central hypothesis that the ΔΨ-dependency of mitochondrial H2O2 production could provide a means of communicating the magnitude of the potential energy gradient across the inner membrane, estimated as ΔΨ, to the extramitochondrial compartment via alterations in the [H2O2]ss established by mitochondria.
2. Materials and methods
Animals and mitochondrial isolation. Male Sprague-Dawley rats, approximately 180 g, were euthanized using carbon dioxide and hind leg musculature was dissected for isolation of mitochondria according to [1]. Following isolation mitochondrial protein concentration was determined using the Biuret assay with bovine serum albumin (BSA) as a standard. All animal use was approved by the University of Manitoba Fort Garry Animal Care Committee.
Mitochondrial assays. All experiments were performed at 37 °C in a medium containing 120 mM KCl, 20 mM HEPES, 5.0 mM KH2PO4, 2.5 mM MgCl2, 1.0 mM EGTA and 0.3% (w/v) of BSA with pH set at 7.2 at 37 °C. Respiration was measured in an Oroboros O2K. When applicable, mitochondrial membrane potential (ΔΨ) and apparent H2O2 production, measured as H2O2 efflux, were measured simultaneously along with respiration using the Oroboros fluorescence module. Briefly, in one chamber the ΔΨ was estimated using the quenching of 2 µM tetramethylrhodamine methylester (TMRM), dissolved in DMSO. At the same time the other chamber received equivalent substrate, ADP or inhibitor additions as the ΔΨ chamber but also had an H2O2 detection system consisting of 5 IU ml−1 of horseradish peroxidase, 25 IU ml−1 of superoxide dismutase (SOD) and 2 µM Amplex UltraRed (dissolved in DMSO). The TMRM signal was calibrated against the fluorescence prior to addition of TMRM as 0% and fully depolarized mitochondria, either fully uncoupled with FCCP or once mitochondria had fully depleted oxygen from the chamber combined with the presence of FCCP, in the presence of TMRM as 100% of the fluorescence signal. The rate of H2O2 efflux was quantitated by addition of known amounts of H2O2 to the chamber at the end of each experimental run. Representative experiments can be found elsewhere [31]. Of note, we found this level of TMRM did inhibit respiration by approximately 20%, which we assumed was not sufficient to invalidate comparisons with assays in the absence of TMRM. Presented respiration rates are the average between the presence and absence of TMRM for the stated assay condition.
Mitochondrial H2O2 balance experiments were conducted as described elsewhere [32] with the exception of using 2 µM Amplex UltraRed instead of 10 µM used in the early work. A typical experiment is shown in Fig. 1. Briefly, multiple cuvettes are set up in an Agilent Eclipse spectrofluorometer with 2 ml of medium (described above) with horseradish peroxidase and SOD at the same levels as would be used in the measurements of H2O2 efflux and monitored at 37 °C with excitation and emission wavelengths of 560 nm and 590 nm respectively. All cuvettes are monitored, however only one of the cuvettes received 2 µM Amplex UltraRed, thereby completing the H2O2 detection system, before the preincubation period prior to adding respiratory substrate. After approximately 3 min all cuvettes have respiratory substrate added (as indicated in Figures) and the one with a complete H2O2 detection system acts as a monitor of total H2O2 that has escaped the mitochondria. At different times following the addition of respiratory substrate 2 µM Amplex UltraRed is added to the other cuvettes, completing the H2O2 detection system, which determines how much of the H2O2 that escaped mitochondria remains in the cuvette based on the difference in fluorescence and a calibration curve to determine the relationship between H2O2 concentration and fluorescence. Typically, experiments were conducted with skeletal muscle mitochondrial concentration set at 0.2 mg protein ml−1.
Fig. 1.

Assays for accumulation of H2O2in mitochondrial respiration medium. Mitochondria are added, at 0.2 mg protein ml−1, to cuvettes with linking enzymes already present and fluorescence increase is monitored over time. Increases in fluorescence is a function of H2O2 outside of the mitochondrion. * indicate the addition of Amplex Ultrared, which completes the H2O2 consuming fluorescence detection system. Mitochondria incubated with rotenone (4 µM) and then energized with succinate (5 mM) have measurable rates of electron leak (combined superoxide and H2O2 production), measured as H2O2 efflux, but do not accumulate measureable H2O2 in the medium (A.). Inclusion of 1 µM auranofin (B.) leads to an increase in apparent electron leak rate and a time-dependent accumulation of H2O2 in the medium (indicated by the arrows). The appearance of H2O2 is a function of auranofin concentration (C.) Fitting data to Eq. (1) (see Materials and Methods) determines VP and k which can then be used to calculate [H2O2]ss (C.inset). Data are mean±SEM for n = 3–4 independent experiments. For clarity the SEM is omitted in the inset figure.
2.1. Statistics and analysis
Data from the mitochondrial balance experiments were fitted assuming a first-order rate of reaction for the consumers (see [30], [32] for the derivation, demonstration and explanation of this rationale). The following equation was used to fit the accumulation of H2O2 in the medium:
| (1) |
Where t = time (s), pr = mitochondrial protein concentration in g·l−1, VP = the rate of actual H2O2 production in nmol H2O2·g protein−1·s−1and k = the first order rate constant for mitochondrial H2O2 consumption in s−1·g protein −1 l. As t→∞ the [H2O2]ss can be estimated because Eq. (1) simplifies to:
| (2) |
All values are presented as mean±SEM unless otherwise indicated. When comparisons were made they were done by t-test (paired or unpaired as appropriate) using a Bonferroni correction for multiple pairwise comparisons and p < 0.05 being considered significant.
3. Results and discussion
3.1. Method and assay development
One challenge with using the succinate fuelled system used previously [30], [32] is that the very high rates of superoxide production seen in the absence of rotenone or other complex I Q-site inhibitors, termed site IQ in other works [26], [33], [7], declines rapidly post-isolation [31]. We found a similar problem of declining production post-isolation with mixes of substrates like glutamate, malate and succinate, which provide electrons via complex I and II, presumably for the same reason of a time-dependent decline in site IQ superoxide production (data not shown).
Previously we have shown that the thioredoxin reductase inhibitor auranofin led to measurable appearance of H2O2 in the medium when under the same conditions, but without auranofin, negligible H2O2 accumulation was observable [32]. Using succinate plus rotenone to energize mitochondria leads to a relatively low rate of H2O2 production but this does not show the time-dependent decline seen in the absence of rotenone [31].
The assay system used to measure H2O2 accumulation in the medium relies on the complete detection system destroying extramitochondrial H2O2 thereby preventing back diffusion and consumption within the mitochondrion [30], [32]. In the absence of auranofin, succinate plus rotenone leads to negligible accumulation of H2O2 in the medium when mitochondria are incubated in the absence of the complete detection system (Fig. 1A). However, addition of auranofin leads to a clear, time-dependent, accumulation of H2O2 in the medium even when the detection system is incomplete (Fig. 1B). Fitting the accumulation of H2O2 over time to Eq. (1) allows determination of VP and k, which based on the rationale established elsewhere [30] can be used to determine the steady-state concentration of H2O2 reached ([H2O2]ss). The [H2O2]ss is a function of the concentration of auranofin (Fig. 1C) with 1 µM appearing to approach a maximum (Fig. 1C inset). This is important because we have previously shown that concentrations of auranofin up to 2 µM do not alter mitochondrial respiration or ΔΨ with rat skeletal muscle mitochondria under the assay conditions used [24]. Therefore we assume the response we see to auranofin is a function of declining H2O2 consumption capacity and not because of altered mitochondrial bioenergetics per se. Of note, based on our previous work [24], [32] the increase in apparent production rates of H2O2 in response to auranofin are from changes in the underestimation of production because of declining competition between the intramitochondrial consumers of H2O2 and the extramitochondrial H2O2 detection system.
3.2. Integrating H2O2 production and mitochondrial energetics
Having shown that the auranofin inhibited system could reliably accumulate H2O2 in the medium we needed a means of manipulating the rate of H2O2 production. We simultaneously monitored mitochondrial ΔΨ via TMRM quenching and respiration while making parallel measurements of H2O2 efflux and respiration under conditions where the respiratory flux was manipulated. In all cases mitochondria were energized with succinate in the presence of rotenone (4 µM) and auranofin (1 µM). After stabilizing in State 2 (mitochondria with respiratory substrate alone), addition of ADP (State 3) lead to a drop in ΔΨ and marked increase in oxygen consumption (Fig. 2A). Transition to State 4o, by adding oligomycin (1 µg/ml), led to a slight inhibition of respiration and a hyperpolarization relative to the State 2 condition (Fig. 2A). Stepwise additions of the uncoupler FCCP increased respiration rate while decreasing ΔΨ.
Fig. 2.

Relationships between membrane potential (ΔΨ) and mitochondrial respiration (A.) or electron leak (B.). Isolated rat skeletal muscle mitochondria (0.2 mg ml−1) were assayed for membrane potential or electron leak as described in the Material and methods with 5 mM succinate and 4 µM rotenone plus 1 µM auranofin (State 2). Other conditions include, addition of 0.5 mM ADP (State 3) followed sequentially by 1 µg/ml oligomycin (State 4o), and two step-wise additions of FCCP (Sate 4o + FCCP). In a separate series of experiments, mitochondria were assayed under State 2 condition with either 0, 0.5, 1.0 or 5.0 mM malonate (Grey symbols). Data are mean±SEM (n = 4–5). If error bars are not visible they are obscured by the symbol.
In a separate series of experiments the substrate oxidation capacity was titrated down with the competitive inhibitor malonate while mitochondria were in State 2. Increasing malonate led to the expected declining respiration rate and ΔΨ Fig. 2A, grey symbols). These results, typical for this type of experimental manipulation of mitochondrial energetics, show that respiration rate can be altered by manipulation of the ΔΨ with ADP or uncoupler or by titrating substrate oxidation capacity. In other words, the respiratory rate and ΔΨ are not linked by a simple functional relationship. However, when the rate of electron leak is estimated as H2O2 efflux (Fig. 2B) there is a generally consistent pattern that arises wherein the apparent rate of H2O2 production declines with declining ΔΨ. The pattern between H2O2 efflux and ΔΨ occurs irrespective of the ΔΨ being decreased by addition of ADP, uncoupler or diminished substrate oxidation capacity. While some subtle differences may occur between these different conditions the salient point is that overall it did not matter how we manipulated ΔΨ to test the relationship between mitochondrial energetic state and [H2O2]ss. For this reason we chose to use the manipulation via addition of malonate which is a logistically simpler experimental design.
3.3. The influence of ΔΨ on [H2O2]ss
Using our standard assay condition of mitochondria in State 2, energized with succinate in the presence of rotenone and auranofin, we found that increasing malonate concentration led to a decline in the accumulation of H2O2 in the medium (Fig. 3) as well as a malonate concentration-dependent decline in [H2O2]ss (Fig. 3, inset).
Fig. 3.

The effect of increasing malonate concentration on the appearance of H2O2in the assay medium from mitochondria respiring on succinate. Assay conditions include auranofin in all cases and see Fig. 1 legend for further details on the assay. Data are mean±SEM (n = 5–6 except for 5 mM malonate where n = 3). The inset omits error bars for clarity. See text for additional details.
Focusing on the most responsive region of the dose-response curve for [H2O2]ss and malonate concentration (1.0 mM and less) we tested if the modelled fits using Eq. (1) were consistent with the change in [H2O2]ss being due to changes in production (VP) or consumption (k). Of note, the 5 mM malonate condition only yielded values for VPand k in half of the experiments tried, the others have too poor of a fit to be usable. For this reason we do not include any further data for 5 mM malonate in the following analysis but do include the mean result for the experiments (n = 3) that did resolve parameters to demonstrate the general pattern on H2O2 appearance in the medium in Fig. 3. Increasing malonate leads to a pattern of declining VP (Fig. 4A.) while, as assumed in the model, k remains unchanged over this range of decreasing substrate oxidation (Fig. 4B), and subsequently lower ΔΨ (Fig. 2). Therefore we can conclude that the system is behaving consistent with a core model assumption, that consumption is relatively high and stable in energized mitochondria, and the declining [H2O2]ss appears to be largely explainable by malonate altering VP.
Fig. 4.

The effect of malonate inhibition on the parameters of the kinetic model of mitochondrial H2O2metabolism. Data are the estimated parameters from the data presented in Fig. 3 and are mean+SEM (n = 5–6). The rate of H2O2 production (A.) is strongly inhibited. Values with different letters differ from each other (paired T-test with Bonferonni Post-hoc correction, p < 0.05 being considered significant). The first-order rate constant estimated for consumption (B.) is unchanged by the inhibition of substrate oxidation (p > 0.2 for all comparisons).
Plotting the malonate induced changes for [H2O2]ss in combination with our estimates of relative ΔΨ demonstrates a clear relationship between declining ΔΨ and the concentration of H2O2 that the mitochondria regulate towards (Fig. 5). Given that the rate of H2O2 formation from many respiratory substrates has a dependency on ΔΨ or Δp [17], [18], [20], [26], [29], [34] it seems possible that the pattern resolved here (Fig. 5) may have relevance to mitochondrial H2O2 metabolism in general. Albeit this will need to be evaluated using mitochondria from other tissues, species and respiring on various substrates before such pattern can be seen as truly generalizable.
Fig. 5.
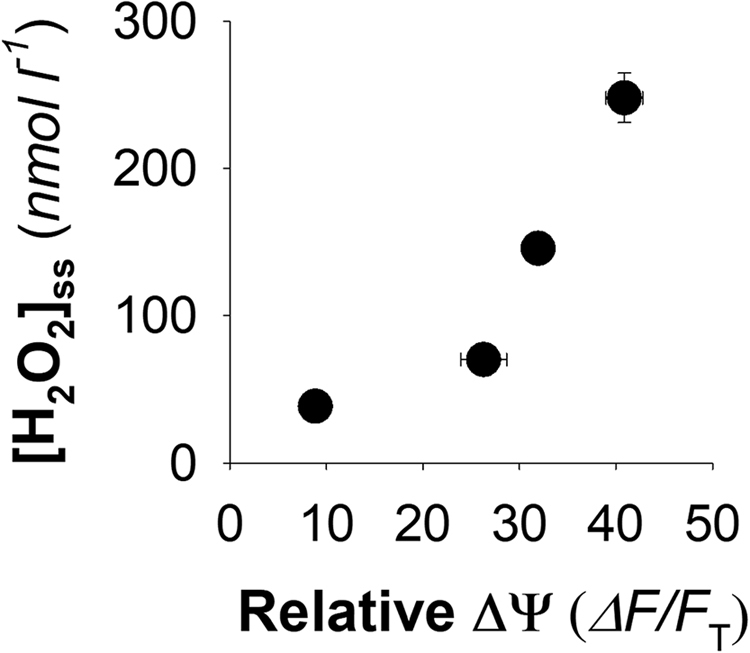
The dependency of [H2O2]sson mitochondrial ΔΨ. Data are from Fig. 2, Fig. 3 for conditions of 0, 0.5, 1.0 and 5.0 mM malonate and are mean ± SEM (n = 3–6).
4. Conclusions
Our pharmacological manipulations in this study provide essential proof of concept that, when considered an integrated whole of H2O2 production and consumption, mitochondria can act as energy-sensing regulators of H2O2. Here we have taken the model of mitochondria as regulators of H2O2 concentration proposed by Starkov and colleagues [30] and tested key core assumptions, namely: i) the [H2O2]ss is a function of the mitochondrial H2O2 consumption capacity and ii) manipulation of VP while consumption remains stable also alters [H2O2]ss. Combining i) and ii) we also establish that the [H2O2]ss can be a function of the influence ΔΨ has on VP (production) under the conditions tested, with consumption (k) being largely unaffected. The latter point could be of crucial physiological significance because this would provide a clear mechanism linking H2O2 signalling and mitochondrial energetic state. In other words, this could allow mitochondria to communicate aspects of their internal state, such as substrate availability or Δp, with the extramitochondrial compartment(s). Also noteworthy, this signalling could be independent of the already well established AMPK pathway which communicates the cytosolic energy charge but may not be directly responsive to H2O2 [13]. A mechanism that can act separately from the cellular adenylate pool supports the notion of multiple pathways of energy sensing within the cell, some reporting the adenylate potential or other reporters from this pool with other alterantive pathways using redox sensing to monitor the status of electrochemical gradients in the mitochondria. There could of course be cross-talk between these systems, for example via the influence of ADP availability on the Δp, but nevertheless an additional redox-based system may facilitate differential regulation between mitochondrial substrate demands and cytosolic ATP requirements.
An additional consideration needs to be made prior to extending the rationale of the current experiment to whole cell studies, which will be an important future step. The intermembrane space has its own H2O2 scavenging including a peroxiredoxin system [11]. While isolated mammalian muscle mitochondria should not have a means of maintaining electron supply to intermembrane space peroxiredoxins, shuttling of electrons from the cytosol to the intermembrane space NADPH pool would be expected, meaning that this would provide another sink for H2O2 localized at the mitochondrion in functioning cells.
A final caveat should also be stressed, the range of [H2O2]ss in the current study is likely far above what would be expected under physiological conditions, which are thought to be well below 10 nmol l−1 under most conditions [28]. Thus, our pharmacological manipulations provide essential proof of concept that, when considered an integrated whole of H2O2 production and consumption, mitochondria can act as energy-sensing regulators of H2O2. However, our current approaches have yet to be able to evaluate if mitochondria regulate H2O2 at physiologically relevant concentrations, which will be an important step for future research in this area.
Acknowledgements
We thank the staff of the Biological Sciences Animal Housing Facility for animal care and husbandry. Lilian Wiens and Nahid Tamanna provided helpful technical assistance during the early stages of these experiments.
Acknowledgments
Funding
This work was funded by a Canadian Natural Sciences and Engineering Research Council (NSERC) Discovery Grant (#418503) and funds from the University of Manitoba, Faculty of Science (Fund# 319254) and JRT is the Canada Research Chair in Environment Dynamics and Metabolism (Grant # 223744). This research was also supported by awards to KB, who was the recipient of a University of Manitoba, Faculty of Science Undergraduate Summer Research Award and PS, who received an NSERC Undergraduate Summer Research Award.
References
- 1.Affourtit C., Quinlan C., Brand M. Measurement of proton leak and electron leak in isolated mitochondria. In: Palmeira C.M., Moreno A.N.J., editors. Mitochondrial Bioenergetics. Humana Press; 2012. pp. 165–182. [DOI] [PubMed] [Google Scholar]
- 2.Andreyev A.Y., Kushnareva Y., Starkov A.A. Mitochondrial metabolism of reactive oxygen species. Biochemistry. 2005;70:200–214. doi: 10.1007/s10541-005-0102-7. [DOI] [PubMed] [Google Scholar]
- 3.Banh S., Treberg J.R. The pH sensitivity of H2O2 metabolism in skeletal muscle mitochondria. FEBS Lett. 2013;587:1799–1804. doi: 10.1016/j.febslet.2013.04.035. [DOI] [PubMed] [Google Scholar]
- 4.Bienert G.P., Møller A.L.B., Kristiansen K.A., Schulz A., Møller I.M., Schjoerring J.K., Jahn T.P. Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J. Biol. Chem. 2007;282:1183–1192. doi: 10.1074/jbc.M603761200. [DOI] [PubMed] [Google Scholar]
- 5.Bienert G.P., Schjoerring J.K., Jahn T.P. Membrane transport of hydrogen peroxide. Biochim. Biophys. Acta (BBA) - Biomembr. 2006;1758:994–1003. doi: 10.1016/j.bbamem.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 6.Boveris A., Oshino N., Chance B. The cellular production of hydrogen peroxide. Biochem. J. 1972;128:617–630. doi: 10.1042/bj1280617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brand M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016;100:14–31. doi: 10.1016/j.freeradbiomed.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 8.Chouchani E.T., Pell V.R., Gaude E., Aksentijevic D., Sundier S.Y., Robb E.L., Logan A., Nadtochiy S.M., Ord E.N.J., Smith A.C., Eyassu F., Shirley R., Hu C.H., Dare A.J., James A.M., Rogatti S., Hartley R.C., Eaton S., Costa A.S.H., Brookes P.S., Davidson S.M., Duchen M.R., Saeb-Parsy K., Shattock M.J., Robinson A.J., Work L.M., Frezza C., Krieg T., Murphy M.P. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014;515:431–435. doi: 10.1038/nature13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dey S., Sidor A., O'Rourke B. Compartment-specific control of reactive oxygen species scavenging by antioxidant pathway enzymes. J. Biol. Chem. 2016;291:11185–11197. doi: 10.1074/jbc.M116.726968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Drechsel D.A., Patel M. Respiration-dependent H2O2 removal in brain mitochondria via the thioredoxin/peroxiredoxin system. J. Biol. Chem. 2010;285:27850–27858. doi: 10.1074/jbc.M110.101196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gomes F., Palma F.R., Barros M.H., Tsuchida E.T., Turano H.G., Alegria T.G., Demasi M., Netto L.E. Proteolytic cleavage by the inner membrane peptidase (IMP) complex or Oct1 peptidase controls the localization of the yeast peroxiredoxin Prx1 to distinct mitochondrial compartments. J. Biol. Chem. 2017;292:17011–17024. doi: 10.1074/jbc.M117.788588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goncalves R.L.S., Quinlan C.L., Perevoshchikova I.V., Hey-Mogensen M., Brand M.D. Sites of superoxide and hydrogen peroxide production by muscle mitochondria assessed ex vivo under conditions mimicking rest and exercise. J. Biol. Chem. 2015;290:209–227. doi: 10.1074/jbc.M114.619072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hawley S.A., Ross F.A., Chevtzoff C., Green K.A., Evans A., Fogarty S., Towler M.C., Brown L.J., Ogunbayo O.A., Evans A.M., Hardie D.G. Use of cells expressing γ subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab. 2010;11:554–565. doi: 10.1016/j.cmet.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holian A., Wilson D.F. Relationship of transmembrane pH and electrical gradients with respiration and adenosine 5'-triphosphate synthesis in mitochondria. Biochemistry. 1980;19:4213–4221. doi: 10.1021/bi00559a012. [DOI] [PubMed] [Google Scholar]
- 15.Jensen P.K. Antimycin-insensitive oxidation of succinate and reduced nicotinamide-adenine dinucleotide in electron-transport particles I. pH dependency and hydrogen peroxide formation. Biochim. Biophys. Acta (BBA) - Enzymol. Biol. Oxid. 1966;122:157–166. doi: 10.1016/0926-6593(66)90057-9. [DOI] [PubMed] [Google Scholar]
- 16.Kamunde C., Sharaf M., MacDonald N. H2O2 metabolism in liver and heart mitochondria: low emitting-high scavenging and high emitting-low scavenging systems. Free Radic. Biol. Med. 2018;124:135–148. doi: 10.1016/j.freeradbiomed.2018.05.064. [DOI] [PubMed] [Google Scholar]
- 17.Korshunov S.S., Skulachev V.P., Starkov A.A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997;416:15–18. doi: 10.1016/s0014-5793(97)01159-9. [DOI] [PubMed] [Google Scholar]
- 18.Lambert A.J., Brand M.D. Superoxide production by NADH: ubiquinone oxidoreductase (complex I) depends on the pH gradient across the mitochondrial inner membrane. Biochem. J. 2004;382:511. doi: 10.1042/BJ20040485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lennicke C., Rahn J., Lichtenfels R., Wessjohann L.A., Seliger B. Hydrogen peroxide - production, fate and role in redox signaling of tumor cells. Cell Commun. Signal. 2015;13:39. doi: 10.1186/s12964-015-0118-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu S.S. Generating, partitioning, targeting and functioning of superoxide in mitochondria. Biosci. Rep. 1997;17:259–272. doi: 10.1023/a:1027328510931. [DOI] [PubMed] [Google Scholar]
- 21.Loschen G., Flohé L., Chance B. Respiratory chain linked H2O2 production in pigeon heart mitochondria. FEBS Lett. 1971;18:261–264. doi: 10.1016/0014-5793(71)80459-3. [DOI] [PubMed] [Google Scholar]
- 22.Mailloux R.J. Mitochondrial antioxidants and the maintenance of cellular hydrogen peroxide levels. Oxid. Med. Cell Longev. 2018;2018:7857251. doi: 10.1155/2018/7857251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marchissio M.J., Francés D.E.A., Carnovale C.E., Marinelli R. l.A. Mitochondrial aquaporin-8 knockdown in human hepatoma HepG2 cells causes ROS-induced mitochondrial depolarization and loss of viability. Toxicol. Appl. Pharmacol. 2012;264:246–254. doi: 10.1016/j.taap.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 24.Munro D., Banh S., Sotiri E., Tamanna N., Treberg J.R. The thioredoxin and glutathione-dependent H2O2 consumption pathways in muscle mitochondria: involvement in H2O2 metabolism and consequence to H2O2 efflux assays. Free Radic. Biol. Med. 2016;96:334–346. doi: 10.1016/j.freeradbiomed.2016.04.014. [DOI] [PubMed] [Google Scholar]
- 25.Munro D., Treberg J.R. A radical shift in perspective: mitochondria as regulators of reactive oxygen species. J. Exp. Biol. 2017;220:1170–1180. doi: 10.1242/jeb.132142. [DOI] [PubMed] [Google Scholar]
- 26.Quinlan C.L., Treberg J.R., Perevoshchikova I.V., Orr A.L., Brand M.D. Native rates of superoxide production from multiple sites in isolated mitochondria measured using endogenous reporters. Free Radic. Biol. Med. 2012;53:1807–1817. doi: 10.1016/j.freeradbiomed.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sies H. Role of metabolic H2O2 generation: redox signaling and oxidative stress. J. Biol. Chem. 2014;289:8735–8741. doi: 10.1074/jbc.R113.544635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sies H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: oxidative eustress. Redox Biol. 2017;11:613–619. doi: 10.1016/j.redox.2016.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Starkov A.A., Fiskum G. Regulation of brain mitochondrial H2O2 production by membrane potential and NAD(P)H redox state. J. Neurochem. 2003;86:1101–1107. doi: 10.1046/j.1471-4159.2003.01908.x. [DOI] [PubMed] [Google Scholar]
- 30.Starkov A., Andreyev A., Zhang S., Starkova N., Korneeva M., Syromyatnikov M., Popov V. Scavenging of H2O2 by mouse brain mitochondria. J. Bioenerg. Biomembr. 2014;46:471–477. doi: 10.1007/s10863-014-9581-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Treberg J.R., Braun K., Zacharias P., Kroeker K. Multidimensional mitochondrial energetics: application to the study of electron leak and hydrogen peroxide metabolism. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2018;224:121–128. doi: 10.1016/j.cbpb.2017.12.013. [DOI] [PubMed] [Google Scholar]
- 32.Treberg J.R., Munro D., Banh S., Zacharias P., Sotiri E. Differentiating between apparent and actual rates of H2O2 metabolism by isolated rat muscle mitochondria to test a simple model of mitochondria as regulators of H2O2 concentration. Redox Biol. 2015;5:216–224. doi: 10.1016/j.redox.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Treberg J.R., Quinlan C.L., Brand M.D. Evidence for two sites of superoxide production by mitochondrial NADH-ubiquinone oxidoreductase (complex I) J. Biol. Chem. 2011;286:27103–27110. doi: 10.1074/jbc.M111.252502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Votyakova T.V., Reynolds I.J. ΔΨ-Dependent and -independent production of reactive oxygen species by rat brain mitochondria. J. Neurochem. 2008;79:266–277. doi: 10.1046/j.1471-4159.2001.00548.x. [DOI] [PubMed] [Google Scholar]
- 35.Zoccarato F., Cavallini L., Alexandre A. Respiration-dependent removal of exogenous H2O2 in brain mitochondria: inhibition by Ca2+ J. Biol. Chem. 2004;279:4166–4174. doi: 10.1074/jbc.M308143200. [DOI] [PubMed] [Google Scholar]